

Abstract
Selenium is present in proteins in the form of selenocysteine, where this amino acid serves catalytic oxidoreductase functions. The use of selenocysteine in nature is strongly associated with redox catalysis. However, selenium is also found in a 2-selenouridine moiety at the wobble position of tRNAGlu, tRNAGln and tRNALys. It is thought that the modifications of the wobble position of the tRNA improves the selectivity of the codon-anticodon pair as a result of the physico-chemical changes that result from substitution of sulfur and selenium for oxygen. Both selenocysteine and 2-selenouridine have widespread analogs, cysteine and thiouridine, where sulfur is used instead. To examine the role of selenium in 2-selenouridine, we comparatively analyzed the oxidation reactions of sulfur-containing 2-thiouracil-5-carboxylic acid (s2c5Ura) and its selenium analog 2-selenouracil-5-carboxylic acid (se2c5Ura) using 1H-NMR spectroscopy, 77Se-NMR spectroscopy, and liquid chromatography-mass spectrometry. Treatment of s2c5Ura with hydrogen peroxide led to oxidized intermediates, followed by irreversible desulfurization to form uracil-5-carboxylic acid (c5Ura). In contrast, se2c5Ura oxidation resulted in a diselenide intermediate, followed by conversion to the seleninic acid, both of which could be readily reduced by ascorbate and glutathione. Glutathione and ascorbate only minimally prevented desulfurization of s2c5Ura, whereas very little deselenization of se2c5Ura occurred in the presence of the same antioxidants. In addition, se2c5Ura but not s2c5Ura showed glutathione peroxidase activity, further suggesting that oxidation of se2c5Ura is readily reversible, while oxidation of s2c5Ura is not. The results of the study of these model nucleobases suggest that the use of 2-selenouridine is related to resistance to oxidative inactivation that otherwise characterizes 2-thiouridine. As the use of selenocysteine in proteins also confers resistance to oxidation, our findings suggest a common mechanism for the use of selenium in biology.
Selenium is an essential trace element for many organisms due to its occurrence in selenoproteins in the form of selenocysteine (Sec, U), the 21st amino acid in the genetic code [1–3]. In humans, Sec replaces cysteine (Cys) in 25 proteins and enzymes [4]. Sec-containing enzymes are primarily oxidoreductases where selenium is involved in thiol/disulfide exchange reactions [5]. The reason given most often for its use in enzymes is that as a superior nucleophile relative to Cys it accelerates the rate of enzymatic reactions and provides a type of catalytic advantage [6].
Selenium is also present in tRNA in the form of 5-methylaminomethyl-2-selenouridine (mnm5se2U) and 5-carboxymethylaminomethyl-2-selenouridine (mcm5se2U) [7], where it is found in position 34 (the wobble position) of the anticodon stem loop of three tRNA species: tRNAGln UUG, tRNALys UUU, and tRNAGlu UUC. Its function in tRNA has been attributed to its different base-pairing affinity in the anticodon loop of the tRNA in comparison to its oxygen-containing analog, uridine [8–10]. Specifically, this modification has been shown to destabilize the U/G wobble pair and increase the affinity of the U/A base pair [10], leading to greater translational fidelity during protein synthesis. Fine-tuning of translational fidelity is also the function that has been assigned to the analogous sulfur-containing nucleosides 5-methylaminomethyl-2-thiouridine (mnm5s2U) and 5-carboxymethylaminomethyl-2-thiouridine (mcm5s2U) [11–13].
A recent study has raised the possibility that the redox chemistry of sulfur could provide an additional rationale for 2-thio substitution in uridine as it showed that 2-thiouridine desulfurizes when subjected to excess H2O2, resulting in two distinct products whose formation is pH dependent [14–16]. This study introduced the idea that desulfurization of 2-thiouridine may be part of a redox signaling pathway in response to oxidative stress [17]. The redox chemistry of selenium is much different than that of sulfur [18], so if 2-thiouridine is part of a redox signaling then substitution of sulfur with selenium should alter this potential signaling pathway in some way.
While there have been multiple studies analyzing the differences in the redox properties of selenium-containing enzymes and their sulfur orthologs [19–23], there has been no such study comparing the redox properties of 2-thiouracil and 2-selenouracil. In the current study, we synthesized water-soluble sulfur- and selenium-containing analogs of uracil for the purposes of making direct comparisons of their redox chemistry. These analogs included uracil-5-carboxylic acid (c5Ura, 1), 2-thiouracil-5-carboxylic acid (s2c5Ura, 2), and 2-selenouracil-5-carboxylic acid (se2c5Ura, 3) as shown in Scheme 1.
Scheme 1.
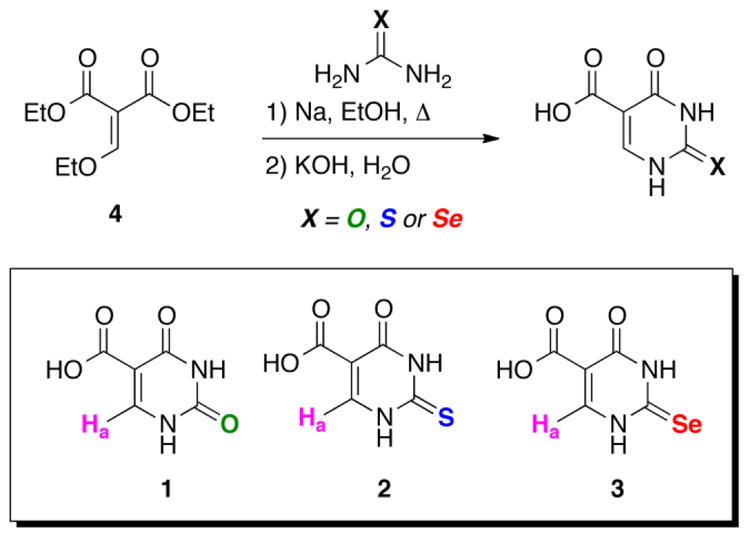
Synthesis of uracil 5-carboxylic acid (c5Ura, 1), 2-thiouracil 5-carboxylic acid (s2c5Ura, 2) and 2-selenouracil 5-carboxylic acid (se2c5Ura, 3). The derivatives contain a single, non-exchangable proton (Ha) visible by 1H-NMR spectroscopy.
These molecules were subjected to oxidation by H2O2 and the time course of the reaction was monitored principally by 1H-NMR, and in the case of se2c5Ura (3), by 77Se-NMR. The identities of the oxidation products were determined by liquid chromatography-mass spectrometric (LCMS) analysis. Biologically relevant reducing agents, glutathione (GSH) and ascorbate (Asc) were added to the assay either before, or during the course of the oxidation in order to determine the extent of the reversibility of the reaction. Our results show significant differences in the redox chemistry of s2c5Ura (2) compared to se2c5Ura (3) when they are oxidized by H2O2, as well as large differences in the reversibility of the oxidation reaction upon the addition of reducing agents. These findings raise the possibility that 2-selenouridine has a redox function in the organisms in which it is found in addition to having a role in translational fidelity. If true, it would be a unifying rationale for the occurrence of selenium in proteins and tRNAs, suggesting a common reason for the use of this element in nature.
MATERIALS AND METHODS
Materials
All materials were purchased from either Sigma-Aldrich (Milwaukee, WI) or ThermoFisher Scientific (Pittsburgh, PA). Reactions were monitored by 1H, 77Se-NMR and high performance liquid chromatography-mass spectrometry (LCMS). 1H, 13C and 77Se-NMR were performed on either a Varian or Bruker ARX 500 MHz spectrometer at 25 °C. LCMS was executed using an ABI Sciex 4000QTrap Pro LCMS equipped with a C18 column in positive-ESI mode. Chemical shifts for 1H and 77Se-NMR are reported in parts per million (ppm) relative to water at 25 °C (δ = 4.79 ppm for 1H-NMR) or KSeCN (δ = −273 ppm for 77Se-NMR). All reactions were performed using oven-dried glassware, and all reagents were used as received.
Synthesis of uracil-5-carboxylic acid (1)
The syntheses of 1 and 2 are essentially identical to the synthesis reported by Ballard and Johnson, with some slight modifications (see Scheme 1) [24]. In an oven dried flask, sodium (1.06 g, 46.3 mmol) was dissolved in anhydrous ethanol (55.5 mL) under an argon atmosphere at room temperature. Upon complete dissolution of the sodium, urea (2.56 g, 42.7 mmol) was added. The solution was then heated (45 °C) to aid in dissolution. Once completely dissolved, diethyl ethoxymethylenemalonate (8.59 mL, 42.5 mmol) was added drop wise to maintain internal temperature of 45 °C. After complete addition the solution was heated (100 °C) and stirred for 1.5 h, then stirred overnight at room temperature. The reaction was vacuum filtered and the white solid obtained was dissolved in 100 mL of water (90 °C) and then slowly acidified with 12 M HCl (5 mL, 60.0 mmol). The solution was cooled overnight (5 °C) then vacuum filtered and triturated with cold H2O to reveal a white solid. The solid was dissolved in 2 M aqueous potassium hydroxide (50 mL, 100.0 mmol), then heated at 95 °C for 2.5 h under an argon atmosphere to saponify the ethyl ester. The sample was slowly acidified with 12 M HCl, washed with cold H2O, and thoroughly dried to yield 6.21 g (39.8 mmol, 93.6% overall yield) of 1 as a white solid. 1H-NMR (500 MHz, D2O): δ (ppm) 8.21 (s, 1H); 13C-NMR (500 MHz, D2O): δ (ppm) 111.2 (C), 160.3 (C) 162.3 (CH) 168.9 (C) 175.1 (C); Melting Point: 270 °C; MS (NEI) m/z 155.14 [(M-), calculated for C5H4O4N2: 156.10].
Synthesis of 2-thiouracil-5-carboxylic acid (2)
In an oven dried flask, sodium (1.06 g, 46.2 mmol) was dissolved in anhydrous ethanol (55.5 mL) under an argon atmosphere at room temperature. Upon complete dissolution of the sodium, thiourea (3.25 g, 42.7 mmol) was added. The solution was then heated (45 °C) to aid in dissolution. Once completely dissolved, diethyl ethoxymethylenemalonate (8.59 mL, 42.50 mmol) was added drop wise to maintain internal reaction temperature of 45 °C. After complete addition the solution was heated (100 °C) and stirred for 1.5 h, then stirred overnight at room temperature. The reaction was vacuum filtered and the white solid obtained was dissolved in 100 mL of H2O (90 °C) and then slowly acidified with 12 M HCl (5 mL, 60.0 mmol). The solution was cooled overnight (5 °C) then vacuum filtered and triturated with cold H2O to reveal a white solid. The solid was dissolved in 2 M aqueous potassium hydroxide (100.00 mmol, 50 mL), then heated at 95 °C for 2.5 h under an argon atmosphere to saponify the ethyl ester. The sample was slowly acidified with 12 M HCl, washed with cold H2O, and thoroughly dried to yield 7.05 g (40.9 mmol, 96.4% overall yield) of 2 as a white solid. 1H-NMR (500 MHz, D2O): δ (ppm) 8.07 (s, 1H); 13C-NMR (500 MHz, D2O): δ (ppm) 115.7 (C), 158.7 (C), 174.3 (CH), 178.3 (C), 184.7 (C); Melting Point: 280 °C; MS (NEI) m/z 171.09 [(M-), calculated for C5H4O3N2S: 172.17].
Synthesis of 2-selenouracil-5-carboxylic acid (3)
For the synthesis of 3, the procedure reported by Ballard and Johnson was modified to substitute selenourea in place of urea/thiourea [24]. Our modified procedure is reported as follows: In an oven dried flask, sodium (0.10 g, 4.35 mmol) was dissolved in anhydrous ethanol (5 mL) under an argon atmosphere at room temperature. Once the sodium was completely dissolved, selenourea (0.54 g, 4.36 mmol) was added. The solution was then heated (70 °C) to aid in dissolution. Once completely dissolved, diethyl ethoxymethylenemalonate (0.89 mL, 4.40 mmol) was added dropwise, maintaining an internal reaction temperature of ~70 °C. After complete addition, the solution was heated (70 °C) and stirred for 1.5 h, then stirred overnight at room temperature. The reaction was filtered and the yellow solid was dissolved in 10 mL of H2O at 90 °C and then slowly acidified with 12 M HCl (0.50 mL, 6.0 mmol). The sample was left stirring overnight at room temperature under an argon atmosphere. The next day, the sample was vacuum filtered and triturated with cold H2O to reveal a yellow solid. The solid was dissolved in 2 M aqueous potassium hydroxide (4.0 mL, 8.0 mmol), then heated at 95 °C for 2.5 hrs under an argon atmosphere. The sample was acidified, washed with cold H2O, and thoroughly dried to yield 0.59 g (2.70 mmol, 62% overall yield) of 3 as a yellow solid. Melting Point decomp > 230 °C; 1H-NMR (500 MHz, D2O) δ (ppm) 7.95 (s, 1H); 13C-NMR (500 MHz, D2O): δ (ppm) 116.1, 144.8, 160.6, 169.3, 173.7; 77Se-NMR (500 MHz, D2O) δ (ppm) 188.50; MS (ESI) m/z 221.2 [(M+H)+, calculated for C5H5O3N2Se: 220.95] [24].
Analysis of 2 and 3 by UV-vis spectroscopy and determination of pKa
UV-vis spectral scans (from 200 nm to 800 nm) of 50 μM solutions of 2 or 3 in 100 mM potassium phosphate buffer, pH 7.0 were performed using a Cary-50 UV-vis spectrophotometer from Varian (Walnut Creek, CA) at 25 °C. For pKa determination, spectral scans of 25 μM solutions of 2 and 3 were taken in the pH range of 4–10 using a tripartite buffer solution consisting of 100 mM sodium acetate, 100 mM sodium phosphate, and 100 mM sodium borate at 25 °C. The spectrophotometer was set to take an absorbance reading every 0.5 nm from 200 nm to 800 nm after it was blanked with the buffer. A plot of λmax versus pH was constructed and the resulting titration curve was fitted to the equation of a sigmoidal curve. Determination of the inflection point yielded the pKa value.
Measurement of glutathione-peroxidase activity
A glutathione reductase (GR) coupled assay was used to measure the glutathione-peroxidase (Gpx) activity of each uracil derivative or ebselen, a well known Gpx-mimic [25]. Assays contained 5 units of GR, 1 mM GSH, 200 μM NADPH, and either 10 μM ebselen, 50 μM 3, or 500 μM 2 in 50 mM potassium phosphate, pH 7.6, in a final volume of 1 mL. The assay was initiated by addition of H2O2 in the range of 0–1 mM and the absorbance at 340 nm was monitored for 2 min with a Cary 50 UV-vis spectrophotometer. The activity was calculated using the extinction coefficient of NADPH (6220 M−1 cm−1).
Oxidation assay of 2 and 3 using 77Se-NMR and 1H-NMR
A 100 mM solution of either 2, or 3 was prepared in 100 mM phosphate buffer (using 100% D2O) at pH 7.4. As 100% D2O was used in the preparation of the buffer, the pD of the solution was 7.81 [26]. An initial 77Se-NMR or 1HNMR spectrum was acquired to establish the initial time point, then one equivalent of H2O2 was added to the solution and the reaction was monitored by either 77Se-NMR (3 only) or 1H-NMR (2 and 3). Spectra were acquired automatically every hour using 77Se-NMR, or every 30 seconds for 10 min using 1H-NMR.
LCMS analysis of the oxidation assays of 2 and 3
A solution of either 2 or 3 at a concentration of 10 mM in 100 mM phosphate buffer at pH 7.4 was prepared in H2O. To the solution was added one equivalent of H2O2 then the reaction was allowed to proceed for 2 min at room temperature. The solution was then flash frozen in a dry ice/isopropanol bath, lyophilized, and the resulting solid was submitted for LCMS analysis.
Rescue assay conditions of 2 and 3 for 1H-NMR analysis
In order to find out whether the oxidation products of 2 and 3 were reversible upon the addition of reducing agents, a “rescue assay” was designed such that 2 and 3 were initially oxidized, after which one equivalent of reducing (“rescue”) agent was added and then the result of the reaction was monitored by both 1H-NMR and LCMS.
A solution of either 2 or 3 at a concentration of 100 mM in 100 mM phosphate buffer at pH 7.4 (pD = 7.81) was prepared in D2O. An initial 1H-NMR spectrum was acquired to establish the initial time point, then one equivalent of H2O2 was added to the solution and the reaction was monitored by 1H-NMR every 30 seconds for 2 min. After acquiring the spectra at t = 2 min, the sample was ejected from the spectrometer and one equivalent of either dithiothreitol (DTT) or ascorbic acid (dissolved in D2O) was added and the reaction was monitored by 1H-NMR every 30 seconds for an additional 8 min.
Rescue assay conditions of 2 and 3 for LCMS analysis
A solution of either 2 or 3 at a concentration of 10 mM in 100 mM phosphate buffer at pH 7.4 was prepared in H2O. To the solution was added one equivalent of H2O2 then the reaction was allowed to proceed for 2 min at room temperature. After 2 min, one equivalent of either dithiothreitol (DTT) or Asc was added and the reaction was allowed to proceed for an additional 2 min. The solution was flash frozen in a dry ice/isopropanol bath, lyophilized, and the obtained solid was submitted for LCMS analysis.
1H-NMR and 77Se-NMR oxidation assay of 2 and 3 in the presence of reducing agent
In order to determine how 2 and 3 would respond to oxidation by H2O2 in the presence of biologically relevant reducing agents, a second rescue experiment was performed in which 2 and 3 were preincubated with reducing agent before the addition of H2O2. A solution of either 2 or 3 at a concentration of 100 mM in 100 mM phosphate buffer at pH 7.4 (pD = 7.81) was prepared in D2O. One equivalent of either Asc or GSH was added to the solution. An initial 1H-NMR spectrum was acquired to establish the initial time point, then one equivalent of H2O2 was added to the solution and the reaction was monitored by 1H-NMR every 30 seconds for 10 min. In the case of 3, identical reaction assays were also monitored by 77Se-NMR for 10 min.
Oxidation assay of 2 and 3 in the presence of reducing agent: analysis by LCMS
A solution of either 2 or 3 at a concentration of 10 mM in 100 mM phosphate buffer at pH 7.4 was prepared in H2O. One equivalent of either Asc or GSH was added to the solution followed by one equivalent of H2O2 and the reaction was allowed to proceed at room temperature for 2 min. The solution was flash frozen in a dry ice/isopropanol bath, lyophilized, and the obtained solid was submitted for LCMS analysis.
Determination of the ratio of 2:1 and 3:1 via 1H-NMR after oxidation
The ratios of 2:1 and 3:1 were determined by integrating the area of each resonance in 1H-NMR in order to quantify the amount of desulfurization (or deselenization) in the reaction after 10 min of oxidation. This ratio was determined by integrating the resonances corresponding to 2 or 3 (or 3d) and dividing the obtained values by the integration for the resonance corresponding to 1.
RESULTS
Preparation of water-soluble nucleotide derivatives
In order to study the intermediate oxidation products of the sulfur- and selenium-containing nucleobases by 1H-NMR in short time frames, we synthesized water-soluble uracil-derivatives containing a carboxylic acid at the 5-carbon position as depicted in Scheme 1. The syntheses of these uracil 5-carboxylates uses a cyclocondensation process between diethyl ethoxymethylenemalonate (4) and urea or the chalcogen derivatives of urea. This approach was first reported by Ballard and Johnson, who had synthesized c5Ura (1), and s2c5Ura (2) [24]. We were able to synthesize se2c5Ura (3), by substituting selenourea for urea in the reaction. The yield of se2c5Ura was lower (62%) in comparison to c5Ura and s2c5Ura, 93% and 96%, respectively. Oxidation of s2c5Ura and se2c5Ura may yield the products as shown in Scheme 2.
Scheme 2.

Proposed oxidation products of 2 and 3. The oxidation products 2a–c and 3a–c refer to the sulfenic/selenenic, sulfinic/seleninic and/or sulfonic/selenonic acid species, respectively.
UV-vis spectral characterization and pKa determination
The UV-vis spectra of s2c5Ura and se2c5Ura are shown in Figure 1. There are 3 maxima visible for s2c5Ura at 214 nm, 270 nm, and 305 nm, which agrees with the previous analysis by Masoud and coworkers [27]. The electronic absorption band at 270 nm was interpreted to be due to a π-π* transition of the carbonyl and thiocarbonyl groups along with hydrogen bonding between N(3)-H and C=O and N(1)-H and C=S [27, 28]. The extinction coefficient of this band increases at acidic pH, but decreases at basic pH and is blue shifted to ~262 nm (Figure S1 of the Supporting Information). This behavior was also previously noted and was interpreted as being due to an equilibrium between thione and thiol tautomers [27]. In contrast, this middle absorption band is missing in the case of se2c5Ura, which shows absorption bands only at 220 and 325 nm. We interpret this behavior as an absence of tautomerization with se2c5Ura existing solely in the selenol form. This interpretation is supported by the 77Se-NMR spectrum as discussed later.
Figure 1.

UV-vis spectra of 50 μM s2c5Ura (left) and 50 μM se2c5Ura (right) in 100 mM potassium phosphate buffer, pH 7.0.
Next, we determined the pKa of s2c5Ura and se2c5Ura by plotting the change in λmax versus pH as shown in Figure 2, which in each case yielded a sigmoidal curve. The pKa values of s2c5Ura and se2c5Ura, determined from the inflection points, were 7.68 and 7.11, respectively. As a point of comparison, pKa values of unsubstituted 2-thiouracil and 2-selenouracil were previously determined to be 7.75 and 7.18 respectively [29], and the pKa of 2-selenouridine (the nucleoside) was previously found to be 7.29 [10].
Figure 2.

Plots of λmax versus pH for s2c5Ura (left) and se2c5Ura (right). The position of the inflection point, denoting the pKa, is indicated by the open blue circle. The pKa values were determined to be 7.68 and 7.11, respectively.
Glutathione-peroxidase activity of se2c5Ura (3)
Because many selenium-containing compounds have Gpx activity, we next set out to determine if se2c5Ura could function as a Gpx-mimic, similar to ebselen, a well-studied small molecule selenium-containing Gpx-mimic [25]. The activity was measured using a GR coupled assay (pH 7.6) in which the mimic initially reacts with H2O2, after which the oxidized mimic is reduced by GSH yielding oxidized glutathione (GSSG). GR reduces the GSSG produced in the reaction using NADPH as a coenzyme, and the activity can then be calculated by measuring the change in absorbance at 340 nm using the extinction coefficient of NADPH (6220 M−1 cm−1). The activity was calculated as the concentration of NADPH consumed (μM) per min per μM of mimic. The activity was then plotted as a function of H2O2 concentration and this plot is shown in Figure 3. As can be seen in the figure, sulfur-containing s2c5Ura has no Gpx-activity, even at high concentrations of H2O2, while the selenium-containing mimics ebselen and se2c5Ura do display activity in our assay. Ebselen is a significantly better Gpx-mimc than se2c5Ura at lower concentrations of H2O2. For example, at 100 μM H2O2, ebselen has a specific activity of 1.41 μM NADPH/min/μM ebselen, while se2c5Ura has a specific activity of 0.13 μM NADPH/min/μM se2c5Ura, an 11-fold difference in activity.
Figure 3.
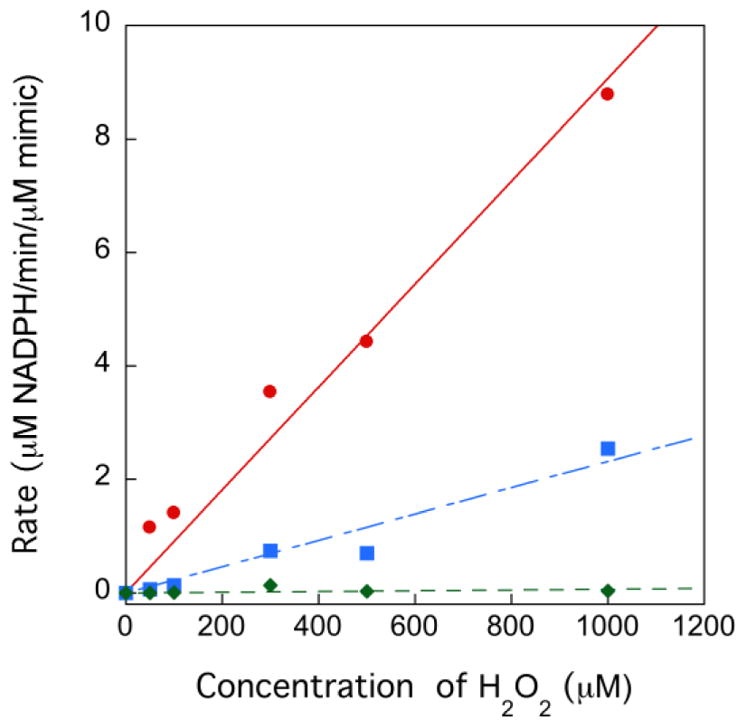
A plot of the consumption of NADPH (μM) per min per μM of mimic versus H2O2 concentration at pH 7.6 in 50 mM potassium phosphate buffer. Ebselen is represented by (●), se2c5Ura by (■), and s2c5Ura(◆). In this assay, the concentration of GSH was fixed at 1 mM.
Analysis of the oxidation of se2c5Ura (3) by 77Se-NMR
The oxidation of se2c5Ura with H2O2 can be monitored by 77Se-NMR because 77Se is an NMR-active nuclide. This experiment provides direct information about both the oxidation state and the electronic configuration of the selenium atom. As shown in Figure 4, we followed the time course of the reaction of se2c5Ura with one equivalent of H2O2 by 77Se-NMR spectroscopy. At the initial time point, a resonance corresponding to se2c5Ura is visible at 188.5 ppm. The position of this resonance indicates that se2c5Ura is exclusively in the selenol form as selones have chemical shifts much further downfield near ~1200 ppm [30]. After the addition of H2O2 we took a spectrum averaged over 12 h (i.e. we acquired the data over a 12 h time period). This was done to improve the signal to noise ratio of the spectrum due to a large amount of noise present from the broadcast of a local FM radio station. At this time point, the resonance corresponding to se2c5Ura disappeared and two new resonances appeared, one at 1273 ppm corresponding to the seleninic acid form of se2c5Ura, and another at 457 ppm, corresponding to the diselenide form of se2c5Ura.
Figure 4.

Time course of the oxidation of 100 mM se2c5Ura by one equivalent of H2O2 monitored by 77Se-NMR spectroscopy at pH 7.4.
After another 6 h, an additional spectrum was taken and only the seleninic acid form of se2c5Ura was present. Our assignment of the oxidation states of se2c5Ura oxidized by H2O2 agrees well with work done by Prabhu and coworkers who followed the oxidation of 2,2′-diselenobis(3-amidopyridine), a compound related to se2c5Ura as both contain a selenoamide [31].
Because we had taken time points over long time periods, we then attempted the same experiment, but took spectra at shorter time intervals in order to elucidate the path of the redox states during the reaction. Figure 5 shows an initial spectrum with resonance at 188.5 ppm, followed by a spectrum averaged over 1.5 h.
Figure 5.

Time course of the oxidation of 100 mM se2c5Ura by one equivalent of H2O2 monitored by 77Se-NMR spectroscopy at pH 7.4. After 1.5 h of reaction, one equivalent of Asc was added to the NMR tube and a new 77Se-NMR was acquired (shown by the top spectrum). As is evident, Asc is able to reduce the diselenide form as well as the selenenic acid form back to the original se2c5Ura form.
Two resonances are visible at the 1.5 h time point: a resonance at 1152 ppm presumably corresponding to the selenenic acid form and the diselenide form of se2c5Ura at 457 ppm. After this spectrum was acquired, the NMR tube was ejected from the spectrometer and 1 equivalent of Asc was added to the reaction. The tube was placed back into the spectrometer and a spectrum was acquired for 30 min. The results show that Asc was able to reduce all of the oxidized 19 intermediates back to the original se2c5Ura form. Moreover, the experiment shows that the redox pathway of the reaction is as follows: se2c5Ura is first oxidized to the selenenic acid form (3a), which can then be converted to the diselenide (3d) by multiple pathways (further elaborated in the Discussion). The diselenide form is further oxidized to the selenoseleninate, which is then hydrolyzed to yield the seleninic acid form.
Analysis of the oxidation of s2c5Ura (2) and se2c5Ura (3) by 1H-NMR
Analysis by 1H-NMR allows for the direct comparison of the oxidation time course of s2c5Ura and se2c5Ura using H2O2 as the oxidant. Within thirty seconds of s2c5Ura being subjected to one equivalent of H2O2, oxidation products were observed downfield in the 1H-NMR spectrum, as shown in the left panel of Figure 6. We have identified these oxidation products as c5Ura (1) (δ = 8.21 ppm; m/z = 157.1), the sulfenic acid 2a (δ = 8.12 ppm; m/z = 189), and the disulfide 2d (δ = 8.15 ppm; m/z = 343.1). The identification of c5Ura is unambiguous due to the matching resonance at δ = 8.2 ppm. Oxidation products 2a and 2d were identified by LCMS as shown in Figure S2 of the Supporting Information. At the 30 s mark, the ratio of s2c5Ura to c5Ura was 0.67:1, which indicates substantial desulfurization of s2c5Ura in the presence of H2O2. This ratio decreased to 0.4 to 1 after 10 min as summarized in Table 1. This result is consistent with previous studies where desulfurization of 2-thiouridine (a nucleoside) to uridine occurred at alkaline pH [14–16]. The sulfinic (RSO2−, 2b) and sulfonic acid (RSO3−, 2c) redox forms, were not identified in our LCMS experiment. This suggests that when s2c5Ura is oxidized progressively to the sulfinic and sulfonic acid redox states, these intermediates are short-lived and eliminate either sulfur dioxide or sulfur trioxide to form c5Ura.
Figure 6.

Time course of the oxidation of 100 mM s2c5Ura (left) and 100 mM se2c5Ura (right) by one equivalent of H2O2 monitored by 1H-NMR spectroscopy at pH 7.4.
Table 1.
Ratio of x2c5Ura:c5Ura in the presence or absence of reducing agent after exposure to one equivalent H2O2 for 10 min.a
| Rescue Agent | s2c5Ura:c5Ura | se2c5Ura:c5Ura |
|---|---|---|
| None | 0.4 | 7.1b |
| DTTc | 0.6 | 8.3 |
| Ascc | 1.1 | 6.3 |
| GSHd | 2.9 | > 25e |
| Ascd | 1.1 | > 25e |
x2c5Ura = s2c5Ura or se2c5Ura.
All se2c5Ura was converted to the diselenide (3d), so this value is reported as the ratio of the 3d to c5Ura.
Reducing agent added after 2 minutes of exposure to H2O2.
s2c5Ura:c5Ura or se2c5Ura was incubated with reducing agent prior to addition of H2O2.
The ratio was very difficult to determine because there was so little c5Ura present.
When se2c5Ura was subjected to one equivalent of H2O2, the resonance at 8.00 ppm in the 1H-NMR shifted slightly downfield to 8.03 ppm (right panel of Figure 6). Besides this change in the 1H-NMR spectrum, three other resonances appeared in the time course. The resonance at 8.20 ppm corresponds to c5Ura and a second new resonance peak appeared at 8.10 ppm. While we cannot be certain of the identity of this resonance, it may be the selenenic acid form (3a). This resonance disappeared as the time course progressed and is consistent with conversion of this form to the diselenide (3d) as observed in the 77Se-NMR time course experiment. Both the selenenic acid form (3a) and the diselenide form (3d) were detected in the LCMS experiment (Figure S3 of the Supporting Information). We believe that the major product of the oxidation is the diselenide form, and that the broad resonance at 8.03 ppm corresponds to this oxidation state. Further supporting evidence of this identification is provided by 77Se-NMR discussed earlier in the Results section. After 10 min of reaction time, a third resonance appeared at 8.31 ppm, which may be the seleninic acid form (3b).
After 10 min, the sample was ejected from the spectrometer and a red precipitate was observed, which is an allotrope of elemental selenium. This indicates that se2c5Ura undergoes minor deselenization in the presence of H2O2 upon conversion to c5Ura, which has not been previously reported. Overall, the oxidation of se2c5Ura with H2O2 was markedly different than the same oxidation of s2c5Ura. This difference is reflected in the ratio of 3d to c5Ura, 7.1:1 (Table 1), which was drastically different from the ratio of s2c5Ura to c5Ura (0.4:1) in the same experiment, an 18-fold difference. Stated another way, oxidation of s2c5Ura with H2O2 largely results in desulfurization to c5Ura under these experimental conditions, while oxidation of se2c5Ura with H2O2 largely results in formation of the diselenide.
Rescue assays with DTT and Asc after addition of H2O2
In order to determine whether the intermediate oxidation products of s2c5Ura and se2c5Ura could be “rescued” (i.e. reduced) by mild reducing agents, we subjected s2c5Ura and se2c5Ura to one equivalent of H2O2 for 2 min then added one equivalent of either Asc or DTT. The results of these rescue experiments with DTT are shown in Figure 7. In the case of the oxidation of s2c5Ura (left panel of Figure 7), oxidation products 1, 2a, and 2d were again observed at the two-minute mark. When one equivalent of DTT was added to the reaction mixture at this time interval, intermediate products 2a and 2d were resolved to form 2 and the mixed disulfide 2e (δ = 8.09 ppm), which was also evident in the LCMS experiment (m/z = 306.9, Figure S4 of the Supporting Information). The addition of DTT made a small difference in the loss of sulfur from s2c5Ura as the ratio of s2c5Ura to c5Ura improved to 0.6:1 as compared to 0.4:1 without the addition of DTT (Table 1).
Figure 7.

Time course of the oxidation of 100 mM s2c5Ura (left) or 100 mM se2c5Ura (right) by one equivalent of H2O2 and subsequent rescue by one equivalent of DTT (added after 2 min of data acquisition) monitored by 1H-NMR spectroscopy at pH 7.4.
Our evidence for the formation of the sulfenic acid and disulfide forms of s2c5Ura come from our LCMS analysis as well as the fact that the peaks we assign to these forms, 2a and 2d in Figure 7, are resolved upon the addition of DTT. Only these forms are known to be able to be reduced back to the thiol/thione form of s2c5Ura. The higher oxidation states of s2c5Ura, the sulfinic acid form (2b) and the sulfonic acid form (2c) are chemically inert to reduction by thiols.
We then repeated this rescue experiment with DTT and se2c5Ura. The result of this experiment is shown in the right panel of Figure 7 and is significantly different in comparison to the same rescue experiment with DTT and s2c5Ura. As before, oxidation products 1, 3a and 3d were observed in the 1H-NMR spectrum at the two-minute mark of the reaction, and were confirmed by LCMS analysis (Figure S5 of the Supporting Information). Note that the resonance at 8.00 ppm corresponding to se2c5Ura shifted slightly downfield to 8.02 ppm as before. When one equivalent of DTT was added after 2 min of reaction time, intermediates 3a and 3d were resolved back to se2c5Ura to near completion. The peak shift from 8.02 ppm back to 8.00 ppm evidences this reduction. There was less red precipitate in the NMR tube after completion of the assay, indicating that less deselenization occurred when DTT was added after the addition of H2O2. This effect can be quantified by the increase in the ratio of se2c5Ura to sc5Ura (8.3:1) compared to when DTT was absent (7.1:1, Table 1). We were also able to detect the presence of the mixed selenosulfide intermediate between DTT and se2c5Ura by LCMS analysis (m/z = 355.1) as shown in Figure S5 of the Supporting Information.
Next we compared the ability of Asc to reduce s2c5Ura and se2c5Ura after exposure to one equivalent of H2O2. In this experiment s2c5Ura was oxidized as before with H2O2 for 2 min, after which one equivalent of Asc was added to the reaction. As shown in the left panel of Figure 8, the addition of Asc did not resolve the resonances corresponding to structures 2a and 2d, in contrast to the rescue experiment with DTT. Less desulfurization occurred in this experiment, as evidenced by the increase in the ratio of s2c5Ura to c5Ura (1.1:1, Table 1), compared to when Asc was absent (0.4:1). This is most likely due to the ability of Asc to scavenge some of the H2O2 in the reaction.
Figure 8.

Time course of the oxidation of 100 mM s2c5Ura (left) or 100 mM se2c5Ura(right) by one equivalent of H2O2 and subsequent rescue by one equivalent of Asc (added after 2 min of data acquisition) monitored by 1H-NMR spectroscopy at pH 7.4.
The rescue of the oxidation of se2c5Ura by Asc is in stark contrast to the rescue of the oxidation of s2c5Ura by Asc as shown by the comparison of the two experiments in Figure 8 (compare the left and right hand panels). The selenenic acid form 3a, and the diselenide form (3d) were reduced back to se2c5Ura, as evidenced by the disappearance of the resonance at 8.11 ppm (3a) and the broad resonance at 8.03 (a mixture of 3 and the 3d) migrating back to 8.00 ppm and becoming distinctly narrower. A new resonance also appeared at 8.08 ppm that is distinct from c5Ura. One possible explanation for this new resonance is that it represents the formation of a c5Ura-Asc adduct that is catalyzed by se2c5Ura. The LCMS data corresponding to the Asc rescue experiments for s2c5Ura and se2c5Ura is shown in Figures S6 and S7 of the Supporting Information. Note that the diselenide form of se2c5Ura is an especially labile diselenide that is capable of being reduced by Asc, as is also evident by the 77Se-NMR data.
Oxidation assays in the presence of reducing agent
We next sought to determine the effect of adding H2O2 to a mixture of reducing agent and either s2c5Ura or se2c5Ura, a condition that resembles that what would occur inside a cell. For this experiment we chose to use the intracellular reducing agents GSH and Asc.
The result of adding one equivalent of H2O2 to a 1:1 mixture of s2c5Ura (100 mM) and GSH (100 mM) is shown in the left panel of Figure 9. We note that in the presence of 100 mM GSH, the resonance frequency of Ha of s2c5Ura is shifted slightly from 8.07 ppm to 8.09 ppm. This is most likely due to a small change in the pH of the solution. The pKa of the thiol of s2c5Ura is 7.68, which is close to the reaction pH. Hence small changes in reaction pH will affect the electron density at the 2-carbon position. At the 30 s time point, c5Ura was present indicating that desulfurization results even in the presence of GSH. This desulfurization was less evident than when GSH was absent from the reaction as the s2c5Ura:c5Ura ratio increased from 0.4:1 to 2.9:1 (Table 1). A resonance at 8.11 ppm (2f) is apparent at the 30 s time point at a ratio of 0.3:1 relative to s2c5Ura. This resonance could potentially be assigned to either the sulfenic acid form 2a, the disulfide form 2d, or to the mixed disulfide between GSH and s2c5Ura. LCMS analysis of the corresponding reaction shows mass signature of the first two forms, but not the mixed disulfide form (Figure S8 of the Supporting Information). This redox form was resolved back to s2c5Ura after 18 h.
Figure 9.

Time course of the oxidation of 100 mM s2c5Ura (left) or 100 mM se2c5Ura (right) preincubated with one equivalent of GSH, by one equivalent of H2O2 monitored by 1H-NMR spectroscopy at pH 7.4.
One possible mechanism to explain the resolution of this redox form back to the original state is the following: s2c5Ura is oxidized by H2O2 to the sulfenic acid form (2a), which is then attacked by a second molecule of s2c5Ura to form the disulfide (2d). This would now mean that the ratio of GSH to 2d is 2:1. The excess GSH would then be able to reduce the disulfide back to s2c5Ura. This is only one possible scenario and multiple other pathways exist that can explain the observation that the oxidation products of s2c5Ura are reduced back to the original state.
During the time course of the reaction of s2c5Ura with H2O2 in the presence of GSH, the resonance corresponding to s2c5Ura broadens significantly. This phenomenon was observed in all of the spectra after the initial time point. The most likely explanation for this is that slight loss of signal lock occurred once the NMR tube was reinserted back into the spectrometer after oxidant was added to the tube.
The reaction of se2c5Ura with H2O2 in the presence of an equimolar amount of GSH is similar to the same reaction with s2c5Ura (compare the right and left panels of Figure 9) with the exception that no c5Ura is visible. This is reflected in the ratio of se2c5Ura to c5Ura increasing to >25:1 (Table 1). In fact, the appearance of c5Ura is barely visible in the right panel of Figure 9 compared to the same reaction with s2c5Ura shown in the left panel. There was a new resonance at 8.09 ppm (3g) that by analogy to the same reaction with s2c5Ura could potentially be assigned to either the selenenic acid form 2a, the diselenide form 2d, or to the mixed selenosulfide between GSH and se2c5Ura. LCMS analysis of the corresponding reaction shows that the mass signature of the first two forms, but not the mixed selenosulfide form (Figure S9 of the Supporting Information). This redox form was resolved back to se2c5Ura after 18 h. We note that the resonance corresponding to se2c5Ura shifted slightly upfield after 18 h of reaction time. This is most likely due to a change in the solution pH as the reaction has progressed.
We then repeated the experiments discussed above with s2c5Ura and se2c5Ura, but replaced GSH with Asc. As shown in Figure 10, the differences between the oxidations of s2c5Ura and se2c5Ura in the presence of Asc are dramatic. The presence of Asc made only a very small difference in the amount of desulfurization that occurred with s2c5Ura as shown by the small increase in the s2c5Ura:c5Ura ratio from 0.4:1 to 1.1:1 (Table 1). Asc also did not prevent the appearance of the sulfenic acid form (2a) and disulfide form (2d) of s2c5Ura as shown by the appearance of these resonances in the 1H-NMR spectrum (left panel of Figure 10). The mass signatures of these redox forms were also present in the LCMS analysis (Figure S10 of the Supporting Information).
Figure 10.

Time course of the oxidation of 100 mM s2c5Ura (left) and 100 mM se2c5Ura (right) preincubated with one equivalent of Asc, by one equivalent of H2O2 monitored by 1H-NMR spectroscopy at pH 7.4.
The reaction of H2O2 with an equimolar amount of se2c5Ura and Asc very strongly contrasts with the same reaction with s2c5Ura. There was almost no detectable c5Ura present. There were also no other redox states present as evidenced by the absence of other resonances, and the resonance position of se2c5Ura did not change throughout the reaction (right panel of Figure 10). Although no other resonances were present in the 1H-NMR spectrum, our LCMS analysis detected the presence of the selenenic acid and diselenide forms of se2c5Ura (Figure S11 of the Supporting Information). One explanation of the 1H-NMR spectrum showing no additional resonances is that a peroxidase cycle occurs with se2c5Ura, H2O2, and Asc (as well as GSH) as shown in Figure 11. This interpretation is bolstered by additional 77Se-NMR analysis described below.
Figure 11.
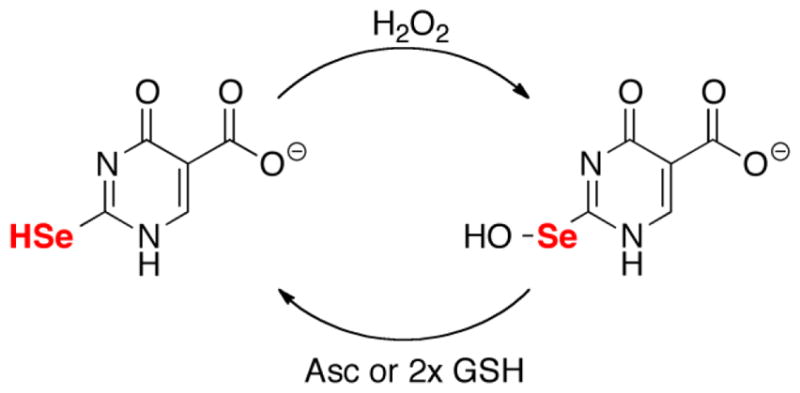
Peroxidase cycle of se2c5Ura with H2O2 and either Asc or GSH.
The results shown in Figures 9 and 10 in which an equimolar amount of reducing agent was present with se2c5Ura before H2O2 was added to the reaction, show that once the selenium atom of se2c5Ura is oxidized it is rapidly reduced back to the original state with little loss of selenium. In order to provide further evidence that se2c5Ura is not destroyed by H2O2 in the presence of reducing agent, we repeated the experiment, but monitored the oxidation reaction by 77Se-NMR instead of 1H-NMR as shown in Figure 12. A resonance at 188.5 ppm is visible in the initial spectrum in the presence of either GSH (left panel) or Asc (right panel) before the addition of H2O2. After 18 h of reaction, the position of the resonance did not change, although the resonance showed broadening in both cases (GSH or Asc). This is likely due to fast exchange that is occurring between redox states even at this point in the reaction due to small amounts of reducing agent still present. No other peak in the 77Se-NMR spectrum is visible (Figure S12 of the Supporting Information).
Figure 12.

Time course of the oxidation of 100 mM se2c5Ura preincubated with one equivalent of GSH (left), or preincubated with one equivalent of Asc (right) by one equivalent of H2O2 monitored by 77Se-NMR spectroscopy at pH 7.4.
DISCUSSION
Characterization of se2c5Ura and its use as a model to study the chemistry of seleniumcontaining nucleosides
The design advantages of s2c5Ura (2) and se2c5Ura (3) shown in Scheme 1 are that they: (i) are water-soluble model compounds which can be assayed by spectroscopic methods including 1H-NMR, (ii) allow for the first direct comparison of the oxidation reactions of sulfur- and selenium-containing uracil derivatives, and (iii) may provide insight into the chemistry that the naturally occurring sulfur- and selenium-containing nucleosides undergo in vivo. We do note that the concentrations of our reactants, 100 mM s2c5Ura or se2c5Ura with 100 mM H2O2, do not mimic physiological conditions. However, these high concentrations allowed us to study the intermediate oxidation products of these compounds as 1H-NMR allowed for spectra to be acquired every 30 seconds under these conditions. Detection of these intermediates would be difficult otherwise.
Previous analysis of the UV/vis spectrum of s2c5Ura showed that it exists as an equilibrium mixture of two tautomeric forms, thione and thiol. We now report that the UV/vis spectrum of se2c5Ura shows only two peaks at 220 and 325 nm (Figure 1), suggesting that no equilibrium of tautomeric forms occurs, as the peak that should correspond to the selone form is missing in the spectrum. Definitive evidence of this comes from the 77Se-NMR spectrum showing a single resonance at 188.5 ppm that corresponds to the selenol form. Selenols have chemical shifts in the range of −300 ppm to +300 ppm in a 77Se-NMR spectrum, while selones have chemical shifts in the range of 1700 ppm to 2100 ppm [30]. This is very strong evidence that 2-selenouridine and its derivatives exist in the selenol form as 77Se-NMR is very sensitive to the electronic structure of the selenium atom. It is interesting that s2c5Ura has less of the active thiol form needed to react with H2O2, yet it is more sensitive to oxidation than se2c5Ura, which exists solely in the reactive selenol form.
In order to assess how the 5-carboxyl group affects the reactivity of the selenium atom at the 2-position, we performed a titration analysis using UV/vis spectroscopy and determined the pKa of the selenol to be 7.11. In comparison, the unsubstituted 2-selenouracil has a pKa of 7.18. Naturally occurring 2-selenouridines have either a methylaminomethyl or carboxymethylaminomethyl substituent at the 5-carbon position. Takai and Yokoyama, making extensive use of Hammett analysis, noted that both the 5-methylaminomethyl and 5-carboxymethylaminomethyl substituents are electron withdrawing and are therefore expected to slightly lower the pKa of the thiol or selenol at the 2-position [32]. Thus the pKa of 5-methylaminomethyl-2-selenouracil and 5-carboxymethylaminomethyl-2-selenouracil should be slightly lower than 7.18. Based on our experimentally determined value of 7.11, we conclude that se2c5Ura is a reasonable model for either naturally occurring selenium-containing nucleobase. This same rationale is true for the use of s2c5Ura as a model for the naturally occurring sulfur-containing nucleobases mcm5s2Ura and mnm5s2Ura.
Differences in the oxidation of sulur- and selenium-containing nucleobases
Our data shows that there are large differences in the redox chemistry between se2c5Ura and s2c5Ura under our chosen reaction conditions. The 77Se-NMR oxidation assay in Figure 4 shows that se2c5Ura is initially oxidized to the diselenide (3d), which is then further oxidized to the seleninic acid (RSeO2−, 3b). In contrast, s2c5Ura is oxidized such that it is significantly desulfurized. It is likely oxidized first to the sulfenic acid form (RSOH, 2a), and then further oxidized to either the sulfinic (RSO2−, 2b) and sulfonic (RSO3−, 2c) acid forms, with the latter most likely resulting in elimination of sulfur.
The mechanism by which sulfur is lost and selenium is retained is chemically interesting and may be biologically relevant. It has been shown that under acidic conditions oxidation of cyclic thioureas to the sulfinic acid results in an elimination reaction, releasing sulfur dioxide in the process [33]. This mechanism would lead to the conversion of 2-thiouridines to a 4-pyrimidinone riboside under oxidative conditions [16, 17]. The conversion of 2-thiouridine to 4-pyrimidinone riboside results in a conformational change of the nucleoside from 3′-endo to 2′-endo [14]. As a result of this conformational change 4-pyrimidinone riboside shows a preference for pairing with G instead of A [15, 34]. Perhaps more importantly the formation of 4-pyrimidinone riboside can lead to cleavage of the glycosidic bond resulting in an abasic lesion [15]. This abasic lesion can then undergo further cleavage resulting in RNA fragments, and these fragments have been proposed to serve a signaling function [15, 17].
At basic pH, similar to the conditions used here, it has been proposed that oxidation of thioureas to the sulfinic or sulfonic acid forms would lead to substitution, replacing sulfur with oxygen [33, 35]. This mechanism can explain the production of c5Ura from s2c5Ura. Our data suggests that it is the sulfonic acid form that is required for the substitution reaction to occur. This is because our data using 77Se-NMR shows that the pathway of oxidation of se2c5Ura is most likely to be: RSeH → RSeOH → RSe-SeR → RSeO2− (Figures 4 and 5). If the RSeO2− form leads to deselenization by substitution, we would not have been able to detect it by 77Se-NMR after 18 h of reaction time. It is therefore reasonable to believe that it is the selenonic acid form (RSeO3−, 3c) that results in substitution at basic pH, and thus in the analogous oxidation of s2c5Ura it must be the sulfonic acid form (RSO3−, 2c) that leads to substitution. This is supported by the fact that oxidation of (RSeO2− to RSeO3− is slow, while the oxidation of RSO2− to RSO3− is fast [18, 36].
To further clarify the mechanism of oxidation, we show the possible oxidation pathways of se2c5Ura and s2c5Ura in Figure 13. Our 77Se-NMR data indicates that oxidation of the diselenide to the selenoseleninate (green pathway) is the path that leads to formation of the seleninc acid (3b). It is logical to think that s2c5Ura is oxidized by the same pathway. This agrees with the proposed mechanism of the oxidation of ergothioneine, a thiourea-containing compound related to thiouracil [37], although the direct oxidation pathway (red) could not be ruled out for ergothioneine and it cannot be ruled out here either. Oxidation of the parent compound to the sulfenic/selenenic acid is the first step in all three pathways shown in Figure 13. The sulfenic/selenenic acid can be further oxidized to the sulfinic/seleninic acid via the “disulfide” (green) pathway, or the “direct” (red) pathway. Sulfenic and selenenic acids are unstable species [38, 39] and are susceptible to disproportionation [40–43], which is a third route by which the sulfinic/seleninic acid can form. We note that we are proposing that s2c5Ura is desulfurized when it is converted to the sulfonic acid, which is different from the desulfurization mechanism proposed for ergothioneine [37].
Figure 13.

Oxidation pathways of s2c5Ura and se2c5Ura (Y = S or Se). Here each derivative is abbreviated as UraYH. The uracil derivatives could be oxidized via a direct oxidation pathway (red) to produce either the sulfinic/seleninic acids or the sulfonic/selenonic acids, which could eliminate sulfur dioxide/selenium dioxide or sulfite/selenite, respectively, to produce uracil-5-carboxylate (Ura). A second pathway (green) involves oxidation of the disulfide/diselenide to form a thiosulfinate/selenoseleninate, which can undergo hydrolysis to the parent compound and the sulfinic/seleninic acid. The parent uracil derivative can then reenter the cycle from multiple entry points. Alternatively, the sulfenic acid/selenenic acid could disproportionate (blue), resulting in production of the disulfide/diselenide and the sulfinic/seleninic acid. This figure is adapted from [37].
Glutathione peroxidase activity of se2c5Ura and the reversibility of the oxidation reaction
While the time courses of the oxidations of s2c5Ura and se2c5Ura by H2O2 alone were very different, the oxidation reactions of s2c5Ura and se2c5Ura in the presence of biologically relevant antioxidants were even more pronounced. Although the experimental conditions used here cannot mimic actual conditions inside a bacterial or mammalian cell, we used a 1:1 ratio of GSH:H2O2, a condition that might be considered to be an extreme case of oxidative stress inside these cells. The intracellular concentration of GSH in E. coli has been experimentally determined to be 17 mM, while in mammals it is in the range of 1–8 mM [44, 45]. Under conditions of oxidative stress, the intracellular concentration of GSH decreases, but is still present in excess relative to H2O2. As the data in Figure 9 shows, s2c5Ura loses sulfur when oxidized with H2O2 in the presence of an equimolar concentration of GSH while se2c5Ura does not lose much selenium.
The reason that GSH protects se2c5Ura from loss of selenium is because once the Seoxide is formed, it is highly electrophilic and is immediately attacked by GSH, releasing water and forms a mixed selenosulfide bond. A second equivalent of GSH then attacks the mixed selenosulfide to produce GSSG, reforming se2c5Ura. This analysis of the redox cycle was confirmed by the measurement of Gpx-activity in a GR coupled assay (Figure 3). While our 36 experiments involve an individual nucleobase, and not in the context of a stacked anticodon loop of a tRNA molecule, the Gpx-activity of the selenium-nucleobase leads us to hypothesize that rapid attack by glutathione with concomitant formation of a glutathionylated 2-selenouridine is possible (and perhaps even likely) in a bacterial cell. Glutathionylation would result in protection against loss of selenium in tRNA.
In direct contrast with the results discussed above, s2c5Ura is significantly desulfurized in the presence of equimolar amounts of GSH and H2O2 (left panel of Figure 9). This is because once s2c5Ura is oxidized to the sulfenic acid (2a) form, it is less likely to be attacked by GSH due to the S-oxide being significantly less electrophilic than the corresponding Se-oxide (reviewed in 18). Consequently, further oxidation to the sulfinic (2b) and sulfonic (2c) acid forms occurs, leading to the elimination of sulfur. This analysis is consistent with the observed absence of Gpx-activity of s2c5Ura.
This difference in the rate of reduction of the sulfenic acid form of thiouridine in comparison to the rate of reduction of the selenenic acid form of selenouridine may be further pronounced in the context of a stacked anticodon loop of a tRNA molecule. Even if the thiouridine moeity of a tRNA remains reduced in the presence of excess glutathione, a significant advantage that selenouridine has relative to thiouridine with respect to reversibility of oxidation is that the seleninic acid form (which results when oxidant is in excess) can still be reduced back to the original selenol form, while the corresponding sulfinic acid form of thiouridine will lead to irreversible desulfurization and impaired function of the tRNA molecule.
A very pronounced difference in the oxidation reactions of s2c5Ura and se2c5Ura is observed when an equimolar amount of Asc is included in the reaction (Figure 8). Very significant desulfurization of s2c5Ura occurs upon reaction with an equimolar amount of H2O2 in the presence of Asc, as evidenced by the calculated ratio of s2c5Ura to c5Ura after oxidation (Table 1). In stark contrast, very little deselenization resulted under the same reaction conditions with se2c5Ura. It is interesting to note that the gene required for the conversion of 2-thiouridine to 2-selenouridine is present mostly in bacteria [46, 47], but bacteria lack Asc. This observation may be relevant to a possible redox/signaling function of 2-selenouridine.
We can envision two scenarios in which 2-selenouridine may attenuate redox signaling as part of an oxidative stress response in bacterial cells based on our in vitro results. First, a larger percentage of 2-thiouridine may be converted into 2-selenouridine in order to prevent desulfurization of 2-thiouridine with concomitant conversion to 4-pyrimidinone riboside or uridine. However, the opposite scenario may also be true; in response to oxidative stress bacterial cells prevent the conversion of 2-thiouridine to selenouridine so as to promote desulfurization. Our data presented here cannot differentiate these two biological responses. We can say that our selenouracil derivative, se2c5Ura, did not undergo conversion to c5Ura when treated with H2O2 at basic pH in the presence of biologically relevant reducing agents in comparison to the rapid conversion of s2c5Ura to c5Ura under the same conditions.
Permanent oxidation of sulfur-containing uracil derivatives versus reversible oxidation of selenium-containing uracils: Comparisons to other systems
A recent study by Toscano and coworkers of the oxidation of thiols and selenols with nitroxyl (HNO) showed that oxidation of a model thiol compound led to the formation of a sulfinamide, considered to be an irreversible oxidative modification, while the same oxidation of a model selenol led to the formation of the diselenide, a reversible oxidative modification [48]. From this study it was concluded that: “…Nature may have chosen to use selenium instead of sulfur in certain biological systems for its enhanced resistance to electrophilic and oxidative modification.” [48]. A near identical hypothesis has been recently reviewed [18]. Our results are in line with those of Toscano and coworkers and show that oxidation of s2c5Ura leads to irreversible modification, even in the presence of biological reducing agents, while the same oxidation of se2c5Ura leads first to diselenide formation and then to the seleninic acid (3c), both reversible modifications for selenium-containing compounds [18].
While it is reasonable to suppose that the results of the oxidation reactions performed here would be quite similar in the context of either a nucleoside or oligonucleoside, it remains to be seen whether the same result would be obtained in the context of a native tRNA. If our results can be extended to sulfur- and selenium-containing natural tRNAs, then incorporation of selenium into a tRNA would be part of an oxidative stress response in the organisms where it is found. A potential scenario is given in Figure 14.
Figure 14.
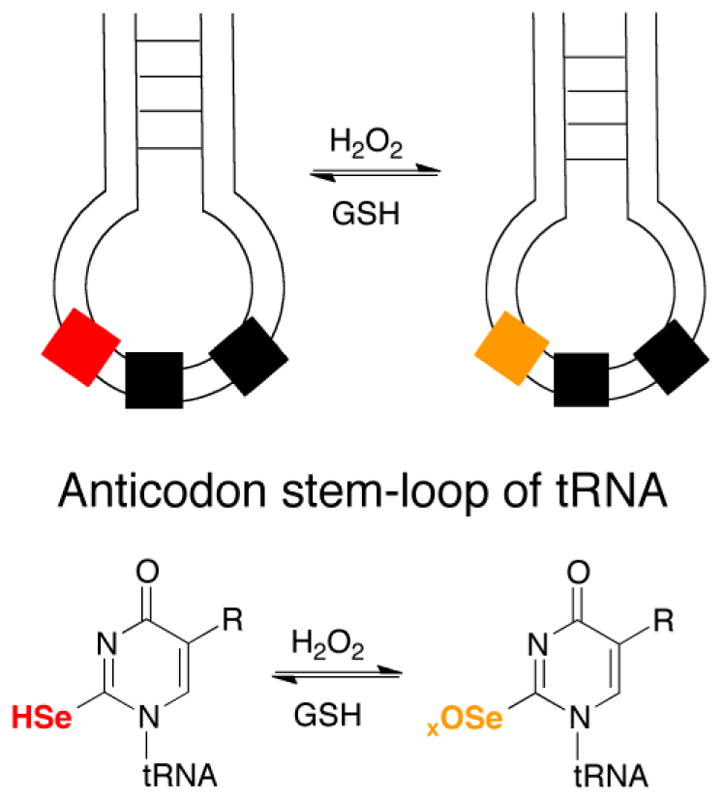
Reversible oxidative modification of a selenium-containing nucleoside in a tRNA molecule. Oxidation of 2-selenouridine in a tRNA would most likely not lead to a diselenide as initially occurred in our experiments. However, oxidation of selenouridine to either the selenenic acid or seleninic acid could result in a reversible oxidative modification since GSH would readily attack either redox form, eventually restoring the oxidized selenium-nucleoside back to its original form [49]. This is underscored by our finding that se2c5Ura has Gpx-activity in vitro. If 2-thiouridine were present instead, significant desulfurization could result, leading to loss of function of the tRNA.
The scenario presented in Figure 14, if it occurs inside a cell, suggests that the use of selenium in a tRNA molecule allows it to retain function during oxidative stress. This would be an advantage relative to the sulfur-containing tRNA, which would desulfurize in response to oxidative stress and result in a loss of function. It is now well established the modifications to tRNA, especiallly the wobble position, are important for translation during oxidative stress [50–54]. An important study by Hidalgo and coworkers showed that the 2-thio modification of tRNALys UUU was important for yeast cells to resist oxidative stress due to the importance of this modification in translating stress proteins [55]. The use of selenium in selenouridine, instead of sulfur in thiouridine, may help bacterial cells cope with oxidative stress by a similar mechanism.
CONCLUSIONS
Oxidation of our model sulfur-containing nucleobase at basic pH, s2c5Ura, leads to desulfurization and irreversible conversion to a uracil analog, c5Ura. Significant desulfurization of s2c5Ura occurs even in the presence of GSH and Asc. In contrast, our model seleniumcontaining nucleobase, se2c5Ura, is reversibly converted into the diselenide and seleninic acid. The key to this reversibility is the much higher electrophilic character of Se-oxides in comparison to S-oxides, which allow for rapid attack by reducing agents, leading to restoration of the original redox state. This property allows selenium-containing biomolecules to resist permanent oxidation, and this trait is possibly a unifying reason for the use of selenium in both 2-selenouridine and Sec-containing proteins.
Supplementary Material
Highlights.
Redox chemistry of sulfur- and selenium-nucleobases is remarkably different
Oxidation of 2-thiouracil-5-carboxylic acid leads to irreversible desulfurization
Oxidation of 2-selenouracil-5-carboxylic acid is reversible
Incorporation of selenium into tRNA may allow it to resist irreversible oxidation
Selenium in tRNA may help cells resist oxidative stress
Acknowledgments
We would like to acknowledge Bruce O’Rourke for acquiring the LCMS data, and Dr. Monika Ivanic for helping acquire NMR spectra, both of the UVM Department of Chemistry. We also thank Dr. Christopher Francklyn of the UVM Department of Biochemistry for providing a critical reading of this manuscript. This work was supported in part by National Institutes of Health Grant GM094172 to RJH.
ABBREVIATIONS
- A
adenine
- Asc
ascorbate
- Cys
cysteine
- DTT
dithiothreitol
- E.coli
Escherichia coli
- GSH
reduced glutathione
- GSSG
glutathione disulfide
- Gpx
glutathione peroxidase
- GR
glutathione reductase
- G
guanine
- LCMS
liquid chromatography-mass spectrometry
- NEI
negative electron ionization
- NMR
nuclear magnetic resonance
- mcm5s2U34
5-carboxymethylaminomethyl-2-thiouridine
- mcm5se2U
5-carboxymethylaminomethyl-2-selenouridine
- mnm5s2U
5-methylaminomethyl-2-thiouridine
- mnm5se2U
5-methylaminomethyl-2-selenouridine
- NADPH
β-nicotinamide adenine dinucleotide phosphate-reduced
- ppm
parts per million
- RSOH
sulfenic acid
- RSO2−
sulfinic acid
- RSO3−
sulfonic acid
- RSeOH
selenenic acid
- RSeO2−
seleninic acid
- RSeO3−
selenonic acid
- s2c5Ura
2-thiouracil-5-carboxylic acid
- se2c5Ura
2-selenouracil-5-carboxylic acid
- Sec
selenocysteine
- U
the one letter code for Sec
- c5Ura
uracil-5-carboxylic acid
Footnotes
These studies were supported by National Institutes of Health Grant GM094172 to RJH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mertz W. The essential trace elements. Science. 1981;213:1332–1338. doi: 10.1126/science.7022654. [DOI] [PubMed] [Google Scholar]
- 2.Böck A, Forchhammer K, Heider J, Leinfelder W, Sawers G, Veprek B, Zinoni F. Selenocysteine: the 21st amino acid. Mol Microbiol. 1991;5:515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- 3.Atkins JF, Gesteland RF. The twenty-first amino acid. Nature. 2000;407:463–464. doi: 10.1038/35035189. [DOI] [PubMed] [Google Scholar]
- 4.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigó R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 5.Hondal RJ, Marino SM, Gladyshev VN. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid Redox Signal. 2013;18:1675–1689. doi: 10.1089/ars.2012.5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnér ES. Selenoproteins: What unique properties can arise with selenocysteine in place of cysteine? Exp Cell Res. 2010;316:1296–1303. doi: 10.1016/j.yexcr.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 7.Stadtman TC. Specific occurrence of selenium in enzymes and amino acid tRNAs. FASEB J. 1987;1:375–379. doi: 10.1096/fasebj.1.5.2445614. [DOI] [PubMed] [Google Scholar]
- 8.Ching WM. Characterization of selenium-containing tRNAGlu from Clostridium sticklandii. Arch Biochem Biophys. 1986;244:137–146. doi: 10.1016/0003-9861(86)90102-5. [DOI] [PubMed] [Google Scholar]
- 9.Ching WM, Tsai L, Wittwer AJ. Selenium-containing transfer RNAs. Curr Top Cell Regul. 1985;27:497–507. doi: 10.1016/b978-0-12-152827-0.50050-5. [DOI] [PubMed] [Google Scholar]
- 10.Sun H, Sheng J, Hassan A, Jiang S, Gan J, Huang Z. Novel RNA base pair with higher specificity using single selenium atom. Nucl Acids Res. 2012;40:5171–5179. doi: 10.1093/nar/gks010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carbon J, David H, Studier MH. Thiobases in Escherchia coli transfer RNA: 2-thiocytosine and 5-methylaminomethyl-2-thiouracil. Science. 1968;161:1146–1147. doi: 10.1126/science.161.3846.1146. [DOI] [PubMed] [Google Scholar]
- 12.Yamada Y, Murao K, Ishikura H. 5-(carboxymethylaminomethyl)-2-thiouridine, a new modified nucleoside found at the first letter position of the anticodon. Nucleic Acids Res. 1981;9:1933–1939. doi: 10.1093/nar/9.8.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gustilo EM, Vendeix FA, Agris PF. tRNA’s modifications bring order to gene expression. Curr Opin Microbiol. 2008;11:134–140. doi: 10.1016/j.mib.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraszewska K, Kaczyńska I, Jankowski S, Karolak-Wojciechowska J, Sochacka E. Desulfurization of 2-thiouracil nucleosides: conformational studies of 4-pyrimidinone nucleosides. Bioorg Med Chem. 2011;19:2443–2449. doi: 10.1016/j.bmc.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Sochacka E, Kraszewska K, Sochacki M, Sobczak M, Janicka M, Nawrot B. The 2-thiouridine unit in the RNA strand is desulfured predominantly to 4-pyrimidinone nucleoside under in vitro oxidative stress conditions. Chem Commun. 2011;47:4914–4916. doi: 10.1039/c1cc10973a. [DOI] [PubMed] [Google Scholar]
- 16.Sochacka E, Bartos P, Kraszewska K, Nawrot B. Desulfuration of 2-thiouridine with hydrogen peroxide in the physiological pH range 6.6–7.6 is pH-dependent and results in two distinct products. Bioorg Med Chem Lett. 2013;23:5803–5805. doi: 10.1016/j.bmcl.2013.08.114. [DOI] [PubMed] [Google Scholar]
- 17.Nawrot B, Sochacka E, Düchler M. tRNA structural and functional changes induced by oxidative stress. Cell Mol Life Sci. 2011;68:4023–4032. doi: 10.1007/s00018-011-0773-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reich HJ, Hondal RJ. Why Nature chose selenium. ACS Chem Biol. 2016;11:821–841. doi: 10.1021/acschembio.6b00031. [DOI] [PubMed] [Google Scholar]
- 19.Rocher C, Lalanne JL, Chaudière J. Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase. Eur J Biochem. 1992;205:955–960. doi: 10.1111/j.1432-1033.1992.tb16862.x. [DOI] [PubMed] [Google Scholar]
- 20.Bell IM, Fisher ML, Wu ZP, Hilvert D. Kinetic studies on the peroxidase activity of selenosubtilisin. Biochemistry. 1993;32:3754–3762. doi: 10.1021/bi00065a030. [DOI] [PubMed] [Google Scholar]
- 21.Choudhury SB, Pressler MA, Mirza SA, Day RO, Maroney MJ. Structure and redox chemistry of analogous nickel thiolato and selenolato complexes: Implications for the nickel sites in hydrogenases. Inorg Chem. 1994;33:4831–4839. [Google Scholar]
- 22.Snider GW, Ruggles EL, Khan N, Hondal RJ. Selenocysteine confers resistance to inactivation by oxidation in thioredoxin reductase: Comparison of selenium and sulfur enzymes. Biochemistry. 2013;52:5472–5481. doi: 10.1021/bi400462j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nauser T, Steinmann D, Grassi G, Koppenol WH. Why selenocysteine replaces cysteine in thioredoxin reductase: a radical hypothesis. Biochemistry. 2014;53:5017–5022. doi: 10.1021/bi5003376. [DOI] [PubMed] [Google Scholar]
- 24.Ballard E, Johnson T. Synthesis of derivatives of pyrimidine-5-carboxylic acid. J Am Chem Soc. 1942;64:794–98. [Google Scholar]
- 25.Müller A, Cadenas E, Graf P, Sies H. A novel biologically active selenoorganic compound--I. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen) Biochem Pharmacol. 1984;33:3235–3239. doi: 10.1016/0006-2952(84)90083-2. [DOI] [PubMed] [Google Scholar]
- 26.Glasoe PK, Long FA. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem. 1960;64:188–189. [Google Scholar]
- 27.Masoud MS, Abd El Zaher Mostafa M, Ahmed RH, Abd El Moneim NH. Structures and chemical equilibria of some N-heterocycles containing amide linkages. Molecules. 2003;8:430–438. [Google Scholar]
- 28.Petel DR, Choxi CS, Petel SP. Pyranoquinolines. Part I Syntheses and ultraviolet absorption characteristics of 6-chloro-4 H-pyrano [3, 2-h] quinoline-4-ones. Can J Chem. 1969;47:105–112. [Google Scholar]
- 29.Mautner HG. The synthesis and properties of some selenopurines and selenopyrimidines. J Am Chem Soc. 1955;78:5292–5294. [Google Scholar]
- 30.Klapötke TM, Broschag M. Compilation of reported 77Se NMR chemical shifts. John Wiley and Sons; Chichester, UK: 1996. [Google Scholar]
- 31.Prabhu P, Beena G, Singh M, Noguchi PP, Phadnis VK, Jain MI, Priyadarsini KI. Stable selones in glutathione-peroxidase-like catalytic cycle of selenonicotinamide derivative. Org Biomol Chem. 2014;12:2404–2412. doi: 10.1039/c3ob42336k. [DOI] [PubMed] [Google Scholar]
- 32.Takai K, Yokoyama S. Roles of 5-substituents of tRNA wobble uridines in the recognition of purine-ending codons. Nucleic Acids Res. 2003;31:6383–6391. doi: 10.1093/nar/gkg839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grivas S, Ronne E. Facile desulfurization of cyclic thioureas by hydrogen peroxide in acetic acid. Acta Chem Scand. 1995;49:225–229. doi: 10.3891/acta.chem.scand.49-0225. [DOI] [PubMed] [Google Scholar]
- 34.Sochacka E, Szczepanowski R, Cypryk M, Sobczak M, Janicka M, Kraszewska K, Bartos P, Chwialkowska A, Nawrot B. 2-Thiouracil deprived of thiocarbonyl function preferentially base pairs with guanine rather than adenine in RNA and DNA duplexes. Nucl Acids Res. 2015;43:2499–2512. doi: 10.1093/nar/gkv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheena M, Loosmorea SM, McKinnona DM. Oxidation products of cyclic thiones. Phosphorus Sulfur Silicon Relat Elem. 1976;1:185–209. [Google Scholar]
- 36.Gancarz RA, Kice JL. The reaction of sulfinic acids with benzeneseleninic acid. Tetrahedron Lett. 1980;21:1697–1700. [Google Scholar]
- 37.Servillo L, Castaldo D, Casale R, D’Onofrio N, Giovane A, Cautela D, Balestrieri ML. An uncommon redox behavior sheds light on the cellular antioxidant properties of ergothioneine. Free Radic Biol Med. 2015;79:228–236. doi: 10.1016/j.freeradbiomed.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura N. A stable sulfenic acid, 9-triptycenesulfenic acid: its isolation and characterization. J Am Chem Soc. 1983;105:7172–7173. [Google Scholar]
- 39.Goto K, Nagahama M, Mizushima T, Shimada K, Kawashima T, Okazaki R. The first direct oxidative conversion of a selenol to a stable selenenic acid: experimental demonstration of three processes included in the catalytic cycle of glutathione peroxidase. Org Lett. 2001;3:3569–3572. doi: 10.1021/ol016682s. [DOI] [PubMed] [Google Scholar]
- 40.Jasperse CP, Reich HJ. Organoselenium chemistry. Preparation and reactions of 2,4,6-tri-tert –butylbenzeneselenenic acid. J Org Chem. 1988;53:2390–2392. [Google Scholar]
- 41.Gancarz RA, Kice JL. The oxidation of aryl diselenides with an equimolar amount of tert-butyl hydroperoxide: Evidence against the formation of significamt amounts of a selenenic anhydride. Tetrahedron Lett. 1981;22:1661–1662. [Google Scholar]
- 42.Abraham RT, Benson LM, Jardine I. Synthesis and pH-dependent stability of purine-6-sulfenic acid, a putative reactive metabolite of 6-thiopurine. J Med Chem. 1983;26:1523–1526. doi: 10.1021/jm00364a031. [DOI] [PubMed] [Google Scholar]
- 43.Acharjee SR, Bhattacharjee SK. Formation and disproportionation of arene sulfenic acids. IOSR J Appl Chem. 2013;3:40–45. [Google Scholar]
- 44.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic Biol Med. 1999;27:922–935. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 46.Wolfe MD, Ahmed F, Lacourciere GM, Lauhon CT, Stadtman TC, Larson TJ. Functional diversity of the rhodanese homology domain: the Escherichia coli ybbB gene encodes a selenophosphate-dependent tRNA 2-selenouridine synthase. J Biol Chem. 2004;279:1801–1809. doi: 10.1074/jbc.M310442200. [DOI] [PubMed] [Google Scholar]
- 47.Romero H, Zhang Y, Gladyshev VN, Salinas G. Evolution of selenium utilization traits. Genome Biol. 2005;6:R66. doi: 10.1186/gb-2005-6-8-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bianco CL, Moore CD, Fukuto JM, Toscano JP. Selenols are resistant to irreversible modification by HNO. Free Radic Biol Med. 2016;99:71–78. doi: 10.1016/j.freeradbiomed.2016.07.008. [DOI] [PubMed] [Google Scholar]
- 49.Ruggles EL, Snider GW, Hondal RJ. Chemical basis for the use of selenocysteine. In: Hatfield DL, Berry MJ, Gladyshev VN, editors. Selenium: Its Molecular Biology and Role in Human Health. 3. New York: Springer; 2012. pp. 73–83. [Google Scholar]
- 50.Gu C, Begley TJ, Dedon PC. tRNA modifications regulate translation during cellular stress. FEBS Lett. 2014;588:4287–4296. doi: 10.1016/j.febslet.2014.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan CT, Pang YL, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat Commun. 2012;3:937. doi: 10.1038/ncomms1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kruger MK, Pedersen S, Hagervall TG, Sorensen MA. The modification of the wobble base of tRNAGlu modulates the translation rate of glutamic acid codons in vivo. J Mol Biol. 1998;284:621–631. doi: 10.1006/jmbi.1998.2196. [DOI] [PubMed] [Google Scholar]
- 53.Endres L, Dedon PC, Begley TJ. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015;12:603–614. doi: 10.1080/15476286.2015.1031947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rezgui VA, Tyagi K, Ranjan N, Konevega AL, Mittelstaet J, Rodnina MV, Peter M, Pedrioli PG. tRNA tKUUU, tQUUG, and tEUUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding. Proc Natl Acad Sci U S A. 2013;110:12289–12294. doi: 10.1073/pnas.1300781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernández-Vázquez J, Vargas-Pérez I, Sansó M, Buhne K, Carmona M, Paulo E, Hermand D, Rodríguez-Gabriel M, Ayté J, Leidel S, Hidalgo E. Modification of tRNA(Lys) UUU by elongator is essential for efficient translation of stress mRNAs. PLoS Genet. 2013;9:e1003647. doi: 10.1371/journal.pgen.1003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.