

Abstract
The recent occurrence of deaths associated with the psychostimulant cis-4,4′-dimethylaminorex (4,4′-DMAR) in Europe indicated the presence of a newly emerged psychoactive substance on the market. Subsequently, the existence of 3,4-methylenedioxy-4-methylaminorex (MDMAR) has come to the authors’ attention and this study describes the synthesis of cis- and trans-MDMAR followed by extensive characterization by chromatographic, spectroscopic, mass spectrometric platforms and crystal structure analysis. MDMAR obtained from an online vendor was subsequently identified as predominantly the cis-isomer (90%). Exposure of the cis-isomer to the mobile phase conditions (acetonitrile/water 1:1 with 0.1% formic acid) employed for high performance liquid chromatography analysis showed an artificially induced conversion to the trans-isomer, which was not observed when characterized by gas chromatography. Monoamine release activities of both MDMAR isomers were compared with the non-selective monoamine releasing agent (+)-3,4-methylenedioxymethamphetamine (MDMA) as a standard reference compound. For additional comparison, both cis- and trans-4,4′-DMAR, were assessed under identical conditions. cis-MDMAR, trans-MDMAR, cis-4,4′-DMAR and trans-4,4′-DMAR were more potent than MDMA in their ability to function as efficacious substrate-type releasers at the dopamine (DAT) and norepinephrine (NET) transporters in rat brain tissue. While cis-4,4′-DMAR, cis-MDMAR and trans-MDMAR were fully efficacious releasing agents at the serotonin transporter (SERT), trans-4,4′-DMAR acted as a fully efficacious uptake blocker. Currently, little information is available about the presence of MDMAR on the market but the high potency of ring-substituted methylaminorex analogues at all three monoamine transporters investigated here might be relevant when assessing the potential for serious side-effects after high dose exposure.
Keywords: new psychoactive substances; aminorex; psychostimulants; 4,4′-DMAR; forensic
Introduction
Since the end of 2012, several member states of the European Union have reported the identification of 4,4′-dimethylaminorex (4,4′-DMAR) (Figure 1A) via the European Union’s Early Warning System on New Psychoactive Substances (EU Early Warning System).[1] In addition, the recent emergence of at least 27 deaths associated with the consumption of 4,4′-DMAR[1,2] led to a request by the European Commission to carry out a risk assessment in order to assess control measures across EU member states.[3] 4,4′-DMAR is a ring-substituted analogue of 4-methylaminorex (4-MAR or U4Euh), a stimulant drug of abuse that was popular in the 1980s.[4] Like 4-methylaminorex, 4,4′-DMAR contains two chiral centres which yield the potential for four stereoisomers and two racemic mixtures (i.e., cis- and trans-racemates). A study carried out recently involving the characterization of 4,4′-DMAR obtained from online vendors and case samples revealed the presence of cis-4,4′-DMAR.[2,5]
Figure 1.
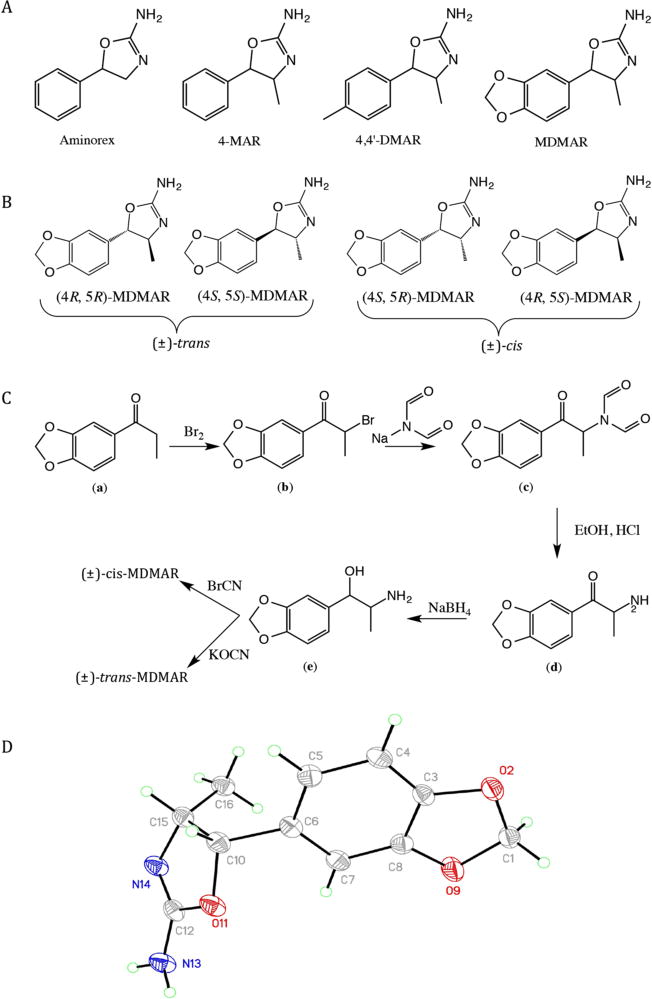
(A) Chemical structures of the three psychostimulants aminorex, 4-methylaminorex (4-MAR), para-methyl-4-methylaminorex (4,4′-DMAR) and the new psychoactive substance 3′,4′-methylenedioxy-4-methylaminorex (MDMAR). (B) Structural representation of all four MDMAR enantiomers. (C) Synthetic route to both (±)-cis- and (±)-trans- MDMAR. Both isomers were prepared from the same 3′,4′-methylenedioxynorephedrine prescursor (e) using cyanogen bromide or potassium cyanate to yield the (±)-cis and (±)-trans-MDMAR product, respectively. (D) Molecular structure of synthesized cis-MDMAR (50% displacement ellipsoids).
Aminorex (2-amino-5-phenyl-oxazoline) and its analogue 4-MAR (Figure 1A) were both first synthesized by McNeil Laboratories in the 1960s to evaluate their potential as appetite suppressants.[6,7] Aminorex was introduced as a prescription drug for weight loss in Europe in 1965 under the trade names Menocil and Apiquel but withdrawn shortly afterwards due to fatal complications related to pulmonary hypertension.[8,9] While 4,4′-DMAR is currently not a controlled substance, 4-MAR (cis-racemate) is classified as a Schedule I substance under the United Nations Convention on Psychotropic Substances 1971, while aminorex is classified as a Schedule IV substance.[10]
Clinical observations related to 4,4′-DMAR intoxication included a range of adverse effects such as agitation, hyperthermia, foaming at the mouth, breathing problems, and cardiac arrest.[11] Little information is available regarding the biological mechanism of action for these substances. The related compound aminorex is a substrate for monoamine transporter proteins, which evokes transporter-mediated release of the monoamine neurotransmitters dopamine, norepinephrine, and serotonin (5-HT) in the central nervous system.[12–14] A previous study revealed that cis-4,4′-DMAR is a powerful monoamine-releasing agent that displays low nanomolar potency at all three monoamine transporters.[5]
The work presented in this study describes the synthesis and analytical characterization of 3′,4′-methylenedioxy-4-methylaminorex (MDMAR) (Figure 1A). Currently, chemical, pharmacological, and clinical information on MDMAR appears to be absent in the scientific literature. Information as to whether MDMAR is currently circulating as a new psychoactive substance (NPS) is not available. However, similar to the work described on 4,4′-DMAR previously, this investigation was also instigated by a donation from an online vendor. Therefore, its appearance on the recreational market could not be fully excluded. Both cis and trans isomeric forms of MDMAR (Figure 1B) were synthesized and subjected to analytical characterization using liquid chromatography-mass spectrometry (LC-MS), gas chromatography-mass spectrometry (GC-MS), nuclear magnetic resonance (NMR) and high resolution-mass spectrometry (HR-MS) platforms. This was followed by the characterization of the donated MDMAR sample and the identification of the corresponding isomer. Previously published work on the releasing effects of the cis-4,4′-DMAR included the comparison with d-amphetamine, aminorex and cis-4-MAR.[5] In the present study, the monoamine transporter activity of cis-MDMAR, trans-MDMAR, cis-4,4′-DMAR and trans-4,4′-DMAR have been evaluated. In these experiments, the non-selective monoamine releasing agent (+)-3,4-methylenedioxymethamphetamine (MDMA) was included as a standard reference compound with known pharmacology.[15]
Experimental
Reagents and standards
All reagents and dry solvents used in the syntheses were obtained from Sigma Aldrich Ltd (Arklow, Co. Wickley, Ireland). LC-MS grade solvents were obtained from Fisher Scientific (Dublin, Ireland). A sample subsequently characterized as (±)-cis-3′,4′-methylenedioxy-4-methylaminorex (cis-MDMAR) was donated by Scientific Supplies Ltd (London, UK).
Syntheses
Unless otherwise stated, the synthesis procedures reported here were essentially adapted from the method reported previously by Brandt et al. for the preparation of cis- and trans-4,4′-DMAR isomers (Figure 1C).[5]
3′,4′-Methylenedioxynorephedrine
3′,4′-Methylenedioxynorephedrine was prepared from 3,4-methylenedioxypropiophenone.[5] The reaction yielded 3′,4′-methylenedioxynorephedrine as a beige powder (7.64 g, 39 mmol, 39%): m. p. 112–114 °C. 1H NMR (DMSO) δ 6.86 (doublet; J = 7.4 Hz; 1H; Ar-H), 6.76 (double doublet; J = 8.0, 1.6 Hz; 1H; Ar-H), 5.98 (Singlet; J = 7.5 Hz; 2H; CH2), 4.21 (doublet; J = 5.4 Hz; 1H; CH(OH)), 2.83 (doublet quartet; J = 12.2, 6.2 Hz; 1H; CH(CH3)), 0.90 (doublet; J = 6.8 Hz; 3H; CH(CH3)); 13C NMR (DMSO) δ 146.77 (Ar C4), 145.67 (Ar C3), 137.69 (Ar C1), 119.73 (Ar C6), 107.41 (Ar C5), 106.99 (Ar C2), 100.55 (CH2), 77.44 (CH(OH)), 52.32 (CH(CH3)), 18.45 (CH(CH3)); HR-ESIMS found 196.0965 (theory [M + H]+: C10H13NO3. 196.0929)
(±)-cis-3′,4′-Methylenedioxy-4-methylaminorex
Cyanogen bromide (0.583 g, 5.5 mmol) in methanol (2 mL) was added dropwise to a solution of 3′,4′-methylenedioxynorephedrine (0.975 g, 5.0 mmol) in methanol (5 mL). The mixture was stirred for 2 h in an ice bath. Following removal of solvent, saturated sodium carbonate (25 mL) was added and this was shaken until a white precipitate formed. The mixture was filtered to yield a colorless powder. Purification by recrystallization in methanol, followed by a further recrystallization step with ethanol, afforded a colorless powder (78 mg, 0.35 mmol, 7%): m.p. 198–200 °C. 1H NMR (DMSO) δ 6.90 (doublet; J = 7.9 Hz; 1H; Ar-H6′), 6.72 (doublet; J = 7.9 Hz; 1H; Ar-H2′);6.69 (doublet; J = 1.8 Hz; 1H; Ar-H5′), 5.91 (singlet; J = 7.5 Hz; 2H; CH2); 5.43 (doublet; J = 8.5 Hz; 1H; H-5), 4.14 (double quartet; J = 8.5, 6.8 Hz; 1H; H-4), 0.60 (doublet; J = 6.8Hz; 3H; CH3); 13C NMR (DMSO) δ 159.59 (C-2); 147.39 (Ar C3′); 146.69 (Ar C4′); 132.36 (Ar C1′); 119.48 (Ar C6′); 108.19 (Ar C2′); 101.23 (CH2); 82.52 (C-5); 63.00 (C-4); 18.90 (CH3); HR-ESIMS found 221.0916 (theory [M + H]+: C11H12N2O3, 221.0881).
(±)-trans-3′,4′-Methylenedioxy-4-methylaminorex
A mixture of 3′,4′-methylenedioxynorephedrine hydrochloride (prepared from 0.96 g (4.92 mmol) of the free base and ethereal hydrogen chloride) and potassium cyanate (410 mg) and ethanol (50 mL) was refluxed for 10 h. Removal of solvent was followed by the addition of thionyl chloride (500 μL), dichloromethane (10 mL) and a stirring period of 4 h at room temperature. Following removal of solvent, ethanol (5 mL) was added and the mixture was refluxed for 4 h. Solvent removal yielded crude trans-MDMAR followed by the addition of distilled water and diethyl ether (50:50). The aqueous layer was collected and made alkaline with 25% sodium hydroxide. This mixture was shaken until a white precipitate formed and then stirred for a further 20 min. The mixture was filtered yielding a beige solid, which was left to air dry. Purification by flash chromatography (dichloromethane/methanol, 7/3) afforded a colorless powder (21 mg, 0.1 mmol, 2 %): m.p. 148–150 °C. 1H NMR (DMSO) δ 6.88 (doublet; J = 1.9 Hz; 1H; Ar-H6′); 6.82 (doublet; J = 8.0Hz; 1H; Ar-H2′); 6.77 (doublet; J = 1.9 Hz; 1H; Ar-H5′); 5.93 (singlet; J =7.5 Hz; 2H; C H2); 4.75 (doublet; J = 6.5 Hz; 1H; H-5); 4.16 (double quartet; J = 6.5, 6.4 Hz; 1H; H-4); 1.12 (doublet; J = 6.4 Hz; 3H; C H3); 13C NMR (DMSO) δ 158.87 (C-2); 147.46 (Ar C3′); 146.85 (Ar C4′); 134.85 (Ar C1′); 119.08 (Ar C6′); 108.16 (Ar C5′); 105.81 (Ar C2′); 100.98 (CH2); 86.33 (C-5); 68.08 (C-4); 21.93 (CH3); HR-ESIMS found 221.0915 (theory [M + H]+: C11H12N2O3, 221.0881).
Instrumental analysis
Gas chromatography quadrupole mass spectrometry
Samples were prepared to give a 1 mg/mL solution in methanol and analyzed on an Agilent 6890 N GC coupled to 5975 Mass Selective Detector. A HP-ULTRA 1 column (12 m × 0.2 mm × 0.33 μm) was used with helium carrier gas at a constant flow of 1 mL/min and a split ratio of 50:1. The injector was set at 250 °C and the transfer line at 280 °C. The initial oven temperature was 60 °C, held for 2 min then ramped at 25 °C/min to 295 °C with a hold time of 3 min. The mass spectra were collected after a 1.5 min solvent delay time. The ionization energy was set at 70 eV and the mass range was m/z 40–450. The total run time was 14.40 min.
Liquid chromatography electrospray single quadrupole mass spectrometry
LC-MS analyses were performed on an Agilent 1100 LC system. Separation was obtained on an Allure PFP Propyl column (5 μm, 50 mm × 2.1 mm) Restek (Bellefonte, PA, USA). Mobile phase A consisted of 0.1% formic acid in water, whereas, mobile phase B consisted of 0.1% formic acid in acetonitrile. The Aligent LC-MSD settings were as follows: positive electrospray mode, capillary voltage 3500 V, drying gas (N2) 12 L/min at 350 °C, nebulizer gas (N2) pressure 60 psi, SIM m/z 221 and 178, fragmentor voltage 70 V. Samples for LC-MS analysis were dissolved in acetonitrile/water (1:1, containing 0.1% formic acid) at a concentration of 10 μg/mL. The injection volume was 1 μL, flow rate was 1 mL/min and the column temperature was 30 °C. The total run time was 40 min. The following gradient elution program was used: 0 4 min 2% B, followed by an increase to 30% within 30 min, reaching 80% within 33 min before returning to 2% within 40 min.
Nuclear magnetic resonance spectroscopy
For NMR analysis, the MDMAR standards and the vendor sample were prepared in deuterated dimethyl sulfoxide (DMSO-d6). 1H(600 MHz) and 13C NMR spectra were recorded on a Bruker AV600 NMR spectrometer using a 5 mm TCI cryoprobe. 1H NMR spectra were referenced to an external TMS reference at δ =0 ppm.
High resolution electrospray mass spectrometry
HR-ESI mass spectra were recorded by direct injection into a LTQ Orbitrap Discovery (Thermo Fisher, Loughborough, UK). Samples were dissolved in acetonitrile/water (1:1, containing 0.1% formic acid) and infused at a rate of 5 μL/min. Full accurate high-resolution (30000) mass scans were performed in positive electrospray mode. Measured accurate masses were within ±5 ppm of the theoretical masses. The following conditions were used: drying gas (N2) 10 L/min, capillary temperature 310 °C, spray voltage 4 V, capillary voltage 22 V and tube lens 77 V.
X-ray crystallography
Intensity data were collected at 100(2) K using a MiTeGen micromount on a Bruker APEX Duo CCD diffractometer equipped with an Oxford Cobra cryosystem. Data were collected using ω and ϕ scans, corrected for Lorentz and polarization effects, and integrated using the Bruker APEX program suite.[16] Structures were solved by direct methods and refined with least squares procedures.[17] All non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed geometrically in the calculated positions using a riding model except for the amino group where hydrogen atoms were located and refined.
Data collected using Cu Kα radiation (1.54178 Å) for a colorless plate crystal 0.35 × 0.16 × 0.02 mm3, C11H12N2O3, M = 220.23, triclinic, Pī, a = 6.1800(2), b = 6.4291(3), c = 14.0726(6)Å, α = 80.360(2), β = 78.857(2) γ = 72.431(2)°, V = 519.43(4) Å3, Z = 2, μ = 0.868mm−1, 1932 unique data (θmax = 69.91°), S = 1.058, R1 = 0.0369 (1730 reflections with I >2σ(I)), wR2 = 0.0960.* CCDC deposition number 1006741. (* R1=Σ||Fo|−|Fc|| / Σ |Fo| and Wr2 = Σw(|Fo|2−|Fc|2)2) / Σw|Fo |2)1/2)
Monoamine transporter assays
Male Sprague-Dawley rats (250–300 g, Charles River Laboratories, Wilmington, MA, USA) were housed 2 per cage and maintained on a 12-h light-dark cycle. Food and water were provided ad libitum. Animal use procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and the Animal Care and Use Committee of the Intramural Research Program of the National Institute on Drug Abuse (Baltimore, MD, USA). Rats were euthanized by CO2 narcosis and brains were processed to yield synaptosomes as previously described.[18,19] For release assays, 9 nM [3H]-1-methyl-4-phenylpyridinium ([3H] MPP+) was used as the radiolabelled substrate for dopamine transporters (DAT) and norepinephrine transporters (NET), while 5 nM [3H]5-HT was used as the radiolabelled substrate for 5-HT transporters (SERT). All buffers used in the release assay methods contained 1 μM reserpine to block vesicular uptake of substrates. The selectivity of release assays was optimized for a single transporter by including unlabelled blockers to prevent the uptake of [3H]MPP+ or [3H]5-HT by competing transporters. Synaptosomes were preloaded with radiolabelled substrate in Krebs-phosphate buffer for 1 h (steady state). Release assays were initiated by adding 850 μL of preloaded synaptosomes to 150 μL of test drug. Release was terminated by vacuum filtration and retained radioactivity was quantified by scintillation counting. Time-course evaluation revealed that cis-MDMAR was subject to isomerization in aqueous assay buffer after approximately 40 min. Thus, for this compound, drug dilutions were prepared immediately prior to assay initiation to avoid the potential for isomerization.
Results and discussion
The presence of (±)-cis-4,4′-DMAR on the European market became apparent following a number of notifications to the EU Early Warning System and from reports published in the scientific literature,[1,2] which triggered an in-depth chemical and analytical characterization and evaluation of its monoamine transporter activity.[5] While cis-4,4′-DMAR has also been advertised under the name Serotoni,[20] data about availability, prevalence of use and properties of MDMAR are currently unavailable. However, since this substance was donated from an online vendor it was deemed important to chemically profile this substance as a potential new psychoactive substance as it seemed possible that it might also potentially become available on the drug market. A number of ring-substituted aminorex analogues have been occasionally discussed, for example on the now defunct Hive forum or in the literature,[21] and it appeared that some discussion has also occurred on MDMAR (MDMAminorex) on an online forum.[22] So far, however, it is unclear whether MDMAR is available for purchase from online retailers.
Synthesis
The synthesis employed for the preparation of the racemic isomers of MDMAR was based on the synthetic pathways outlined in the literature.[5,6,23,24] The synthesis involved bromination of the 3,4-methylenedioxypropiophenone precursor (a), yielding α-bromo-3,4-methylenedioxypropiophenone (b). This was reacted with sodium diformylamide (Gabriel Reaction) to give the methylenedioxy-N,N-diformylamide derivative (c). Hydrolysis under acidic conditions provided the methylenedioxycathinone species (d) and reduction to the alcohol provided the methylenedioxynorephedrine intermediate (e). This was subsequently converted to either (±)-cis-, via the cyanogen bromide route, or (±)-trans-, via the potassium cyanate route, 3′,4′-MDMAR (Figure 1C). The preliminary synthesis of the trans isomer, using the literature based methods, yielded a cyanamide intermediate and not the trans MDMAR isomer (supplementary data). This compound failed to undergo cyclization to form the oxazoline ring, which is necessary for the formation of the MDMAR isomer. The synthesis of the trans-MDMAR isomer was then conducted using an alternative synthetic route. This synthetic route involved reacting 3′,4′-methylenedioxynorephedrine hydrochloride with potassium cyanate in the presence of ethanol. The urea intermediate formed was then reacted with thionyl chloride, which spontaneously cyclized[25] to form the desired trans-MDMAR isomer.
X-ray crystallography
The structure of the synthesized cis-MDMAR isomer is shown in Figure 1D. After repeated crystallization from propanol, a single colorless very thin plate crystal was used for analysis. The structure in the triclinic space group Pī, confirms the 4S,5R-cis arrangement of the oxazoline moiety. Bond lengths and angles in the oxazoline moiety are similar to those found in (4S,5R)-(−)-cis-4-methyl-5-phenyloxazoline-2-amine.[25] The angle of the oxazoline plane to benzodioxole plane (64°, torsion angle C7-C6-C10-O11 = 29.54(16)°) is more acute than in the phenyloxazoline (104°, torsion angle C9-C4-C3-O1 = 34.4°). cis-MDMAR is linked in the solid state by hydrogen bonds between the amino group and neighboring oxazoline groups (supplementary data). Short contacts are also formed between neighboring dioxoles.
Gas chromatography-mass spectrometry
The chromatographic method used was able to achieve separation between the two isomers obtaining a retention time of 7.97 min for the cis isomer (Figure 2A) and a retention time of 7.88 min for the trans isomer (Figure 2B). The EI mass spectra obtained for the synthesized cis and trans racemates were similar as expected. The base peak was observed at m/z 70 and indicated the loss of 3,4-methylenedioxybenzaldehyde, which gave rise to a radical cation. The suggested species, with molecular formula C3H6N2, which may be the 3-methylaziridin-2-imine fragment, was consistent with the EI-MS data reported for 4-MAR[26] and 4,4′-DMAR.[5] The loss of a methyl group (m/z 205) from the molecular ion was observed while the presence of an acylium ion was represented by the ion at m/z 149, followed by a loss of CO to give m/z 121. The observed species at m/z 176 was due to the presence of an aziridinium ion. Overall, it was found that the main principles of fragmentation were in agreement with the EI spectra reported previously on 4,4′-DMAR.[5] The proposed electron ionization fragmentation pattern for 3′,4′-methylenedioxy-4-methylaminorex by GC-MS is outlined in Figure 2C.
Figure 2.
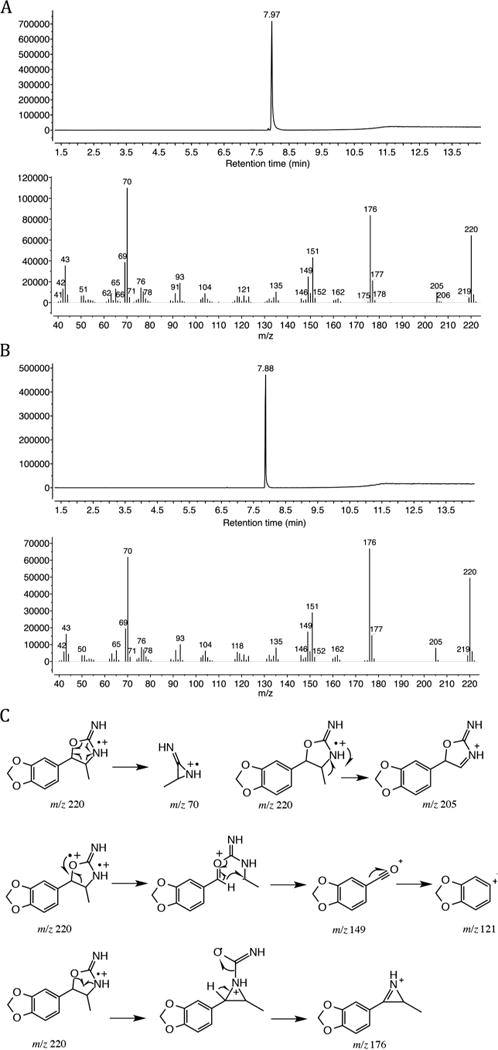
(A) GC-MS data obtained for the (±)-cis-MDMAR isomer. (B) GC-MS data obtained for the (±)-trans-MDMAR isomer. (C) A proposed EI-MS fragmentation pattern for (±)-cis- and (±)-trans-MDMAR.
Liquid chromatography-mass spectrometry
The HPLC method successfully separated and distinguished between both isomers of MDMAR. A retention time of 15.25 min was observed for the cis isomer, while a retention time of 16.29 min was observed for the trans isomer. The EI mass spectra of the synthesized cis and trans racemates were identical, sharing highly abundant fragments, i.e. m/z 221 for the protonated molecule and m/z 178 (supplementary data). The mass spectrum indicated the loss of 43 Da, which represented HNCO, resulting in a species with m/z 178 (Figure 3A). The loss of 43 Da (HNCO) was described previously in studies involving mass spectrometric analysis of 4,4′-DMAR[5] and 4-MAR.[27] However, due to the mass difference between MDMAR, 4,4′-DMAR and 4-MAR, the equivalent loss of 43 Da resulted in ions at m/z 148 and m/z 134, respectively. An identical fragmentation pattern was obtained from high resolution mass spectrometric analysis (supplementary data). An unexpected phenomenon occurred when analyzing the cis isomer by LC, i.e. the cis isomer underwent an isomerization process in solution (acetonitrile/water 1:1 with 0.1% formic acid) that led to the increasing formation of the trans isomer. The cis isomer was analyzed twenty times in succession to monitor the cis to trans conversion. A ratio of cis and trans peak area was calculated and is presented graphically in Figure 3B. It was considered possible that the cis isomer reacted with the H2O in the LC mobile phase, hence, resulting in the change of configuration from the cis isomer to its trans equivalent (Figure 3C). The isomerization of cis-MDMAR to trans-MDMAR was examined under three conditions: acetonitrile only, LC grade water only and aqueous assay buffer conditions. Isomerization did not occur in acetonitrile only conditions. Isomerization began to occur in water only conditions and the aqueous assay buffer conditions after approximately 40 min (supplemental data). This cis to trans conversion was unique to cis-MDMAR and had not been encountered in previous analyses of 4-MAR or 4,4′-DMAR.
Figure 3.
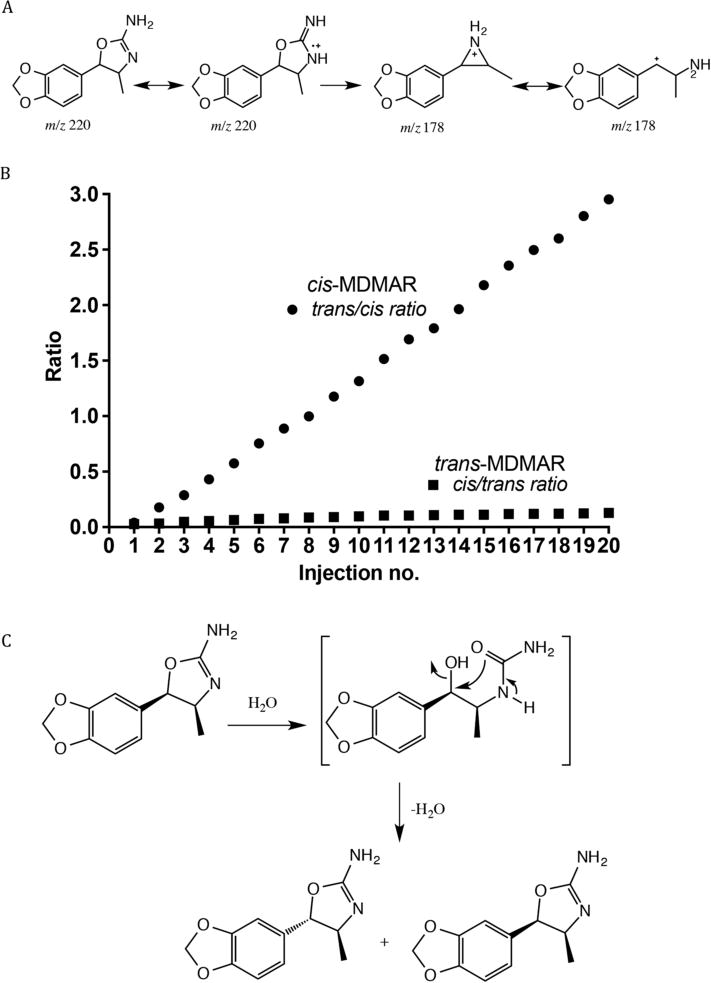
(A) Suggested mechanism for the formation of the m/z 178 ion following loss of HCNO. (B) Graph representing the cis to trans conversion overtime. (C) Suggested mechanism to explain the cis to trans conversion where the cis isomer reacts with the H2O in the LC mobile phase which resulted in a change of configuration.
Nuclear magnetic resonance spectrometry
The NMR spectra associated with both racemic MDMAR isomers shared some key characteristics, which were consistent with those reported previously for cis and trans 4,4′-DMAR and 4-MAR.[5] However, there were also differences in chemical shifts present that facilitated the differentiation between the cis and trans MDMAR isomers. The significant differences occurred at the chemical environments around the chiral carbons. In the 1H NMR, the protons of the CH3 attached to the carbon at position four of the oxazoline ring in the trans isomer produced a signal at 1.12 ppm while the protons on the CH3 at the same position in the cis isomer was shifted upfield and produced a signal at 0.60 ppm. Again, the proton attached to the carbon at position five of the oxazoline ring in the trans isomer was represented by a signal at 4.75 ppm whereas the same proton was shifted downfield in the cis isomer and was represented by a signal at 5.43 ppm. The chiral carbons in both isomers were in different environments and produced different resonances. The carbon at position four of the oxazoline ring in the trans isomer produced a signal at 68.08 ppm, while the same carbon in the cis isomer was represented by a signal at 63.00 ppm. The second chiral carbon at position five of the oxazoline ring in the trans isomer produced a signal at 86.33 ppm and a signal at 82.52 ppm in the cis isomer. The carbon of the methyl group gave a signal at 21.93 ppm in the trans isomers compared to a signal at 18.90 ppm in the cis isomer.
The cis to trans isomerization conversion highlighted by LC-MS analysis was further investigated using NMR. The composition of the LC mobile phase was recreated using deuterated solvents (acetonitrile-d3:D2O [1:1] with 0.1% formic acid-d2). Both, cis and trans isomers were dissolved in the deuterated mobile phase and analyzed at t = 0 h, t = 6 h, t = 12 h and t = 24 h. From the 1H NMR time study, it was evident that the cis to trans conversion process was occurring when the compound was in solution (Figure 4). The signal representing the protons on the methyl group in the cis isomer (1.28 ppm) reduced over time whereas the signal representing the same protons in the trans isomer (1.78 ppm) increased. The signal response related to the proton attached to the carbon at position four of the oxazoline ring (4.94 ppm) was seen to decrease in the cis isomer while increasing in the case of the trans isomer (4.60 ppm). The signal representing the proton attached to the carbon at position five of the oxazoline ring in the trans isomer (5.85 ppm) was increasingly formed while signal intensity representing the same proton in the cis isomer (6.48 ppm) decreased.
Figure 4.
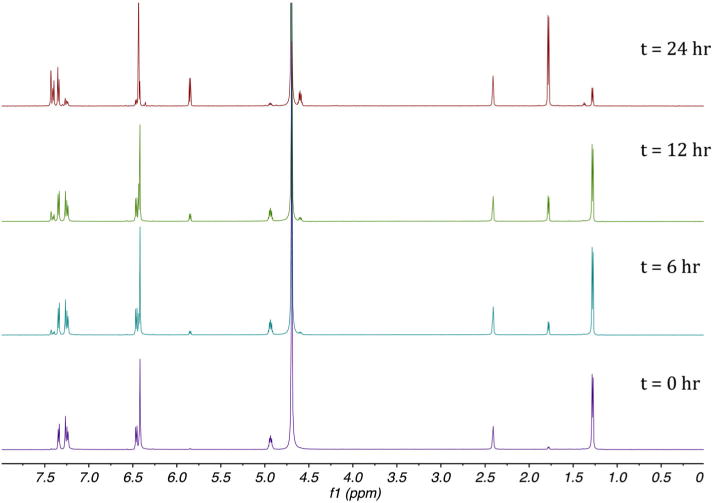
1H NMR data obtained for the cis to trans conversion of MDMAR monitored over 24 h.
Monoamine transporter activity
Figure 5 shows the dose-response effects of cis-DMAR, trans-DMAR, cis-MDMAR and trans-MDMAR on transmitter release at DAT, NET, and SERT. Table 1 summarizes potency values at concentrations producing 50% of maximal release (EC50 concentration) for the test drugs based on data depicted in Figure 5. All of the ring-substituted 4-methylaminorex analogues displayed potent substrate-type releasing activity at DAT, with EC50 values ranging from 10.2 ± 1.2 nM for cis-MDMAR to 36.2 ± 3.6 nM for trans-MDMAR. The drugs were also potent releasers at NET, with EC50 values ranging from 11.8 ± 2.0 nM for cis-DMAR to 38.9 ± 4.7 nM for trans-MDMAR. All of the 4-methylaminorex analogues were more potent than MDMA as substrates at DAT and NET (Table 1). Activity at SERT varied from 17.7 ± 2.3 nM for cis-DMAR to 73.4 ± 12.0 nM for trans-MDMAR. The test drugs were fully efficacious in their ability to evoke release at DAT, NET and SERT (drug effects achieved 100% of maximal release), with the exception of trans-DMAR, which functioned as a ‘partial’ releaser at SERT (75% maximal release). Subsequent experiments revealed that trans-DMAR is fully efficacious as an uptake blocker at SERT (data not shown), suggesting this drug displays ‘hybrid’ activity across monoamine transporters. It is proposed that trans-DMAR acts as a substrate-type releaser at DAT and NET, but an uptake blocker at SERT. When considering the transporter selectivity of the test compounds, all were essentially non-selective, with DAT/SERT ratios ranging from 0.6 for MDMA to 4.3 for cis-DMAR.
Figure 5.
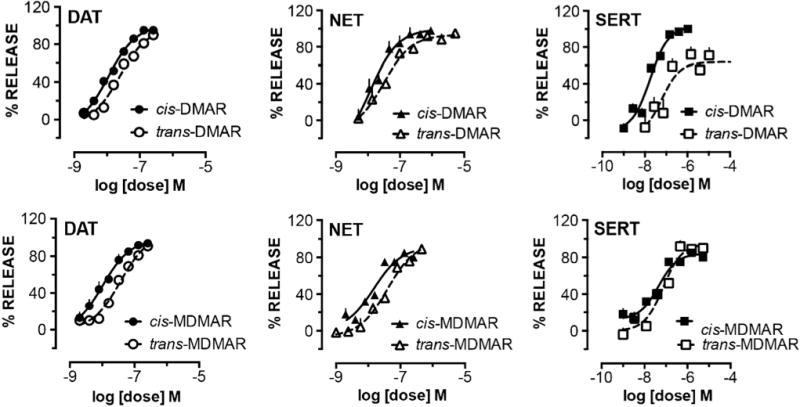
Dose-response effects of cis-DMAR, trans-DMAR, cis-MDMAR and trans-MDMAR on transmitter release at DAT, NET, and SERT. Data are mean ± SD for N = 3–4 experiments performed in triplicate.
Table 1.
Summary of the potency values (EC50 concentration) for the test drugs based on data depicted in Figure 5
| Drug | a Release at DAT EC50 (nM) | a Release at NET EC50 (nM) | a Release at SERT EC50 (nM) | b DAT/SERT ratio |
|---|---|---|---|---|
| (+)-MDMA | 143 ± 16 | 98.3 ± 15.0 | 85.0 ± 13.3 | 0.6 |
| Cis-DMAR | 10.9 ± 0.7 | 11.8 ± 2.0 | 17.7 ± 2.3 | 1.6 |
| Trans-DMAR | 24.4 ± 2.7 | 31.6 ± 4.6 | 59.9 ± 17.2 | 2.5 |
| Cis-MDMAR | 10.2 ± 1.2 | 14.8 ± 2.7 | 43.9 ± 6.7 | 4.3 |
| Trans-MDMAR | 36.2 ± 3.6 | 38.9 ± 4.7 | 73.4 ± 12.0 | 2.0 |
Data are expressed as mean ± SD for N = 3–4 experiments performed in triplicate
DAT/SERT ratio calculated by (EC50 at DAT)−1/(EC50 at SERT)−1; higher value indicates greater DAT selectivity
cis-DMAR, trans-DMAR, cis-MDMAR and trans-MDMAR were potent and efficacious substrate-type releasers at DAT and NET in rat brain tissue. Importantly, all of the ring-substituted 4-methylaminorex analogues were more potent than MDMA as catecholamine releasers. cis-DMAR, cis-MDMAR and trans-MDMAR were fully efficacious releasing agents at SERT as well, while trans-DMAR displayed partial releasing activity at this transporter. It is hypothesized that trans-DMAR displays the unusual profile of a catecholamine releaser with 5-HT uptake blocking properties, but further experiments are needed to confirm this proposal. The potencies for cis and trans racemates of DMAR and MDMAR were generally similar at each transporter, indicating a minimal influence of stereoselectivity in determining drug-transporter interactions across this group of structures. It seems worth mentioning that all of the 4-methylaminorex analogues tested here had a profile of transporter releasing activity that mimics the effects of MDMA, which is a non-selective transporter releaser, but the methylaminorex compounds were more potent.
Confirmation of cis-MDMAR in vendor sample
The sample donated from an online vendor and thought to contain MDMAR was subjected to identical analytical conditions. GC-MS studies showed that the online vendor product consisted predominantly of the cis MDMAR isomer (90%). The retention time (7.97 min) and fragmentation pattern of the vendor sample was congruent with that of the synthesized cis MDMAR standard. LC-MS studies, which included the comparison with the synthesized cis standard, also confirmed the presence of cis MDMAR in the vendor sample. Inspection of the LC chromatogram derived from the donated sample also suggested that the product contained a significant amount of the trans isomer. However, based on the observations described and investigated above, it was determined that this detection of the trans isomer was formed artificially following exposure to the water-containing mobile phase. Analysis by GC-MS, on the other hand, did not result in isomerization and detected the trans species at a much less significant level (10%). The 1H and 13C NMR spectra were in agreement with the GC and LC data confirming the presence of cis MDMAR in the vendor product.
Conclusion
The preparation and analytical characterization of cis- and trans-3′,4′-methylenedioxy-4-methylaminorex (MDMAR) demonstrated facile differentiation between both isomers but also showed a potential for misinterpretation under HPLC conditions when it was observed that the aqueous mobile phase caused isomerization of the cis- to the trans form. The high potency of ring-substituted methylaminorex analogues and their ability to be fully efficacious substrate-type releasers might contribute to the possibility of a range of serious side effects after high dose exposure and/or when combined with other substances that act on similar targets. Psychotic symptoms, agitation and hyperthermia could result from overstimulation of central dopamine and 5-HT systems, whereas dangerous cardiovascular effects could be produced by excessive norepinephrine release in the periphery.[28,29] The comparison between the monoamine transporter activity of cis- and trans-4,4′-dimethylaminorex pointed towards the possibility that the trans-species might display ‘hybrid’ activity across monoamine transporters. This is the first report on the characterization of MDMAR, which demonstrates the continuous need to remain vigilant on the availability of newly emerging psychoactive substances.[30]
Supplementary Material
Acknowledgments
Portions of this research were generously supported by the National Institute on Drug Abuse, National Institutes of Health, USA.
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article at the publisher’s web site.
References
- 1.EMCDDA–Europol. Joint Reports. Publications Office of the European Union; Luxembourg: 2014. Joint Report on a new psychoactive substance: 4,4′-DMAR (4-methyl-5-(4-methylphenyl)-4,5- dihydrooxazol-2-amine) Available at: http://www.emcdda.europa.eu/attachements.cfm/att_229825_EN_TDAS14006ENN.pdf [21 July 2014] [Google Scholar]
- 2.Cosbey S, Kirk S, McNaul M, Peters L, Prentice B, Quinn A, Elliott SP, Brandt SD, Archer RP. Multiple fatalities involving a new designer drug: para-methyl-4-methylaminorex. J Anal Toxicol. 2014;38:383. doi: 10.1093/jat/bku031. [DOI] [PubMed] [Google Scholar]
- 3.Council of the European Union. Request for risk assessment on new psychoactive substance 4,4′-DMAR (4-methyl-5-(4-methylphenyl)-4,5-dihydrooxazol-2-amine) 2014 Available at: http://register.consilium.europa.eu/doc/srv?l=EN&f=ST106192014INIT [21 July 2014]
- 4.Davis FT, Brewster ME. A fatality involving U4Euh, a cyclic derivative of phenylpropanolamine. J Forensic Sci. 1988;33:549. [PubMed] [Google Scholar]
- 5.Brandt SD, Baumann MH, Partilla JS, Kavanagh PV, Power JD, Talbot B, Twamley B, O’Brien J, Mahony O, Elliott SP, Archer RP, Patrick J, Singh K, Dempster NM, Cosbey SH. Characterization of a novel and potentially lethal designer drug, (±)-cis-para-methyl-4-methylaminorex (4,4′-DMAR, or ’Serotoni’) Drug Test Anal. 2014;6:684. doi: 10.1002/dta.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poos GI, Carson JR, Rosenau JD, Roszkowski AP, Kelley NM, McGowin J. 2-Amino-5-aryl-2-oxazolines. Potent new anorectic agents. J Med Chem. 1963;6:266. doi: 10.1021/jm00339a011. [DOI] [PubMed] [Google Scholar]
- 7.Carson JR, Poos GI, Almond HR., Jr 2-Amino-5-aryl-2-oxazolines. Tautomerism, stereochemistry, and an unusual reaction. J Org Chem. 1965;30:2225. [Google Scholar]
- 8.Follath F, Burkart F, Schweizer W. Drug-induced pulmonary hypertension? Br Med J. 1971;1:265. doi: 10.1136/bmj.1.5743.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurtner HP. Aminorex and pulmonary hypertension. A review. Cor Vasa. 1985;27:160. [PubMed] [Google Scholar]
- 10.United Nations. Convention on Psychotropic Substances. 1971 Available at: http://www.unodc.org/pdf/convention_1971_en.pdf [25 March 2014]
- 11.EMCDDA-Europol. Dangerous synthetic drugs hit the EU market. Available at: http://www.emcdda.europa.eu/news/2014/europol-emcdda1 [05 March 2014]
- 12.Rothman RB, Ayestas MA, Dersch CM, Baumann MH. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates. Implications for primary pulmonary hypertension. Circulation. 1999;100:869. doi: 10.1161/01.cir.100.8.869. [DOI] [PubMed] [Google Scholar]
- 13.Rothman RB, Baumann MH. Monoamine transporters and psychostimulant drugs. Eur J Pharmacol. 2003;479:23. doi: 10.1016/j.ejphar.2003.08.054. [DOI] [PubMed] [Google Scholar]
- 14.Hofmaier T, Luf A, Seddik A, Stockner T, Holy M, Freissmuth M, Ecker GF, Schmid R, Sitte HH, Kudlacek O. Aminorex, a metabolite of the cocaine adulterant levamisole, exerts amphetamine like actions at monoamine transporters. Neurochem Int. 2014;73:32. doi: 10.1016/j.neuint.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumann MH, Wang X, Rothman RB. 3,4-Methylenedioxymetham phetamine (MDMA) neurotoxicity in rats: A reappraisal of past and present findings. Psychopharmacology (Berl) 2007;189:407. doi: 10.1007/s00213-006-0322-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.APEX2 program suite. Bruker AXS Inc.; Madison, Wisconsin, USA: 2012. [Google Scholar]
- 17.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 18.Rothman RB, Vu N, Partilla JS, Roth BL, Hufeisen SJ, Compton-Toth BA, Birkes J, Young R, Glennon RA. In vitro characterization of ephedrine-related stereoisomers at biogenic amine transporters and the receptorome reveals selective actions as norepinephrine transporter substrates. J Pharmacol Exp Ther. 2003;307:138. doi: 10.1124/jpet.103.053975. [DOI] [PubMed] [Google Scholar]
- 19.Baumann MH, Ayestas MA, Jr, Partilla JS, Sink JR, Shulgin AT, Daley PF, Brandt SD, Rothman RB, Ruoho AE, Cozzi NV. The designer methcathinone analogs, mephedrone and methylone, are substrates for monoamine transporters in brain tissue. Neuropsychopharmacology. 2012;37:1192. doi: 10.1038/npp.2011.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serotoni. Available at: http://serotoni.info [22 July 2014]
- 21.Cooper DA. In: Proceedings of the international symposium on the forensic aspects of controlled substances: March 28 – April 1, 1988. Castonguay RT, editor. Laboratory Division, Federal Bureau of Investigation, U.S. Dept of Justice; Washington, DC: 1988. p. 79. [Google Scholar]
- 22. (Thread: Substituted Aminorex Series).Bluelight. 2008 Available at: http://www.bluelight.org/vb/threads/367053-Substituted-Aminorex-Series [28 May 2014]
- 23.Fodor G, Koczka K. 155. The stereochemical course of the conversion of 2-ureidoalcohols into oxazolidines. Part I. J Chem Soc. 1952:850. [Google Scholar]
- 24.Rodriguez WR, Allred RA. Synthesis of trans-4-methylaminorex from norephedrine and potassium cyanate. Microgram J. 2005;3:154. [Google Scholar]
- 25.Cruz A, Padilla-Martínez II, García-Báez EV, Contreras R. Reactivity of chlorodeoxypseudoephedrines with oxo-, thio-, and selenocyanates. Tetrahedron: Asymmetry. 2007;18:123. [Google Scholar]
- 26.Klein RFX, Sperling AR, Cooper DA, Kram TC. The stereoisomers of 4-methylaminorex. J Forensic Sci. 1989;34:962. [Google Scholar]
- 27.Henderson GL, Harkey MR, Chueh YT. Metabolism of 4-methylaminorex (“EU4EA”) in the rat. J Anal Toxicol. 1995;19:563. doi: 10.1093/jat/19.7.563. [DOI] [PubMed] [Google Scholar]
- 28.Hill SL, Thomas SH. Clinical toxicology of newer recreational drugs. Clin Toxicol (Phila) 2011;49:705. doi: 10.3109/15563650.2011.615318. [DOI] [PubMed] [Google Scholar]
- 29.Schep LJ, Slaughter RJ, Beasley DM. The clinical toxicology of metamfetamine. Clin Toxicol (Phila) 2010;48:675. doi: 10.3109/15563650.2010.516752. [DOI] [PubMed] [Google Scholar]
- 30.Brandt SD, King LA, Evans-Brown M. The new drug phenomenon. Drug Test Anal. 2014;6:587. doi: 10.1002/dta.1686. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.