

Abstract
It has been suggested that compounds affecting glycosylphosphatidylinositol (GPI) biosynthesis in bloodstream form Trypanosoma brucei should be trypanocidal. We describe cell-permeable analogues of a GPI intermediate that are toxic to this parasite but not to human cells. These analogues are metabolized by the T. brucei GPI pathway, but not by the human pathway. Closely related nonmetabolizable analogues have no trypanocidal activity. This represents the first direct chemical validation of the GPI biosynthetic pathway as a drug target against African human sleeping sickness. The results should stimulate further inhibitor design and synthesis and encourage the search for inhibitors in natural product and synthetic compound libraries.
Keywords: de-N-acetylase, glycosylphosphatidylinositol, inositol acyltransferase, mannosyltransferase, Trypanosoma
Introduction
A significant number of eukaryotic cell-surface glycoproteins are attached to the plasma membrane by covalent linkage to a glycosylphosphatidylinositol (GPI) anchor. The structure and biosynthesis of GPI membrane anchors have been reviewed (Ferguson et al, 1999; Kinoshita and Inoue, 2000; McConville and Menon, 2000; Morita et al, 2000a; de Macedo et al, 2003). The basic GPI core structure attached to protein is NH2CH2CH2PO4H-6Manα1-2Manα1-6Manα1-4GlcNα1-6D-myo- inositol-1-HPO4-lipid (EtN-P-Man3GlcN-PI), where the lipid can be diacylglycerol, alkylacylglycerol or ceramide. This core structure may be embellished with additional ethanolamine phosphate groups and/or carbohydrate side chains, and/or by inositol acylation in a species- and tissue-specific manner (Ferguson et al, 1999).
Protozoa tend to express significantly higher densities of GPI-anchored proteins than do higher eukaryotes. In particular, Trypanosoma brucei, the tsetse fly-transmitted agent of African sleeping sickness, expresses a cell-surface coat of 5 × 106 dimers of a GPI-anchored variant surface glycoprotein (VSG), which protects the parasite from the alternative complement pathway and, through antigenic variation, from specific immune responses (Cross, 1996). There are 0.3–0.5 million cases of sleeping sickness each year and current therapies are toxic and difficult to administer in the field. More effective therapies are desperately needed to stem this human disease and the related cattle disease nagana that severely limits farming in sub-Saharan Africa. The GPI biosynthetic pathway may be a target for the development of new therapeutics. This notion has been validated genetically by knockout and RNA-interference knockdown studies of genes encoding GPI biosynthetic enzymes (Nagamune et al, 2000; Chang et al, 2002; Lillico et al, 2003).
GPI biosynthesis has been studied in several organisms (Ferguson et al, 1999; Kinoshita and Inoue, 2000; McConville and Menon, 2000; Morita et al, 2000a; de Macedo et al, 2003). It involves the addition of GlcNAc to phosphatidylinositol (PI) to give GlcNAc-PI, which is de-N-acetylated to GlcN-PI. These events occur on the cytoplasmic face of the endoplasmic reticulum (ER), whereas subsequent events occur on the lumenal face (McConville and Menon, 2000). Differences between the T. brucei and mammalian GPI biosynthetic pathways occur from GlcN-PI onwards, including the timing of inositol acylation and deacylation (Güther and Ferguson, 1995; Chen et al, 1998), the addition of extra ethanolamine phosphate groups to mammalian GPI anchors (Hirose et al, 1992; Puoti and Conzelmann, 1993) and fatty-acid remodelling of T. brucei GPI anchors (Masterson et al, 1990; Morita and Englund, 2001). In T. brucei, the GPI precursor glycolipid A′ (EtN-P-Man3GlcN-PI) undergoes fatty-acid remodelling, via glycolipid θ, to yield the dimyristoyl-PI-containing version called glycolipid A. Glycolipid A is the mature GPI precursor that is added to nascent VSG molecules. It is held in equilibrium with glycolipid C via inositol acylation and inositol deacylation (Güther and Ferguson, 1995).
Using synthetic analogues of GlcN-PI, and cell-free systems (Masterson et al, 1989), we have investigated the substrate specificities of T. brucei and human (HeLa cell) GPI biosynthesis enzymes with respect to the stereochemistry and anomeric configuration of the amino sugar, the absolute configuration of and substitution in the myo-inositol residue and the nature of the lipid moiety of the PI component (Smith et al, 1996, 1997, 1999, 2000, 2001, 2002). These studies have produced inhibitors of the T. brucei GPI pathway. However, these analogues do not affect living parasites because the negative charge on the phosphodiester group of the PI moiety renders them cell impermeable.
Here, we describe cell-membrane-permeable GlcN-PI and GlcNAc-PI analogues utilizing the temporary acetoxymethyl-protecting group (Farquhar et al, 1983; Schultz, 2003). These neutral phosphotriester compounds diffuse through the plasma membrane and revert to charged phosphodiesters, through the action of resident cytosolic esterase, and are metabolized by the parasite GPI pathway. The accumulation of these novel metabolites affects endogenous GPI biosynthesis and leads to cell death.
Results
The design of cell-permeable GPI analogues
Analogues of GlcN-PI and GlcNAc-PI lacking the phosphodiester component or containing a neutral methylphosphonate were not recognized by GPI pathway enzymes in the T. brucei cell-free system (Supplementary Figure S1), suggesting that, in designing inhibitors, a charged phosphodiester is indispensable.
Studies with p-nitrophenylacetate and p-nitrophenylphosphate showed that the bloodstream from T. brucei cytosol and cell-free system were rich in esterase activities that were not inhibited by TLCK, leupeptin, PMSF, DFP or NEM (data not shown). We anticipated that utilizing a neutral and esterase-labile phosphotriester would allow masked analogues to diffuse through the plasma membrane and subsequently revert to the charged phosphodiester through the action of resident cellular esterase. Precedents (Farquhar et al, 1983; Schultz, 2003) indicated that the introduction of a temporary acetoxymethyl (CH2OAc=AcM) protecting group would fulfil this purpose. To this end, we synthesized analogues of GlcN-PI and GlcNAc-PI with and without an AcM-protected phosphotriester (Figure 1A; compounds 1, 2 and 3, 4, respectively) having a C18 alkyl component in place of phospholipase-sensitive diacylglycerol (Smith et al, 2002) and a methyl ether at C-2 of the D-myo-inositol residue to prevent substrate recognition by human GPI mannosyltransferases (Smith et al, 1997). We also prepared nonmetabolizable control analogues containing galactosamine (GalN) and N-acetyl-galactosamine (GalNAc) in place of GlcN and GlcNAc (Figure 1A; compounds 5 and 6) and an N-hexanoyl group (GlcNHex) in place of the N-acetyl group of GlcNAc (Figure 1A; compound 7).
Figure 1.
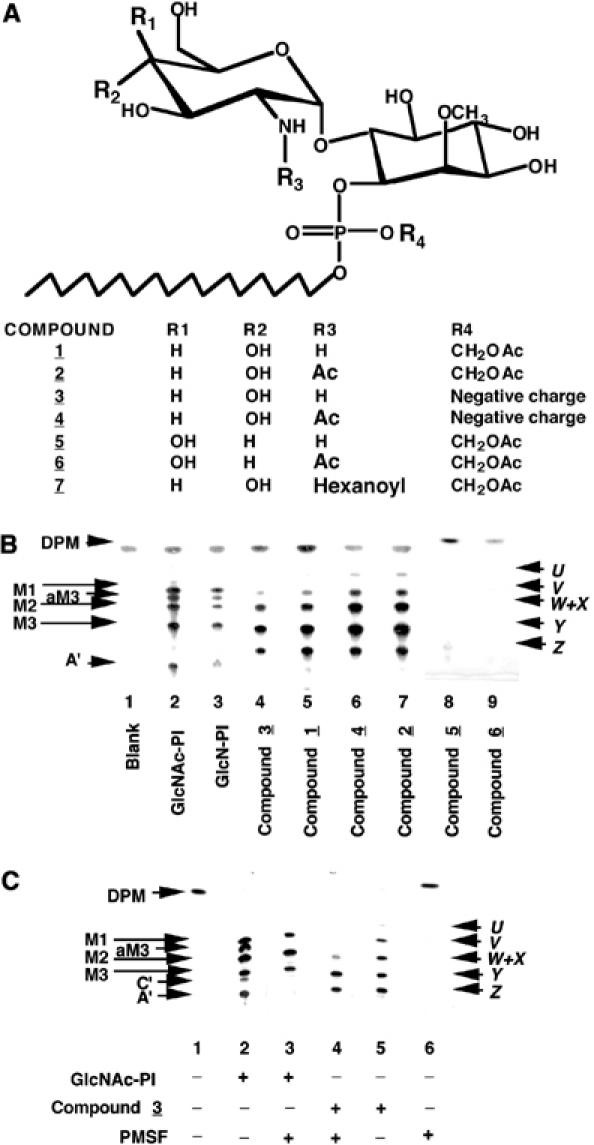
Novel analogues of GlcN-PI and their metabolism by a trypanosome cell-free system. (A) Structures of the synthetic GlcN-PI analogues used in this study. (B) Synthetic GlcNAc-PI and GlcN-PI (positive controls, lanes 2 and 3) and analogues thereof (compounds 1–6, lanes 4–9) were incubated with a modified trypanosome cell-free system and GDP-[3H]Man. Radiolabelled glycolipid products were analysed by HPTLC and fluorography. The products were characterized by enzymatic and chemical digestions (Table I) and are: DPM, dolichol-P-mannose; M1-3, Man1–3GlcNα1-6PI; aM3, Man3GlcNα1-6(2-O-acyl)PI; A′, EtNP-Man3GlcNα1-6PI; C′, EtNP-Man3GlcNα1-6(2-O-acyl)PI; X–Z, Man1–3GlcNα1-6(2-O-methyl)D-myo-inositol1-P-octadecanol; U–W, Man1–3GlcNα1-6(2-O-methyl) (?-O-acyl)D-myo-inositol1-P-octadecanol. (C) Synthetic GlcNAc-PI (control, lanes 2 and 3) and compound 3 were incubated with the trypanosome cell-free system in the presence (+) and absence (−) of PMSF. Radiolabelled glycolipid products were analysed by HPTLC and fluorography.
GlcNAc- and GlcN-containing AcM-protected GPI analogues are deprotected and metabolized by the T. brucei cell-free system
We tested these various analogues with the T. brucei cell-free system and found that N-[3H]acetylated compounds 2 and 4 were substrates for GlcNAc-PI de-N-acetylase, releasing [3H]acetate at rates similar (102 and 111%, respectively) to that from a GlcN[3H]Ac-PI control substrate, whereas N-[3H]acetylated compound 6 was a very poor substrate (about 1% of the control). The latter activity was similar to that observed with GalN[3H]Ac-PI (Smith et al, 2001). These data show that the deprotection of the AcM-modified substrates is not rate limiting in the T. brucei cell-free system and that the use of the C18 alkyl chain in place of an sn-1,2-dipalmitoylglycerol moiety does not affect the nonmetabolizable nature of GalNAc- and GlcNHex-containing GPI analogues (Smith et al, 2001). None of the aforementioned compounds, apart from the control, were de-N-acetylated by the HeLa cell-free system. Based on an indirect assay (Smith et al, 2001), compound 7 (containing an N-hexanoyl group) was not N-deacylated by the GlcNAc-PI de-N-acetylase, nor did it inhibit the de-N-acetylation and subsequent processing of GlcNAc-PI (data not shown).
In the presence of GDP-[3H]Man, compounds 1, 2, 3 and 4 were processed to a set of six identical products (Table I), two of which co-migrate (Figure 1B, lanes 4–7). The products were identified as Man1–3GlcN-(2-O-Me)myo-inositol-1-P-C18 (Figure 1B, bands X–Z) and inositol-acylated versions thereof (Figure 1B, bands U–W). The formation of the inositol-acylated products of the GlcNAc-PI control (Figure 1C, compare lanes 2 and 3) and of compound 3 (Figure 1C, compare lanes 4 and 5) was inhibited by phenylmethylsulphonyl fluoride (PMSF), a known inhibitor of T. brucei inositol acyltransferase (Güther et al, 1994; Güther and Ferguson, 1995). Thus, in the presence of PMSF, (a) glycolipid bands aM3 and C′ are absent (Figure 1C, lane 3) and the glycolipid A′ band disappears because it is the product of glycolipid C′ (Güther and Ferguson, 1995) and (b) inositol-acylated glycolipid bands U, V and W are absent (Figure 1C, lane 4), while nonacylated glycolipids X, Y and Z persist. Acylation of compound 3 was unexpected and must occur at the 3-, 4- or 5-position of the inositol instead of at the usual 2-position that is blocked with an O-methyl group. Presumably, conformational flexibility of the C18 lipid, compared with diacylglycerol, allows access of the inositol acyltransferase to other position(s). Unlike 2-acylation, this aberrant inositol acylation does not allow the addition of ethanolamine phosphate to form a glycolipid A′ equivalent (compare Figure 1B, lanes 4–7 with lanes 2 and 3 and Figure 1C, lane 5 with lane 2). As expected (Smith et al, 2002), nonmetabolizable analogues containing galactosamine (compounds 5 and 6) instead of glucosamine were not processed to [3H]mannosylated products (Figure 1B, lanes 8 and 9). We also showed that compounds 3 and 4 are neither substrates nor inhibitors in the HeLa cell-free system (Supplementary Figure S2). Taken together, these data suggest that if compounds 1 and 2 could enter cells, they should lose the AcM phosphate-protecting group and enter the T. brucei (but not the human) GPI pathway via the cytoplasmic leaflet of the endoplasmic reticulum.
Table 1.
Characterization of the [3H]mannosylated glycolipids formed in the trypanosomal cell-free system with various GlcN-PI analogues
| Abbreviationsa | JBαMb | PI-PLC | Basec | Glycan sized | Assignment |
|---|---|---|---|---|---|
| M1 | +e | + | + | 2.3 | Man1GlcN-PI |
| M2 | + | + | + | 3.1 | Man2GlcN-PI |
| M3 | + | + | + | 4.2 | Man3GlcN-PI |
| aM3 | + | − | + | 4.1 | Man3GlcN-(acyl)PI |
| C′ | − | − | + | 4.1 | EtNP-Man3GlcN-(acyl)PI |
| A′ | − | + | + | 4.1 | EtNP-Man3GlcN-PI |
| C | − | − | + | 4.2 | EtNP-Man3GlcN-(acyl)PI(dimyristyl) |
| A | − | + | + | 4.2 | EtNP-Man3GlcN-PI(dimyristyl) |
| M1-GlcN(2-O-Me)I-P-C18) | + | − | − | 2.4 | Man1-GlcN(2-O-Me)I-P-C18) |
| M2-GlcN(2-O-Me)I-P-C18) | + | − | − | 3.2 | Man2-GlcN(2-O-Me)I-P-C18) |
| M3-GlcN(2-O-Me)I-P-C18) | + | − | − | 4.2 | Man3-GlcN(2-O-Me)I-P-C18) |
| aM1-GlcN(2-O-Me)I-P-C18) | + | − | +(M1-GlcN(2-O-Me)I-P-C18) | 2.3 | Man1-GlcN(2-O-Me)(acyl)I-P-C18) |
| aM2-GlcN(2-O-Me)I-P-C18) | + | − | +(M2-GlcN(2-O-Me)I-P-C18) | 3.1 | Man2-GlcN(2-O-Me)(acyl)I-P-C18) |
| aM3-GlcN(2-O-Me)I-P-C18) | + | − | +(M3-GlcN(2-O-Me)I-P-C18) | 4.3 | Man3-GlcN(2-O-Me)(acyl)I-P-C18) |
| See Figures 1 and 2 for corresponding bands. All of the glycolipids listed here were sensitive to GPI-PLD and deamination. | |||||
| JBαM=jack bean α-mannosidase. | |||||
| The products of base treatment, if sensitive, are given in brackets. | |||||
| The size of the neutral glycans terminating in 2,5-anhydromannitol (AHM) were determined as described in Materials and methods, and are expressed in glucose units. The published values for authentic Manα1–4AHM, Manα1–6Manα1–4AHM and Manα1–2Manα1–6Manα1–4AHM are 2.4, 3.2 and 4.2 glucose units, respectively (Ferguson, 1992). | |||||
| ‘+' indicates a positive digestion, that is, a loss (or shift) of the radiolabelled band; ‘−' indicates a negative digestion, that is, no loss or shift of band. | |||||
AcM-protected GPI analogues are taken up, deprotected and metabolized by living trypanosomes
We pre-incubated living trypanosomes with 50 μM of each of compounds 1 and 5 and labelled the cells with [3H]Man and [3H]myristate. The well-described labelling of glycolipids A and C with these precursors in the absence of inhibitors (Masterson et al, 1989; Güther and Ferguson, 1995) was unaffected by pre-incubation with compound 1 for 30 min (Figure 2A, lane 1 and B, lane 1). However, pre-incubation with compound 1 for 40–60 min caused significant perturbations of the labelling patterns (Figure 2A and B, compare lanes 2–4 with lane 1). In contrast, pre-incubation with compound 5 (the nonmetabolizable GalN-containing analogue) for 1 h had no effect on the labelling of glycolipids A and C (Figure 2A and B, lane 5). These labelling experiments show a pre-incubation time-dependent inhibition of glycolipid C synthesis by compound 1. [3H]Man labelling also showed the accumulation of additional bands (Figure 2A, lanes 2–4); structural analyses of these metabolites (Table I) revealed them to be Man3GlcN-(2-O-Me)myo-inositol-1-P-C18 (product Z) and its inositol-acylated version (product W). The radiolabelling of these metabolites clearly showed that compound 1 enters the trypanosomes, wherein after loss of the AcM group it is metabolized by GPI pathway enzymes up to and including the third α-mannosyltransferase. Furthermore, the data indicate that compound 1 and/or its metabolite(s) is/are inhibiting the conversion of glycolipid A to glycolipid C. By contrast, pre-incubation of the HeLa cells with compound 1 had no effect on [3H]Man labelling of GPI intermediates (data not shown). [3H]Man labelling of trypanosomes was repeated using a range of concentrations of compound 1 with 60 min pre-incubation. The results showed that the labelling pattern was significantly perturbed at 10 μM in compound 1 and that labelling of the endogenous glycolipid C band is abolished by 30 μM (Figure 2C, lanes 3 and 4). Identical data were obtained using the N-acetylated version (compound 2), whereas cell-impermeable analogues lacking the AcM group (compounds 3 and 4) did not affect GPI synthesis (data not shown). Despite the perturbations to the GPI pathway, pre-incubation with compound 1 or 2 did not have any significant effect on the labelling of membrane from VSG with [3H]myristate over the time-frame of the experiment (Figure 2D).
Figure 2.
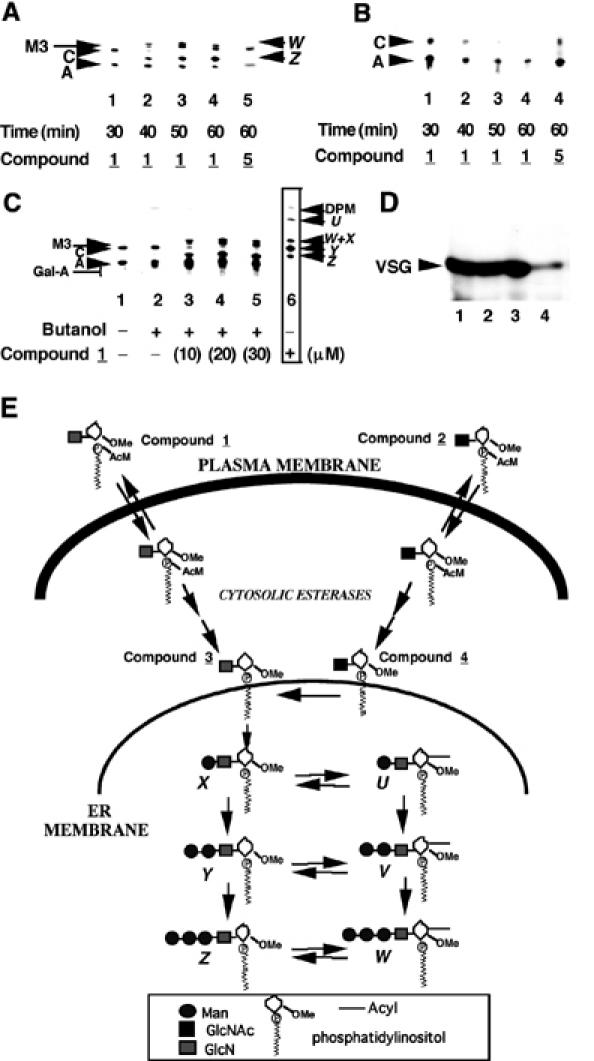
Compound 1 is metabolized by the trypanosome GPI pathway in vivo and inhibits the formation of glycolipid C. (A) Living trypanosomes were pre-incubated with 50 μM compound 1 or compound 5 for 30–60 min, as indicated, and then labelled for 60 min with [3H]Man. Extracted glycolipids were analysed by HPTLC and fluorography. The products, which were characterized by enzymatic and chemical digestions (Table I), are as described in the legend to Figure 1. (B) As for (A), but labelling with [3H]myristate. (C) As for (A), but with a fixed pre-incubation time of 60 min and varying concentrations of compound 1. Compound 1 was added as a butanol solution and a control with butanol alone is included (lane 2). The boxed data (lane 6) are from a cell-free system labelling experiment to provide markers for glycolipids U–Z. (D) Trypanosomes were pre-incubated without any additive (lane 1), or with 30 μM compound 1 (lane 2), 30 μM compound 2 (lane 3) or 60 μg/ml cycloheximide (lane 4) for 60 min, labelled with [3H]myristate for 60 min, lysed in boiling sample buffer and analysed by SDS–PAGE and fluorography. The radiolabel incorporated in the presence of the protein synthesis inhibitor cycloheximide represents that due to myristate exchange on pre-existing VSG rather than de novo synthesis of VSG. (E) Summary of the fates of compounds 1 and 2, as indicated by the in vivo labelling data (see text).
We also investigated the uptake and metabolism of radiolabelled (N-[3H]acetylated) compounds 2 and 4 by living trypanosomes and HeLa cells. After 2 h, negligible amounts of negatively charged compound 4 were cell-associated. By contrast, compound 2 was taken up by HeLa cells and by trypanosomes. However, whereas the radiolabel was neither concentrated nor metabolized by the HeLa cells, the radiolabel was concentrated about 3.5-fold by the parasites. Extraction and HPTLC analysis of the labelled trypanosomes revealed that about 96% of the label was found as N-[3H]acetylated compound 4 (suggesting rapid and complete removal of the AcM group) and that most of the released [3H]acetate (4% of the radiolabel) had been incorporated into other lipids (presumably via fatty-acid elongation; Buxbaum et al, 1996). These data support the supposition that the AcM group is necessary for compound 2 to be taken up by the parasites, and suggest that loss of the AcM group in the cytosol allows concentration of the resulting metabolite in the cells. The relatively small proportion of this metabolite (i.e. compound 4) that becomes de-N-acetylated (4% in 2 h) presumably reflects the fact that it will randomly insert into the intracellular membranes of the parasite and that only a small proportion of these membranes, probably a subcompartment of the ER (McConville and Menon, 2000), contains the GlcNAc-PI de-N-acetylase. By measuring the incorporation of [3H]Man into mannosylated metabolites of compound 4, we estimate that the GPI pathway metabolizes essentially all of the de-N-acetylated material during the second hour of incubation. We also investigated the uptake of other N-[3H]acetylated compounds. The uptake of GlcN[3H]Ac-(2-O-methyl)-I-P-C18, GlcN[3H]Ac-I-C16 and GlcN[3H]Ac-PI(Me-P) was negligible, despite the fact that the latter two are neutral and amphipathic. This suggests that the efficient uptake of compound 2 is due to its trapping as compound 4 once inside the cell.
The results of the in vivo labelling experiments are summarized in Figure 2E. Both compounds 1 and 2 are able to pass though the parasite membrane, thanks to the charge-masking AcM group. Once inside the cell, a two-step process removes the AcM group, triggered by esterase action (Farquhar et al, 1983; Schultz, 2003), to yield compounds 3 and 4, respectively. De-N-acetylation of compound 4 to compound 3 can then occur on the cytoplasmic face of the ER and, after translocation to the luminal face (McConville and Menon, 2000), compound 3 is metabolized by the GPI pathway to yield products up to and including W and Z.
Glycolipid C synthesis is inhibited by metabolite(s) of the GPI analogues
To ascertain whether it is compound 3 (the deprotected form of compound 1) or one of its metabolites that inhibits the formation of glycolipid C, we pre-labelled a trypanosome cell-free system with GDP-[3H]Man (to form labelled glycolipid A′ and θ) and chased with cold GDP-Man and a fatty-acid remodelling mix (a myristoyl-CoA-generating system) in the presence of either PMSF or compound 3. A control incubation led to the remodelling of labelled glycolipids A′ and θ to form glycolipid A and subsequent inositol acylation of glycolipid A to glycolipid C (Figure 3A) (Masterson et al, 1990; Güther et al, 1994; Güther and Ferguson, 1995). The inclusion of PMSF, a compound that has no effect on fatty-acid remodelling, but that inhibits inositol acylation (Güther et al, 1994), led to the accumulation of glycolipid A (Figure 3B). The inclusion of compound 3 had no effect on the conversion of pre-existing glycolipids A and θ to glycolipids A and C (Figure 3C). From these data, we conclude that a metabolite of compound 3, rather than compound 3 itself, inhibits the conversion of glycolipid A to glycolipid C.
Figure 3.
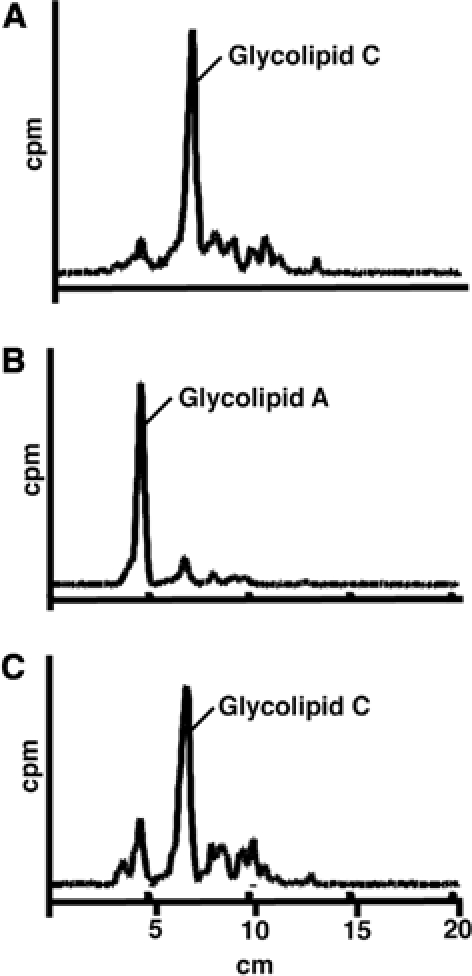
Compound 3 does not directly inhibit the formation of glycolipid C. (A) The trypanosome cell-free system was pulse-labelled with GDP-[3H]Man, to form labelled glycolipids A′ and θ, and then chased with cold GDP-Man and a fatty-acid remodelling mix. The remodelled (glycolipid A) and inositol acylated (glycolipid C) products were extracted and analysed by HPTLC and by scanning with a linear analyser. (B) As for (A), except that PMSF was added to the chase. (C) As for (A), except that compound 3 was added to the chase.
Cell-permeable GPI analogues kill T. brucei parasites
Longer incubation of trypanosomes with compounds 1 and 2 led to their death in a time- and concentration-dependent manner (Figure 4). At 40 μM, compound 1 killed all trypanosomes in about 6 h (and >90% in 24 h at 20 μM), whereas at 100 μM it had no noticeable effect on HeLa cell cultures over 72 h (data not shown). In addition to compounds 1–7, the following compounds were tested for cytotoxicity with T. brucei and HeLa cell cultures at 50 μM: GlcN-I-C16, GlcNAc-I-C16, GlcNAc-PI(Me-P), D-myo-inositol-1-(acetoxymethyl)PO4-octadecyl, D-GlcNMe2α1-6D-2-O-methyl- myo-inositol-1-(acetoxymethyl)PO4-octadecyl and per-O-acetylated compound 2. None of them showed cytotoxicity towards trypanosomes or HeLa cells for up to 72 h, except for the latter compound which was made to see if O-acetylation (also reversible by cellular esterases) might enhance its uptake and potency. We found it to be only equally as trypanocidal as compound 2 and, like compound 2, it showed no apparent toxicity to HeLa cells. Taken together, these results establish that the killing of trypanosomes by compounds 1 and 2 was dependent on: (i) the presence of the AcM group (since compounds 3 and 4 had no effect); (ii) the presence of the GlcN residue (since compounds 5 and 6 and an analogue without an amino sugar had no effect); (iii) the presence of a free amino group on the GlcN residue or an N-acyl group that can be removed by the GlcNAc-PI de-N-acetylase (since compound 7 had no effect).
Figure 4.
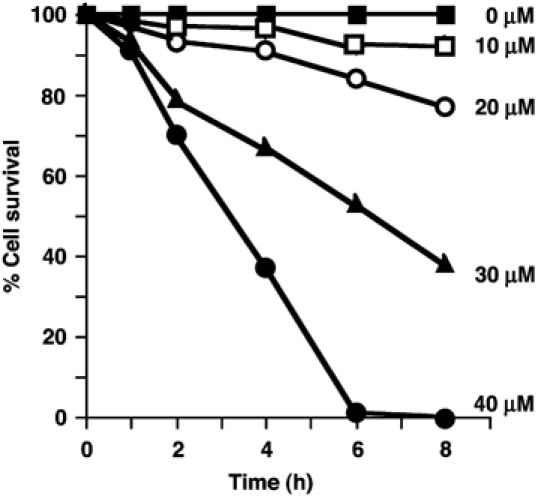
Selective toxicity of a GPI analogue (compound 1) to bloodstream from T. brucei. Parasites were cultured in the presence of 0 (control), 10, 20, 30 and 40 μM compound 1. Viable cells were estimated after 2, 4, 6 and 8 h and normalized to a percentage of the control cultures (values are means of at least nine separate determinations and standard errors are ⩾±5%).
Discussion
The mechanism of killing by metabolites of compounds 1 and 2 is not clear. The presence of these metabolites does not appear to prevent the synthesis and transfer of glycolipid A to nascent VSG molecules (Figure 2D) but, rather, to inhibit the synthesis of glycolipid C from glycolipid A. Thus, the [3H]Man and [3H]myristate labelling of glycolipid A in living trypanosomes persists in the presence of compound 1, while the labelling of glycolipid C is inhibited in a time-dependent (Figure 2A) and concentration-dependent (Figure 2C) manner. The formation of glycolipid W (Figure 2A and C), an inositol-acylated product of compound 1, suggests that it may be the accumulation of this metabolite that inhibits the acylation of glycolipid A to glycolipid C. The role of glycolipid C itself in GPI biosynthesis in the trypanosome is not fully understood. It may act as reservoir, providing glycolipid A via the action of inositol deacylase (Güther and Ferguson, 1995), and/or it may be the main metabolite destined for catabolism of excess GPIs (Milne et al, 1999). GPI catabolism is necessary because T. brucei synthesizes GPI precursors at a rate >10-fold higher than that required to sustain VSG processing (Masterson and Ferguson, 1991; Ralton et al, 1993). The aforesaid compounds lead to an accumulation of their own metabolites and to the accumulation of polar endogenous GPI species that are most likely galactosylated versions of glycolipid A (Gal-A; see Figure 2C, lanes 3–5). We have previously noted the accumulation of Gal-A species in inositol deacylase (GPIdeAc) knockout parasites (Güther et al, 2001, 2003). However, GPIdeAc knockout trypanosomes are viable, suggesting that metabolites of compound 1 are the principal toxic agents.
The compounds described herein provide an important proof-of-concept; that is, that it is possible to make parasite-selective cell-permeable trypanocidal molecules that exploit differences in the specificities of the trypanosomal and human GPI pathway enzymes. Previously, myristate analogues and inhibitors of myristate biosynthesis were shown to be selectively toxic to bloodstream from T. brucei (Doering et al, 1991, 1994; Morita et al, 2000b). They appear to act by incorporation into the VSG GPI anchor or by starvation of GPI fatty-acid remodelling, respectively. However, as the authors point out, alternative mechanisms of killing (e.g. affecting lipid metabolism and/or protein acylation) cannot be excluded. In this case, the lack of toxicity of nonmetabolizable, but virtually identical, analogues (compounds 5 and 6) allows unambiguous chemical validation of the GPI pathway as a drug target. Antimicrobial drug development programmes generally require both genetic and chemical validation of a target because not all essential gene products are amenable to selective chemical inhibition. Thus, together with previous genetic validation of the GPI pathway as a drug target (Nagamune et al, 2000; Chang et al, 2002; Lillico et al, 2003), the chemical validation presented in this paper should stimulate the search for new therapeutic agents against African human sleeping sickness. In this context, while compounds 1 and 2 are primarily chemical validation tools, they also represent potential drug leads. The AcM group, that renders the molecules cell-permeable, is labile to serum esterases. Nevertheless, AcM esters of phosphates have been shown to be sufficiently stable in human serum to allow bioavailability of, for example, antiviral nucleotide analogues (reviewed in Schultz, 2003). Nevertheless, the potency of compounds 1 and 2 will need to be optimized by, for example, removing unnecessary hydroxyl groups (e.g. the 3-, 4-, and 5-hydroxyls of the myo-insoitol and the 6′-hydroxyl of the GlcN residue (Smith et al, 2002)) and adjusting the nature of the lipid group. In addition, compounds 1 and 2 are complex molecules, requiring enantiomeric resolution and regioselective synthetic steps, and we are considering ways to make less synthetically complex mimetic scaffolds.
Materials and methods
Materials
GDP-[2-3H]mannose (14.9–17.8 Ci/mmol), [9,10-3H]myristic acid (49 Ci/mmol) and [3H]acetic anhydride (50.0 Ci/mmol) were purchased from NEN; jack bean α-mannosidase, Aspergillus pheonicus α-mannosidase and GPI-specific phospholipase D from Roche and Bacillus thuringiensis PI-specific phospholipase C from Glyko. All other reagents were purchased from Merck-BDH or Sigma.
Substrates and substrate analogues
D-GlcNα1-6D-myo-inositol-1-HPO4-sn-1,2-dipalmitoylglycerol (GlcN-PI) was synthesized as described previously (Cottaz et al, 1993). The lipid-modified substrate analogues D-GlcNα1-6D-myo-inositol-1-HPO4-sn-1,2-dioctylglycerol (GlcN-PI(diC8)) and D-GlcNα1-6D-myo-inositol-1-HPO4-octadecyl (GlcN-I-P-C18) and the phosphate-free analogue D-GlcNα1-6D-myo-inositol-1-hexadecyl (GlcN-I-C16) were synthesized as described (Crossman et al, 1999, 2002). The preparation of the C-phosphonate analogue D-GlcNα1-6D-myo-inositol-1-PO3(CH3)-sn-1,2-dioctylglycerol (GlcN-PI(Me-P)) will be described elsewhere (Brimacombe and Borissow, unpublished data). The preparation of D-GlcNα1-6D-2-O-methyl-myo-inositol-1-HPO4-octadecyl (compound 3), D-GlcNα1-6D-2-O-methyl-myo-inositol-1-(acetoxymethyl)PO4-octadecyl (compound 1) and D-GalNα1-6D-2-O-methyl-myo-inositol-1-(acetoxymethyl)PO4-octadecyl (compound 5) will be published elsewhere. Compounds 2, 4 and 6 were prepared by N-acetylation, as described previously (Smith et al, 1996). Compound 7 was prepared by N-acylation of compound 1 with hexanoic anydride (Sharma et al, 1997). D-GlcNMe2α1-6D-2-O-methyl-myo-inositol-1-(acetoxymethyl)PO4-octadecyl was prepared from compound 1, as described for the preparation of GlcNMe2-PI from GlcN-PI (Smith et al, 2001). D-myo-Inositol-1-(acetoxymethyl)PO4-octadecyl was prepared by nitrous acid deamination of compound 1. 3,4,5-Tri-O-acetyl-D-GlcNAcα1-6D-2-O-methyl-3,4,5-tri-O-acetyl-myo-inositol-1-(acetoxymethyl)PO4-octadecyl was formed on peracetylation of compound 1 with acetic anhydride/pyridine (1:1). The identities and purities of the synthetic analogues were assessed by negative and/or positive-ion electrospray mass spectrometry, and the concentration of each stock solution was determined by measuring the inositol content by selected ion-monitoring gas chromatography-mass spectrometry (Smith et al, 1996).
N-[3H]acetylation and purification of GlcN-PI analogues
Synthetic GlcN-PI and analogues thereof were treated with [3H]acetic anhydride and the N-[3H]acetylated products were purified by reverse-phase chromatography as described previously (Smith et al, 2001). The GlcN[3H]Ac-PI analogues were diluted with the corresponding nonradioactive compound to give a final specific activity of 18.2 mCi/mmol.
Preparation of trypanosomal and HeLa membranes
T. brucei and HeLa cell membranes (cell-free systems) were prepared as described previously (Smith et al, 1997). Trypanosome membranes were washed twice and suspended at 5 × 108 cell equivalents/ml in 2 × concentrated incorporation buffer supplemented with freshly prepared N-ethylmaleimide and n-octyl β-D-glucopyranoside (Smith et al, 1996), unless stated otherwise. The lysate was sonicated briefly and added to dry GDP-[3H]Man (0.3 μCi/2.5 × 107 cell equivalents). After brief sonication, aliquots of 20 μl (2.5 × 107 cell equivalents) were added to reaction tubes containing an equal volume of the various GlcN-PI analogues in 10 mM n-octyl β-D-glucopyranoside and were then incubated at 30°C for 1 h. The glycolipid products were recovered and analysed by HPTLC before and after enzymatic and chemical treatments.
For those experiments involving PMSF, the inhibitor was dissolved at 100 μM in dry propan-2-ol immediately prior to use and was used at 1 mM final concentration. PMSF was pre-incubated with the membranes prior to the addition of the GlcN-PI analogues.
In the experiment described in Figure 3, washed membranes were suspended in incorporation buffer supplemented with 1 mM dithiothreitol (DTT). The lysate was pulsed with GDP-[3H]Man (0.3 μCi/2.5 × 107 cell equivalents) and UDP-GlcNAc (1 mM) for 10 min at 30°C and chased for 10 min by the addition of GDP-Man (1 mM). The primed membranes were washed to remove unincorporated nucleotides, re-suspended in incorporation buffer without MnCl2 and incubated in the absence (control) or presence of either compound 3 (50 μM) or PMSF (1 mM) for 5 min on ice. Subsequently, fatty-acid remodelling was initiated by the addition of DTT (1 mM), ATP (200 μM), CoA (200 μM) and myristoyl-CoA (100 μM), brief sonication and incubation at 30°C for 30 min. The glycolipid products were extracted as above and analysed by HPTLC using solvent system B.
HeLa cell lysate was thawed, supplemented with 2.5 mM MnCl2, 2 mg/ml leupeptin, 0.1 mM TLCK, 1 μg/ml tunicamycin, 1 mM DTT, 100 μM CoA and ATP-regenerating system (100 μM ATP, 5 mM phosphocreatine and 5 U of creatine phosphokinase) and then added to dry GDP-[3H]Man (1 μCi/1 × 108 cell equivalents) (Smith et al, 2000). After brief sonication, 100 μl aliquots were added to reaction tubes containing the GlcN-PI analogues and incubated at 35°C for 1.5 h. The glycolipid products were recovered and analysed by HPTLC before and after enzymatic and chemical treatments.
De-N-acetylase assay
De-N-acetylase assays using T. brucei and HeLa cell-free systems were carried out as described previously (Smith et al, 2001). Each GlcN[3H]Ac-PI substrate analogue was studied in triplicate at each time interval (0, 15, 30, 60 min) and its initial rate of de-N-acetylation was determined.
Enzymatic and chemical treatments of radiolabelled glycolipids
Digestions with jack bean α-mannosidase (JBαM), GPI-specific phospholipase D (GPI-PLD) and PI-specific phospholipase C (PI-PLC) and base hydrolysis, deamination and N-acetylation were performed as described previously (Güther et al, 1994; Smith et al, 1996, 1997).
Glycan headgroup analysis
The HPTLC-purified radiolabelled glycolipids were delipidated, deaminated, reduced, dephosphorylated with aqueous HF and desalted by passage through AG50X12(H+) and AG3X4 (OH−) ion-exchange resins. The resulting neutral glycan headgroups were analysed by Bio-Gel P4 gel filtration (Ferguson, 1992).
HPTLC
Samples and glycolipid standards were applied to 10 cm aluminium-backed silica gel 60 HPTLC plates, which were developed using either solvent system A: chloroform/methanol/1 M NH4Ac/13 M NH4OH/water (180/140/9/9/23, v/v) or B: chloroform/methanol/water (10/10/3, v/v). Radiolabelled components were detected by fluorography at −70°C, after spraying with En3Hance™, using Kodak XAR-5 film with an intensifying screen.
Metabolic labelling of trypanosomes
Trypanosomes were pre-incubated with test compounds in HMI-9 at 1 × 106 parasites/ml, as indicated in the figures. The test compounds were dissolved in butan-1-ol and added to the cell cultures such that the final butan-1-ol concentration was ⩽0.2% v/v. The parasites were centrifuged, washed and re-suspended (at 1 × 107 parasites/ml) in either glucose- or myristate-free minimal essential media containing tunicamycin (1 μg/ml) and the relevant compound in the absence (control) or presence of either PMSF (1 mM) or cycloheximide (60 μg/ml). The cells were subsequently labelled for 1 h at 37°C with either 25 μCi/ml [3H]Man or [3H]myristate. Aliquots were centrifuged and the trypanosomes were washed in ice-cold medium. Aliquots of the pelleted cells were extracted for with 400 μl of chloroform/methanol/water (10:10:3 v/v), and processed for glycolipid analysis by HPTLC (as above) or re-suspended in SDS sample buffer at 100°C for 10 min and analysed for labelled protein by SDS–PAGE on a 10% gel. The gel was stained with Coomassie blue, soaked in En3Hance, dried and subjected to fluorography.
Uptake and processing of N-[3H]acetyl compounds
N-[3H]acetylated compounds (15 or 30 μM) were incubated with trypanosomes (2 × 106/ml) in 10 ml fresh HMI-9. At 30, 60, 90 and 120 min, duplicate aliquots (1 ml) were centrifuged. Aliquots of the supernatants were counted for radioactivity (unincorporated radiolabel). The cells washed twice with ice-cold trypanosome dilution buffer (TDB) and the cell pellets were extracted with 400 μl of chloroform/methanol/water (10:10:3 v/v). After drying, the total cell-associated [3H]acetate radiolabel was partitioned between butan-1-ol (lipid-associated [3H]acetate radiolabel) and water (free [3H]acetate radiolabel) and each phase was counted for radioactivity.
Cytotoxicity studies
Trypanosomes (1 × 106 parasites/ml) were incubated with the test compounds at different concentrations in HMI-9 medium. At various intervals, triplicate aliquots (2 ml) were centrifuged and the cells were washed and resuspended in 1 ml TDB and 100 μl 5 mg/ml methylthiazoletetrazolium (MTT). After 30 min at 37°C, the cells were centrifuged and the supernatant was removed. The cell pellet was dissolved in 100 μl of DMSO and the absorbance at 570 nm was measured.
HeLa cells (2 × 105 in 2 ml) were grown in 12-well plates for 2 days and then incubated with the test compounds at different concentrations. At designated intervals, the cells were washed with PBS and thereafter incubated in 1 ml TDB and 100 μl MTT (5 mg/ml) for 30 min at 37°C. The cells were washed with PBS, treated with EDTA and centrifuged. The cell pellet was dissolved in 100 μl DMSO and the absorbance at 570 nm was measured.
Absorbances were adjusted for the increase in cell numbers at each time interval. Both the trypanosome and HeLa cytotoxicity studies were conducted at least of four times at each test compound concentration.
Supplementary Material
Online Supplementary Data
Acknowledgments
This work was supported by Wellcome Trust Programme Grants 054491 and 071463. TKS is presently a Wellcome Trust Senior Research Fellow 067441.
References
- Buxbaum LA, Milne KG, Werbovetz KA, Englund PT (1996) Myristate exchange on the Trypanosoma brucei variant surface glycoprotein. Proc Natl Acad Sci USA 93: 1178–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang T, Milne KG, Güther ML, Smith TK, Ferguson MAJ (2002) Cloning of Trypanosoma brucei and Leishmania major genes encoding the GlcNAc-phosphatidylinositol de-N-acetylase of glycosylphosphatidylinositol biosynthesis that is essential to the African sleeping sickness parasite. J Biol Chem 277: 50176–50182 [DOI] [PubMed] [Google Scholar]
- Chen R, Walter EI, Parker G, Lapurga JP, Millan JL, Ikehara Y, Udenfriend S, Medof ME (1998) Mammalian glycophosphatidylinositol anchor transfer to proteins and posttransfer deacylation. Proc Natl Acad Sci USA 95: 9512–9517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottaz S, Brimacombe JS, Ferguson MAJ (1993) Parasite glycoconjugates. Part 1. Synthesis of some early and related intermediates in the biosynthetic pathway of glycosyl-phosphatidylinositol membrane anchors. J Chem Soc Perkin Trans 1: 2945–2951 [Google Scholar]
- Cross GAM (1996) Antigenic variation in trypanosomes: secrets surface slowly. BioEssays 18: 283–287 [DOI] [PubMed] [Google Scholar]
- Crossman A, Brimacombe JS, Ferguson MAJ, Smith TK (1999) Synthesis of some second-generation substrate analogues of early intermediates in the biosynthetic pathway of glycosylphosphatidylinositol membrane anchors. Carbohydr Res 321: 42–51 [DOI] [PubMed] [Google Scholar]
- Crossman A, Paterson MJ, Ferguson MAJ, Smith TK, Brimacombe JS (2002) Further probing of the substrate specificities and inhibition of enzymes involved at an early stage of glycosylphosphatidylinositol (GPI) biosynthesis. Carbohydr Res 337: 2049–3055 [DOI] [PubMed] [Google Scholar]
- Doering TL, Lu T, Werbovetz KA, Gokel GW, Hart GW, Gordon JI, Englund PT (1994) Toxicity of myristic acid analogs toward African trypanosomes. Proc Natl Acad Sci USA 91: 9735–9739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering TL, Raper J, Buxbaum LU, Adams SP, Gordon JI, Hart GW, Englund PT (1991) An analog of myristic acid with selective toxicity for African trypanosomes. Science 252: 1851–1854 [DOI] [PubMed] [Google Scholar]
- Farquhar D, Srivastva KN, Saunders PJ (1983) Biologically reversible phosphate-protective groups. J Pharm Sci 72: 324–331 [DOI] [PubMed] [Google Scholar]
- Ferguson MAJ (1992) The chemical and enzymic analysis of GPI fine structure. In Lipid Modifications of Proteins: A Practical Approach, Hooper NM, Turner AJ (eds), pp 191–230. Oxford, UK: IRL Press [Google Scholar]
- Ferguson MAJ, Brimacombe JS, Brown JR, Crossman A, Dix A, Field RA, Güther MLS, Milne KG, Sharma DK, Smith TK (1999) The GPI biosynthetic pathway as a therapeutic target for African sleeping sickness. Biochim Biophys Acta 1455: 327–340 [DOI] [PubMed] [Google Scholar]
- Güther ML, Ferguson MAJ (1995) The role of inositol acylation and inositol deacylation in GPI biosynthesis in Trypanosoma brucei. EMBO J 14: 3080–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güther ML, Leal S, Morrice NA, Cross GA, Ferguson MAJ (2001) Purification, cloning and characterization of a GPI inositol deacylase from Trypanosoma brucei. EMBO J 20: 4923–4934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güther ML, Masterson WJ, Ferguson MAJ (1994) The effects of phenylmethylsulfonyl fluoride on inositol-acylation and fatty acid remodeling in African trypanosomes. J Biol Chem 269: 18694–18701 [PubMed] [Google Scholar]
- Güther ML, Prescott AR, Ferguson MAJ (2003) Deletion of the GPIdeAc gene alters the location and fate of glycosylphosphatidylinositol precursors in Trypanosoma brucei. Biochemistry 42: 14532–14539 [DOI] [PubMed] [Google Scholar]
- Hirose S, Prince GM, Sevlever D, Ravi L, Rosenberry TL, Ueda E, Medof ME (1992) Characterization of putative glycoinositol phospholipid anchor precursors in mammalian cells. Localization of phosphoethanolamine. J Biol Chem 267: 16968–16974 [PubMed] [Google Scholar]
- Kinoshita T, Inoue N (2000) Dissecting and manipulating the pathway for glycosylphosphatidylinositol-anchor biosynthesis. Curr Opin Chem Biol 4: 632–638 [DOI] [PubMed] [Google Scholar]
- Lillico S, Field MC, Blundell P, Coombs GH, Mottram JC (2003) Essential roles for GPI-anchored proteins in African trypanosomes revealed using mutants deficient in GPI8. Mol Biol Cell 14: 1182–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Macedo CS, Shams-Eldin H, Smith TK, Schwarz RT, Azzouz N (2003) Inhibitors of glycosyl-phosphatidylinositol anchor biosynthesis. Biochimie 85: 465–472 [DOI] [PubMed] [Google Scholar]
- Masterson WJ, Doering TL, Hart GW, Englund PT (1989) A novel pathway for glycan assembly: biosynthesis of the glycosyl-phosphatidylinositol anchor of the trypanosome variant surface glycoprotein. Cell 56: 793–800 [DOI] [PubMed] [Google Scholar]
- Masterson WJ, Ferguson MAJ (1991) Phenylmethanesulphonyl fluoride inhibits GPI anchor biosynthesis in the African trypanosome. EMBO J 10: 2041–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterson WJ, Raper J, Doering TL, Hart GW, Englund PT (1990) Fatty acid remodeling: a novel reaction sequence in the biosynthesis of trypanosome glycosyl-phosphatidylinositol membrane anchors. Cell 62: 73–80 [DOI] [PubMed] [Google Scholar]
- McConville MJ, Menon AK (2000) Recent developments in the cell biology and biochemistry of glycosylphosphatidylinositol lipids. Mol Membr Biol 17: 1–16 [DOI] [PubMed] [Google Scholar]
- Milne KG, Ferguson MAJ, Englund PT (1999) A novel glycosylphosphatidylinositol in African trypanosomes. A possible catabolic intermediate. J Biol Chem 274: 1465–1472 [DOI] [PubMed] [Google Scholar]
- Morita YS, Acosta-Serrano A, Englund PT (2000a) The biosynthesis of GPI anchors. In Oligosaccharides in Chemistry and Biology—A Comprehensive Handbook, Ernst P, Sinay P, Hart G (eds), pp 417–433. Weinheim, Germany: Wiley-VCH [Google Scholar]
- Morita YS, Englund PT (2001) Fatty acid remodeling of glycosyl phosphatidylinositol anchors in Trypanosoma brucei: incorporation of fatty acids other than myristate. Mol Biochem Parasitol 115: 157–164 [DOI] [PubMed] [Google Scholar]
- Morita YS, Paul KS, Englund PT (2000b) Specialized fatty acid synthesis in African Trypanosomes: myristate for GPI anchors. Science 288: 140–143 [DOI] [PubMed] [Google Scholar]
- Nagamune K, Nozaki T, Maeda Y, Ohishi K, Fukuma T, Hara T, Schwarz RT, Sutterlin C, Brun R, Riezman H, Kinoshita T (2000) Critical roles of glycosylphosphatidylinositol for Trypanosoma brucei. Proc Natl Acad Sci USA 97: 10336–10341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puoti A, Conzelmann A (1993) Characterization of abnormal free glycophosphatidylinositols accumulating in mutant lymphoma cells of classes B, E, F and H. J Biol Chem 268: 7215–7224 [PubMed] [Google Scholar]
- Ralton JE, Milne KG, Güther M, Field RA, Ferguson MAJ (1993) The mechanism of inhibition of glycosylphosphatidylinositol anchor biosynthesis in Trypanosoma brucei by mannosamine. J Biol Chem 268: 24183–24189 [PubMed] [Google Scholar]
- Schultz C (2003) Prodrugs of biologically active phosphate esters. Bioorg Med Chem 11: 885–891 [DOI] [PubMed] [Google Scholar]
- Sharma DK, Smith TK, Crossman A, Brimacombe JS, Ferguson MAJ (1997) Substrate specificity of the N-acetylglucosaminyl-phosphatidylinositol de-N-acetylase of glycosylphosphatidylinositol membrane anchor biosynthesis in African trypanosomes and human cells. Biochem J 328: 171–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TK, Cottaz S, Brimacombe JS, Ferguson MAJ (1996) Substrate specificity of the dolichol phosphate mannose: glucosaminyl phosphatidylinositol α1–4 mannosyltransferase of the glycosylphosphatidylinositol biosynthetic pathway of African trypanosomes. J Biol Chem 271: 6476–6482 [DOI] [PubMed] [Google Scholar]
- Smith TK, Crossman A, Borissow CN, Paterson MJ, Dix A, Brimacombe JS, Ferguson MAJ (2001) Specificity of GlcNAc-PI de-N-acetylase of GPI biosynthesis and synthesis of parasite-specific suicide substrate inhibitors. EMBO J 20: 3322–3332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TK, Crossman A, Paterson MJ, Borissow CN, Brimacombe JS, Ferguson MAJ (2002) Specificities of enzymes of glycosylphosphatidylinositol biosynthesis in Trypanosoma brucei and HeLa cells. J Biol Chem 277: 37147–37153 [DOI] [PubMed] [Google Scholar]
- Smith TK, Paterson MJ, Crossman A, Brimacombe JS, Ferguson MAJ (2000) Parasite-specific inhibition of the glycosylphosphatidylinositol biosynthetic pathway by stereoisomeric substrate analogues. Biochemistry 39: 11801–11807 [DOI] [PubMed] [Google Scholar]
- Smith TK, Sharma DK, Crossman A, Brimacombe JS, Ferguson MAJ (1999) Selective inhibitors of the glycosylphosphatidylinositol biosynthetic pathway of Trypanosoma brucei. EMBO J 18: 5922–5930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TK, Sharma DK, Crossman A, Dix A, Brimacombe JS, Ferguson MAJ (1997) Parasite and mammalian GPI biosynthetic pathways can be distinguished using synthetic substrate analogues. EMBO J 16: 6667–6675 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Supplementary Data