

Abstract
Translation of the genetic code requires attachment of tRNAs to their cognate amino acids. Errors during amino-acid activation and tRNA esterification are corrected by aminoacyl-tRNA synthetase-catalyzed editing reactions, as extensively described for aliphatic amino acids. The contribution of editing to aromatic amino-acid discrimination is less well understood. We show that phenylalanyl-tRNA synthetase misactivates tyrosine and that it subsequently corrects such errors through hydrolysis of tyrosyl-adenylate and Tyr-tRNAPhe. Structural modeling combined with an in vivo genetic screen identified the editing site in the B3/B4 domain of the β subunit, 40 Å from the active site in the α subunit. Replacements of residues within the editing site had no effect on Phe-tRNAPhe synthesis, but abolished hydrolysis of Tyr-tRNAPhe in vitro. Expression of the corresponding mutants in Escherichia coli significantly slowed growth, and changed the activity of a recoded β-galactosidase variant by misincorporating tyrosine in place of phenylalanine. This loss in aromatic amino-acid discrimination in vivo revealed that editing by phenylalanyl-tRNA synthetase is essential for faithful translation of the genetic code.
Keywords: editing, genetic code, phenylalanine, transfer RNA, translation
Introduction
The functioning of living systems requires maintaining a certain level of fidelity in all processes dealing with the transfer of information. The fidelity of translation during protein synthesis is determined at two major points: selection of aminoacyl-tRNA by ribosomes and aminoacylation of tRNA with the cognate amino acid by aminoacyl-tRNA synthetases (aaRSs). The aaRSs define the genetic code by attaching tRNAs to the corresponding amino acids (aa). When the cognate aa displays high structural similarities to other isosteric or slightly smaller compounds, the aaRS is not able to distinguish between the cognate and the noncognate substrates with sufficient specificity to prevent mischarging. Under these circumstances, accurate aminoacyl-tRNA synthesis often requires an additional editing activity intrinsic to many aaRSs (reviewed in Jakubowski and Goldman, 1992; Hendrickson and Schimmel, 2003). Inaccuracies are much less frequent with noncognate tRNA, the diverse combinations of bases ensuring that cognate molecules are specifically selected by aaRSs (Ebel et al, 1973; Fersht, 1979).
AaRS-catalyzed aminoacylation occurs via a universally conserved two-step reaction: (i) aa activation with ATP to form an adenylate (aa∼AMP), and (ii) transfer of the activated aa to the tRNA to form the aa-tRNA. The two-step reaction contributes to aa discrimination, since insufficient selectivity at the activation step can still be resolved at subsequent points in aminoacylation. Pre-transfer editing occurs by hydrolysis of the noncognate aa∼AMP, while post-transfer editing relies on the ability of the aaRS to hydrolyze the noncognate aa-tRNA bound to the enzyme. In a number of aaRSs, another tRNA-independent editing mechanism accounts for misactivated homocysteine and other amino acids by cyclization (reviewed in Jakubowski, 2004), while certain D-aminoacyl-tRNAs are cleared by specific deacylases (Ferri-Fioni et al, 2001).
The structural basis of editing is best understood for aliphatic amino-acid discrimination for both class I and II aaRSs, despite the fact that the two classes are unrelated. Class I aaRSs such as isoleucyl- (IleRS) (Nureki et al, 1998; Silvian et al, 1999), leucyl- (LeuRS) (Lincecum et al, 2003) and valyl-tRNA synthetase (ValRS) (Fukai et al, 2000) edit noncognate aa∼AMP and aa-tRNA by means of the conserved CP1 domain inserted in the catalytic domain of the enzyme. By contrast, class II aaRSs display more diversity in the structure and the phylogenetic distribution of their known editing domains. Three structurally unrelated domains functional in post-transfer editing have so far been described in class II aaRSs; the ‘HxxxH' domain found in both alanyl- (AlaRS) (Beebe et al, 2003) and threonyl-tRNA synthetase (ThrRS) (Dock-Bregeon et al, 2000), an unrelated domain in archaeal ThrRS (Beebe et al, 2004; Korencic et al, 2004), and the Ybak-like domain in prolyl-tRNA synthetase (ProRS) (Beuning and Musier-Forsyth, 2000; Ahel et al, 2003; Wong et al, 2003). In ThrRS and AlaRS, the HxxxH domain is responsible for editing Ser by a post-transfer mechanism. This domain is universally present in the C-terminal region of AlaRS, whereas it is localized near the N-terminus of most ThrRSs (Sankaranarayanan et al, 1999), the only exceptions being the archaeal ThrRSs that contain an alternate editing domain. Functional genomics analyses have uncovered freestanding editing-competent versions of all three class II domains (Ahel et al, 2003; Wong et al, 2003; Korencic et al, 2004), supporting the hypothesis that such modules arose late in aaRSs evolution to enhance the accuracy of aminoacylation and protein synthesis (Pezo et al, 2004).
In contrast to the extensive use of editing to maintain fidelity in protein synthesis with aliphatic amino acids, aromatic amino acids are chiefly discriminated during their recognition and activation. No editing mechanisms have been postulated or described for the closely related class I tryptophanyl- and tyrosyl-tRNA synthetases (TyrRS), and it has been explicitly shown that the specificity of the latter for Tyr is sufficient to maintain fidelity during translation (Fersht et al, 1980). Phenylalanyl-tRNA synthetase (PheRS), a class II aaRS, is one of the largest and most complex aaRSs (Mosyak et al, 1995; Goldgur et al, 1997), but lacks any structural features reminiscent of known editing domains. Editing activity directed towards Tyr has been reported for yeast cytoplasmic PheRS (Lin et al, 1983, 1984). These experiments suggested both pre- and post-transfer mechanisms for the editing of Tyr∼AMP and Tyr-tRNA, respectively, although the technology employed did not allow isolation and subsequent monitoring of the direct hydrolysis of Tyr-tRNAPhe. More recent studies of an Escherichia coli PheRS active site variant also indicated an editing activity directed towards tyrosine. This activity was highly specific as other para-substituted Phe analogues were stably attached to tRNAPhe, producing noncognate aminoacyl-tRNAs readily utilized in protein synthesis (Ibba et al, 1994; Sharma et al, 2000). Here, we show that E. coli PheRS specifically hydrolyzes Tyr-tRNAPhe, and that the editing site is located in the B3/B4 domain of the β subunit of the protein. Replacements of key residues at this site abolished editing activity, allowing Tyr-tRNAPhe to be stably synthesized both in vitro and in vivo.
Results
E. coli PheRS possesses an editing activity against Tyr-tRNAPhe
An A294G replacement at the active site of the α subunit of E. coli PheRS yields an enzyme significantly enhanced in Tyr activation but unable to stably transfer the noncognate aa to tRNA, suggesting that an editing mechanism might prevent accumulation of Tyr-tRNAPhe (Ibba et al, 1994). To further investigate the existence of such a mechanism, we followed ATP consumption by wild-type and αA294G PheRS in the presence of labeled γATP and Tyr. ATP consumption was comparable for wild type (k−tRNA=2.2±0.3 min−1) and the αA294G variant (k−tRNA=4±0.12 min−1; Table I and inset Figure 1). Addition of in vitro transcribed E. coli tRNAPhe enhanced ATP hydrolysis 14-fold with αA294G PheRS, but only 1.5-fold for the wild type (Figure 1 and Table I). These findings indicate that, like yeast PheRS, the E. coli enzyme possessed tRNA-dependent editing activity. To directly investigate possible post-transfer editing activity, hydrolysis of preformed Tyr-tRNAPhe by wild-type and αA294G PheRS was followed. Both enzymes rapidly deacylated Tyr-tRNAPhe but not Phe-tRNAPhe (Figure 2), indicating that a specific post-transfer editing activity is present in PheRS.
Table 1.
Tyr-dependent ATP hydrolysis by E. coli PheRS
| PheRS |
k (apparent rate of ATP hydrolysis, μM/min)a |
||
|---|---|---|---|
| −tRNAPhe | +tRNAPhe | k+tRNA/k−tRNA | |
| Wild type | 2.2±0.34 | 3.4±0.68 | 1.5 |
| αA294G |
4±0.12 |
55±0.25 |
13.7 |
| The apparent rate of ATP hydrolysis was calculated as the initial velocity of the decrease of the concentration of ATP divided by the concentration of enzyme used. | |||
Figure 1.
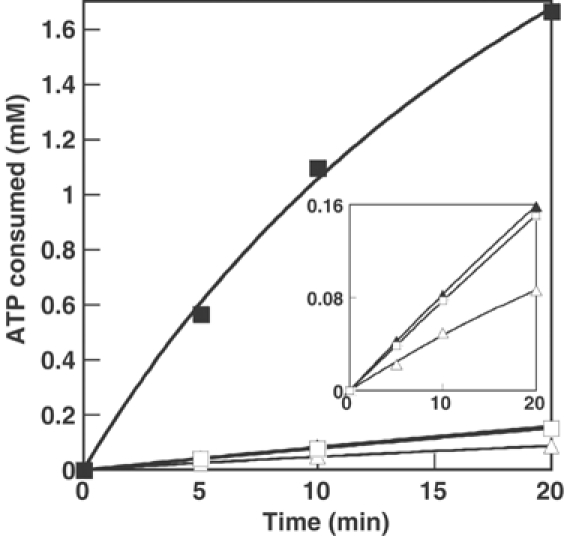
Tyr-dependent ATP hydrolysis by E. coli PheRS. Reactions were performed as described (15 μl samples) in the presence of 2 μM PheRS, with addition of tRNAPhe (2 μM) as indicated. Wild-type PheRS (▵), wild-type PheRS and tRNAPhe (▴), αA294G PheRS, (□), αA294G PheRS and tRNAPhe (▪). Inset, as main chart.
Figure 2.
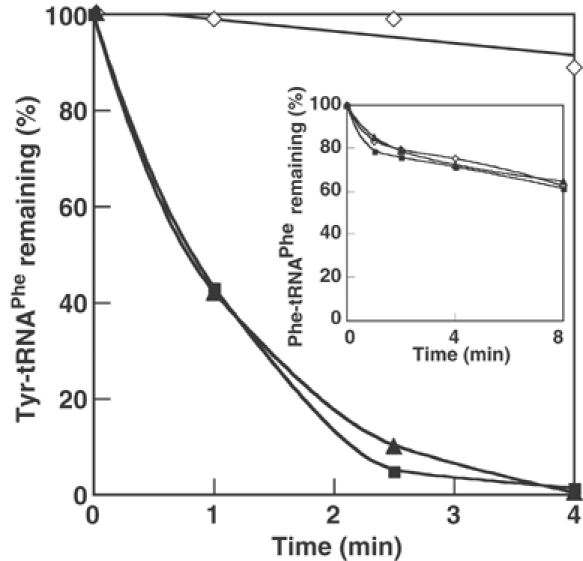
Specific deacylation of Tyr-tRNAPhe by E. coli PheRS. Reactions were performed as described (15 μl samples) in the presence of 2 nM PheRS and 1 μM Tyr-tRNAPhe. Wild-type PheRS (▴), αA294G PheRS (▪), no enzyme (◊). Inset, as main chart, except Phe-tRNAPhe (1 μM) was used instead of Tyr-tRNAPhe.
The editing activity of PheRS is not triggered by 3′-end modified tRNAPhe
To further investigate the role of tRNA in the editing of Tyr, tRNAPhe transcripts were modified at their 3′-ends using terminal tRNA nucleotidyl transferase. The 3′-terminal adenosine (A76) was replaced by a 2′-deoxy-, 3′-deoxy- or dideoxy-adenosine, and the resulting species purified. These tRNAPhe variants were examined for aminoacylation with Phe or Tyr and activation of Tyr editing using PheRS αA294G, which provides an effective amplification of the tRNA-dependent reaction (Figure 1). Only 3′-deoxy tRNAPhe retained phenylalanylation activity, in agreement with the observation that E. coli PheRS aminoacylates tRNA on the 2′OH of A76 (Sprinzl and Cramer, 1975). Unlike tRNAPhe, none of the modified tRNAs enhanced Tyr editing and none were tyrosylated. These results suggest that the editing activity of PheRS against Tyr may occur predominantly at the post-transfer stage.
Identification of putative editing sites by conservation mapping on PheRS·tRNAPhe
Residues potentially involved in PheRS editing were searched for by mapping conservation data from a sequence alignment of 104 eubacterial PheRS α and β subunits onto the 3-D structure of the Thermus thermophilus PheRS·tRNAPhe complex (Goldgur et al, 1997). This analysis was initially performed manually, and subsequently automated to provide a generally applicable algorithm (Alimap 3D, C++ source code available on request). Alimap 3D visualizes the level of conservation of an input sequence on the surface of a corresponding 3-D structure and automatically generates scripts for visualization using Swiss pdb viewer (Kaplan and Littlejohn, 2001), MOLMOL (Koradi et al, 1996) and PyMol (DeLano, 2002). A color code is attributed to each residue depending on its level of conservation derived from the sequence alignment. The conservation rate deduced for each residue can be calculated in two different ways: from the best-represented identical residue (the approach used here) or from the most conserved residues that belong to a user-defined similarity group.
Several conserved patches were identified on the surface of the PheRS·tRNAPhe complex (Figure 3): one near the active site on the α subunit, and others centered on six universally conserved residues on the surface of the β subunit (D33, E124, N254, H265 and E334, E. coli sequence numbering). Two conserved patches on the β subunit were particularly noteworthy. The first is located on the surface of the B1/B2 domain (E124, Figure 3), the second on the B3/B4 domain (H265, E334, Figure 3). Both these conserved patches were approximately 20 Å from the key flexible nucleotide group (A73) of tRNAPhe, which is ∼20 Å from the active site on the α subunit. Thus, it was postulated that the aminoacylated 3′-end of the tRNA could readily translocate from the active site to either of the two conserved patches in the β subunit. Such a structural juxtaposition of aminoacylation and editing domains was previously observed in ThrRS, another class II aaRS. In this case, post-transfer editing is accomplished by the translocation of the flexible 3′-end without the dissociation of Ser-tRNAThr from the enzyme (Dock-Bregeon et al, 2000).
Figure 3.
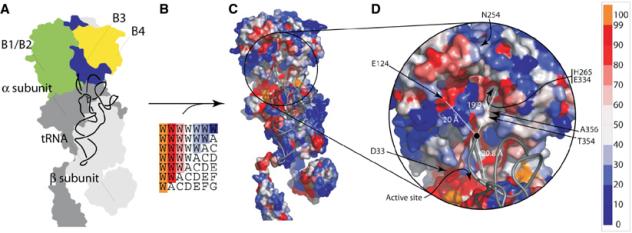
Mapping of conserved residues at putative editing sites in the β subunit of PheRS. (A) Schematic representation of the structure of T. thermophilus (αβ)2 PheRS complexed with tRNAPhe. (B) Representation of the coloring scheme used for mapping conservation onto the surface of the structure. (C) Surface map of eubacterial PheRS sequence conservation. (D) Enlarged section from C showing putative editing sites and their distances from A73 of tRNAPhe. The distance to the active site is also shown for comparison. Residue numbering for the corresponding positions in the E. coli enzyme is shown.
The editing site of PheRS is located in the B3/B4 domain
To determine if the editing site resided in either of the candidate-conserved patches on the surface of the β subunit, residues at these sites were replaced by either Ala, an isosteric aa or a bulkier aa. In order to enhance the potential accumulation of Tyr-tRNAPhe, these variants were combined with the promiscuous active site replacement αA294G. E. coli strain CC503 (Cupples and Miller, 1988) was then co-transformed with plasmids containing corresponding constructs for the production of particular PheRS variants and a second plasmid for the production of the amber suppressor tRNAPheCUA. CC503 (lacZ503 amber) contains a chromosomal lacZ allele with an amber (UAG) codon encoding Tyr503. Tyr503 of β-galactosidase (encoded by lacZ) is essential for catalysis (Penner et al, 1999), and any amino-acid substitution leads to >99.9% reduction in activity (Cupples and Miller, 1988). Thus, insertion of tyrosine at 503 results in blue colonies on X-gal indicator plates; in CC503, blue colonies are only expected in the event that Tyr-tRNAPheCUA stably accumulated, leading to UAG 503 being translated as tyrosine (Figure 4A). When this procedure was used to screen replacements at both putative editing sites, the B3/B4 domain changes gave rise to an editing-defective phenotype on indicator plates (Table II; see, for example, Figure 4B). These and other editing site variants were then further characterized in vitro.
Figure 4.
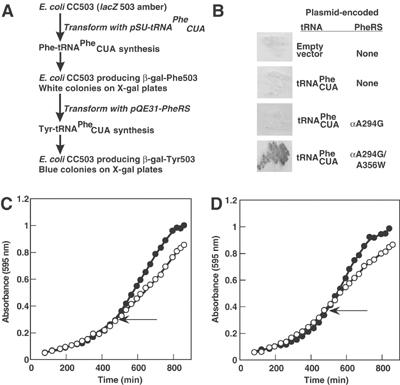
In vivo phenotypes of editing defective PheRS. (A) Schematic representation of the restoration of recoded β-galactosidase by editing defective PheRS. (B) Restoration of β-galactosidase activity in CC503 by PheRSαA294G/βA356W. (C, D) Editing-defective PheRS slows E. coli growth. E. coli containing plasmids encoding PheRSαA294G (•) or PheRSαA294G/βA356W (○) were grown until A595 nm was approximately 0.3, and then IPTG (1 mM) was added (indicated by ←) to increase the production of plasmid-encoded PheRS. Either complete minimal medium (C) or minimal medium lacking Phe and enriched in Tyr (D) were employed.
Table 2.
Kinetics of steady-state phenylalanylation of tRNA, and tRNAPhe enhancement of editing by wild-type E. coli PheRS and variants
| PheRS | CC503/X-gala | KM (L-Phe, μM) | kcat (min−1) | kcat/KM (min−1 μM−1) | k+tRNA/k−tRNAb | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Wild type | White | 10.3±1.0 | 103.3±3.7 | 10.0±0.1 | 1.5±0.1 | ||||||
| αA294G | White | 21.2±2.8 | 122.6±7.8 | 5.8±0.1 | 13.7±0.5 | ||||||
| αA294G, βH265A | Blue | 33.1±2.7 | 172.8±7.2 | 5.2±0.1 | 3.5±0.8 | ||||||
| αA294G, βH265L | Blue | 31.2±2.9 | 198.2±9.2 | 6.3±0.1 | 4.6±0.1 | ||||||
| αA294G, βE334A | Blue | 26.1±1.6 | 158.7±4.9 | 6.0±0.1 | 1.2±0.1 | ||||||
| αA294G, βT354W | Blue | 22.5±5.9 | 58.5±7.2 | 2.6±0.3 | 1.1±0.01 | ||||||
| αA294G, βA356W |
Blue |
28.2±1.3 |
187.0±4.4 |
6.6±0.1 |
1.3±0.1 |
||||||
| Color of colony upon expression of the corresponding PheRS-encoding gene in CC503-based indicator strains (see Figure 4B). | Fold enhancement of Tyr-dependent ATP hydrolysis upon addition of tRNA (see Table I). | ||||||||||
In vitro characterization of Tyr-tRNAPhe editing determinants
Substitutions βD33A and βE124L in the B1/B2 domain and βN254A in B3/B4, all in an αA294G background, do not have any effect on editing activity with or without addition of tRNA (data not shown). However, B3/B4 domain substitutions at the entrance to the proposed editing site (βT354W, βA356W) and within the binding pocket itself (βH265A or L, βE334A), all resulted in significant losses in editing activity (Figure 5A and Table II) and increased Tyr-tRNAPhe synthesis (Figure 5B). The variants had essentially wild-type phenylalanylation efficiency (kcat/KM, Table II), indicating that the editing function in the B3/B4 domain of the β subunit is independent of the aminoacylation activity in the α subunit. The ability of isolated wild-type β subunits to hydrolyze Tyr-tRNAPhe was also investigated in vitro, and modest editing activity was detected (Figure 6A). Control aminoacylation assays indicated that the observed editing activity of isolated β subunits could not be attributed to trace contaminants of native PheRS (Figure 6B). These findings indicate that the editing site resides in the B3/B4 domain of the β subunit, and suggest that the corresponding activity is only fully realized in the context of the native enzyme containing the α subunit.
Figure 5.
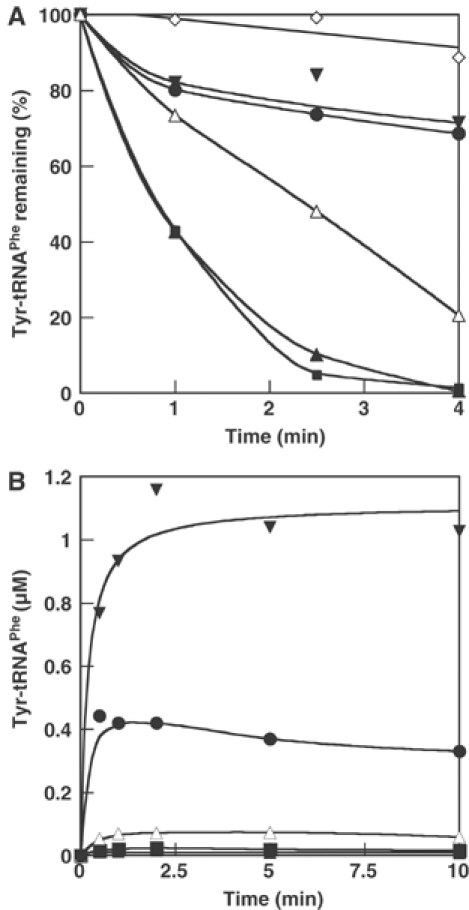
In vitro phenotypes of editing defective PheRS. (A) Hydrolysis of Tyr-tRNAPhe (1 μM) by PheRS (2 nM). (B) Aminoacylation of tRNAPhe (2.7 μM) with Tyr (30 μM) by PheRS (250 nM). Wild-type PheRS (▴), αA294G PheRS (▪), αA294G-βH265A (▵), αA294G-βE334A (•), αA294G-βA356W (▾), no enzyme (◊).
Figure 6.
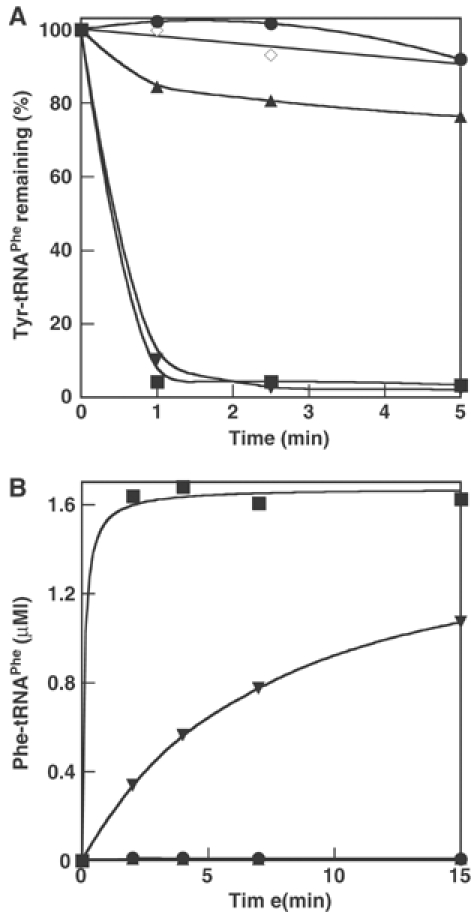
In vitro phenotypes of individual PheRS subunits. (A) Hydrolysis of Tyr-tRNAPhe (0.5 μM) by PheRS (12 nM). (B) Aminoacylation of tRNAPhe (2.4 μM) with Phe (20 μM) by PheRS (50 nM). Native αA294G/wild-type β subunit PheRS (▪), reconstituted αA294G/wild-type β subunit PheRS (▾), A294G α subunit (•), wild-type β subunit (▴), no enzyme (◊).
Editing-defective PheRS retards E. coli growth
The growth of E. coli XL-1 blue cells transformed with plasmids encoding PheRSαA294G and PheRSαA294G/βA356W was compared on various media before and after induction of expression of the mutant pheST genes (encoding PheRS). On complex medium (LB), there was no significant difference in growth of the strain encoding PheRSαA294G before or after induction of expression, while production of PheRSαA294G/βA356W slowed growth (data not shown). To investigate whether this reflected a general defect or a specific response to certain Tyr:Phe ratios, growth was investigated on minimal medium supplemented either with a complete set of L-amino acids or with a mixture lacking Phe but enriched in Tyr (supplemented M9, see Materials and methods for details). Upon induction, no change was seen for the control, while growth of the strain producing PheRSαA294G/βA356W slowed significantly on both media (Figure 4C and D). These results indicate that Tyr-tRNAPhe editing is required for optimal growth, consistent with previous reports of defective growth associated with disruptions in aaRS editing (Hendrickson et al, 2002; Nangle et al, 2002).
Specificity of editing towards Phe analogues
To explore the specificity of the proofreading activity of PheRS, several Phe analogues with an intact α-COOH group were tested for editing. These included intermediates of aromatic amino-acid metabolism (phenylpyruvate, p-hydroxy-phenylpyruvate and prephenate), D-Phe and several unnatural aa known to be activated by PheRSαA294G (p-fluoro, -chloro, -bromo and amino-Phe, benzofuranylalanine and triazole; Ibba et al, 1994; Bentin et al, 2004). Neither the metabolic intermediates nor D-Phe stimulated ATP hydrolysis to any significant degree, while the unnatural amino acids tested were all attached to tRNAPhe but not edited. These data indicate that while numerous noncognate amino acids are substrates for PheRS, the editing activity of the enzyme is specific for Tyr.
Discussion
Post-transfer editing by PheRS
Conservation mapping suggested that the editing site of PheRS was localized 40 Å from the active site on the surface of the B3/B4 domain of the β subunit of PheRS (positions 211–396 of the β subunit in the T. thermophilus PheRS 3-D structure (Mosyak et al, 1995)). This idea was supported by the observation that substitutions of His 261, Glu 334 or Ala 356 in the β subunit of PheRS, all impaired post-transfer editing of Tyr-tRNAPhe but did not affect Phe-tRNAPhe synthesis. The observation that tRNAs with modified termini could accept Phe but no longer accepted Tyr or enhanced editing provided further indirect evidence for post-transfer editing by PheRS. While these findings indicate the presence of a post-transfer mechanism in PheRS, the relative contribution of pre-transfer editing requires further clarification. Attempts to address this question in other systems have proven difficult and the contribution of the pre-transfer mechanism to synthetase editing in general remains controversial. The isolation here of both tRNAs and proteins with specific editing defects will allow renewed mechanistic studies, which should provide insights into the relative importance of the pre- and post-transfer routes.
Model of the bacterial PheRS editing site
All residues that when replaced yielded enzymes defective in editing are located inside, or in the close vicinity of, a pocket on the surface of the B3/B4 domain (see Figure 3). B4 is an inserted domain within the larger structural fragment B3, and the surface of the editing pocket is formed at the interface of these two subdomains. The large bifurcated editing pocket, observed in all available T. thermophilus PheRS structures, could accommodate C74 and C75 of the tRNA in its main cavity and A76 and the acylated Tyr in the two edges of the pocket. On the basis of these observations and our experimental assignment of roles in editing to certain residues, we used the structure of the T. thermophilus PheRS·tRNAPhe complex to build a model by manual fitting of the CCA-Tyr moiety into the B3/B4 pocket (Figure 7). Other parts of the complex were not modified and the proportion of side chains that contribute to pocket formation remained the same as in the native structure. The complete CCA-Tyr is accommodated simply by displacement of side chains, with a calculated RMS of 0.4 Å for the backbone of the B3/B4 domain before and after fitting. This model shows how Tyr could be specifically recognized by the editing site found in the eubacterial B3/B4 domain. Tyr is laid on the conserved motif ‘GVMGGxxS/T' (positions 315–322 in T. thermophilus PheRS) of the B4 domain and stacked by the highly conserved Phe 263 of B3. The α carbonyl and amino groups of Tyr interact with the well-conserved Glu334. The position of Tyr is maintained laterally by interactions with His 261 of the conserved motif ‘QPxHxFD' (residues 258–264) and the well-conserved hydrophobic residue Leu 311. The specific recognition of Tyr-tRNAPhe, preventing editing of Phe-tRNAPhe, might be achieved through interaction of the para-hydroxyl group of Tyr with the main chain carbonyl O of Leu 311 and/or the hydroxyl function of the conserved Ser/Thr residue in the ‘GVMGGxxS/T' motif.
Figure 7.
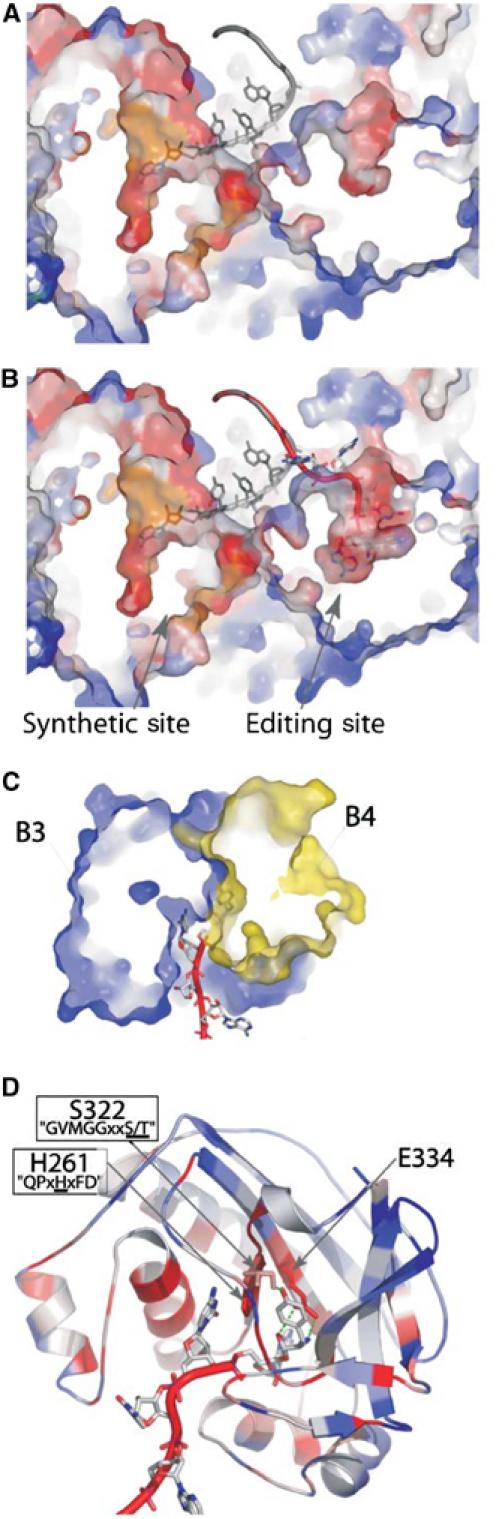
Model of the post-transfer editing site of PheRS. (A) Cross section of T. thermophilus PheRS in complex with tRNAPhe (1EIY). The CCA moiety of tRNAPhe (grey) is bound into the synthetic site of the α subunit of the enzyme. (B) Model for Tyr-tRNAPhe (red) binding in the editing site of the B3/B4 domain of the β subunit (see text, geometry of the model was optimized with DSviewer pro 5.0 (Accelrys)). tRNAPhe (grey) as found in the original structure is superimposed. (C) Cross section of the model of the B3/B4 domain bound to Tyr-tRNAPhe. The editing site is localized at the interface of domains B3 (in blue) and B4 (in yellow). (D) Ribbon representation of the model of the B3/B4 domain bound to Tyr-tRNAPhe. A76-Tyr is maintained between the two conserved motifs ‘GVMGGxxS/T' and ‘QPxHxFD'. Conserved residues in close contact with the Tyr moiety are displayed. Except for tRNAs and part (C), colors represent the percentage of identity for each position in an alignment of 103 eubacterial PheRSs (see Figure 3).
Evolution and retention of PheRS editing activity
Phylogenetic analyses of PheRS amino-acid sequence alignments showed two distinct groups, an archaeal/eukaryotic cluster (also including spirochete PheRSs) and a second eubacterial/mitochondrial grouping, which includes the E. coli protein investigated here (Woese et al, 2000; Brown, 2001). The B3/B4 domain residues that form the editing site are highly conserved in bacterial PheRSs, suggesting that editing function is ubiquitous in this group. Whether similar editing sites are also present in eukaryotic and archaeal PheRSs is not obvious, domains B3 and B4 of the β subunit being very different in sequence to the bacterial consensus. Nevertheless, the similar pattern of tRNAPhe CCA end binding in bacterial and eukaryotic PheRSs (Moor et al, 2003) suggests that the B3/B4 region of the enzyme would at least provide a good candidate for the location of an editing site in archaea and eukaryotes. The genomes of some archaea also encode a paralog of the α subunit of PheRS (Das and Vothknecht, 1999), raising the possibility of trans-editing of Tyr-tRNAPhe. Although this PheRS paralog is produced in vivo in Methanothermobacter thermautotrophicus, editing activity was not detected in vitro (H Roy and M Ibba, unpublished results).
An interesting anomaly is presented by the mitochondrial enzymes, which do not have the conventional (αβ)2 oligomeric form and are instead monomers (Sanni et al, 1991; Bullard et al, 1999). Mitochondrial PheRS is a chimera of the canonical α subunit with a C-terminal appendage containing the tRNA anticodon-binding domain (B8) of the canonical β subunit, but no putative editing domain. It was originally proposed that mitochondrial PheRSs are active in editing (Gabius et al, 1983), although later studies questioned the quality of the enzymes used in these studies (Sanni et al, 1991). Mitochondrial amino-acid pools are believed to be similar to those in the cytoplasm (Costantino and Attardi, 1973), suggesting that the need for editing mischarged Tyr would be similar in both compartments. Renewed investigations are now needed to determine whether mitochondrial PheRS possesses proofreading activity and, if this is the case, to localize the site responsible.
Can PheRS edit other noncognate aminoacyl-tRNAs?
Numerous synthetic Phe analogues are activated by PheRS and stably attached to tRNAPhe, both in vitro and in vivo (Ibba et al, 1994; Kirshenbaum et al, 2002). While many of these resemble Tyr in that they contain para-substitutions, none are substrates for editing. This implies that other substrates cannot establish interactions comparable to those proposed above between the editing site and the p-hydroxyl group of Tyr. The specificity of Tyr recognition over other analogues is further enhanced by the requirement that the editing site discriminates against Phe binding. While our present findings do not allow quantification of the specificity of the editing site in PheRS, comparisons to TyrRS provide some indication of the specificity necessary for Tyr discrimination. TyrRS specifically binds the p-hydroxyl group of tyrosine and is about 105 more specific for Tyr than for Phe (Fersht et al, 1980). Replacements in the active site of TyrRS can increase or decrease specificity approximately 10-fold (Fersht et al, 1985; de Prat Gay et al, 1993), resulting in a discrimination range of 104–106 while retaining binding and activity. If the editing site of PheRS also specifically binds the p-hydroxyl group of tyrosine, then it is reasonable to expect discrimination for Tyr over Phe of at least 104. Such a level of specificity has two important functional implications for the editing activity of PheRS. Firstly, it ensures that there is virtually no hydrolysis of cognate Phe-tRNAPhe. Secondly, it suggests that if PheRS is able to edit noncognate amino acids other than those already tested here, they will likely not be aromatic. This last point is supported by the observation that engineered enlargement of the active site of PheRS allows Trp (but not Tyr) misincorporation at Phe codons under certain starvation conditions, suggesting that Trp-tRNAPhe is synthesized but not edited (Datta et al, 2002).
Tyr-tRNAPhe editing contributes to quality control during protein synthesis
With wild-type PheRS no β-galactosidase activity was detected in E. coli CC503, but when a PheRS variant was introduced, which mischarged tRNACUAPhe with tyrosine but failed to correct the mistake, functional β-galactosidase was made. This clearly demonstrates that without editing an enzyme can result with an activity different from that genetically encoded. While Phe to Tyr is generically considered to be a conservative replacement, the example described here shows a dramatic change. In other cases less desirable functional changes are associated with noncoded Phe to Tyr changes, such as the introduction of an unwanted phosphorylation site.
Our findings suggest that steps in protein synthesis following aminoacylation do not always provide effective points of quality control for noncognate aminoacyl-tRNAs. The first possible point of quality control is deacylation of Tyr-tRNAPhe by the chromosomally encoded wild-type PheRS acting in trans, the same reaction as we followed in vitro. The ineffectiveness of this reaction in vivo probably reflects the rapid sequestration of aminoacyl-tRNAPhe by elongation factor Tu (EF-Tu), which binds these molecules several orders of magnitude more tightly than does PheRS (Nazarenko et al, 1994; Ibba et al, 1995). This also suggests that, in other studies where trans-editing of noncognate aminoacyl-tRNAs has been proposed (Ahel et al, 2003; Wong et al, 2003; Korencic et al, 2004), the success of such a mechanism may require substrate channeling between the charging and editing proteins (Stapulionis and Deutscher, 1995). Given that EF-Tu does outcompete PheRS for Tyr-tRNAPhe, the elongation factor apparently does not discriminate against this mischarged tRNA, as it does in other cases (Stanzel et al, 1994; Becker and Kern, 1998; LaRiviere et al, 2001). The final step at which Tyr-tRNAPhe might be discriminated is on the ribosome (Fahlman and Uhlenbeck, 2004), which does not appear to be the case here. Overall, the ineffectual discrimination of Tyr-tRNAPhe after its release emphasizes the critical role of PheRS editing in maintaining quality control during protein synthesis at Phe codons.
Materials and methods
Strains, plasmids and general methods
E. coli XL1-Blue/pQE31-FRS producing His6-tagged E. coli PheRS was a gift from DA Tirrell. E. coli BL21/pQE30 producing His6-tagged E. coli terminal tRNA nucleotidyl transferase was a gift from T Ueda (Tomari et al, 2000), and E. coli CC503 a gift from CG Cupples (Nghiem et al, 1988). All His6-tagged proteins were purified on Ni-NTA agarose (Qiagen) by standard procedures. Point mutations were introduced into the pheST gene (encoding PheRS) by PCR with two self-complementary 33-mer oligonucleotides that carried the appropriate mutations. The reaction was performed with the Quickchange Site-Directed Mutagenesis Kit (Stratagene). Introduction of the desired mutations was monitored by sequencing of the resulting genes. Commercial L-Tyr (Sigma) was shown to be free of Phe contamination by pyrophosphate exchange before and after re-crystallization of the amino acid as described previously (Lin et al, 1983). LB and M9 media were prepared as described (Atlas, 1993).
Preparation of in vitro transcribed tRNAPhe
In vitro T7 RNA polymerase runoff transcription reactions were conducted according to standard procedures (Sampson and Uhlenbeck, 1988). tRNA transcript was purified on a denaturing 12% polyacrylamide gel and recovered by passive elution in 0.5 M ammonium acetate, 10 mM MgCl2, 1 mM EDTA and 0.1% SDS for 12 h at 4°C under agitation. tRNA was phenol and chloroform extracted, ethanol precipitated, resuspended in 2 mM MgCl2 and finally re-folded by incubation for 1 min at 75°C, followed by slow cooling (2°C/min) to 25°C.
Preparation of tRNAPhe 2′-deoxy-, 3′-deoxy- and dideoxyadenosine 76
A runoff transcript of tRNAPhe CC75 deprived of 3′-adenosine was prepared (see above) and the addition of the 76th nucleotide was made by using the terminal tRNA nucleotidyl transferase from E. coli under the following conditions. The reaction media contained 50 mM Tris-glycine buffer (pH 9.0), 10 mM MgCl2, 7 mM β-mercaptoethanol, 0.1 μM of E. coli terminal tRNA nucleotidyl transferase and 3 μM of tRNACC75 transcript in the presence of 2 mM ATP, dATP, cordycepine triphosphate or ddATP. After 1 h of incubation at 37°C, the reaction was applied to a G25 gel filtration column and the eluted tRNA was then phenol and chloroform extracted, precipitated and finally resuspended in water prior to re-folding (see above). Trace contamination with full-length native tRNAPhe transcript was removed by oxidation using a solution of 6.5 μM tRNA with 3.3 mM sodium periodate. After 30 min incubation at 20°C in the dark, the tRNA was ethanol precipitated, washed and resuspended in water. The addition of the modified 3′-terminal adenosine was checked by electrophoresis on a 6% acrylamide gel containing 8 M urea in TBE buffer and the Phe acceptor capacity was checked by phenylalanylation with PheRS (see below).
Preparation of aa-tRNAPhe
The aminoacylation mix contained 100 mM Na-Hepes (pH 7.2), 30 mM KCl, 2 mM ATP, 10 mM MgCl2, 15 μM L-[14C]Phe (280 cpm/pmol) or 30 μM [3H]Tyr (160 cpm/pmol) and 5 μM tRNA transcripts. Phenylalanylation of transcript was obtained with 0.5 μM of E. coli PheRSαA294G and tyrosylation with the same amount of PheRSαA294G-βA356W. After 4 min of incubation at 37°C, the reaction was stopped by addition of 60 mM potassium acetate (pH 4.5) and 250 mM KCl. The aa-tRNA was extracted with acid-buffered phenol (60 mM sodium acetate (pH 4.5)), followed by chloroform extraction and ethanol precipitation. The aa-tRNA pellet was dried, resuspended in water and stored at −20°C. Aminoacyl-tRNA concentrations in such preparations were determined prior to use by precipitation with 5% TCA on 3MM filter disks, followed by extensive washing in 5% TCA to remove unbound radioactive amino acid, drying and scintillation counting. Comparison to the total tRNA concentration then allowed us to estimate that Phe-tRNAPhe yields were about 50% (i.e. these preparations also contained 50% uncharged tRNA), while Tyr-tRNAPhe yields were 35% (65% uncharged), within the typical range expected for aminoacyl-tRNA preparations (H Roy, unpublished results).
Preparation of isolated PheRS β subunits
The E. coli PheRS operon was amplified by PCR using the plasmid pQE31-PheRS as template, and cloned between the NdeI and XhoI sites of the vector pTYB2 (New England Biolabs). This construct was used to transform E. coli BL21 (Stratagene), yielding a strain able to overproduce recombinant PheRS carrying a His6 tag at the N-terminus of the α subunit and a chitin-binding intein tag at the C-terminus of the β subunit. This strain was grown in LB medium (200 mg/l ampicillin) at 37°C, and then expression of the recombinant protein was induced by incubation at room temperature for 6 h with 0.5 mM IPTG. The protocol used hereafter to purify each PheRS subunit relies on their ability to reversibly dissociate in the presence of 0.5 M NaSCN (Ducruix et al, 1983). The cells were disrupted by sonication in a lysis buffer containing 20 mM Tris–HCl (pH 8), 300 mM NaCl, 0.5 M NaSCN and 5 mM imidazole–HCl (pH 8.0) and subsequently centrifuged at 75 000 g (4°C). The resulting supernatant was applied to a column containing BD TALON affinity resin (BD Bioscience) connected in series with a column containing chitin beads (New England Biolabs). The extract was applied and washed extensively with the lysis buffer. The α subunit was first eluted from the TALON resin with the lysis buffer supplemented with 300 mM of imidazole and the β subunit was subsequently eluted from the Chitin beads by washing and incubation overnight in the lysis buffer containing 50 mM of β-mercaptoethanol. The fractions containing each subunit were separately pooled and exhaustively dialyzed against 20 mM Tris–HCl (pH 7.5), 150 mM KCl and 5 mM β-mercaptoethanol and subsequently stored at −20°C in the same buffer with 50% glycerol. The cross-contamination of each subunit preparation was assayed by aminoacylation and reconstitution of the active enzyme was achieved upon mixing of both subunit preparations as described previously (Ducruix et al, 1983).
Steady-state aminoacylation kinetics
Kinetics parameters (kcat and KM) for L-Phe were determined using the conditions described above for aa-tRNA synthesis, in the presence of a saturating concentration of tRNA (4 mg/ml of total tRNA from E. coli MRE 600, Roche), a range of 2–50 μM of L-[14C]Phe (280 cpm/pmol) and 2 nM PheRS. The choice of total tRNA was based upon previous studies by us and others (Peterson and Uhlenbeck, 1992; Ibba et al, 1994) that showed only modest differences in the steady-state kinetic parameter kcat when comparing total tRNA, purified tRNAPhe, and in vitro transcribed tRNAPhe. Kinetics parameters were deduced by nonlinear regression of the Michaelis–Menten curve to the experimental data. The displayed values of kcat and KM are the means of at least two independent experiments that showed a standard deviation inferior to 15%.
Post-transfer editing assay
Reaction mixtures contained 1 μM of [14C]- or [3H]-labeled aa bound to tRNA, a catalytic amount of PheRS (2 nM), 100 mM Na-Hepes (pH 7.2), 30 mM KCl and 10 mM MgCl2. The mixture was incubated at 37°C and the post-transfer editing reaction followed by measuring the remnant radiolabeled aa-tRNA in aliquots of 15 μl after 0–10 min of incubation as described previously (Kern et al, 1977).
ATP consumption assay
Aa editing was measured as ATP consumption catalyzed by PheRS in the presence or absence of 2 μM of tRNAPhe. A 15 μl reaction contained 2 mM aa, 2 mM [γ-32P]ATP (5 cpm/pmol), 100 mM Hepes-Na (pH 7.2), 30 mM KCl, 10 mM MgCl2 and 2 U/ml of yeast pyrophosphatase (Roche). The reaction was conducted at 37°C and was initiated by addition of PheRS to 2 μM. At variable times, ranging from 0 to 40 min, the reaction was quenched by mixing 2 μl of sample with 2 μl of glacial acetic acid. The remaining [γ-32P]ATP and the [γ-32P]Pi formed during the reaction were separated by TLC on a PEI cellulose plate (Sigma) prewashed with water. The TLC was subsequently developed in 0.7 M potassium phosphate (pH 3.5) and the labeled products were visualized and quantified on a Storm phosphorimager (Amersham Biosciences). The concentration of Pi (mM) formed during the time-course reaction was calculated by multiplying the measured Pi/ATP ratio by the initial concentration of ATP (2 mM).
Assay for Tyr-tRNAPhe synthesis in vivo
In order to express the amber suppressor tRNAPhe (tRNAPheCUA) and pheST mutants in CC503, we cloned the tRNAPheCUA-encoding gene into pKK223 (Amersham Biosciences). The tac promoter, the tRNAPhe gene and the transcription terminator rrnB were amplified by PCR and subcloned into the vector pSU2719 (Martinez et al, 1988), yielding pSU-tRNAPheCUA. CC503 was transformed by the vectors pSU-tRNAPhe(CUA) and pQE31-PheRS and spread on LB media plates supplemented with 200 mg/l of ampicillin and 30 mg/l of chloramphenicol. Transformants were subsequently spread on M9 media plates supplemented with antibiotics, 1.5 mM IPTG, 60 mg/ml of 5-bromo-4-chloro-3-indolyl-galactopyrano-side (X-Gal) and 0.2% (w/v) glucose or 1% (w/v) lactose as unique carbon source. The clones were then grown for 60 h at 37°C.
Determination of growth phenotypes
E. coli XL-1 blue cells transformed with pQE-FRS-PheRSαA294G or pQE-FRS-PheRSαA294G/βA356W were grown aerobically in LB medium (200 mg/l ampicillin) overnight at 37°C, and 1 ml was then used to inoculate 50 ml of either LB (200 mg/l ampicillin) or M9 medium supplemented with glucose (2 g/l), thiamine (1 mg/l), MgSO4 (1 mM), CaCl2 (0.1 mM), 20 amino acids (40 mg/l; when Phe was omitted, Tyr was increased to 0.2 g/l) and ampicillin (200 mg/l). Cultures were then grown aerobically at 37°C and their growth monitored spectrophotometrically as absorbance at 595 nm. When the absorbance reached approximately 0.3, IPTG (1 mM) was added and monitoring of growth was then continued.
Acknowledgments
We thank K Fredrick and J Reeve for critical reading of the manuscript, and C Cupples, D Tirrell and T Ueda for gifts of strains and plasmids. This work was supported by grant MCB-0344002 from the National Science Foundation.
References
- Ahel I, Korencic D, Ibba M, Söll D (2003) Trans-editing of mischarged tRNAs. Proc Natl Acad Sci USA 100: 15422–15427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas RM (1993) Handbook of Microbiological Media. Boca Raton: CRC Press [Google Scholar]
- Becker HD, Kern D (1998) Thermus thermophilus: a link in evolution of the tRNA-dependent amino acid amidation pathways. Proc Natl Acad Sci USA 95: 12832–12837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe K, Merriman E, Ribas De Pouplana L, Schimmel P (2004) A domain for editing by an archaebacterial tRNA synthetase. Proc Natl Acad Sci USA 101: 5958–5963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe K, Ribas De Pouplana L, Schimmel P (2003) Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J 22: 668–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentin T, Hamzavi R, Salomonsson J, Roy H, Ibba M, Nielsen PE (2004) Photoreactive bicyclic amino acids as substrates for mutant Escherichia coli phenylalanyl-tRNA synthetases. J Biol Chem 279: 19839–19845 [DOI] [PubMed] [Google Scholar]
- Beuning PJ, Musier-Forsyth K (2000) Hydrolytic editing by a class II aminoacyl-tRNA synthetase. Proc Natl Acad Sci USA 97: 8916–8920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR (2001) Genomic and phylogenetic perspectives on the evolution of prokaryotes. Syst Biol 50: 497–512 [DOI] [PubMed] [Google Scholar]
- Bullard JM, Cai YC, Demeler B, Spremulli LL (1999) Expression and characterization of a human mitochondrial phenylalanyl-tRNA synthetase. J Mol Biol 288: 567–577 [DOI] [PubMed] [Google Scholar]
- Costantino P, Attardi G (1973) Atypical pattern of utilization of amino acids for mitochondrial protein synthesis in HeLa cells. Proc Natl Acad Sci USA 70: 1490–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupples CG, Miller JH (1988) Effects of amino acid substitutions at the active site in Escherichia coli beta-galactosidase. Genetics 120: 637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Vothknecht UC (1999) Phenylalanyl-tRNA synthetase from the archaeon Methanobacterium thermoautotrophicum is an (alphabeta)2 heterotetrameric protein. Biochimie 81: 1037–1039 [DOI] [PubMed] [Google Scholar]
- Datta D, Wang P, Carrico IS, Mayo SL, Tirrell DA (2002) A designed phenylalanyl-tRNA synthetase variant allows efficient in vivo incorporation of aryl ketone functionality into proteins. J Am Chem Soc 124: 5652–5653 [DOI] [PubMed] [Google Scholar]
- de Prat Gay G, Duckworth HW, Fersht AR (1993) Modification of the amino acid specificity of tyrosyl-tRNA synthetase by protein engineering. FEBS Lett 318: 167–171 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL molecular graphics system http://www.pymol.org
- Dock-Bregeon A, Sankaranarayanan R, Romby P, Caillet J, Springer M, Rees B, Francklyn CS, Ehresmann C, Moras D (2000) Transfer RNA-mediated editing in threonyl-tRNA synthetase. The class II solution to the double discrimination problem. Cell 103: 877–884 [DOI] [PubMed] [Google Scholar]
- Ducruix A, Hounwanou N, Reinbolt J, Boulanger Y, Blanquet S (1983) Purification and reversible subunit dissociation of overproduced Escherichia coli phenylalanyl-tRNA synthetase. Biochim Biophys Acta 741: 244–250 [DOI] [PubMed] [Google Scholar]
- Ebel JP, Giegé R, Bonnet J, Kern D, Befort N, Bollack C, Fasiolo F, Gangloff J, Dirheimer G (1973) Factors determining the specificity of the tRNA aminoacylation reaction. Non-absolute specificity of tRNA-aminoacyl-tRNA synthetase recognition and particular importance of the maximal velocity. Biochimie 55: 547–557 [DOI] [PubMed] [Google Scholar]
- Fahlman RP, Uhlenbeck OC (2004) Contribution of the esterified amino acid to the binding of aminoacylated tRNAs to the ribosomal P- and A-sites. Biochemistry 43: 7575–7583 [DOI] [PubMed] [Google Scholar]
- Ferri-Fioni ML, Schmitt E, Soutourina J, Plateau P, Mechulam Y, Blanquet S (2001) Structure of crystalline D-Tyr-tRNATyr deacylase: a representative of a new class of tRNA-dependent hydrolase. J Biol Chem 276: 47285–47290 [DOI] [PubMed] [Google Scholar]
- Fersht AR (1979) Editing mechanisms in the aminoacylation of tRNA. In Transfer RNA: Structure, Properties and Recognition, Schimmel PR, Söll D and Abelson JN (eds), pp 247–254. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, Blow DM, Brick P, Carter P, Waye MM, Winter G (1985) Hydrogen bonding and biological specificity analysed by protein engineering. Nature 314: 235–238 [DOI] [PubMed] [Google Scholar]
- Fersht AR, Shindler JS, Tsui WC (1980) Probing the limits of protein-amino acid side chain recognition with the aminoacyl-tRNA synthetases. Discrimination against phenylalanine by tyrosyl-tRNA synthetases. Biochemistry 19: 5520–5524 [DOI] [PubMed] [Google Scholar]
- Fukai S, Nureki O, Sekine S, Shimada A, Tao J, Vassylyev DG, Yokoyama S (2000) Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell 103: 793–803 [DOI] [PubMed] [Google Scholar]
- Gabius HJ, Engelhardt R, Schroder FR, Cramer F (1983) Evolutionary aspects of accuracy of phenylalanyl-tRNA synthetase. Accuracy of fungal and animal mitochondrial enzymes and their relationship to their cytoplasmic counterparts and a prokaryotic enzyme. Biochemistry 22: 5306–5315 [DOI] [PubMed] [Google Scholar]
- Goldgur Y, Mosyak L, Reshetnikova L, Ankilova V, Lavrik O, Khodyreva S, Safro M (1997) The crystal structure of phenylalanyl-tRNA synthetase from Thermus thermophilus complexed with cognate tRNAPhe. Structure 5: 59–68 [DOI] [PubMed] [Google Scholar]
- Hendrickson TL, Nomanbhoy TK, De Crécy-Lagard V, Fukai S, Nureki O, Yokoyama S, Schimmel P (2002) Mutational separation of two pathways for editing by a class I tRNA synthetase. Mol Cell 9: 353–362 [DOI] [PubMed] [Google Scholar]
- Hendrickson TL, Schimmel P (2003) Transfer RNA-dependent amino acid discrimination by aminoacyl-tRNA synthetases. In Translation Mechanisms, Lapointe J and Brakier-Gingras L (eds), pp 34–64. Dordrecht: Kluwer Academic/Plenum Publishers [Google Scholar]
- Ibba M, Johnson CM, Hennecke H, Fersht AR (1995) Increased rates of tRNA charging through modification of the enzyme–aminoacyl-adenylate complex of phenylalanyl-tRNA synthetase. FEBS Lett 358: 293–296 [DOI] [PubMed] [Google Scholar]
- Ibba M, Kast P, Hennecke H (1994) Substrate specificity is determined by amino acid binding pocket size in Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry 33: 7107–7112 [DOI] [PubMed] [Google Scholar]
- Jakubowski H (2004) Accuracy of aminoacyl-tRNA synthetases: proofreading of amino acids. In Aminoacyl-tRNA Synthetases, Ibba M, Francklyn CS, Cusack S (eds), pp 384–396. George town, Texas: Landes Bioscience [Google Scholar]
- Jakubowski H, Goldman E (1992) Editing of errors in selection of amino acids for protein synthesis. Microbiol Rev 56: 412–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan W, Littlejohn TG (2001) Swiss-PDB viewer (deep view). Brief Bioinform 2: 195–197 [DOI] [PubMed] [Google Scholar]
- Kern D, Dietrich A, Fasiolo F, Renaud M, Giegé R, Ebel JP (1977) The yeast aminoacyl-tRNA synthetases. Methodology for their complete or partial purification and comparison of their relative activities under various extraction conditions. Biochimie 59: 453–462 [DOI] [PubMed] [Google Scholar]
- Kirshenbaum K, Carrico IS, Tirrell DA (2002) Biosynthesis of proteins incorporating a versatile set of phenylalanine analogues. Chembiochem 3: 235–237 [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14: 32–51 [DOI] [PubMed] [Google Scholar]
- Korencic D, Ahel I, Schelert J, Sacher M, Ruan B, Stathopoulos C, Blum P, Ibba M, Söll D (2004) A freestanding proofreading domain is required for protein synthesis quality control in Archaea. Proc Natl Acad Sci USA 101: 10260–10265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRiviere FJ, Wolfson AD, Uhlenbeck OC (2001) Uniform binding of aminoacyl-tRNAs to elongation factor Tu by thermodynamic compensation. Science 294: 165–168 [DOI] [PubMed] [Google Scholar]
- Lin SX, Baltzinger M, Remy P (1983) Fast kinetic study of yeast phenylalanyl-tRNA synthetase: an efficient discrimination between tyrosine and phenylalanine at the level of the aminoacyladenylate–enzyme complex. Biochemistry 22: 681–689 [DOI] [PubMed] [Google Scholar]
- Lin SX, Baltzinger M, Remy P (1984) Fast kinetic study of yeast phenylalanyl-tRNA synthetase: role of tRNAPhe in the discrimination between tyrosine and phenylalanine. Biochemistry 23: 4109–4116 [DOI] [PubMed] [Google Scholar]
- Lincecum TL, Tukalo M, Yaremchuk A, Mursinna RS, Williams AM, Sproat BS, Van Den Eynde W, Link A, Van Calenbergh S, Grøtli M, Martinis SA, Cusack S (2003) Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol Cell 11: 951–963 [DOI] [PubMed] [Google Scholar]
- Martinez E, Bartolome B, de la Cruz F (1988) pACYC184-derived cloning vectors containing the multiple cloning site and lacZ alpha reporter gene of pUC8/9 and pUC18/19 plasmids. Gene 68: 159–162 [DOI] [PubMed] [Google Scholar]
- Moor N, Lavrik O, Favre A, Safro M (2003) Prokaryotic and eukaryotic tetrameric phenylalanyl-tRNA synthetases display conservation of the binding mode of the tRNA(Phe) CCA end. Biochemistry 42: 10697–10708 [DOI] [PubMed] [Google Scholar]
- Mosyak L, Reshetnikova L, Goldgur Y, Delarue M, Safro MG (1995) Structure of phenylalanyl-tRNA synthetase from Thermus thermophilus. Nat Struct Biol 2: 537–547 [DOI] [PubMed] [Google Scholar]
- Nangle LA, De Crécy-Lagard V, Döring V, Schimmel P (2002) Genetic code ambiguity. Cell viability related to the severity of editing defects in mutant tRNA synthetases. J Biol Chem 277: 45729–45733 [DOI] [PubMed] [Google Scholar]
- Nazarenko IA, Harrington KM, Uhlenbeck OC (1994) Many of the conserved nucleotides of tRNA(Phe) are not essential for ternary complex formation and peptide elongation. EMBO J 13: 2464–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nghiem Y, Cabrera M, Cupples CG, Miller JH (1988) The mutY gene: a mutator locus in Escherichia coli that generates G.C–T.A transversions. Proc Natl Acad Sci USA 85: 2709–2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nureki O, Vassylyev DG, Tateno M, Shimada A, Nakama T, Fukai S, Konno M, Hendrickson TL, Schimmel P, Yokoyama S (1998) Enzyme structure with two catalytic sites for double-sieve selection of substrate. Science 280: 578–582 [DOI] [PubMed] [Google Scholar]
- Penner RM, Roth NJ, Rob B, Lay H, Huber RE (1999) Tyr-503 of beta-galactosidase (Escherichia coli) plays an important role in degalactosylation. Biochem Cell Biol 77: 229–236 [DOI] [PubMed] [Google Scholar]
- Peterson ET, Uhlenbeck OC (1992) Determination of recognition nucleotides for Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry 31: 10380–10389 [DOI] [PubMed] [Google Scholar]
- Pezo V, Metzgar D, Hendrickson TL, Waas WF, Hazebrouck S, Döring V, Marlière P, Schimmel P, De Crécy-Lagard V (2004) Artificially ambiguous genetic code confers growth yield advantage. Proc Natl Acad Sci USA 101: 8593–8597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JR, Uhlenbeck OC (1988) Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc Natl Acad Sci USA 85: 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan R, Dock-Bregeon AC, Romby P, Caillet J, Springer M, Rees B, Ehresmann C, Ehresmann B, Moras D (1999) The structure of threonyl-tRNA synthetase-tRNA(Thr) complex enlightens its repressor activity and reveals an essential zinc ion in the active site. Cell 97: 371–381 [DOI] [PubMed] [Google Scholar]
- Sanni A, Walter P, Boulanger Y, Ebel JP, Fasiolo F (1991) Evolution of aminoacyl-tRNA synthetase quaternary structure and activity: Saccharomyces cerevisiae mitochondrial phenylalanyl-tRNA synthetase. Proc Natl Acad Sci USA 88: 8387–8391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Furter R, Kast P, Tirrell DA (2000) Efficient introduction of aryl bromide functionality into proteins in vivo. FEBS Lett 467: 37–40 [DOI] [PubMed] [Google Scholar]
- Silvian LF, Wang J, Steitz TA (1999) Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science 285: 1074–1077 [PubMed] [Google Scholar]
- Sprinzl M, Cramer F (1975) Site of aminoacylation of tRNAs from Escherichia coli with respect to the 2′- or 3′-hydroxyl group of the terminal adenosine. Proc Natl Acad Sci USA 72: 3049–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanzel M, Schön A, Sprinzl M (1994) Discrimination against misacylated tRNA by chloroplast elongation factor Tu. Eur J Biochem 219: 435–439 [DOI] [PubMed] [Google Scholar]
- Stapulionis R, Deutscher MP (1995) A channeled tRNA cycle during mammalian protein synthesis. Proc Natl Acad Sci USA 92: 7158–7161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomari Y, Suzuki T, Watanabe K, Ueda T (2000) The role of tightly bound ATP in Escherichia coli tRNA nucleotidyltransferase. Genes Cells 5: 689–698 [DOI] [PubMed] [Google Scholar]
- Woese CR, Olsen GJ, Ibba M, Söll D (2000) Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev 64: 202–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong FC, Beuning PJ, Silvers C, Musier-Forsyth K (2003) An isolated class II aminoacyl-tRNA synthetase insertion domain is functional in amino acid editing. J Biol Chem 278: 52857–52864 [DOI] [PubMed] [Google Scholar]