

Controlling the MOF pore geometry, size and functionality is of prime importance for the effective and selective gas adsorption. Gas adsorption studies on ultra-microporous MOFs were conducted to gain insight on the pore geometry–sorption energetics relationship.
Keywords: tailored pore geometry, metal–organic frameworks, MOFs, hydrogen storage, dispersive interactions
Abstract
This study aims to assess the possibility of improving H2 and CH4 binding affinity to the aromatic walls of a designed new Metal–Organic Framework (MOF) through simultaneous dispersive interactions. It is suggested here that desirable H2 and CH4 storage media at low pressures require narrow uniform pores associated with large surface area, a trade-off that is challenging to achieve.
1. Introduction
Metal–Organic Frameworks (MOFs), an emerging class of functional solid-state materials, continue to receive wide scientific interest due to their potential applications in hydrogen storage, gas separation, carbon dioxide capture, enhanced catalysis and drug delivery (Eddaoudi et al., 2001 ▸; Moulton & Zaworotko, 2001 ▸; Kitagawa et al., 2004 ▸; Férey et al., 2005 ▸; Horcajada et al., 2006 ▸; Cho et al., 2007 ▸). MOFs possess unique structural attributes including dual composition, crystallinity and a modular pore system (Rowsell & Yaghi, 2006 ▸; Belof et al., 2007 ▸; Cho et al., 2007 ▸; Dincă et al., 2007 ▸; Hayashi et al., 2007 ▸; Liu et al., 2007 ▸; Alkordi et al., 2008 ▸; Banerjee et al., 2008 ▸; Llewellyn et al., 2008 ▸; Nugent et al., 2013 ▸; Belmabkhout et al., 2014 ▸; Shekhah et al., 2014 ▸). Markedly, these attributes are ideal for the assessment and the establishment of the requisite structure–function relationship toward the construction of made-to-order MOFs for a targeted application. In particular, porous MOFs are regarded as prospective adsorbents that can offer practical solutions to the enduring challenges pertaining to the safe storage and efficient use of H2 in mobile applications. Conceivably, MOFs are widely investigated for hydrogen storage due to the ability to control their pore system (functionality and volume) and subsequently impact the H2–MOF interactions and the total H2 uptake (Rowsell & Yaghi, 2005 ▸; Collins & Zhou, 2007 ▸; Lin et al., 2007 ▸; Chen et al., 2008 ▸; Dincă & Long, 2008 ▸; Nouar et al., 2008 ▸; Kishan et al., 2010 ▸; Zhou et al., 2012 ▸). Our group, among others, continue to explore the modularity of MOFs in order to gain better insights on the structure–property relationship and subsequently construct a made-to-order MOF with the ideal gas–MOF interactions and suitable gas uptake for given gas separation/storage applications (Nugent et al., 2013 ▸; Belmabkhout et al., 2014 ▸; Shekhah et al., 2014 ▸). Our study on the soc-MOF platform with the underlying square-octahedral (soc) topology (Belof et al., 2007 ▸; Liu et al., 2007 ▸; Alezi et al., 2015 ▸; Cairns et al., 2016 ▸) indicated that a made-to-order MOF suitable for hydrogen storage at relatively moderate pressures has to be highly porous (high surface area) and concomitantly encompass narrow pores (< 1 nm) and a high localized charged density (polarizable field charges). Notably, a large number of studies on CH4 and H2 storage by MOFs delineated the requirement of high surface area and high heat of adsorption for gas storage (Zhou et al., 2012 ▸). Hydrogen interactions with metal complexes, clusters or ions, within the inorganic part of a given MOF, are dominated by electrostatic forces between the quadrupole moment of the hydrogen molecule and the inorganic complex. Specifically, such H2-MOF interactions’ strengths play a major role in determining the H2 uptake characteristics and hence are the subject of considerable theoretical and experimental investigations (Rowsell & Yaghi, 2005 ▸; Collins & Zhou, 2007 ▸; Hirscher & Panella, 2007 ▸; Lin et al., 2007 ▸; Dincă & Long, 2008 ▸; Nouar et al., 2008 ▸; Murray et al., 2009 ▸; Zhou et al., 2012 ▸). In particular, the weaker, dispersive interactions between H2 molecules and the organic linkers in MOFs, best represented by benzene ring moieties, have been theoretically investigated (Hübner et al., 2004 ▸; Bhatia & Myers, 2006 ▸; Lochan & Head-Gordon, 2006 ▸; Düren et al., 2009 ▸; Han et al., 2009 ▸) and experimentally documented (Rosi et al., 2003 ▸). Recent studies demonstrate that such interactions could, in principle, be enhanced through chemical modifications of the organic linkers, providing a prospective strategy for a material designer to fine-tune the organic building blocks and subsequently enhance H2 sorption characteristics of the MOF (Lochan & Head-Gordon, 2006 ▸). Nevertheless, to the best of our knowledge, no experimental synthetic studies have been published to address the potential to improve the H2 and CH4 binding affinity to the walls of MOFs through simultaneous dispersive interactions, acting additively, between the gas molecules and multiple aromatic rings placed at optimal interaction distance(s) within a specific geometry. Therefore, we opted to explore this approach separately, regardless of the degree of porosity, which could potentially pave the way for the rational design of MOF adsorbents as suitable and effective gas storage media. Computational studies revealed moderate binding affinities for the H2 molecule towards benzene and various aromatic rings (Hübner et al., 2004 ▸; Bhatia & Myers, 2006 ▸; Lochan & Head-Gordon, 2006 ▸; Düren et al., 2009 ▸; Han et al., 2009 ▸). Such interactions are mostly dispersive and within the range of 3.4–4.0 kJ mol−1 for a H2 molecule interacting with the benzene ring of terephthalic acid (Lochan & Head-Gordon, 2006 ▸). The aforementioned binding enthalpy is remotely below the estimated and debated target for efficient H2 storage materials, range of 15–20 kJ mol−1 (room temperature at pressures up to 30 bar; Han et al., 2009 ▸) or 21–32 kJ mol−1 (−20°C and pressure range 1–100 bar; Lochan & Head-Gordon, 2006 ▸). Herein, we set to investigate if such interactions could be additive and hence can lead to enhanced interactions between a H2 molecule and multiple aromatic rings in a tailored MOF adsorbent. As a test model, we envision a molecular square constructed of four benzene rings interacting simultaneously with a single H2 molecule, residing in the center of the square, as a potential model for a material with enhanced H2 binding affinity.
2. Experimental
The solvothermal reaction of Pb(NO3)2 and 4,4′-sulfonyldibenzoic acid in N,N-dimethylformamide (DMF) yields colorless crystals of 1 (Fig. 1 ▸). The as-synthesized compound was characterized and formulated by single-crystal X-ray diffraction studies as [Pb2(C14H8O6S)2]·DMF (1). The phase purity of 1 was confirmed by similarities between its calculated and experimental powder X-ray diffraction patterns (PXRD, supporting information).
Figure 1.

Crystal structure of 1 (top), the Pb—CO2 rod-shaped infinite SBU (middle), and the coordination mode of the carboxylic linker and the Pb(II) ion (below). Pb (green), C (gray), S (yellow), O (red), H (white).
A sample of 1 was activated for sorption studies by solvent exchange in acetonitrile, where complete removal of the DMF guest molecules was confirmed by IR spectroscopy, see the supporting information. The activated sample was found to be stable up to 400°C as confirmed by TGA studies, see the supporting information.
3. Results and discussion
The crystal structure of 1 revealed square-like channels running through the a-axis. The distinctive shape of the ditopic ligand molecule (dihedral Ph—SO2—Ph angle of 103.93°) complemented by the coordination sphere around Pb(II) (CO2—Pb—CO2 dihedral angle of 78.73°) facilitated the construction of the MOF containing square-like channels (Table 1 ▸). In the crystal structure of 1, infinite CO2—Pb(II) secondary building units (SBUs) (Rosi et al., 2005 ▸) are observed and resulting from coordination of the carboxylate linkers in the bridging bis-bidentate mode to Pb(II) ions. Each carboxylate group is coordinated to three Pb(II) ions, enabling the formation of the CO2—Pb(II) infinite coordination chains, along the a-axis. Each Pb(II) ion is coordinated to six oxygen atoms from bridging carboxylate groups (O—Pb bond distances of 2.434–2.815 Å). Additionally, each Pb(II) ion is coordinated to an oxygen atom from the nearest sulfone group (O—Pb bond distance of 2.865 Å).
Table 1. Selected geometric parameters (Å, °).
| Pb1—O3i | 2.434 (6) | Pb1—O4i | 2.441 (6) |
| O3—Pb1—O4i | 72.0 (2) | O2—S3—C6ii | 108.1 (3) |
| O3i—Pb1—O4i | 117.1 (2) | O1—S3—C6ii | 107.6 (4) |
Symmetry codes: (i) 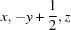 ; (ii)
; (ii) 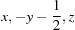 .
.
The resultant connectivity of the Pb ions by the organic linkers facilitated the construction of parallelogram, square-like, shaped channels running along the a-axis, held together in the bc-plane through the sulfone–Pb coordination. Such orthogonal bridging interactions resulted in square-like, guest-accessible, channels in 1. The surface area of 1 as probed by N2 and Ar at 77 K and 87 K (supporting information) were estimated to be 165 and 169 m2 g−1, respectively. The resulting square-like channels in 1 encompass a periodic array of aromatic rings with a relatively short interplanar distance between opposing rings (8.448 Å, centroid-to-centroid). Essentially, the periodically aligned aromatic rings delimiting the pore system dictate the pore aperture size and its maximum opening to be around ∼ 4 Å (excluding the nearest van der Waals surfaces). These special structural features encountered in 1 (narrow one-dimensional channels aligned with a periodic array of aromatic rings) inspired us to explore and further investigate the potential effect(s) of the pore system (size, geometry and functionality) on the H2 interactions with the aromatic walls. Indeed, the observed H2 adsorption properties of the present MOF are remarkable and unique. Of special note are the observed H2 adsorption isotherms with a sharp steepness (type I isotherm shape particularly at 77 K), which, to the best of our knowledge, are scarce for physical adsorbents. Equally interesting is the observed steady H2 isosteric heat of adsorption (Q st) at 9 kJ mol−1 (Fig. 2 ▸). In fact, the H2 adsorption isotherms for 1 demonstrate rapid saturation at early dosing stages and nearly linear behaviour for heats of adsorption throughout the entire H2 adsorption loading, two highly desirable features for H2 storage applications. The sharp step in the H2 adsorption isotherm can be translated to sorption sites saturation by H2 molecules at moderate pressures. This behaviour could be attributed to the equal distribution of H2 sorption sites with uniform binding affinities within the framework, most probably on the surfaces of aromatic rings present in 1. Interestingly, such uniform interactions were also observed for methane adsorption in 1 (Fig. 3 ▸), with a relatively high and steady Q st of 25 kJ mol−1 over the entire loading range, thus confirming the interesting structural aspects of 1 for enhanced gas–solid adsorbent interactions.
Figure 2.

(a) Variable-temperature H2 adsorption isotherms and (b) Q st of H2 adsorption in 1.
Figure 3.

(a) Variable-temperature CH4 adsorption isotherms and (b) Q st of CH4 adsorption in 1.
Furthermore, high-pressure gas adsorption studies conducted on 1 revealed interesting adsorption behaviour of selected gases (Fig. 4 ▸). The recorded uptakes for O2 and N2 in 1 were comparable. This is in contrast to commonly observed preferential N2 uptake, compared with O2, in numerous examples of zeolites (Talu et al., 1996 ▸; Hutson et al., 1999 ▸; Agha et al., 2005 ▸). In the case of most MOFs and zeolites, preferential N2 sorption is attributed to stronger interactions between N2 molecules and the material surface due to a larger quadrupole moment of N2. The interaction/adsorption sites in 1 are dominated by the periodic array of aromatic rings aligned in the one-dimensional channels, thus explaining the weak quadrupole interactions between the gas molecules and the framework and the subsequent comparable N2 and O2 uptakes. Similarly, the nature of the gas/framework interactions is also reflected in the observed comparable uptake of CH4 and CO2 (two distinct molecules with and without quadrupole moment) in 1 (Fig. 4 ▸), emphasizing the dominance of dispersive interactions between adsorbed gas molecules and the aromatic walls of the MOF porous material. It is noteworthy that porous materials having similar uptakes for CO2 and CH4 in a wide range of pressure is uncommon behaviour in adsorption on porous materials (Nugent et al., 2013 ▸). Additionally, the adsorption of C2H6 and C3+ on 1 (supporting information) showed to some extent the same behaviour, uncommon for most MOF materials. The aforementioned results demonstrate that molecules with various degrees of high polarizabilities probe the surface of 1 in the same way. Specifically, the interaction potential of 1 with different molecules, having different chemical–physical properties like CO2, CH4, C2H6 and C3+, is governed mainly by dispersive (non-electrostatic) interactions.
Figure 4.
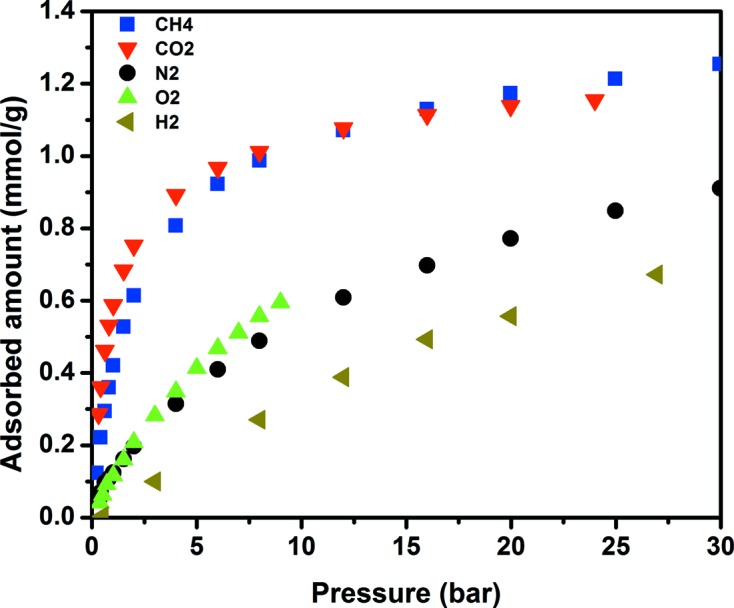
High-pressure sorption isotherms for different gases at 298 K in 1.
4. Conclusions
In conclusion, we present an unprecedented experimental illustration of uptake–energetics relationships for H2 and CH4 adsorption in a novel MOF with narrow one-dimensional channels aligned with a periodic array of aromatic rings. The newly synthesized material represents a model material for pinpointing the interplay between porosity/storage capacity and the strength of host–guest interactions. This approach supports the impact of narrow channels with a periodic array of aromatic rings, controlling the access to prospective larger pores within a targeted porous material, for the effective gas adsorption. The present results pave the way to additional experimental and theoretical investigations in order to further assess the extent of additive dispersive interactions in enhancing H2 and CH4 binding affinity in gas storage materials, in general, and MOFs, in particular. Noticeably, the distinct sharp step at relatively low pressures in the H2 adsorption isotherm at 77 K and the relatively high Q st for CH4 storage was achieved despite the detrimental reduction in the MOF overall porosity. Conceivably, from the present study, the optimal combination of narrow pores/windows (< 1 nm diameter) with a suitable and uniform charge density in the pores (coordinatively unsaturated metal sites and polar functional groups) operating synergistically could play a significant role in promoting the storage of H2 and CH4 at moderate pressures and ambient temperatures.
Supplementary Material
Crystal structure: contains datablock(s) kme002_0m. DOI: 10.1107/S2052252516019060/lq5001sup1.cif
Structure factors: contains datablock(s) kme002_0m. DOI: 10.1107/S2052252516019060/lq5001kme002_0msup2.fcf
Supporting adsorption and structural data. DOI: 10.1107/S2052252516019060/lq5001sup3.pdf
CCDC reference: 1528379
Acknowledgments
We acknowledge funding from King Abdullah University of Science and Technology (KAUST).
References
- Agha, R. K., De Weireld, G. & Frère, M. (2005). Adsorption 11, 179–182.
- Alezi, D., Belmabkhout, Y., Suyetin, M., Bhatt, P. M., Weseliński, L. J., Solovyeva, V., Adil, K., Spanopoulos, I., Trikalitis, P. N., Emwas, A. H. & Eddaoudi, M. (2015). J. Am. Chem. Soc. 137, 13308–13318. [DOI] [PMC free article] [PubMed]
- Alkordi, M. H., Liu, Y., Larsen, R. W., Eubank, J. F. & Eddaoudi, M. (2008). J. Am. Chem. Soc. 130, 12639–12641. [DOI] [PubMed]
- Banerjee, R., Phan, A., Wang, B., Knobler, C., Furukawa, H., O’Keeffe, M. & Yaghi, O. M. (2008). Science, 319, 939–943. [DOI] [PubMed]
- Belmabkhout, Y., Mouttaki, H., Eubank, J. F., Guillerm, V. & Eddaoudi, M. (2014). RSC Adv. 4, 63855–63859.
- Belof, J. L., Stern, A. C., Eddaoudi, M. & Space, B. (2007). J. Am. Chem. Soc. 129, 15202–15210. [DOI] [PubMed]
- Bhatia, S. K. & Myers, A. L. (2006). Langmuir, 22, 1688–1700. [DOI] [PubMed]
- Cairns, A. J., Eckert, J., Wojtas, L., Thommes, M., Wallacher, D., Georgiev, P. A., Forster, P. M., Belmabkhout, Y., Ollivier, J. & Eddaoudi, M. (2016). Chem. Mater. 28, 7353–7361.
- Chen, B., Zhao, X., Putkham, A., Hong, K., Lobkovsky, E. B., Hurtado, E. J., Fletcher, A. J. & Thomas, K. M. (2008). J. Am. Chem. Soc. 130, 6411–6423. [DOI] [PubMed]
- Cho, S. H., Gadzikwa, T., Afshari, M., Nguyen, S. T. & Hupp, J. T. (2007). Eur. J. Inorg. Chem. 2007, 4863–4867.
- Collins, D. J. & Zhou, H.-C. (2007). J. Mater. Chem. 17, 3154–3160.
- Dincă, M., Han, W. S., Liu, Y., Dailly, A., Brown, C. M. & Long, J. R. (2007). Angew. Chem. Int. Ed. 46, 1419–1422. [DOI] [PubMed]
- Dincă, M. & Long, J. R. (2008). Angew. Chem. Int. Ed. 47, 6766–6779. [DOI] [PubMed]
- Düren, T., Bae, Y.-S. & Snurr, R. Q. (2009). Chem. Soc. Rev. 38, 1237–1247. [DOI] [PubMed]
- Eddaoudi, M., Moler, D. B., Li, H., Chen, B., Reineke, T. M., O’Keeffe, M. & Yaghi, O. M. (2001). Acc. Chem. Res. 34, 319–330. [DOI] [PubMed]
- Férey, G., Mellot-Draznieks, C., Serre, C. & Millange, F. (2005). Acc. Chem. Res. 38, 217–225. [DOI] [PubMed]
- Han, S. S., Mendoza-Cortés, J. L. & Goddard, W. A. III (2009). Chem. Soc. Rev. 38, 1460–1476. [DOI] [PubMed]
- Hayashi, H., Côté, A. P., Furukawa, H., O’Keeffe, M. & Yaghi, O. M. (2007). Nat. Mater. 6, 501–506. [DOI] [PubMed]
- Hirscher, M. & Panella, B. (2007). Scr. Mater. 56, 809–812.
- Horcajada, P., Serre, C., Vallet-Regí, M., Sebban, M., Taulelle, F. & Férey, G. (2006). Angew. Chem. 118, 6120–6124. [DOI] [PubMed]
- Hübner, O., Glöss, A., Fichtner, M. & Klopper, W. (2004). J. Phys. Chem. A, 108, 3019–3023.
- Hutson, N. D., Rege, S. U. & Yang, R. T. (1999). AIChE J. 45, 724–734.
- Kitagawa, S., Kitaura, R. & Noro, S. (2004). Angew. Chem. Int. Ed. 43, 2334–2375. [DOI] [PubMed]
- Lin, X., Jia, J., Hubberstey, P., Schröder, M. & Champness, N. R. (2007). CrystEngComm, 9, 438–448.
- Liu, Y., Eubank, J. F., Cairns, A. J., Eckert, J., Kravtsov, V. C., Luebke, R. & Eddaoudi, M. (2007). Angew. Chem. Int. Ed. 46, 3278–3283. [DOI] [PubMed]
- Llewellyn, P. L., Bourrelly, S., Serre, C., Vimont, A., Daturi, M., Hamon, L., De Weireld, G., Chang, J.-S., Hong, D.-Y., Kyu Hwang, Y., Hwa Jhung, S. & Férey, G. (2008). Langmuir, 24, 7245–7250. [DOI] [PubMed]
- Lochan, R. C. & Head-Gordon, M. (2006). Phys. Chem. Chem. Phys. 8, 1357–1370. [DOI] [PubMed]
- Moulton, B. & Zaworotko, M. J. (2001). Chem. Rev. 101, 1629–1658. [DOI] [PubMed]
- Murray, L. J., Dincă, M. & Long, J. R. (2009). Chem. Soc. Rev. 38, 1294–1314. [DOI] [PubMed]
- Nouar, F., Eubank, J. F., Bousquet, T., Wojtas, L., Zaworotko, M. J. & Eddaoudi, M. (2008). J. Am. Chem. Soc. 130, 1833–1835. [DOI] [PubMed]
- Nugent, P., Belmabkhout, Y., Burd, S. D., Cairns, A. J., Luebke, R., Forrest, K., Pham, T., Ma, S., Space, B., Wojtas, L., Eddaoudi, M. & Zaworotko, M. J. (2013). Nature, 495, 80–84. [DOI] [PubMed]
- Radha Kishan, M., Tian, J. K., Thallapally, P. K., Fernandez, C. A., Dalgarno, S. J., Warren, J. E., McGrail, B. P. & Atwood, J. L. (2010). Chem. Commun. 46, 538–540. [DOI] [PubMed]
- Rosi, N. L., Eckert, J., Eddaoudi, M. Vodak, D. T., Kim, J., O’Keeffe, M. & Yaghi, O. M. (2003). Science, 300, 1127–1129. [DOI] [PubMed]
- Rosi, N. L., Kim, J., Eddaoudi, M., Chen, B., O’Keeffe, M. & Yaghi, O. M. (2005). J. Am. Chem. Soc. 127, 1504–1518. [DOI] [PubMed]
- Rowsell, J. L. & Yaghi, O. M. (2005). Angew. Chem. Int. Ed. 44, 4670–4679. [DOI] [PubMed]
- Rowsell, J. L. & Yaghi, O. M. (2006). J. Am. Chem. Soc. 128, 1304–1315. [DOI] [PubMed]
- Shekhah, O., Belmabkhout, Y., Chen, Z., Guillerm, V., Cairns, A., Adil, K. & Eddaoudi, M. (2014). Nat. Commun 5, 4428. [DOI] [PMC free article] [PubMed]
- Talu, O., Li, J., Kumar, R., Mathias, P. M., Moyer, J. D. & Schork, J. M. (1996). Gas Sep. Purif. 10, 149–159.
- Zhou, H.-C., Long, J. R. & Yaghi, O. M. (2012). Chem. Rev. 112, 673–674. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) kme002_0m. DOI: 10.1107/S2052252516019060/lq5001sup1.cif
Structure factors: contains datablock(s) kme002_0m. DOI: 10.1107/S2052252516019060/lq5001kme002_0msup2.fcf
Supporting adsorption and structural data. DOI: 10.1107/S2052252516019060/lq5001sup3.pdf
CCDC reference: 1528379