

Abstract
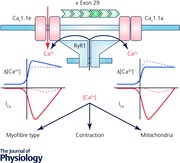
Voltage‐gated calcium channels represent the sole mechanism converting electrical signals of excitable cells into cellular functions such as contraction, secretion and gene regulation. Specific voltage‐sensing domains detect changes in membrane potential and control channel gating. Calcium ions entering through the channel function as second messengers regulating cell functions, with the exception of skeletal muscle, where CaV1.1 essentially does not function as a channel but activates calcium release from intracellular stores. It has long been known that calcium currents are dispensable for skeletal muscle contraction. However, the questions as to how and why the channel function of CaV1.1 is curtailed remained obscure until the recent discovery of a developmental CaV1.1 splice variant with normal channel functions. This discovery provided new means to study the molecular mechanisms regulating the channel gating and led to the understanding that in skeletal muscle, calcium currents need to be restricted to allow proper regulation of fibre type specification and to prevent mitochondrial damage.
Abbreviations
- CICR
calcium‐induced calcium release
- EC
excitation–contraction
- LTCC
L‐type calcium current
- RyR1
type 1 ryanodine receptor
- RyR2
type 2 ryanodine receptor
- SR
sarcoplasmic reticulum
- VSD
voltage sensing domain
Introduction
As indicated by their name, voltage‐gated calcium channels (CaVs) are capable of sensing the depolarization of membrane potentials and in response to this electrical stimulus open a calcium‐selective channel pore. Accordingly, their primary role is in conducting activity‐regulated calcium influx in excitable cells. However, there is one notable exception: the skeletal muscle calcium channel CaV1.1. This member of the CaV channel family has the same structure as all the other CaVs, with four voltage‐sensing domains (VSDs) grouped around a single ion pore, but its activation by skeletal muscle action potentials does not result in substantial calcium influx into muscle cells. Instead, its voltage‐dependent activation is directly transmitted to opening of the calcium release channel (type 1 ryanodine receptor; RyR1) in the sarcoplasmic reticulum (SR). Therefore, in skeletal muscle CaV1.1 primarily functions as a voltage sensor for excitation–contraction (EC) coupling, but not as a calcium channel (Melzer et al. 1995). Indeed, it has long been known that calcium influx through CaV1.1 is dispensable for EC coupling in skeletal muscles of most vertebrates (Armstrong et al. 1972), although during long‐lasting tetanic stimulation a small L‐type calcium current (LTCC) may contribute to refilling of the SR calcium stores and thus help to maintain muscle performance during prolonged activity (Robin & Allard, 2015). Nonetheless, it appears paradoxical that skeletal muscle contraction – the prototypical calcium regulated cell function – involves a member of the CaV family with limited channel function.
This raises the obvious question as to why the skeletal muscle CaV1.1 is such an ineffective calcium channel. Is there a physiological necessity for curtailing its channel function, or are its poor channel properties the result of an evolutionary accident that somehow remained without consequence because calcium release through the RyR1 compensated for the missing channel function? Recent findings by us and others indicate that curtailing voltage‐dependent calcium influx is important for skeletal muscle differentiation, function and health. In particular the discovery of a CaV1.1 splice variant, which functions as a sizeable channel, demonstrated that during skeletal muscle development calcium influx is actively turned off by alternative splicing of CaV1.1 (Tuluc et al. 2009). This raises a second question: How is the calcium current function perturbed without affecting the voltage‐sensing function of CaV1.1? Apparently CaV1.1 channels of different species utilize multiple mechanisms and even alternative strategies to curtail their currents. Importantly, recent structure–function studies on the two CaV1.1 splice variants revealed hitherto unnoticed intramolecular interactions, which are specific for the particular VSDs of CaV channels and provide control over the voltage sensitivity of the channel function without affecting EC coupling (Tuluc et al. 2015). Here we review the recent studies addressing the physiological importance and the biophysical mechanism of the unique gating properties of the skeletal muscle CaV1.1 calcium channel.
Structure and function of CaV1.1 in skeletal muscle
Voltage‐gated calcium channels are multi‐subunit complexes composed of a pore‐forming α1 subunit, an extracellular α2δ subunit, a cytoplasmic β subunit and, at least in skeletal muscle, another integral membrane protein, the γ subunit (Catterall, 2011) (Fig. 1). The mammalian genome contains 10 genes encoding α1 subunits and four each for α1δ and β subunits. The α1 subunit contains four homologous repeats (I–IV) with six transmembrane helixes (S1–S6) each. S1–S4 of each repeat form a voltage‐sensing domain (VSD); S5, S6 and the connecting P‐loops of all four repeats together form the channel pore with its activation gate. The cytoplasmic loop between repeats I and II contains the binding site for the β subunit and the long cytoplasmic C‐terminus contains important sites for current modulation, protein–protein interactions and channel targeting (Walker & De Waard, 1998; Striessnig, 2007). Unique for the skeletal muscle CaV1.1 complex are the presence of the γ1 subunit, the functional interaction of the cytoplasmic loop between repeats II and III with the RyR1, and the recently proposed interaction with the adapter protein STAC3 (Tanabe et al. 1990; Flucher et al. 2005; Horstick et al. 2013; Nelson et al. 2013). β subunits are involved in membrane targeting and endow the channels with specific gating properties (Buraei & Yang, 2010; Campiglio & Flucher, 2015). The α2δ subunits also contribute to normal membrane expression of the channels, but in muscle cells they are important determinants of the channel's specific activation kinetics (Obermair et al. 2005; Tuluc et al. 2007; Savalli et al. 2016). Whereas the α2δ‐1 and γ1 subunits are dispensable for skeletal muscle EC coupling, the β1 subunit and STAC3 are essential components of the skeletal muscle EC coupling apparatus (Campiglio & Flucher, 2015; Polster et al. 2016).
Figure 1. Structure of CaV1.1.
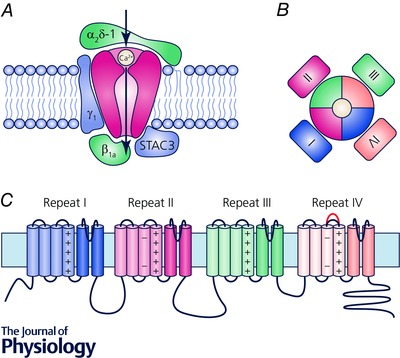
A, the subunit composition of CaV1.1 with the pore‐forming α1S subunit, the auxiliary α2δ‐1, β1a and γ1 subunits and the essential, skeletal muscle‐specific adaptor protein STAC3. B, the α1 subunit of CaV channels has a pseudotetrameric structure with a central pore surrounded by four voltage‐sensing domains (VSDs I–IV). C, the α1S subunit consists of four homologous repeats of six transmembrane helices. The first four helices (S1–S4) of each repeat form a functional VSD with the positively charged S4 helix representing the primary voltage sensor. Helices S5–S6 and the connecting pore loops together form the channel pore. Note that the extracellular S3–S4 loop of the fourth VSD is subject to alternative splicing. Inclusion of exon 29 in this loop (red) has massive consequences for channel gating properties.
In skeletal muscle these components of the calcium channel complex assemble with the RyR1 calcium release channel and several other proteins to form calcium release units (Flucher & Franzini‐Armstrong, 1996). Depending on whether they form junctions between the SR and T‐tubules or between the SR and the plasma membrane, the calcium release units are called triads or peripheral couplings, respectively, although functionally they are equivalent. The specific and probably physical interaction of four CaV1.1 channels with a RyR1 homotetramer is reflected in the regular arrangement of CaV1.1. In freeze‐fracture electron microscopy these can be seen as tetrads of integral membrane particles (Block et al. 1988). This coordinated organization of the two calcium channels is considered to be the structural prerequisite for skeletal muscle EC coupling.
Upon depolarization of the plasma membrane and T‐tubules CaV1.1 senses the change in membrane potential and activates opening of the RyR1 release channel (Ríos & Pizarro, 1991). The exact mode of interaction between the two channels is still elusive. However, it is clear that the specific skeletal muscle sequences of the CaV1.1 II–III loop and the specific skeletal muscle isoform β1a are both essential for the signalling process (Grabner et al. 1999; Schredelseker et al. 2005; Sheridan et al. 2006). Perhaps the recently discovered STAC3 is also involved in the communication of the two channels (Horstick et al. 2013; Nelson et al. 2013; Polster et al. 2016). The functional coupling of CaV1.1 and RyR1 is extremely efficient so that in response to an action potential SR calcium release is activated within milliseconds and repetitive RyR1 activation can be maintained at frequencies of several hundred hertz, resulting in tetanic contractions (Rome, 2006). As mentioned above this process is independent of calcium influx though the CaV1.1 channel.
Upon unphysiologically strong and long depolarizations, CaV1.1a also produces slowly activating LTCC. Importantly, current activation requires up to 30 mV stronger depolarizations than activation of EC coupling by the same CaV1.1 (Tuluc et al. 2009). Thus, the voltage sensors of CaV1.1 trigger two distinct processes at different membrane potentials. In dysgenic (CaV1.1‐null) myotubes reconstituted with only the classical CaV1.1a isoform, half‐maximal activation (V ½) of SR calcium release occurs near +10 mV whereas half‐maximal activation of LTCC requires depolarization to approximately +40 mV (Tuluc et al. 2009). Depending on the cell type and experimental conditions, the absolute V ½ values vary considerably. For example in freshly isolated mouse muscle fibres, V ½ of gating charge movement (Q on) and current activation have been reported as low as –37 mV and 0 mV, respectively (Collet et al. 2003). Nevertheless, the pronounced difference in the voltage dependence of Q on or SR calcium release on the one hand and of LTCCs on the other hand is seen in mature fibres and in myotubes solely expressing CaV1.1a. In primary cultures and C2C12 myotubes this difference is less pronounced, probably because of a significant contribution of the developmental CaV1.1e splice variant (Garcia et al. 1994; Schuhmeier & Melzer, 2004). These, current gating properties make CaV1.1a the slowest activating and the least responsive to physiological depolarizations of all voltage‐gated calcium channels.
Curtailed calcium currents – necessary condition or accidental byproduct of skeletal muscle EC coupling?
Considering the central role of activity‐dependent calcium signalling in the control of muscle contraction, it is unexpected and even counter‐intuitive that CaV1.1 calcium currents should not contribute to it. In fact, in the heart the closely related CaV1.2 channel contributes to cardiac muscle EC coupling in a twofold manner. Voltage‐dependent activation of CaV1.2 causes calcium influx into the nano‐domain of the calcium release units of cardiac myocytes (peripheral couplings), where it acts as trigger‐calcium to activate further calcium release through the type 2 ryanodine receptors (RyR2) by a mechanism termed calcium‐induced calcium release (CICR; Bers, 2008). But in addition to this trigger function, the calcium entering the cytoplasm through CaV1.2 substantially contributes to the calcium signal which regulates contraction. Thus, the magnitude of the LTCC directly and indirectly determines the force of contraction, and modulation of LTCC, for example by cAMP‐dependent phosphorylation of CaV1.2, is a central mechanism for regulating cardiac contraction. This example clearly demonstrates that LTCC can be useful in EC coupling.
Interestingly, this ‘cardiac’ EC coupling mechanism is also used by skeletal muscles of invertebrates and early chordates like amphioxus, indicating that in principle CICR is compatible with the requirements of skeletal muscle function (Schredelseker et al. 2010). This notion is further supported by experiments in myotubes of dysgenic (CaV1.1‐null) mice, where the lacking CaV1.1 was replaced with other members of the CaV1 family. For example when dysgenic myotubes were reconstituted with CaV1.2 or with CaV1.3, cardiac‐type EC coupling could be observed in skeletal myotubes (Kasielke et al. 2003; Tuluc et al. 2007). The only requirement was that the CaV channel needed to be colocalized with RyR1 in the triads. This indicated that expression of the skeletal muscle RyR1 isoform does not necessitate a direct coupling mechanism with the CaV1.1 voltage sensor: it can also be activated by CICR, provided that the trigger calcium is supplied by a CaV channel targeted into the nano‐domain of the calcium release unit.
Thus, the direct coupling mechanism in skeletal muscle is not a necessity because CICR had been abolished, but direct coupling of CaV1.1 and RyR1 functionally replaced the CICR mechanism in skeletal muscle. Moreover, several lines of evidence support the view that suppressing calcium influx during skeletal muscle EC coupling was not only tolerable but even beneficial (Schredelseker et al. 2010). Different vertebrate species utilize distinct strategies to abolish or reduce LTCC through the skeletal muscle CaV1.1. In higher teleost fishes calcium conduction is completely abolished by amino acid substitution in the channel pore. Teleosts have two CaV1.1 genes due to gene duplication. Interestingly, both are non‐conductive, although this is accomplished by distinct amino acid substitutions in and near the calcium selectivity filter (Fig. 2). Moreover, non‐conductivity in one of the two fish CaV1.1 isoforms was achieved by different substitutions in the higher and lower teleost orders, exemplified by zebrafish and medaka, respectively (Schredelseker et al. 2010). Collectively, the use of three different amino acid substitutions to achieve non‐conductivity in both isoforms of all higher teleost is a strong indicator that curtailing calcium currents in skeletal muscle is of physiological importance.
Figure 2. CaV1.1 structures involved in curtailing calcium currents.
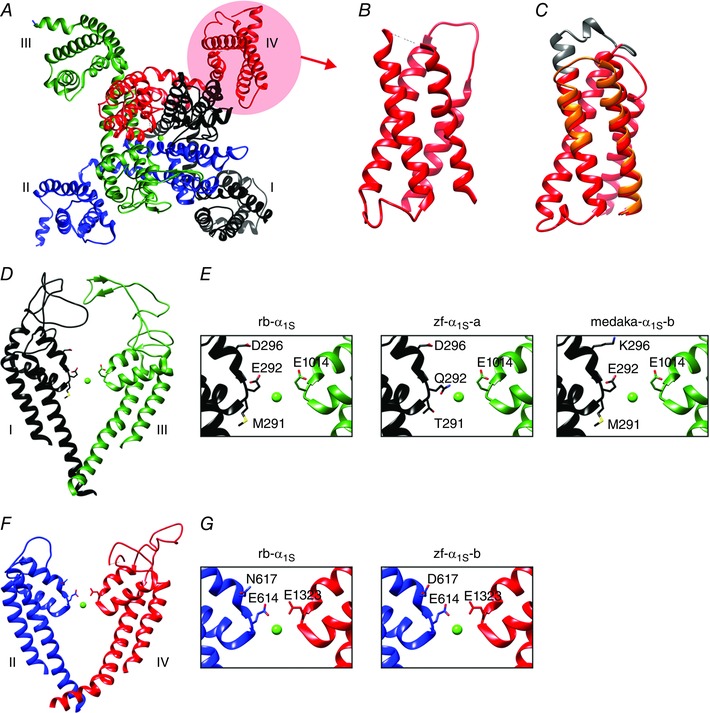
A, top view of the CaV1.1 channel with the four homologous repeats shown in different colours, using the high‐resolution cryo‐EM structure of Wu et al. (2015). B and C, side views of VSD IV models constructed of the high‐resolution cryo‐EM structure (B), and our Rosetta structure models (C), of the two splice variants with and without exon 29 (grey) (Tuluc et al. 2015). Note the overall similarity of the predicted and experimentally obtained structures. Inclusion of exon 29 slightly displaces IVS3 and IVS4 relative to each other, resulting in a channel which fails to activate during physiological depolarizations. D and F, side view structure of the channel pore. E and G, critical pore residues of the calcium selectivity filter of the rabbit channel (rb‐α1S) and the single amino acid substitutions which render the two fish CaV1.1 paralogues (zf‐α1S‐a, ‐b, medaka‐α1S‐b) non‐conductive (Schredelseker et al. 2010).
In tetrapods, CaV1.1 channels are principally calcium conductive, but under physiological conditions CaV1.1a currents are curtailed by other mechanisms. As mentioned above, upon long‐lasting depolarization mammalian CaV1.1a channels conduct LTCC. However, activation requires strong depolarization, activation is very slow and current amplitudes are small. These limiting channel properties are not the consequence of a single mutation in the pore or even a single molecular mechanism. In contrast – as if suppressing the currents were an essential goal to achieve – multiple mechanisms contribute to this effect. First of all, two distinct VSDs determine the slow activation and the right‐shifted voltage sensitivity of CaV1.1a. Sequence substitutions in VSD I affect the kinetic properties without influencing the voltage dependence of activation (Nakai et al. 1994; Tuluc et al. 2016). On the other hand, sequence substitutions and point mutations in VSD IV affect voltage dependence without influencing current kinetics (Tuluc et al. 2015, 2016; Fig. 3). Secondly, the auxiliary α2δ‐1 and γ subunits contribute to the poor current properties (Flucher et al. 2005; Andronache et al. 2007). Knock‐down and targeted deletion of α2δ‐1 in skeletal muscle cells accelerates activation kinetics of CaV1.1 but not of CaV1.2, showing that in skeletal muscle this auxiliary subunit is an important determinant of the specific slow activation of LTCC (Obermair et al. 2005; Tuluc et al. 2007; Fuller‐Bicer et al. 2009). Knockout of the γ subunit causes a right‐shifted voltage dependence of inactivation, indicating that this auxiliary subunit limits the window current of LTCC in skeletal muscle (Ursu et al. 2004). The cooperation of these multiple mechanisms to achieve a single goal, suppression of LTCC, again supports the notion that curtailing calcium currents in skeletal muscle might be physiologically important.
Figure 3. Distinct contributions of the four VSDs to determining the specific gating properties of CaV1.1.
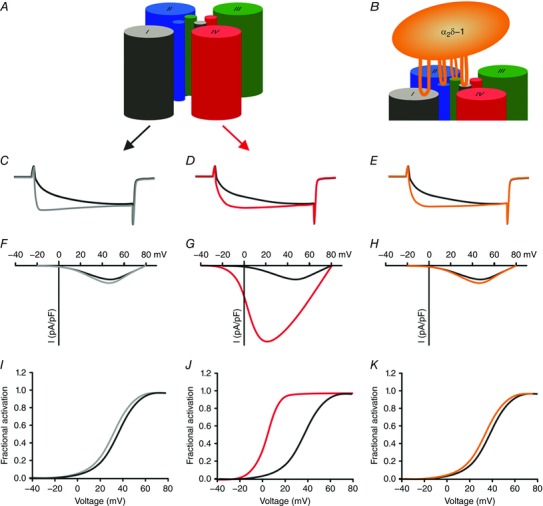
A and B, schematic models showing the role of VSD I in regulating activation kinetics (C) and its modulation by the α2δ‐1 subunit (B and E). Note that changes in current kinetics are not accompanied by changes in voltage sensitivity and current amplitude. D, G and J, VSD IV regulates the voltage dependence of activation (D and J) and current amplitude (G) by alternative splicing of exon 29. C–E, typical calcium currents of CaV1.1a (including exon 29; black), CaV1.1e (lacking exon 29; red), CaV1.1 with IS3+IS3–S4 linker from CaV1.2 (grey), and CaV1.1a without α2δ−1 (orange). F–K, current–voltage relationship (F–H) and conductance–voltage relationships (I–K) of the currents shown above.
However, there is one notable exception to this trend. Skeletal muscle calcium currents are facilitated by specific interactions with the RyR1. In the absence of RyR1, for example in myotubes of dyspedic mice, CaV1.1 currents are very small (Nakai et al. 1996). Only CaV1.1's specific interaction with RyR1 substantially increases CaV1.1a current density to the still comparably small, but measurable normal levels. This would suggest that the currents are important after all; however, the physiological relevance of the restoration of LTCC by this retrograde coupling mechanism is still elusive (Flucher, 2016).
Molecular mechanism for limiting voltage sensitivity of CaV1.1 activation
As mentioned above the classical CaV1.1a isoform can independently activate two voltage‐dependent signalling processes. In dysgenic myotubes reconstituted with CaV1.1a, depolarization to about 0 to +10 mV rapidly activates RyR1‐mediated calcium release (EC coupling), whereas activation of calcium currents through its own channel pore (LTCC) requires approximately +30 mV stronger depolarizations and is extremely slow. How can this be? Our recent discovery of a CaV1.1 splice variant with different current activation properties set off a series of studies which together unravelled this puzzle (Tuluc et al. 2009).
The alternatively spliced exon 29 encodes a 19 amino acid sequence in the extracellular linker connecting the transmembrane helices IVS3 and IVS4. Before birth the majority of CaV1.1 transcripts lack exon 29. However, after birth expression of CaV1.1a is strongly increased and within 3 weeks the great majority of CaV1.1 transcripts include exon 29 (Sultana et al. 2016). Importantly, the developmental splice variant lacking exon 29 activates EC coupling and LTCC at the same voltage and its current amplitude is comparable to that of CaV1.2 (Tuluc et al. 2009). Thus, exclusion of exon 29 in the developmental CaV1.1e splice variant causes the –30 mV shift in voltage dependence of activation and the dramatic increase of current density, but without shifting the voltage sensitivity of gating charges or EC coupling (Tuluc et al. 2009; Fig. 3). Conversely, inclusion of exon 29 in VSD IV is sufficient to specifically right‐shift the voltage dependence of LTCCs, suggesting that activation of this VSD is required for activation of LTCC but not for EC coupling. Parallel analysis of the gating charge movement (Q ON) demonstrated that its voltage dependence coincided with that of EC coupling and was not significantly right‐shifted by the inclusion of exon 29. This indicated that, although voltage dependent, the conformational changes of VSD IV leading to the opening of the channel pore contributed little to the total charges moved upon CaV1.1a activation. Thus, membrane depolarization to about 0 mV causes the bulk movement of the CaV1.1a VSDs, which is sufficient to activate EC coupling. However additional depolarization is necessary to effect final transitions of VSD IV of CaV1.1a that remain below detectability in Q ON but are necessary for opening of the channel pore. Interestingly, when exon 29 is included in VSD IV the activated state of the channel appears to be less stable, as also the channel open probability is dramatically reduced compared to the CaV1.1e splice variant lacking exon 29 (Tuluc et al. 2009, 2015).
How does the 19 amino acid sequence of exon 29 affect these dramatic changes of the gating properties in VSD IV? One possibility, consistent with its position at the extracellular face of the channel, would be that it serves as a protein–protein interaction site for an extracellular modulator. A probable candidate would be the α2δ‐1 subunit, which is located extracellularly and modulates current kinetics in the same direction as inclusion of exon 29. However, siRNA depletion of α2δ‐1 equally affected activation kinetics in CaV1.1 channels with and without exon 29, demonstrating that this sequence is not necessary for the modulatory action of α2δ‐1 on CaV1.1 gating properties (Tuluc et al. 2009). This notion is further supported by the recent cryo‐EM structure of the CaV1.1 complex, according to which VSD IV does not participate in the interaction with α2δ‐1 (Wu et al. 2015), as well as by a very recent voltage‐clamp fluorometry study indicating that in CaV1.2 α2δ‐1 differentially modulates VSDs I, II and III, whereas VSD IV is not affected by the presence or absence of α2δ‐1 (Savalli et al. 2016).
Alternatively, intrinsic structural properties of the exon 29 sequence could alter the function of VSD IV. In fact de novo structure Rosetta modelling and secondary structure prediction tools suggested that inclusion of exon 29 not only alters the length of the linker between IVS3 and IVS4 but also endowed it with distinct structure (helix and/or β‐sheet) and hydrophobicity (Tuluc et al. 2015). Our models further predicted that inclusion of the exon 29 sequence would result in major structural rearrangements of this IVS3–S4 linker during the transitions from the closed state to the activated state. It is likely that any such structural features and dynamic rearrangements will increase the necessary activation energy and thus require stronger depolarization for the activation process. Indeed the substitution of two lysines for asparagines, which abolished secondary structure and reduced the hydrophobicity of the IVS3–S4 linker, caused a substantial left‐shift of the voltage dependence of activation of CaV1.1a (Tuluc et al. 2016). Thus, the structure and not merely the length of the IVS3–S4 linker is critical for its role in regulating the voltage sensitivity of VSD IV.
If the IVS3–S4 linker containing exon 29 by itself is able to right‐shift the voltage sensitivity of a VSD, this function should not be limited to VSD IV. However, inserting the IVS3–S4 linker into the corresponding position of VSD I of CaV1.1e failed to reproduce the right‐shifted voltage dependence of activation of CaV1.1a (Tuluc et al. 2016). Apparently exon 29 exerts its modulatory function in VSD IV but not in VSD I. Where is the difference? Sequence comparison of the two VSDs showed that in S4 the innermost positively charged amino acids (R4) are spaced differently (third position in IS4, fourth position in IVS4) and that IVS3 contains an additional negatively charged residue (aspartate 1196; D4) that is absent in IS3. Homology structure modelling of VSD IV with and without the exon 29 sequence in four different activation states predicted that D4 could act as additional countercharge for the two outermost arginines (R1 and R2) during the late voltage sensor transition (Tuluc et al. 2015). Importantly the structure model further predicted striking differences in the distance and number of bonds between these residues between CaV1.1a and CaV1.1e. In the intermediate state 2 and in the activated state the CaV1.1e splice variant lacking exon 29 had more and stronger (shorter distances) hydrogen bonds between D4 and R1 and R2 compared to CaV1.1a. Considering the biophysical properties of the two splice variants the models suggested that these interactions of oppositely charged residues may facilitate the transition of the voltage sensor into the activated state and its subsequent stabilization in the activated state (Fig. 4).
Figure 4. Model showing the molecular mechanism by which alternative splicing of exon 29 regulates the voltage dependence of activation and open probability of CaV1.1.
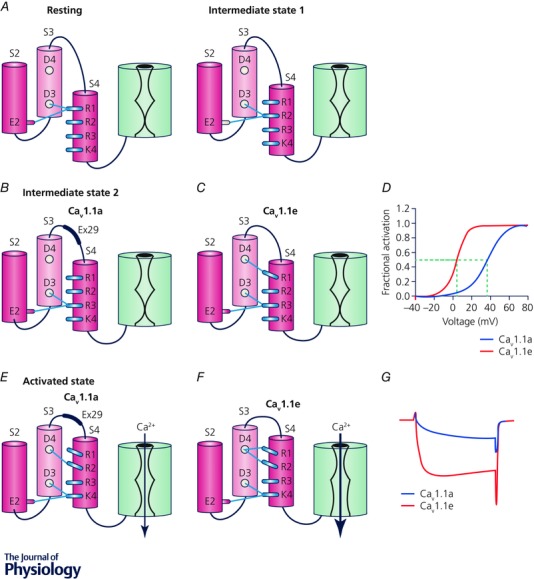
A, in the resting and intermediate state 1, R1 and R2, respectively, interact with two countercharges (glutamate E2 in IVS2 and aspartate D3 in IVS3) forming the charge transfer centre. Consequently there are no interactions of the IVS4 arginines with D4 of IVS3 and also no modulation by insertion/exclusion of exon 29 in the IVS3–S4 linker. B–D, in the intermediate state 2, R3 occupies the charge transfer centre and only in the absence of exon 29 (CaV1.1e) does R1 establish an interaction with D4 that facilitates the transition of the voltage sensor into the active state and therefore shifts the voltage dependence of CaV1.1e to the left. E and F, in the activated state, D4 of CaV1.1e forms several hydrogen bonds with both R1 and R2, thus stabilizing the activated state and increasing the open probability. B and E, inclusion of exon 29 in the IVS3–S4 linker of the adult splice variant CaV1.1a reduces the number and strength of the hydrogen bonds between D4 and R1 and R2. Consequently channel gating is severely perturbed and currents curtailed in the adult splice variant of the skeletal muscle calcium channel.
This model was tested and confirmed using site directed mutagenesis and patch‐clamp analysis of channels reconstituted in dysgenic myotubes (Tuluc et al. 2015). Mutations in CaV1.1e that abolished the charges of D4, R1, or R2 all resulted in right‐shifted voltage dependence and reduced amplitude, comparable to the voltage sensitivity of CaV1.1a. The same mutations in the presence of exon 29 (in CaV1.1a) did not result in a further shift of the voltage dependence. Thus, VSD IV of CaV1.1 comprises a functionally critical intramolecular interaction between oppositely charged residues of the S3 and S4 transmembrane helices that has not been reported in previous studies of VSDs of other voltage‐gated cation channels. This newly identified interaction is important for proper voltage sensitivity of the calcium channel and it makes this VSD amenable to modulation by alternative splicing in the IVS3–S4 linker. When the exon 29 sequence is included in the IVS3–S4 linker, S3 and S4 are displaced from each other and the important interactions between D4 and R1/R2 are weakened or lost, resulting in similar perturbations of channel voltage sensitivity and open probability as mutations of any one of the participating amino acids.
Moving exon 29 into VSD I does not reconstitute this modulatory mechanism because it does not contain D4 in IS3 (Tuluc et al. 2016). Surprisingly, also the transfer of the IVS3–S4 linker plus IVS3 (containing D4) was not sufficient to restore the mechanism in the first repeat. Conversely, moving IS3 and the IS3–S4 linker into VSD IV perturbed channel gating, even though this loop does not contain exon 29. Apparently the critical difference between the two VSDs resides in the sequence of the IVS4 helix, which requires the D4 countercharge in S3 for proper function.
Interestingly, the charged aspartate (D4) in IVS3 is highly conserved among the members of high voltage‐activated calcium channels (CaV1, CaV2). Moreover, alternative splicing in the IVS3–S4 linker is a common feature among the CaV family members (Perez‐Reyes et al. 1990; Lin et al. 1997, 1999; Bourinet et al. 1999; Liao et al. 2007), and in some cases it has been shown to affect voltage dependence of activation (Tang et al. 2004). Therefore it is possible that this mechanism, which is responsible for the dramatic differences in the gating properties of the two skeletal muscle CaV1.1 splice variants, actually might represent a more general mechanism for regulating the voltage sensitivity and open probability of voltage‐gated calcium channels. However, voltage‐clamp fluorometry study of CaV1.2 indicated that in this CaV isoform VSD IV does not contribute to channel gating (Pantazis et al. 2014). Future studies will need to examine this possibility.
Structural determinants of slow activation kinetics of CaV1.1
Whereas exclusion of the exon 29 sequences in VSD IV normalizes voltage dependence of activation and the open probability of CaV1.1, it does not alter the slow activation kinetics (Tuluc et al. 2016). Previously work showed that transfer of IS3 and the IS3–S4 linker from CaV1.1 into CaV1.2 could confer slow activation kinetics but did not alter the voltage dependence of activation (Nakai et al. 1994). Together these observations indicate that the two properties are determined by similar sequence domains in two different VSDs. However, contrary to the voltage sensitivity mechanism, slow activation kinetics could not be conferred to fast activating CaV1.1 channel constructs by moving sequences from the first to the fourth repeat (Tuluc et al. 2016). Notably, the same sequences (IS3 and the IS3–S4 linker) which were sufficient to transfer slow activation from CaV1.1 to VSD I of CaV1.2, were not sufficient to transfer slow activation properties from VSD I to VSD IV within CaV1.1. This indicated that VSDs I of CaV1.1 and CaV1.2 resemble each other more closely than VSDs I and IV within CaV1.1. Based on the original domain swapping experiments it had been assumed that the specific slow activation mechanism resides in the IS3 and IS3–S4 linker sequences (Nakai et al. 1994). The recent findings that any changes in VSD I abolish slow activation, but that transfer of the entire sequence containing IS3, the IS3–S4 linker, and IS4 cannot confer slow activation properties to VSD IV demonstrate that regulation of activation kinetics depends on the integrity of the entire VSD I and probably also of the pore region (Tuluc et al. 2016). Thus, determining slow activation kinetics is a property of the first VSD and utilizes a different and more complex molecular mechanism than the focused mechanism controlling voltage sensitivity in VSD IV.
Modular organization of CaV1.1 calcium channels
Together these findings indicate that CaV1.1 is organized in a modular manner. The four VSDs are homologous but not identical in structure and function, enabling them to contribute differentially to channel gating. Specific gating properties seem to be regulated by distinct individual VSDs. We now know that VSD I determines activation kinetics and VSD IV regulates the voltage dependence of activation. Because the bulk of CaV1.1a gating charges moves rapidly and at lower membrane potential than current activation (Tuluc et al. 2009), we assume that all four VSDs need to move to the activated state to initiate pore opening. For a given gating property, a single VSD can be limiting: the slowest VSD might determine the speed of activation, the least voltage‐sensitive VSD the voltage dependence of activation, regardless to how fast and at which voltages the other VSDs are activated. A similar model was suggested 20 years ago to explain the slow activation kinetics of frog muscle fibres (Melzer et al. 1995). Riccardo Olcese's team have recently introduced the voltage‐clamp fluorometry method to study CaV1.2 channel gating and thus provided direct experimental evidence that different VSDs contribute differentially to channel gating (Pantazis et al. 2014). In CaV1.2 VSDs II and III critically determine voltage dependence and kinetics of channel opening. Interestingly, in CaV1.2 VSD I is of minor importance and VSD IV appears not to participate at all in channel opening. Evidently, CaV1.1 and CaV1.2 operate on the same principle that the four VSDs contribute differentially to channel gating, but differ in that they utilize distinct VSDs for this purpose.
This difference may be associated with the specific requirement of CaV1.1 to activate EC coupling. Differential use of VSDs for different gating properties and functions enables CaV1.1 to gate EC coupling and calcium conduction at different voltages and with different kinetics. Because activation of EC coupling occurs at substantially lower membrane potentials than current activation, and because activation of EC coupling is fast, we propose that VSDs I and IV are not critically involved in activation of EC coupling. This hypothesis is further supported by a malignant hyperthermia mutant of the innermost arginine of VSD I (R174W) which abolished LTCC in response to test pulses of up to +60 mV without perturbing EC coupling (Eltit et al. 2012). A primary role of VSDs II and III in EC coupling is consistent with the strategically favourable position of sequences which are critical for the functional interaction with RyR1 in the cytoplasmic loop connecting repeats II and III (Grabner et al. 1999). Whether both or only one of these two VSDs are required for activation of EC coupling is currently unknown.
Importance of curtailing calcium influx for skeletal muscle fibre type specification
So far we have reviewed the evidence indicating that curtailing LTCC in skeletal muscle might be important and we summarized our current knowledge of the molecular mechanisms which determine the poor voltage sensitivity and slow kinetics of CaV1.1 current activation. What remains to be discussed is the physiological importance of curtailing the calcium currents. Mouse models have been generated with mutations in the CaV1.1 channel pore that abolish the calcium conductance (Dayal et al. 2014; Lee et al. 2015). Analyses of mouse motor behaviour and skeletal muscle contraction revealed no differences between wild‐type and non‐conducting CaV1.1 mutants. Nevertheless, the mice of the Hamilton group revealed deficient refilling of SR calcium stores during sustained activity and an increased type IIb fibre content and increased fatigability (Lee et al. 2015), and they suffered from changed calcium‐dependent fatty acid metabolism (Georgiou et al. 2015). This phenotype indicates that LTCCs are dispensable for EC coupling but may play a role in muscle differentiation and during prolonged activity of mammalian skeletal muscle.
Given this limited role of LTCC in skeletal muscle function, how important is it to curtail the currents? During normal development expression of the calcium‐conducting CaV1.1e in the fetal stage is almost completely replaced by the virtually non‐conducting adult CaV1.1a splice variant (Tuluc & Flucher, 2011; Sultana et al. 2016). The consequences of circumventing this alternative splicing event have recently been analysed in a genetic mouse model in which exon 29 was constitutively knocked out (Sultana et al. 2016). Overall the development and motor performance of these exon 29 knockout mice was normal. However they displayed reduced voluntary running and reduced grip force. This was confirmed in isolated muscles, where significantly reduced contractile force, an increased endurance and a shift of tetanic fusion to lower frequencies was observed. Because these effects of permanent exclusion of exon 29 resembled the contractile properties of slow muscle fibres, the fibre type composition has been analysed. Indeed, both fast extensor digitorum longus and slow soleus muscles of exon 29 knockout mice experienced a substantial shift towards slower muscle fibre types compared to wild‐type controls. Consistent with the interpretation that increased LTCC during EC coupling caused a dysregulation of fibre type specification, this shift was accompanied by an up‐regulation of the chief activity‐ and calcium‐dependent regulators of fibre type composition, calcineurin and CaMKII, in exon 29 knockout mice.
As expected from an increased expression of slow oxidative muscle fibres, young exon 29 knockout muscle fibres displayed an increased activity of succinyl dehydrogenase (SDH). Surprisingly, however, in older animals SDH activity was reduced. Electron microscopic analysis of these muscle fibres revealed severe mitochondrial damage and a reduction in total mitochondrial volume that would explain the reduced SDH activity (Sultana et al. 2016). Calcium recording in isolated muscle fibres of exon 29 knockout mice demonstrated increased depolarization‐induced calcium influx, an increased contribution of LTCC to refilling of SR calcium stores, and an increased incidence of calcium sparklets at rest. Evidently the increased depolarization‐induced calcium influx was not reflected by a parallel increase in contractile force. In part this may be counteracted by the expected reduction of maximal force associated with the shift towards slower muscle fibre types. In fact the observed activation of the slow fibre pathway may be part of a compensatory response to the aberrantly increased calcium signals in muscles constitutively expressing the calcium‐conducting CaV1.1e isoform. On the other hand the increased calcium influx during activity or the altered calcium homeostasis probably caused the progressive damage and loss of mitochondria. Together these findings indicate that curtailing calcium influx by developmentally regulated splicing of CaV1.1 is important for the normal regulation of muscle fibre types and to prevent muscle fibres from mitochondrial damage. Apparently, curtailing LTCC in skeletal muscle is of minor importance for calcium's primary signalling function in EC coupling, but is of considerable importance for calcium's second signalling function in activity‐dependent regulation of the fibre type composition.
CaV1.1e calcium influx is linked to muscle disease
The observed detrimental effects of exclusion of exon 29 on mitochondria suggest a contribution of the resulting CaV1.1e LTCC to muscle disease. It is well known that calcium overload can lead to mitochondrial damage in any excitable tissue. Moreover, mitochondrial damage is often the first manifestation of muscle damage in muscle diseases associated with dysregulated calcium homeostasis (Brookes et al. 2004; Boncompagni et al. 2009). Most importantly, muscle weakness in patients with myotonic dystrophy is accompanied by the aberrant expression of the calcium‐conducting developmental CaV1.1e splice variant in adult muscle (Tang et al. 2012; Santoro et al. 2014). Although aberrant expression of CaV1.1e in adult mice by itself did not cause a dystrophic phenotype or severe muscle weakness, the observed mitochondrial damage might be a first sign of muscle damage. During the much longer life span in humans and in combination with other genetic defects in myotonic dystrophy it is likely that the additional calcium influx through CaV1.1e will lead to muscle disease. If this can be confirmed, targeting CaV1.1e LTCC with licensed calcium channel drugs may be a reasonable therapeutic strategy to ease the symptoms of myotonic dystrophy (Benedetti et al. 2015).
Conclusion
Several lines of evidence indicate that the developmental switch from a calcium conducting to an effectively non‐conducting CaV1.1 splice variant is of physiological and pathological relevance for skeletal muscle. The existence of multiple strategies to abolish or reduce calcium conduction in skeletal muscle calcium channels highlights the importance of curtailing calcium currents. In CaV1.1a unique molecular mechanisms in distinct VSDs determine slow activation kinetics, right‐shifted voltage dependence of activation, and low open probability. The latter two gating properties can be regulated by alternative splicing of exon 29. When the splicing event is circumvented the continued expression of the calcium‐conducting CaV1.1e splice variant leads to a dysregulation of muscle fibre type specification. Furthermore, aberrant expression of CaV1.1e causes mitochondrial damage in mice and correlates with the degree of muscle weakness in human myotonic dystrophy patients. The simultaneous use of cytoplasmic calcium signals to control muscle contraction and regulate the specification of fibre type properties, as well as the potential danger to mitochondrial integrity emanating from a high calcium load necessitates a tight control of calcium. Evidently, in skeletal muscle this tightrope act is better achieved when activity‐dependent influx of calcium through CaV1.1 channels has been curtailed.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Research in the laboratory of B.E. Flucher has been funded by grants from the Austrian Science Fund, FWF P23479, P27031, W1101, and F4406.
Biographies
Bernhard Flucher received his PhD in Biology at the University of Salzburg, Austria. For his postdoctoral training he joined Matt Daniels at the Laboratory of Biochemical Genetics, and then launched his research line on voltage‐gated calcium channels in the lab of Brian Andrews at the Laboratory of Neurobiology, both at the National Institutes of Health, Bethesda, MD, USA. In 1994 he received a Fellowship from the Austrian Academy of Sciences to establish his lab at the Department of Biochemical Pharmacology, University of Innsbruck. Since 1997 he has been associate professor in physiology at the Department of Physiology and Medical Physics, Medical University of Innsbruck.

Petronel Tuluc received his BA in Biophysics at the University of Iasi, Romania and then joined the Flucher Lab at the Medical University Innsbruck where he received his PhD in 2009. Since 2013 he has held an assistant professor position at the Institute of Pharmacy, University of Innsbruck.
References
- Andronache Z, Ursu D, Lehnert S, Freichel M, Flockerzi V & Melzer W (2007). The auxiliary subunit γ1 of the skeletal muscle L‐type Ca2+ channel is an endogenous Ca2+ antagonist. Proc Natl Acad Sci USA 104, 17885–17890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla FM & Horowicz P (1972). Twitches in the presence of ethylene glycol bis(β‐aminoethyl ether)‐N,N′‐tetracetic acid. Biochim Biophys Acta 267, 605–608. [DOI] [PubMed] [Google Scholar]
- Benedetti B, Tuluc P, Mastrolia V, Dlaska C & Flucher BE (2015). Physiological and pharmacological modulation of the embryonic skeletal muscle calcium channel splice variant CaV1.1e. Biophys J 108, 1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2008). Calcium cycling and signalling in cardiac myocytes. Annu Rev Physiol 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Block BA, Imagawa T, Campbell KP & Franzini‐Armstrong C (1988). Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol 107, 2587–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompagni S, Rossi AE, Micaroni M, Hamilton SL, Dirksen RT, Franzini‐Armstrong C & Protasi F (2009). Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc Natl Acad Sci USA 106, 21996–22001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J & Snutch TP (1999). Splicing of α1A subunit gene generates phenotypic variants of P‐ and Q‐type calcium channels. Nat Neurosci 2, 407–415. [DOI] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW & Sheu S‐S (2004). Calcium, ATP, and ROS: a mitochondrial love‐hate triangle. Am J Physiol Cell Physiol 287, C817–C33. [DOI] [PubMed] [Google Scholar]
- Buraei Z & Yang J (2010). The β subunit of voltage‐gated Ca2+ channels. Physiol Rev 90, 1461–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiglio M & Flucher BE (2015). The role of auxiliary subunits for the functional diversity of voltage‐gated calcium channels. J Cell Physiol 230, 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2011). Voltage‐gated calcium channels. Cold Spring Harb Perspect Biol 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collet C, Csernoch L & Jacquemond V (2003). Intramembrane charge movement and L‐type calcium current in skeletal muscle fibres isolated from control and mdx mice. Biophys J 84, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayal A, Schroetter K, Melzer W, Schwarzer C & Grabner M (2014). The DHPR calcium current in mammalian skeletal muscle: physiological necessity or tolerated evolutionary remnant? Biophys J 106, 126a. [Google Scholar]
- Eltit JM, Bannister RA, Moua O, Altamirano F, Hopkins PM, Pessah IN, Molinski TF, López JR, Beam KG & Allen PD (2012). Malignant hyperthermia susceptibility arising from altered resting coupling between the skeletal muscle L‐type Ca2+ channel and the type 1 ryanodine receptor. Proc Natl Acad Sci USA 109, 7923–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher BE (2016). Retrograde coupling: muscle's orphan signalling pathway? Biophys J 110, 870–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher BE & Franzini‐Armstrong C (1996). Formation of junctions involved in excitation‐contraction coupling in skeletal and cardiac muscle. Proc Natl Acad Sci USA 93, 8101–8106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flucher BE, Obermair GJ, Tuluc P, Schredelseker J, Kern G & Grabner M (2005). The role of auxiliary dihydropyridine receptor subunits in muscle. J Muscle Res Cell Motil 26, 1–6. [DOI] [PubMed] [Google Scholar]
- Fuller‐Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, Muth JN, Mikala G, Petrashevskaya NN, Jordan MA, Zhang S‐P, Qin N, Flores CM, Isaacsohn I, Varadi M, Mori Y, Jones WK & Schwartz A (2009). Targeted disruption of the voltage‐dependent calcium channel α2/δ‐1‐subunit. Am J Physiol Heart Circ Physiol 297, H117–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J, Tanabe T, Beam KG & García J (1994). Relationship of calcium transients to calcium currents and charge movements in myotubes expressing skeletal and cardiac dihydropyridine receptors. J Gen Physiol 103, 125–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiou DK, Dagnino‐Acosta A, Lee CS, Griffin DM, Wang H, Lagor WR, Pautler RG, Dirksen RT & Hamilton SL (2015). Ca2+ binding/permeation via calcium channel, CaV1.1, regulates the intracellular distribution of the fatty acid transport protein, CD36, and fatty acid metabolism. J Biol Chem 290, 23751–23765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M, Dirksen RT, Suda N & Beam KG (1999). The II‐III loop of the skeletal muscle dihydropyridine receptor is responsible for the bi‐directional coupling with the ryanodine receptor. J Biol Chem 274, 21913–21919. [DOI] [PubMed] [Google Scholar]
- Horstick EJ, Linsley JW, Dowling JJ, Hauser MA, McDonald KK, Ashley‐Koch A, Saint‐Amant L, Satish A, Cui WW, Zhou W, Sprague SM, Stamm DS, Powell CM, Speer MC, Franzini‐Armstrong C, Hirata H & Kuwada JY (2013). Stac3 is a component of the excitation‐contraction coupling machinery and mutated in Native American myopathy. Nat Commun 4, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasielke N, Obermair GJ, Kugler G, Grabner M & Flucher BE (2003). Cardiac‐type EC‐coupling in dysgenic myotubes restored with Ca2+ channel subunit isoforms α1C and α1D does not correlate with current density. Biophys J 84, 3816–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Dagnino‐Acosta A, Yarotskyy V, Hanna A, Lyfenko A, Knoblauch M, Georgiou DK, Poché RA, Swank MW, Long C, Ismailov II, Lanner J, Tran T, Dong K, Rodney GG, Dickinson ME, Beeton C, Zhang P, Dirksen RT & Hamilton SL (2015). Ca2+ permeation and/or binding to CaV1.1 fine‐tunes skeletal muscle Ca2+ signalling to sustain muscle function. Skelet Muscle 5, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao P, Yu D, Li G, Tan FY, Jia LS, Yeow LC & Tuck WS (2007). A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state‐dependent inhibition by nifedipine. J Biol Chem 282, 35133–35142. [DOI] [PubMed] [Google Scholar]
- Lin Z, Haus S, Edgerton J & Lipscombe D (1997). Identification of functionally distinct isoforms of the N‐type Ca2+ channel in rat sympathetic ganglia and brain. Neuron 18, 153–166. [DOI] [PubMed] [Google Scholar]
- Lin Z, Lin Y, Schorge S, Pan JQ, Beierlein M & Lipscombe D (1999). Alternative splicing of a short cassette exon in α1B generates functionally distinct N‐type calcium channels in central and peripheral neurons. J Neurosci 19, 5322–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer W, Herrmann‐Frank A & Lüttgau H‐C (1995). The role of Ca2+ ions in excitation‐contraction coupling of skeletal muscle fibres. Biochim Biophys Acta 1241, 59–116. [DOI] [PubMed] [Google Scholar]
- Nakai J, Adams BA, Imoto K & Beam KG (1994). Critical roles of the S3 segment and S3‐S4 linker of repeat I in activation of L‐type calcium channels. Proc Natl Acad Sci USA 91, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Dirksen RT, Nguyen HT, Pessah IN, Beam KG & Allen PD (1996). Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380, 72–75. [DOI] [PubMed] [Google Scholar]
- Nelson BR, Wu F, Liu Y, Anderson DM, McAnally J, Lin W, Cannon SC, Bassel‐Duby R & Olson EN (2013). Skeletal muscle‐specific T‐tubule protein STAC3 mediates voltage‐induced Ca2+ release and contractility. Proc Natl Acad Sci USA 110, 11881–11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Kugler G, Baumgartner S, Tuluc P, Grabner M & Flucher BE (2005). The Ca2+ channel α2δ‐1 subunit determines Ca2+ current kinetics in skeletal muscle but not targeting of α1S or excitation‐contraction coupling. J Biol Chem 280, 2229–2237. [DOI] [PubMed] [Google Scholar]
- Pantazis A, Savalli N, Sigg D, Neely A & Olcese R (2014). Functional heterogeneity of the four voltage sensors of a human L‐type calcium channel. Proc Natl Acad Sci USA 111, 18381–18386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E, Wei X, Castonello A & Birnbaumer L (1990). Molecular diversity of L‐type calcium channels. J Biol Chem 265, 20430–20436. [PubMed] [Google Scholar]
- Polster A, Nelson BR, Olson EN & Beam KG (2016). Stac3 has a direct role in skeletal muscle‐type excitation–contraction coupling that is disrupted by a myopathy‐causing mutation. Proc Natl Acad Sci USA 113, 10986–10991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ríos E & Pizarro G (1991). Voltage sensor of excitation‐contraction coupling in skeletal muscle. Physiol Rev 71, 849–908. [DOI] [PubMed] [Google Scholar]
- Robin G & Allard B (2015). Voltage‐gated Ca2+ influx through L‐type channels contributes to sarcoplasmic reticulum Ca2+ loading in skeletal muscle. J Physiol 593, 4781–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome LC (2006). Design and function of superfast muscles: new insights into the physiology of skeletal muscle. Annu Rev Physiol 68, 193–221. [DOI] [PubMed] [Google Scholar]
- Santoro M, Piacentini R, Masciullo M, Bianchi MLE, Modoni A, Podda MV, Ricci E, Silvestri G & Grassi C (2014). Alternative splicing alterations of Ca2+ handling genes are associated with Ca2+ signal dysregulation in myotonic dystrophy type 1 (DM1) and type 2 (DM2) myotubes. Neuropathol Appl Neurobiol 40, 464–476. [DOI] [PubMed] [Google Scholar]
- Savalli N, Pantazis A, Sigg D, Weiss JN, Neely A & Olcese R (2016). The α2δ‐1 subunit remodels CaV1.2 voltage sensors and allows Ca2+ influx at physiological membrane potentials. J Gen Physiol 148, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schredelseker J, Di Biase V, Obermair GJ, Felder ET, Flucher BE, Franzini‐Armstrong C & Grabner M (2005). The β1a subunit is essential for the assembly of dihydropyridine‐receptor arrays in skeletal muscle. Proc Natl Acad Sci USA 102, 17219–17224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schredelseker J, Shrivastav M, Dayal A & Grabner M (2010). Non‐Ca2+‐conducting Ca2+ channels in fish skeletal muscle excitation‐contraction coupling. Proc Natl Acad Sci USA 107, 5658–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmeier RP & Melzer W (2004). Voltage‐dependent Ca2+ fluxes in skeletal myotubes determined using a removal model analysis. J Gen Physiol 123, 33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan DC, Takekura H, Franzini‐Armstrong C, Beam KG, Allen PD & Perez CF (2006). Bidirectional signalling between calcium channels of skeletal muscle requires multiple direct and indirect interactions. Proc Natl Acad Sci USA 103, 19760–19765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J (2007). C‐terminal tailoring of L‐type calcium channel function. J Physiol 585, 643–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana N, Dienes B, Benedetti A, Tuluc P, Szentesi P, Sztretye M, Rainer J, Hess MW, Schwarzer C, Obermair GJ, Csernoch L & Flucher BE (2016). Restricting calcium currents is required for correct fibre type specification in skeletal muscle. Development 143, 1547–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe T, Beam KG, Adams Ba, Niidome T & Numa S (1990). Regions of the skeletal muscle dihydropyridine receptor critical for excitation‐contraction coupling. Nature 346, 567–569. [DOI] [PubMed] [Google Scholar]
- Tang ZZ, Liang MC, Lu S, Yu D, Yu CY, Yue DT & Soong TW (2004). Transcript scanning reveals novel and extensive splice variations in human l‐type voltage‐gated calcium channel, Cav1.2 α1 subunit. J Biol Chem 279, 44335–44343. [DOI] [PubMed] [Google Scholar]
- Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT & Thornton CA (2012). Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Cav1.1 calcium channel. Hum Mol Genet 21, 1312–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuluc P, Benedetti B, Bagneaux PC, De Grabner M & Flucher BE (2016). Two distinct voltage‐sensing domains control voltage sensitivity and kinetics of current activation in CaV1.1 calcium channels. J Gen Physiol 147, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuluc P & Flucher BE (2011). Divergent biophysical properties, gating mechanisms, and possible functions of the two skeletal muscle CaV1.1 calcium channel splice variants. J Muscle Res Cell Motil 32, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuluc P, Kern G, Obermair GJ & Flucher BE (2007). Computer modelling of siRNA knockdown effects indicates an essential role of the Ca2+ channel α2δ‐1 subunit in cardiac excitation‐contraction coupling. Proc Natl Acad Sci USA 104, 11091–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuluc P, Molenda N, Schlick B, Obermair GJ, Flucher BE & Jurkat‐Rott K (2009). A CaV1.1 Ca2+ channel splice variant with high conductance and voltage‐sensitivity alters EC coupling in developing skeletal muscle. Biophys J 96, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuluc P, Yarov‐Yarovoy V, Benedetti B & Flucher BE (2015). Molecular interactions in the voltage sensor controlling gating properties of CaV calcium channels. Structure 24, 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursu D, Schuhmeier RP, Freichel M, Flockerzi V & Melzer W (2004). Altered inactivation of Ca2+ current and Ca2+ release in mouse muscle fibres deficient in the DHP receptor γ1 subunit. J Gen Physiol 124, 605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D & De Waard M (1998). Subunit interaction sites in votage‐dependent Ca2+ channels: role in channel function. Trends Neurosci 21, 148–154. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M & Yan N (2015). Structure of the voltage‐gated calcium channel Cav1.1 complex. Science 350, aad2395. [DOI] [PubMed] [Google Scholar]