

Abstract
Vesiculoviruses enter cells by membrane fusion, driven by a large, low‐pH‐induced, conformational change in the fusion glycoprotein G that involves transition from a trimeric pre‐fusion toward a trimeric post‐fusion state via monomeric intermediates. Here, we present the structure of the G fusion protein at intermediate pH for two vesiculoviruses, vesicular stomatitis virus (VSV) and Chandipura virus (CHAV), which is responsible for deadly encephalopathies. First, a CHAV G crystal structure shows two intermediate conformations forming a flat dimer of heterodimers. On virions, electron microscopy (EM) and tomography reveal monomeric spikes similar to one of the crystal conformations. In solution, mass spectrometry shows dimers of G. Finally, mutations at a dimer interface, involving fusion domains associated in an antiparallel manner to form an intermolecular β‐sheet, affect G fusion properties. The location of the compensatory mutations restoring fusion activity strongly suggests that this interface is functionally relevant. This work reveals the range of G structural changes and suggests that G monomers can re‐associate, through antiparallel interactions between fusion domains, into dimers that play a role at some early stage of the fusion process.
Keywords: conformational change, glycoprotein, intermediate structures, membrane fusion, vesiculovirus
Subject Categories: Microbiology, Virology & Host Pathogen Interaction; Structural Biology
Introduction
Entry of enveloped viruses into their host cell requires fusion of the viral membrane with a cellular membrane. This step is mediated by viral glycoproteins, anchored in the viral membrane by a transmembrane (TM) domain, that undergo structural rearrangements from a pre‐ to a post‐fusion state upon interaction with specific triggers (e.g. a low pH environment and/or cellular receptors). During this conformational change, hydrophobic motifs (the so‐called fusion peptides or fusion loops) are exposed and interact with one or both of the participating membranes, resulting in their destabilization and merger. At the end of the refolding process, the fusion proteins are in a hairpin‐like post‐fusion structure, in which the fusion loops (or peptides) and TM domain are at the same extremity of the molecule and in the same membrane (Harrison, 2015). Viral fusion glycoproteins have been classified based on common structural motifs (Li & Modis, 2014; Harrison, 2015). Experimental data suggest that the membrane fusion pathway is very similar for all the enveloped viruses studied so far whatever the organization of their fusion machinery (Chernomordik & Kozlov, 2008).
The Rhabdoviridae are enveloped bullet‐shaped viruses that are widespread among a great variety of organisms including plants, insects, crustaceans, fishes, reptiles, and mammals (Rose & Whitt, 2001). This family includes VSV, the prototype of the vesiculovirus genus, as well as notable human pathogens, such as Chandipura virus (CHAV), a vesiculovirus responsible for deadly encephalopathies (Rao et al, 2004), and rabies virus (RABV), the prototype of the lyssavirus genus. In their membrane, rhabdoviruses have a single glycoprotein (G) that mediates both virus attachment to specific receptors and, after virion endocytosis and acidification of the endosome lumen, fusion between viral and endosomal membranes (Albertini et al, 2012b). It has been demonstrated that the low‐pH‐induced structural transition of rhabdovirus G is reversible (Doms et al, 1987; Gaudin et al, 1993) in contrast to what has been shown for fusion glycoproteins from other viral families (Gaudin, 2000). The reversibility is the consequence of the existence of a pH‐dependent thermodynamic equilibrium between different states of G (pre‐fusion trimer, flexible monomers, and post‐fusion trimer). At pH above 7.5, both pre‐fusion trimers and flexible monomers can be observed at the viral surface, whereas the equilibrium is shifted toward the post‐fusion trimer at low pH (Roche & Gaudin, 2002; Libersou et al, 2010; Albertini et al, 2012a). EM studies performed on VSV revealed that fusion is driven by two successive events involving the glycoprotein. First, fusion is initiated at the flat base of the virion. Second, glycoproteins located in the cylindrical part of the particle reorganize into regular arrays to complete the fusion reaction (Libersou et al, 2010).
The trimeric structures of both the pre‐ and post‐fusion states of a soluble form of the ectodomain of VSV G (VSV Gth, amino acid (aa) residues 1–422, generated by thermolysin‐limited proteolysis of viral particles) (Roche et al, 2006, 2007) and of the post‐fusion state of CHAV G ectodomain (CHAV Gth, aa residues 1–419, generated the same way as VSV Gth) (Baquero et al, 2015b) have been determined. The structures of post‐fusion trimers for herpesvirus glycoprotein gB (Heldwein et al, 2006; Backovic et al, 2009; Burke & Heldwein, 2015; Chandramouli et al, 2015) and baculovirus glycoprotein gp64 (Kadlec et al, 2008) show that they and vesiculovirus G define the class III of viral fusion glycoproteins (Baquero et al, 2015a).
The polypeptide chain of G ectodomain folds into three distinct domains (Baquero et al, 2013) (Table 1 and Fig EV1). The fusion domain (FD, in yellow) is made of an extended β‐sheet structure at the tip of which are located the two fusion loops (in pink). FD is inserted in a loop of a pleckstrin homology domain (PHD, in orange) that is itself inserted into a central domain (CD, in red), which is involved in trimerization of the molecule in both the pre‐ and post‐fusion states. The CD connects to the segment R5 (in purple), which itself connects to the C‐terminal TM domain in the full‐length glycoprotein. Segments R1 and R4 (in cyan) connect CD to PHD, while R2 and R3 (in green) connect PHD to FD.
Table 1.
Domains and segments nomenclature used in the text
| Part of the molecule | Color | VSV residues | CHAV residues |
|---|---|---|---|
| CD | Red | 1–17 and 273–382 | 1–17 and 277–386 |
| PHD | Orange | 36–46 and 181–259 | 36–45 and 183–262 |
| FD | Yellow | 53–172 | 55–170 |
| R1 | Cyan | 18–35 | 18–35 |
| R2 | Green | 47–52 | 46–54 |
| R3 | Green | 173–180 | 171–182 |
| R4 | Cyan | 260–272 | 263–276 |
| R5 | Purple | 383–413 | 387–410 |
Figure EV1. Structure of the pre‐ and post‐fusion states of vesiculovirus G ectodomain.
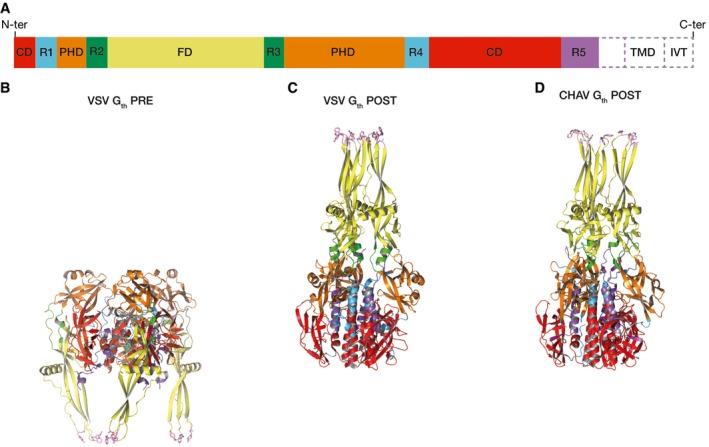
- Domain organization of vesiculovirus G ectodomain polypeptide chain. TMD: Transmembrane domain; IVT: Intraviral tail.
- Ribbon diagram of VSV Gth in its trimeric pre‐fusion conformation.
- Ribbon diagram of VSV Gth in its trimeric post‐fusion conformation.
- Ribbon diagram of CHAV Gth in its trimeric post‐fusion conformation.
In the trimers (Fig EV1), Gth pre‐ and post‐fusion states of a protomer (respectively referred to as PRE and POST hereafter) are related by flipping both FD and the C‐terminal segment around CD (Roche et al, 2007) thanks to refolding of segments R1 to R5. The complex conformational change involves monomeric intermediates (Albertini et al, 2012a), but the pathway of the transition remains unknown.
Here, we report a new crystal structure of CHAV Gth. The crystal contains two distinct conformations, apparently corresponding to early and late refolding states of G, arranged in a flat dimer of heterodimers exposing fusion loops. At the viral surface and in the absence of target membrane, under appropriate pH and temperature conditions, electron microscopy (EM) and tomography revealed monomeric spikes that are in a conformation similar to one of the protomers present in the crystalline structure. Through native mass spectrometry (MS), we provide evidences that dimeric assemblies of both VSV and CHAV Gth are present in solution at intermediate pH. Furthermore, mutations of residues located at a crystalline dimeric interface, formed by two FDs laterally associated in an antiparallel manner, affect the fusion properties. The location of the compensatory mutations restoring the fusion activity strongly suggests that this interface is functionally relevant. Taken together, these data offer a detailed pathway for the conformational transition from pre‐ to post‐fusion trimers. They further suggest that flat dimers, associated through their FDs, play a role at some early stage of the fusion process. Therefore, this work provides the molecular basis of a more detailed model for vesiculovirus membrane fusion.
Results
A new crystal structure of a vesiculovirus glycoprotein
CHAV Gth was generated as previously described (Baquero et al, 2012, 2015b). Crystals of CHAV Gth were grown at pH 7.5, at which VSV Gth was shown to assume a range of intermediate monomeric conformations in solution (Albertini et al, 2012a). The structure of CHAV Gth was determined to near 3‐Å resolution by molecular replacement (see Materials and Methods and Table 2). The asymmetric unit contains four CHAV Gth molecules assembled via twofold noncrystallographic symmetry (Fig 1A and B). The four molecules exhibit two very different conformations (Fig 1A, B, D and E) that are distinct from those of VSV G PRE and CHAV/VSV G POST (Fig 1C and F).
Table 2.
Statistics of data collection and refinement
| Resolution range (Å)a | 50–3.0 (3.1–3.0) |
| Space group and unit cell parameters (Å) |
P21 21 2 a = 150.3, b = 228.2, c = 78.8 |
| No. of unique reflectionsa | 55,165 (5,336) |
| Completeness (%)a | 99.8 (99.0) |
| Redundancya | 6.7 (6.8) |
| <I/σ(I)>a | 16.7 (1.5) |
| R sym (%)a , b | 9.0 (125.3) |
| Wilson B factor (Å2) | 84.6 |
| No. of reflections used in refinement | 55,157 |
| No. of atoms (total/protein/solvent) | 12,825/12,820/5 |
| Final R work a , c | 18.8 (29.6) |
| No. of reflections for R free | 2,759 |
| Final R free a , c | 22.2 (33.6) |
| Rmsd, bond (Å) | 0.004 |
| Rmsd, angle (degrees) | 0.80 |
| B factor (average/min/max, Å2) | 112.2/44.9/360.8 |
| Ramachandran plot analysis | |
| Most favored regions (%) | 97.0 |
| Additionally allowed regions (%) | 2.9 |
| Disallowed regions (%) | 0.1 |
In parentheses, statistics for the highest resolution shell.
Figure 1. Intermediate CHAV Gth conformations and comparison with previous VSV and CHAV Gth crystal structures.
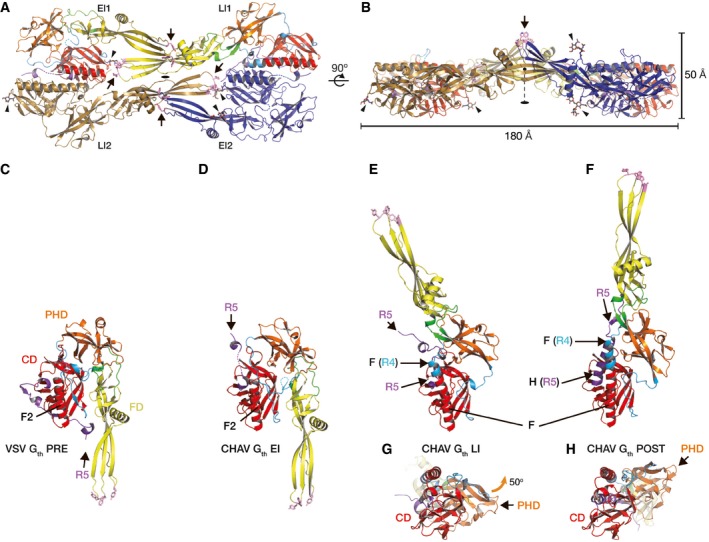
-
A, BTwo views of the tetrameric assembly in the CHAV Gth crystal rotated by 90°. One of the protomers in the early intermediate conformation (CHAV Gth EI2) is colored in blue, and one of the protomers in the late intermediate conformation (CHAV Gth LI2) is colored in brown. The two other protomers are colored by domains and segments as defined in Table 1. The hydrophobic residues of the fusion loops (in pink, indicated by arrows) and the glycan chains (indicated by arrowheads) are in sticks representation. The twofold symmetry axis of the dimer of heterodimers is indicated.
-
C–HOverall structures of VSV Gth protomer in the pre‐fusion conformation PRE (C), CHAV Gth early refolding intermediate EI (D), CHAV Gth late refolding intermediate LI (E, G), and CHAV Gth protomer in the post‐fusion conformation POST (F, H). The proteins are superposed on their CD viewed from the side (C–F) or from the bottom with the FD in the back of the figure (G, H). When present, helices F2, F, and H are indicated as well as the position of segment R5. The orange arrow in (G) indicates the 50° rotation of the PHD upon refolding of G from LI to POST.
The first conformation (Fig 1D) can be traced up to residue 405 (corresponding to residue 401 of VSV G, Appendix Fig S1), with two breaks where density is too weak to be traced (residues 269–270 in R4 and 394–398 in R5, Appendix Fig S2A). This molecule is broadly superimposable with VSV G PRE (Fig 1C) with CD, PHD, and FD in the same relative orientations. This indicates that the CHAV PRE conformation is very likely the same as VSV PRE, just as CHAV POST is the same conformation as VSV POST (Baquero et al, 2015b). There is however a single obvious difference: The R5 segment has left the hydrophobic groove it occupies in VSV G PRE (Fig 1D).
The second conformation (Fig 1E) can be unambiguously traced (Appendix Fig S2B) up to residue 410 (corresponding to residue 406 of VSV G, Appendix Fig S1). It is an elongated hairpin that is similar in shape to CHAV G POST (Fig 1F) but with a ~50° difference in orientation of PHD relative to CD (Fig 1G and H). Indeed, R2 and R3 have the same conformations as in CHAV G POST, but the refolding of R1 and R4 is still incomplete (Appendix Fig S1). Particularly, in segment R4, the central helix of LI (Fig 1E) is longer than F2 of PRE (Fig 1C), but is still shorter than F in POST (Fig 1F). Similarly, R5 is relocated (Fig 1E) but has not refolded into the post‐fusion lateral helix H (Fig 1F).
Those two new conformations of CHAV Gth are structural intermediates of the transition from PRE to POST. The first one (Fig 1D) is very close to PRE and thus is an early refolding intermediate. The second one (Fig 1E), which is closer to POST, is at a more advanced stage of the process and thus is a late refolding intermediate. The two new conformations will be further referred to as EI (early intermediate) and LI (late intermediate), respectively.
In the crystal asymmetric unit, the four CHAV Gth molecules (two EI, two LI) assemble through a combination of five interfaces (Appendix Fig S3) that together bury 10,650 Å2 of the four molecules' surfaces (Fig 1A and Appendix Fig S3A). First, there are two instances of a large interface between EI1 and LI2 and between EI2 and LI1 that buries about 3,400 Å2 of their CD and of segments in intermediate refolding states (R5 and R4 in LI, R5 in EI) (Appendix Fig S3D–F). In contrast, the other three interfaces involve contacts between FDs—one LI1‐LI2 interface (Appendix Fig S3B), two identical EI1‐LI1 and E12‐LI2 interfaces (Appendix Fig S3C)—and bury each about 1,200 Å2 in segments that keep the same structure in all conformations of G. Both LI1‐LI2 and EI‐LI FD interfaces involve strand d (residues 75–80 downstream of the first G fusion loop, Appendix Fig S1). In the EI1‐LI1 and EI2‐LI2 FD interfaces, strand d of EI actually forms an intermolecular β‐sheet with strand e of LI (residues 120–125 downstream of the second fusion loop, Appendix Figs S1 and S3C). The fusion loops of the two EI are exposed on the same side of the assembly and fully solvent accessible in this crystal packing (Fig 1B). Overall, the assembly thus formed is a flat structure with a thickness of 50 Å from the bottom of the four CDs to the top of the protruding EI fusion loops (Fig 1B).
EM reveals G monomers with LI‐like conformation at the viral surface on the path to the post‐fusion trimer
We next analyzed the structure of G at the surface of the virions, finding conditions to isolate structural intermediates during the transition. For this, we used negative staining EM on virions incubated at various pH and 20°C (Fig 2, Appendix Figs S4 and S5). The visualization of the rare intermediates at the viral surface requires a high contrast that is not achieved by cryo‐EM. Due to the weak stability of recombinant VSV‐CHAV G virions after negative staining, particularly at low pH, we chose to further characterize the structure of VSV G. Indeed, we had previously demonstrated that VSV G and CHAV G have the same behavior (Baquero et al, 2015b).
Figure 2. Morphology of VSV and CHAV glycoproteins at the surface of negatively stained virions.

-
A–EVSV particles were incubated at indicated pH and temperature. At pH 8, VSV G forms the characteristic ˜7‐nm‐wide layer (Libersou et al, 2010) (A). At lower pH, both elongated monomers (E) and post‐fusion trimers (B–D) are detected at the viral surface. In panel (D), the higher magnification of the red boxed region shows post‐fusion trimers in side views. In panel (E), the higher magnification of the blue boxed region shows thin elongated structures. Micrographs (A–E) are at the same scale (scale bar for 1,000 Å is shown in panel A).
-
F, GHigher magnification of spikes observed at pH 6.6 and 37°C (F, thick spikes of panel D) or pH 6.6 and 4°C (G, thin spikes of panel E). The length and the width of such spikes are indicated. All the measurements are given ± SD (n = 50).
-
H, ITomogram section of negatively stained VSV (in hydration‐preserving conditions) incubated at pH 5.5 and 20°C (H) or pH 6.6 and 4°C (I). Green arrows indicate post‐fusion trimers. The pink arrow indicates an elongated monomer.
-
J, KTomography reconstructions (gray mesh) of VSV G spikes obtained after negative staining in hydration‐preserving conditions at pH 6.6 and 37°C (J) or pH 6.6 and 4°C (K). In the left panels, the density is oriented such that the viral membrane is at the bottom of the figure. (J) The VSV G post‐fusion trimer was fitted with an R‐factor of 36.7% at 35‐Å resolution (Appendix Table S3). (K), The CHAV G LI was fitted in the VSV G thin spike density with an R‐factor of 35.1% at 35‐Å resolution (Appendix Table S4). The VSV G post‐fusion protomer's best fit (Appendix Fig S6) yielded a worse R‐factor of 43.6% (Appendix Table S4).
At pH 8, VSV G (Fig 2A, Appendix Fig S4A) forms the characteristic ~7‐nm‐wide layer, which has been previously demonstrated to result from the presence of both pre‐fusion trimers and flexible monomers at the viral surface (Libersou et al, 2010; Albertini et al, 2012a). At pH 6, we observed fusion of viral particles (Appendix Fig S4B) and all the spikes had the typical post‐fusion shape (Fig 2B). At pH 5.5, VSV G, in its trimeric post‐fusion conformation, forms a regular helical network (Fig 2C and Appendix Fig S4C).
To slow down the reaction and isolate intermediates, we characterized the structure of VSV G at the viral surface at intermediate pH (6.6). We performed experiments either at 37°C or at 4°C. Whereas after an incubation at 37°C, the post‐fusion trimer was the major species at the viral surface (Fig 2D and F, and Appendix Fig S5A and B), at 4°C, many thin and elongated spikes were observed. These spikes were sometimes oblique to the membrane (Fig 2E and Appendix Fig S5C), and their dimensions were consistent with those of LI (Fig 2G).
To further probe the nature of this elongated spike, we performed electron tomography (Movies EV1 and EV2) on frozen VSV samples for which hydration was partially preserved (see online methods) using cryo‐negative staining. When the virions were incubated at pH 5.5 and 37°C, sub‐volumes corresponding to the post‐fusion form were manually picked at the viral surface (Fig 2H). When aligned and averaged, these sub‐volumes display threefold symmetry and the post‐fusion trimer was fitted inside with good statistics (Fig 2J, Appendix Tables S1–S3).
On the samples that had been incubated at pH 6.6 and 4°C, sub‐volumes corresponding to the thinner spikes (having an elongated shape at the surface of the virus) were picked up (Fig 2I). They were aligned and averaged. In the averaged density, we were able to fit the CHAV LI structure (Fig 2K) with the fusion loops pointing toward the viral membrane. Fitting of VSV POST protomer resulted in significantly worse statistics (see legend of Fig 2, Appendix Fig S6 and Appendix Table S4).
EM thus revealed three different organizations of the spikes at the viral surface, which are (i) pre‐fusion trimers in equilibrium with poorly resolved monomers (above pH 7.5) (Libersou et al, 2010; Albertini et al, 2012a), (ii) LI‐like elongated monomers (at pH 6.6 and 4°C), and (iii) post‐fusion trimers. In these three organizations, viral particles remain separate and do not tend to cluster together. In contrast, in suboptimal fusion conditions known to favor glycoprotein conformations exposing fusion loops (at pH 6.2 and 4°C) (Durrer et al, 1995), aggregation is massively observed (Appendix Fig S5D). However, the size of the aggregates precludes a deeper structural analysis, and we could not identify the fourth organization responsible for the viral aggregation.
Evidence for dimeric species for both CHAV Gth and VSV Gth
In the crystal asymmetric unit, CHAV Gth molecules form a flat dimer of heterodimers (Fig 1A and B, Appendix Fig S3), whereas the oligomeric species of class III fusion proteins identified so far are symmetric protruding trimers (Heldwein et al, 2006; Roche et al, 2006, 2007; Kadlec et al, 2008; Backovic et al, 2009; Baquero et al, 2015b; Burke & Heldwein, 2015; Chandramouli et al, 2015; Zeev‐Ben‐Mordehai et al, 2016). This unexpected crystalline organization prompted us to investigate the oligomeric states of CHAV Gth in our preparation. For this, we used native MS (Boeri Erba & Petosa, 2015; Mehmood et al, 2015). CHAV Gth sample solutions were first desalted and buffer‐exchanged in 200 mM ammonium acetate at pH values 6.0, 7.5, and 8.8 (final protomer concentration: 7 μM). At pH 8.8, only monomers were detected. At pH 6.0, only trimers were observed. However, at pH 7.5, both monomeric and dimeric species were present (Fig 3A).
Figure 3. Evidence for dimers of CHAV Gth and VSV Gth in solution.
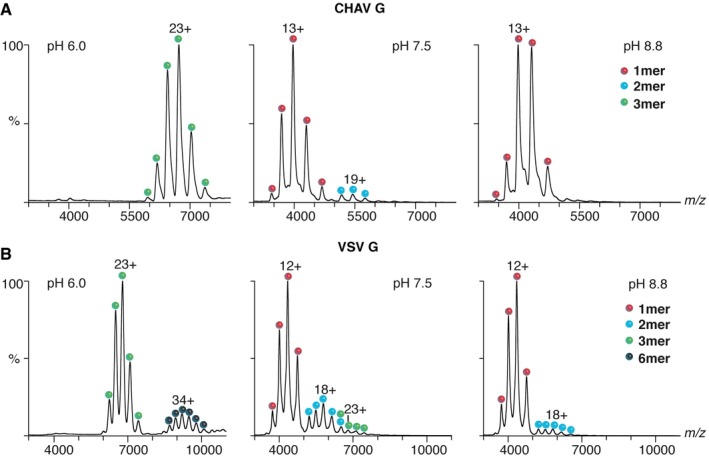
- Native mass spectra of CHAV Gth at three pH values (pH 6.0, 7.5 and 8.8). In acidic conditions, CHAV Gth is a trimer. At pH 7.5, both dimers and monomers were observed; at pH 8.8, only monomers were detected.
- Native mass spectra of VSV Gth. At pH 6.0, VSV Gth is detected as trimers and hexamers. At pH 7.5, trimers, dimers, and monomers are present; the trimeric ions with 24 charges (24+) have the same m/z of dimeric ions with 16 charges (16+) (m/z 6,462.6). At pH 8.8, VSV Gth forms monomers and dimers.
A similar analysis performed on VSV Gth revealed an analogous behavior (Fig 3B). A dimeric species was observed (along with a subpopulation of trimers) together with monomers at pH 7.5 and to a lesser extent at pH 8.8. In contrast, at pH 6.0, only trimers and hexamers (likely corresponding to two post‐fusion trimers associated through their fusion loops) were detected. Therefore, the native MS experiments revealed the ability of the vesiculovirus glycoprotein to form dimeric species around pH 7.5.
Mutations at the FD EI/LI interface of the intermolecular β‐sheet affect VSV G fusion properties and raise compensatory mutations
In the crystal, on one side of the intermolecular β‐sheet formed at the EI1‐LI1 (or EI2‐LI2) FD interface, there is a network of polar interactions, which involves Glu123 and Asp121 in LI and Lys76 in EI (Fig EV2A), three residues that were previously reported as sites of mutations that change the pH threshold for fusion for both VSV (Zhang & Ghosh, 1994; Fredericksen & Whitt, 1995, 1998) and viral hemorrhagic septicemia virus (VHSV, a fish rhabdovirus) (Gaudin et al, 1999) or to compensate for a deleterious mutation in the fusion loops (Stanifer et al, 2011; Table 3). While a molecular explanation for those phenotypes could not be deduced from the analysis of pre‐ and post‐fusion G conformations (Fig EV2B and C), the formation of antiparallel intermolecular contacts between adjacent fusion domains at some stage of fusion provides one. To test the relevance of this interface for fusion, we introduced mutations on either side in a cell–cell fusion assay and in recombinant viruses. We used VSV G to take advantage of the tools available for this virus (commercially available monoclonal antibodies and reverse genetics system). We targeted the conserved acidic residues in positions 121 and 123 on the LI side and the conserved His80 facing them on the EI side (Fig EV2A).
Figure EV2. An antiparallel contact between the EI and LI fusion domains.
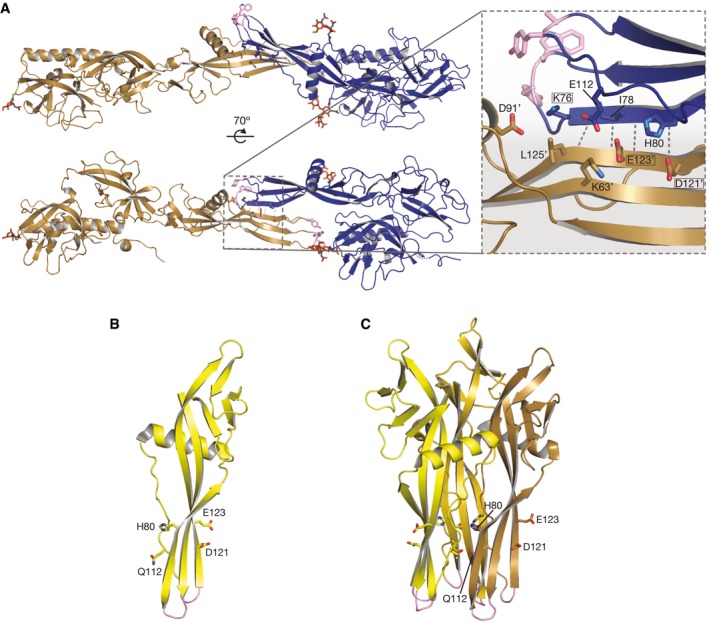
-
AViews of the antiparallel contact between the EI and LI fusion domains in the tetrameric crystal structure. CHAV Gth LI is in brown, and CHAV Gth EI is in blue. Hydrophobic residues in the fusion loops are in pink sticks. Glycan chains are also represented in a sticks model. The inset shows a zoom on one of the intermolecular EI/LI beta sheets. Sites of mutations previously reported to affect the fusion properties are boxed. Side chains that cluster together (including H80, E112, D121, E123) in the interface on the sheet's face opposite the EI's fusion loops are shown as sticks and labeled, with for LI residues.
-
B, CLocalization of residues H80, E112, D121, E123 in the fusion domain in the pre‐fusion (B) and post‐fusion (C) conformations of VSV G.
Table 3.
Fusion properties of mutants in residues bridging the intermolecular EI/LI β‐sheet
| Virus/mutationa | Corresponding CHAV G residuesa | Phenotype |
|---|---|---|
| VSV/D137Lb | D121 | Fusion less efficient than WT |
| VSV/D137Nc | D121 | Fusion poorly efficient, detected only below pH 5.5 |
| VSV/E139Lb | E123 | Fusion less efficient than WT |
| VHSV/Q118Rg, d | K76 | Shift in the pH threshold for membrane fusion |
| VSV/E76Ke | K76 | Compensatory mutation rescuing nonfusogenic VSV G with lethal mutation A117F |
|
VSV/H80Af
VSV/H80Ef |
H80 | No fusion detected |
| VSV D121L/E123Lf | D121/E123 | No fusion detected |
| VSV/H80Kf | H80 | Mutation rescuing nonfusogenic VSV G with lethal mutations H80E |
| VSV/Q112Pf | E112 | Compensatory mutation rescuing nonfusogenic VSV G with lethal mutations H80A or D121L/E123L |
Numbering of residues corresponds to that used by the authors in the original publications in the first column, to CHAV G numbering used in this work in the second column. Inconsistent numbering in places is due to the length of the signal peptide being counted in some of the original publications.
Referenced in Fredericksen and Whitt (1995).
Referenced in Zhang and Ghosh (1994).
Referenced in Gaudin et al (1999).
Referenced in Stanifer et al (2011).
This work.
Three strains sharing only this mutation from the parental VHSV strain (07–71) all show a shift of their fusion pH (Gaudin et al, 1999).
First, we replaced His80 by either an alanine or a glutamic acid residue. Mammalian cells were transfected with pCAGGS plasmids encoding wild‐type or mutant VSV G glycoproteins (WT, H80A or H80E). Protein expression and cell surface localization of G proteins were assessed by indirect immunofluorescence microscopy on BSR cells using Mab 8G5F11 directed against G ectodomain. For both mutants, the glycoprotein was detected at the surface of nonpermeabilized cells (Fig EV3). However, the pattern of fluorescence of permeabilized cells was slightly different, revealing a more intense perinuclear fluorescence pattern for mutant glycoproteins (Fig EV3).
Figure EV3. Effect of mutations of residues H80, D121, and E123 on VSV G transport and surface expression in mammalian cells.
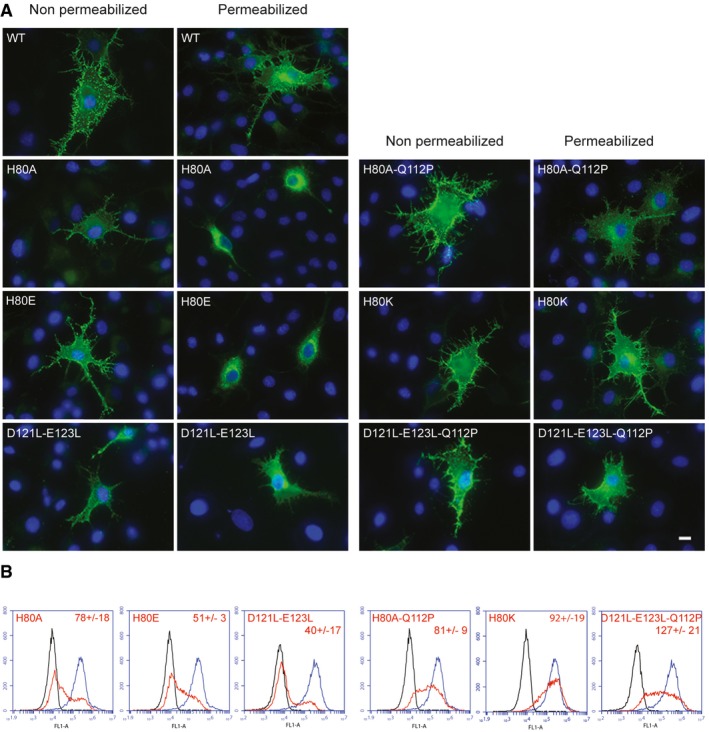
- Expression and subcellular localization of G proteins (WT and mutants) in BSR cells. Epifluorescence images of transfected cells. At 24 h post‐transfection, cells were permeabilized (or not) and fixed and G was detected by immunostaining with a monoclonal antibody to VSV G (KeraFAST, 8G5F11), which was subsequently detected by using a goat anti‐mouse IgG secondary antibody conjugated to Alexa Fluor 488. Nuclei were stained with DAPI. All the images are at the same scale, scale bar (bottom right): 10 μm.
- Surface expression of G in HEK cells. The surface G protein expression was detected with a monoclonal antibody to VSV G (KeraFAST, 8G5F11) and a goat anti‐mouse IgG secondary antibody conjugated to Alexa Fluor 488. A total of 10,000 cells were counted by flow cytometry. In each frame, the black curve corresponds to the fluorescence of untransfected cells, the blue (resp. red) one corresponds to the fluorescence of cells transfected by pCAGGS plasmids encoding WT G (resp. indicated mutant). Experiments were performed three times, and the data were used to determine the average level of surface expression for positive cells (indicated for each mutant ± SD). Although the percentage of cells expressing G at their surface is lower for the mutants, the average levels of surface expression for positive cells are similar to wild type (varying from 40 to 127% of wild type).
The amount of G protein present on the surface of HEK cells was also quantified using flow cytometry. The proportion of cells expressing G at their surface was smaller for both mutants when compared to WT (Fig EV3). When G was detected, the surface expression of mutant glycoproteins H80E and H80A was equal to 51 and 78% of WT VSV G, respectively (Fig EV3). Taken together, these data indicated that the mutant glycoproteins, being able to leave the endoplasmic reticulum (ER), have the characteristics of folded proteins (Hammond & Helenius, 1995).
As previously observed (Stanifer et al, 2011; Ferlin et al, 2014), cells expressing wild‐type G protein formed massive syncytia when exposed to low pH, between 5.5 and 6.3. Only small syncytia were observed at pH up to 7. The replacement of His80 by Glu or Ala led to complete inhibition of cell–cell fusion, even at pH as low as 5 (Fig 4). This phenotype cannot be attributed to the lower expression of G at the cell surface since mutants with similar surface expression levels have been previously shown to keep the fusion activity (Ferlin et al, 2014).
Figure 4. Effect of mutations of residues H80, D121, and E123 on VSV G fusion properties in mammalian cells.
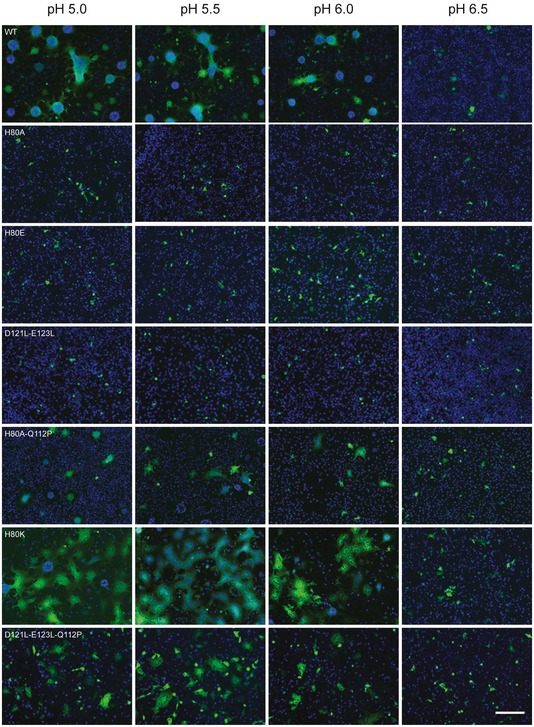
Fusion activity of WT and VSV G mutants analyzed in a cell–cell fusion assay. BSR cells were transfected with plasmids expressing VSV G (either WT or mutant) and P‐GFP (a protein that is exclusively located in the cytoplasm of transfected cells) allowing an easy observation of syncytia. Twenty‐four hours post‐transfection, the cells were exposed for 10 min to DMEM adjusted to the indicated pH, which was then replaced by DMEM at pH 7.4. The cells were then kept at 37°C for 1 h before fixation. Nuclei were stained with DAPI. All the images are at the same scale, scale bar (bottom right): 200 μm.
We then generated recombinant VSV containing mutations H80E and H80A (VSV G.H80E and VSV G.H80A) employing a complementation strategy to support their growth (Jeetendra et al, 2003; Stanifer et al, 2011; Ferlin et al, 2014). Briefly, the recovery of those recombinant viruses was supported by expression of functional WT VSV G protein from a transfected plasmid. To generate virus particles containing only the fusion‐defective VSV G expressed from the viral genome, viruses present in the supernatant were amplified in cells that lack the trans‐complementing VSV G plasmid. Using this approach, we recovered infectious viruses for each mutant. In both cases, sequence analyses revealed a single additional coding change in the genome.
In the case of mutant H80A, the additional coding change resulted in the replacement of Q112 by a proline leading to the double mutant VSV G.H80A/Q112P. In CHAV Gth, the residue corresponding to VSV Q112 is E112, which, in the EI protomer, lies in the middle of the EI‐LI polar network between E123 of LI and K76 of EI (Fig EV2A). In the case of mutant H80E, the additional coding change in the genome resulted in a lysine in position 80, leading to virus VSV G.H80K, which might restore a positive charge at the edge of the EI‐LI polar network.
We analyzed the transport (Fig EV3) and fusion properties (Fig 4) of mutant glycoprotein H80K and of the double mutant H80A/Q112P. The phenotype of mutant H80K was indistinguishable from WT G. On the other hand, the double mutant H80A/Q112P was slightly less efficiently transported to the cell surface (cell surface expression was equal to 81% of WT) but syncytia were clearly observed from pH 5 to pH 6 although they were smaller than those observed with WT G.
In a reciprocal manner, we constructed a double mutant D121L/E123L. The surface expression of this mutant was equal to 40% of WT VSV G (Fig EV3). This mutant was unable to form syncytia even at pH 5 (Fig 4). We also generated a recombinant VSV containing the double mutation D121L/E123L (VSV G.D121L/E123L) employing the same complementation strategy as described above. We recovered infectious viruses, and sequence analysis revealed a single additional coding change in the genome, which, once again, resulted in the replacement of Q112 by a proline leading to the triple mutant VSV G.D121L/E123L/Q112P.
We analyzed the transport (Fig EV3) and fusion properties (Fig 4) of this triple mutant D121L/E123L/Q112P. It was efficiently transported to the cell surface (cell surface expression was equal to 127% of WT) (Fig EV3) and syncytia were clearly observed from pH 5 to pH 6 although they were smaller than those observed with WT G (Fig 4).
Therefore, the same compensatory mutation Q112P rescues mutations of residues located on either side of the EI/LI FD interface. The genetics data thus link D121/E123 to H80, two locations that are clustered in the EI‐LI FD interface (Fig EV2A) but scattered in all previously known oligomeric forms of G (Fig EV2B and C), even in the helical arrays made by the post‐fusion trimer due to the inward facing of H80 (Fig EV2C). This suggests that the antiparallel interaction between FDs leading to the formation of the intermolecular β‐sheet is functionally relevant to fusion.
Discussion
Crystal and EM structures of fusion glycoprotein ectodomains have mainly provided static pictures of pre‐ and post‐fusion conformations. An exception is the recent determination of intermediate conformations for class II viral fusion glycoproteins: an extended monomeric conformation of Rift Valley fever virus fusion glycoprotein (Dessau & Modis, 2013) and the structure of dengue virus glycoprotein E in a late‐stage fusion intermediate (Klein et al, 2013). The new crystal structure of CHAV Gth and the associated EM data, which are presented here, provide atomic views of the structural intermediates formed by the vesiculovirus glycoprotein during its conformational change. They also provide further evidence that the vesiculovirus structural transition from the pre‐fusion to the post‐fusion trimer involves monomeric intermediates (Albertini et al, 2012a). The EI and LI structures enlighten the pathway for G refolding during the pre‐fusion trimer to post‐fusion trimer transition.
A pathway for the pre‐fusion trimer to post‐fusion trimer transition
In the absence of a target membrane, it is reasonable to infer that the pre‐ to post‐fusion trimer transition proceeds through monomeric EI and LI conformations. Changes in the PRE–EI–LI–POST transition involve several successive partial refoldings of the segment connecting the ectodomain to the viral membrane (R5) and of the CD/PHD hinge (R1 and R4), and a single change in the PHD/FD hinge (R2 and R3, in the transition between EI and LI) (Fig 5).
Figure 5. Close‐up views of the hinge regions and refolding segments.
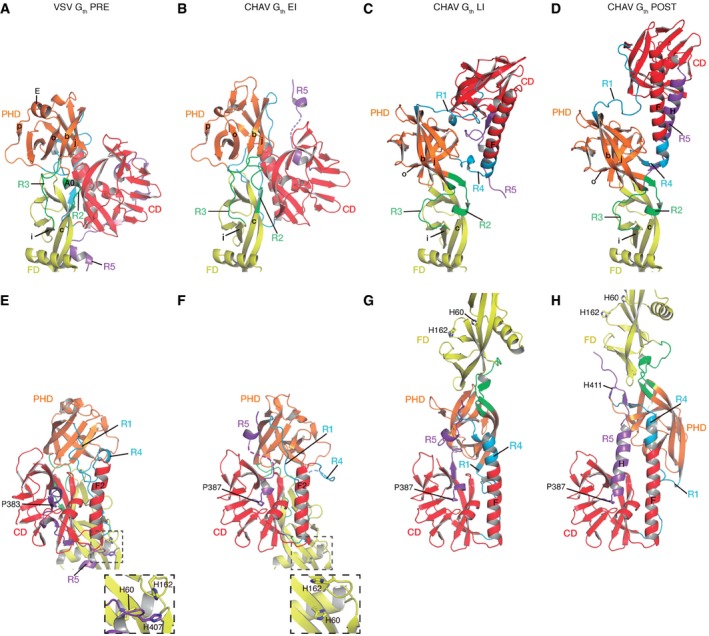
-
A–HVSV Gth protomer in the pre‐fusion conformation (A and E), CHAV Gth EI (B and F), CHAV Gth LI (C and G), and CHAV Gth protomer in the post‐fusion conformation (D and H) are colored by domains and segments as defined in Table 1. Top (A–D): Close‐up views of the hinge regions between PHD and FD (segments R2 and R3). Molecules are superposed on their FD. Secondary structure elements are named and numbered according to Appendix Fig S1. Bottom (E–H): Close‐up views of molecules superposed on their CD. The conserved histidine cluster is enlarged in the insets in (E) and (F). Note that in (F) this cluster is disrupted due to the repositioning of R5. The conformational transition starts with the switch of R5 at P383 (VSV)/P387 (CHAV).
The main change going from PRE (Fig 5A and E) to EI (Fig 5B and F) is the release of R5 segment, which leaves the hydrophobic groove located at the base of CD. This is likely assisted by protonation of the previously identified cluster of histidines (H60, H162, and H407) (Roche et al, 2007), which destabilizes the FD‐R5 interaction in PRE (Fig 5E). It should be noted that R5 is the only segment to adopt a different conformation in all the protomers characterized so far. At this stage, the release of R5 and its intrinsic flexibility confer orientational mobility to the rest of the molecule, even in the absence of any other conformational changes. The subsequent change between EI (Fig 5B and F) and LI (Fig 5C and G) includes refolding of segments R1 through R4 and thus both relocation of FD relative to PHD and relocation of PHD relative to CD. Unlike the former, the latter relocation is not completed at this stage (see above, Fig 1G and H) and involves partial refolding of both R1 and R4. The central helix of LI (Fig 5G) is thus longer than F2 of PRE and EI (Fig 5E and F), but shorter than F in POST (Fig 5H). Partial relocation of PHD, combined with complete relocation of FD, swings FD around CD (Fig 1E and F). As R5 is positioned in an antiparallel orientation relatively to central helix F (Fig 5C and G), LI is already an elongated hairpin. Going from LI to POST involves the final refolding of R1 and R4, together with refolding of R5 into helix H and post‐fusion trimer association (Fig 5C, D, G and H). At an intermediate pH (i.e. 6.6), this last refolding step seems to involve a high activation barrier as it is considerably slowed down at low temperature.
An antiparallel interaction between fusion domains required for fusion
In the crystal, the fusion domains of E11 and LI1 (or EI2 and LI2) are associated in an antiparallel manner to form an intermolecular β‐sheet (Fig EV2A). Residues on either side of this interface were previously reported as sites of mutations that change the pH threshold for fusion for both VSV (Zhang & Ghosh, 1994; Fredericksen & Whitt, 1995, 1998; Stanifer et al, 2011) and VHSV (Gaudin et al, 1999). However, a molecular explanation for the associated phenotypes could not be deduced from the analysis of pre‐ and post‐fusion G conformations.
Here, we show that, in VSV G, lethal mutations located on either side of the crystalline EI/LI FD interface are rescued by the same compensatory mutation Q112P. In a single FD, residue 112, being on the opposite side of the three‐stranded β‐sheet, is far from residues 121 and 123 (Fig EV2B). However, in the CHAV Gth crystal structure, it clusters with those residues (and His80) on the same side of the intermolecular β‐sheet (Fig EV2A). This strongly suggests that this antiparallel association of the fusion domains is functionally relevant. We confirm with native MS that G monomers can indeed associate to form dimeric assemblies. As those dimers are observed above pH 7, they are likely involved at some early stage of the fusion process. In the absence of direct structural data on the organization of G that promotes interaction between fusion loops and the target membrane, a clear‐cut explanation of the effect of the compensatory mutation Q112P is speculative. First, it may modify the orientation of the nearby fusion loops. Second, in the dimer context, it modifies the local hydrophobicity near the EI fusion loops by extending a hydrophobic patch present on the associated LI (Appendix Fig S3C). Third, it may also directly modulate the association between fusion domains.
The antiparallel character of such an assembly implies that at this stage, the fusion domains are parallel to the viral membrane (unlike their orthogonal orientation in pre‐ and post‐fusion trimers). This state is thus different from the elongated trimeric intermediate formed by both class I and class II fusion glycoproteins, which is orthogonal to the viral membrane (Sanchez‐San Martin et al, 2008; Baquero et al, 2013; Ivanovic et al, 2013; Pierson & Kielian, 2013; Chao et al, 2014; Jardetzky & Lamb, 2014).
Thus, the ability to form at least transiently stable monomeric molecules in intermediate conformations opens up a previously unsuspected possibility. Depending on local environment (pH, viral membrane curvature, local glycoprotein density, presence or absence of a cellular target membrane), these molecules may be either shuttled onto the path to the known post‐fusion trimer network, which is required to achieve the fusion process (Libersou et al, 2010), or in some other antiparallel assembly, which plays a role at some early stage of the fusion process.
Materials and Methods
Production, purification, and crystallization of CHAV Gth
Crystals of the ectodomain of CHAV G were obtained as described (Baquero et al, 2012). Briefly, we took advantage of a VSV harboring the CHAV G (Rose et al, 2000) and cleaved the ectodomain from this recombinant virus with thermolysin. This CHAV Gth comprising residues 1–419 (Appendix Fig S1) as assessed by mass spectrometry was further purified by anion exchange and size exclusion chromatographies. Crystals were grown by the hanging drop vapor diffusion method by mixing 1 μl of Gth (4 mg/ml) with 1 μl of the reservoir solution (14% PEG 3350, 0.3 M Na2SO4 and 0.1 M HEPES pH 7.6; final pH 7.5) and equilibrating against 500 μl of reservoir solution.
Structure determination
Crystals were harvested in a cryoprotecting solution containing reservoir solution supplemented with 30% glycerol and flash‐cooled by plunging in liquid nitrogen. Diffraction data were collected at 100 K at PROXIMA 1 and ID‐29 beam lines in SOLEIL and ESRF synchrotrons (France). Data for the best crystal were processed using XDS/XSCALE (Kabsch, 2010). The crystals belong to space group P21212 with cell parameters consistent with three to six molecules in the asymmetric unit. A self‐rotation function computed with MOLREP (Vagin & Teplyakov, 2010) showed twofold noncrystallographic symmetry (NCS) with a split peak (Baquero et al, 2012), indicating the most likely content to be four molecules for 60% solvent content.
The structure was determined by molecular replacement (MR) with the PHENIX suite of programs (Adams et al, 2010). The VSV G post‐fusion ectodomain (PDB 2CMZ, chain A) was first trimmed of nonconserved side chains using the default procedure in phenix.sculptor. The three search models were the CD (144 out of 419 residues with 56% sequence identity between CHAV G and VSV G), FD (99 residues, 67% sequence identity), and PHD (97 residues, 20% sequence identity) further trimmed of carbohydrates and of a couple of residues upstream and downstream of refolding segments R1 to R5. Eight out of the twelve expected domains in the asymmetric unit were placed by phenix.phaser when the search was carried out in the following order: CD, CD, FD, CD, CD, PHD, PHD, FD. The first two CD displayed twofold NCS consistent with the self‐rotation function, confirming the correctness of this initial solution. The eight‐domain partial solution was automatically refined and rebuilt and the missing side chains placed using phenix.autobuild. The refined FD and PHD thus obtained were subsequently used as search models, and two more PHD copies and one FD were placed. The last FD was placed manually by using NCS.
The structure was completed and rebuilt with COOT (Emsley et al, 2010) and refined with phenix.refine to an R free of 22% with excellent stereochemical statistics (Table 2). Tight NCS restraints were enforced throughout between the two EI and the two LI on the one hand, and between the distal parts (including the fusion loops) of all four FD (residues 65–123) on the other hand. Tight stereochemical restraints were used (Table 2) as test calculations indicated that these were best in reducing R free and R free‐R gap. B factors were refined as TLS groups plus tightly restrained individual Bs. There were seven TLS groups per molecule: one each for CD, PHD, and FD, one each for R1, R4, and R5, and one for R2 plus R3. Carbohydrates were checked for stereochemical correctness using the tools of the PDB Carbohydrate Residue Check service http://www.glycosciences.de/tools/pdb-care/ (Lutteke, 2009). The correct alpha 1–6 linkage for fucose residues was enforced by setting the linkage to beta 1–6 due to a bug in the libraries used by PHENIX (version dev‐894). The final model comprises four CHAV G molecules and a single sulfate ion.
Native MS analyses
As buffers, 200 mM ammonium acetate (NH4Ac) at pH values 6.0, 7.5, and 8.8 was used. Sample solutions were desalted and buffer‐exchanged using ultrafiltration with a 30‐kDa cutoff Amicon Ultra‐0.5‐ml centrifugal filters (Millipore). Samples were then diluted in NH4Ac to a concentration of 7 μM. Protein complex ions were generated using a nanoflow electrospray (nano‐ESI) source. MS analyses were carried out on a quadrupole time‐of‐flight mass spectrometer (Q‐TOF Ultima, Waters Corporation, Manchester, UK). The instrument was modified for the detection of high masses. The following instrumental parameters were used: capillary voltage up to 1.2–1.3 kV, cone potential = 40 V, RF lens‐1 potential = 40 V, RF lens‐2 potential = 1 V, aperture‐1 potential = 0 V, collision energy = 20 V, and microchannel plate (MCP) = 1,900 V. All mass spectra were calibrated externally using a solution of cesium iodide (6 mg/ml in 50% isopropanol) and were processed using the Masslynx 4.0 software (Waters Corporation, Manchester, UK) and the software package Massign (free of charge to academic nonprofit institutions, http://massign.chem.ox.ac.uk/).
Electron microscopy
Purified virions and CHAV Gth preparations were diluted in 150 mM NaCl in 50 mM Tris–HCl pH 8.0, or in 50 mM MOPS (3‐(N‐morpholino)propanesulfonic acid) at pH 6.6 or 6.25, or 50 mM acetate buffer at pH 6.0 or 5.5.
Negative staining
Samples were adsorbed onto airglow discharge carbon‐coated grids and stained with sodium phospho‐tungstic acid adjusted to the sample pH (8.0, 6.6, 6.0 or 5.5). For samples maintained at pH 5.5, no major difference was observed when staining was performed with phospho‐tungstic acid (pH 5.5) or uranyl acetate (UA) (pH 4.8). Thus, UA was used in these conditions, including for the recording of the tomograms. Images were recorded in an electron microscope (model CM12; Philips) operated at 80 kV, with a nominal magnification of 35,000.
Cryo‐negative staining
Samples were prepared as for negative staining except that they were let dry in a 76% relative humidity chamber. The humidity was controlled by the presence of a large quantity of water saturated by NaCl. After 30–60 min, the grid was removed from the chamber and rapidly plunged in liquid nitrogen and transferred in a liquid nitrogen‐cooled holder (Gatan 626). Samples were observed in a FEI Tecnai F‐200 electron microscope operated at 80 kV. Images were recorded on Kodak SO163 image. Data were recorded under minimal dose conditions at a 29,000 magnification. Cryo‐ and conventional negative staining gave similar results; however, the success rate as far as virus shape preservation is concerned was larger for cryo‐negative staining, at least for the samples maintained at pH 6.6 and 4°C.
Tomographic data collection
Tomograms were recorded with a FEI Technai 2 with a field emission gun operated at 200 kV. Images were recorded on a 2,048 × 2,048 pixel CCD camera (Gatan) at a nominal magnification of 29,000 (5.2 nm‐pixel) and 1.5–3 μm defocus corresponding to first zeros at (2.0 nm)−1 – (3.0 nm)−1. Data were recorded under low‐dose conditions using the microscope program.
Image processing for 3D reconstructions
Projections in a tilt series were mutually aligned and the tomograms calculated by the weighted back‐projection method, using the FEI Inspect3D package as explained in the manual. UCSF Chimera (Pettersen et al, 2004) was used to visualize volume and extract sub‐tomograms. To increase the signal‐to‐noise ratio of the reconstructions, G spikes were boxed and the sub‐tomograms averaged with Imagic (van Heel et al, 1996). For both conditions (pH 5.5, 20°C and pH 6.6, 4°C), nine reconstructions were averaged. The resolution of the maps was estimated using Fourier shell correlation (FCS) between two independent sets and found to be about 35 Å.
Quantitative fits were performed in reciprocal space using the VEDA software (Navaza et al, 2002). The quality of the fit was evaluated by calculating correlation coefficients and R‐factors. In general, and particularly for negatively stained samples, the threshold at which the densities can be attributed to the studied object in reconstruction is difficult to determine. We assessed the threshold, and the magnification factor on reconstructions obtained at pH 5.5 by fitting the post‐fusion trimeric crystallographic model (Roche et al, 2006). The normalized map obtained at pH 5.5 was filtered so that all points having values less than a threshold were set to this threshold value. The crystallographic post‐fusion trimer was fitted in the reconstruction for different threshold values. R‐factor values were calculated in the resolution range 150–35 Å and 150–45 Å. In both cases, the lowest R‐factor was found for a threshold value of 1 (Appendix Table S2), which was applied to all normalized reconstructions.
To minimize magnification errors, the post‐fusion crystallographic trimer was fitted in the experimental map (obtained at pH 5.5) for different magnification factors (Appendix Table S3). R‐factor values were minimal for a magnification factor of 0.99, a magnification that was applied to all reconstructions.
Note that the lower value of fitting resolution range for which the R‐value is larger than 40% for the post‐fusion trimer fit in the EM map also constitutes an estimate of the reconstruction resolution, which was consistent with FCS.
Cells and antibodies
BSR, clones of BHK‐21 (baby hamster kidney, ATCC CCL‐10), and HEK‐293T (Human Embryonic Kidney expressing SV40T‐Antigen, ATCC CRL‐3216) cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 8% fetal calf serum (FCS). Mouse monoclonal antibody directed against G ectodomain was supplied by KeraFAST (8G5F11).
Indirect immunofluorescence
BSR cells plated on glass coverslips at 70% confluence were transfected with pcDNA3.1 plasmids encoding WT or mutant G, as described above. Twenty‐four hours after transfection, cells were fixed with 4% paraformaldehyde in 1× PBS for 15 min and then permeabilized or not in 0.1% Triton X‐100 in PBS for 5 min. Glycoprotein was detected by using a mouse monoclonal anti‐G ectodomain antibody (KeraFAST, 8G5F11). Goat anti‐mouse Alexa Fluor 488 (Invitrogen) was used as secondary antibodies. Images were acquired with Zeiss Axiovert 200 fluorescence microscope (Carl Zeiss MicroImaging, Germany system) equipped with a 63× lens (oil) connected to a charge‐coupled device camera and a computer equipped with AxioVision software. Excitation was performed at 488 nm (Alexa 488). Cell nuclei were stained with DAPI.
Cell surface expression
In order to quantify the expression of G protein on the cell surface, HEK‐293 cells plated on six‐well dishes at 70% confluence were transfected as described above. Twenty‐four hours after transfection, cells were collected by scraping into 1 mM EDTA–PBS, followed by centrifugation at 600 g for 5 min. Cells were incubated with a 1:2,000 dilution of mouse monoclonal anti‐G ectodomain antibody (KeraFAST, 8G5F11) in PBS on ice for 1 h. Cells were washed twice in PBS, fixed at 4°C in paraformaldehyde, incubated with a 1:100 dilution of goat anti‐mouse Alexa Fluor 488 (Invitrogen) on ice for 1 h, and rinsed in PBS. After resuspension in 500 μl of 0.5 mM EDTA–PBS, the fluorescence of 10,000 cells from each population was determined by flow cytometry using a FACS BD Accuri C6. The mean fluorescence intensity (MFI) of the transfected cells expressing G at their surface was quantified by flow cytometry. The relative cell surface expression of transfected cells was determined as follows: (MFI for mutant)/(MFI for wt). For each mutant, the percentage given in Fig EV3 is the average of three independent experiments.
Cell–cell fusion assay
BSR cells plated on glass coverslips at 70% confluence were cotransfected with pCAGGS plasmids encoding WT G or mutant G, and P‐GFP plasmid encoding the phosphoprotein of Rabies virus fused to GFP. Twenty‐four hours after transfection, cells were incubated with fusion buffer (DMEM+10 mM MES) at various pH values (from 5.0 to 6.5) for 10 min at 37°. Cells were then washed once and incubated with DMEM+10 mM HEPES‐NaOH buffered at pH 7.4, 1% BSA at 37°C for 1 h. Cells were fixed with 4% paraformaldehyde in 1× PBS for 15 min. Cells nuclei were stained with DAPI, and syncytium formation was analyzed with Zeiss Axiovert 200 fluorescence microscope with a 20× lens.
Recovery of recombinant virus
Plasmids pVSV‐FL(+) expressing the 11,161‐nt positive‐strand full‐length VSV RNA sequence, pBS‐N, pBS‐P, and pBS‐L, respectively encoding N, P, and L proteins, have been previously described (Lawson et al, 1995; Schnell et al, 1996; Ferlin et al, 2014). The mutant G genes of the VSV Indiana serotype (Mudd‐Summers strain) were inserted into the original full‐length genomic plasmid pVSV‐FL(+) (Lawson et al, 1995) using two unique sites of pVSVFL(+), MluI in the 5′ noncoding sequence of the glycoprotein (G) gene and NheI present in a sequence introduced between G and L, after removal of the corresponding VSV Indiana Mudd‐Summers G gene. The plasmids were transfected into the cells with Lipofectamine 2000 (Invitrogen) in the presence of 10 μg/ml AraC (Sigma). The recovery of the mutants required expression of functional WT VSV G protein in trans from a plasmid (pcDNA3.1 Gwt), which was cotransfected with plasmids pVSV‐FL(+), pBS‐N, pBS‐P, and pBS‐L (Ferlin et al, 2014). Recombinant VSV was recovered as described by Schnell et al (1996).
Data deposition
The crystal structure from this publication has been submitted to the Protein Data Bank (http://www.wwpdb.org/) and assigned the accession code 5MDM.
Author contributions
EB purified and characterized the protein, grew the crystals, participated in diffraction data collection, in data processing and in the structure determination, refined the structure, and prepared figures and tables; AAA developed the purification protocol, characterized the protein, and participated in diffraction data collection and in data processing; AA‐H performed experiments required for the revised version of the manuscript; HR performed mutagenesis experiments and the functional analysis of the mutants; EBE performed MS and interpreted MS data; MO performed EM, LB and JKR constructed the VSV‐CHAV G recombinant virus; JL designed and performed EM experiments and analyzed the data; SB solved and refined the structure, analyzed the data, and wrote the manuscript; and YG supervised the work, analyzed the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File
Acknowledgements
This work was supported by grants from the EU through the Marie Curie ITN VIRUS ENTRY (Project reference 235649) to YG (with predoctoral funding to EB), from the Fondation pour la Recherche Médicale (FRM DEQ20120323711) to YG and by grants from ANR (ANR‐08‐BLAN‐0256, ANR 11 BSV8 002 01, and ANR CE11 MOBARHE) to YG, JL, and SB (with postdoctoral funding to AA, AAH, and EB). We acknowledge the European Synchrotron Radiation Facility (Grenoble, France) and synchrotron SOLEIL (Saint‐Aubin, France) for provision of radiation facilities. We acknowledge the Structural biology and Proteomics pole of the IMAGIF integrated platform (https://www.imagif.cnrs.fr/?nlang=en) for access to Crystallization and Mass Spectrometry (Manuela Argentini and David Cornu) services. We thank Dr. Luca Signor (IBS, Grenoble) for the technical assistance in the mass spectrometry facility of Grenoble Instruct Centre (ISBG; UMS 3518 CNRS‐CEA‐UJF‐EMBL). It was financially supported by the French Infrastructure for Integrated Structural Biology Initiative (FRISBI, ANR‐10‐INSB‐05‐02) and GRAL (ANR‐10‐LABX‐49‐01) [within the Grenoble Partnership for Structural Biology]. We also thank MS Vision (Almere, NL) for their technical assistance to keep our Q‐TOF Ultima in optimal conditions.
The EMBO Journal (2017) 36: 679–692
Contributor Information
Jean Lepault, Email: jean.lepault@i2bc.paris-saclay.fr.
Stéphane Bressanelli, Email: stephane.bressanelli@i2bc.paris-saclay.fr.
Yves Gaudin, Email: yves.gaudin@i2bc.paris-saclay.fr.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertini AA, Merigoux C, Libersou S, Madiona K, Bressanelli S, Roche S, Lepault J, Melki R, Vachette P, Gaudin Y (2012a) Characterization of monomeric intermediates during VSV glycoprotein structural transition. PLoS Pathog 8: e1002556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albertini AAV, Baquero E, Ferlin A, Gaudin Y (2012b) Molecular and cellular aspects of rhabdovirus entry. Viruses 4: 117–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backovic M, Longnecker R, Jardetzky TS (2009) Structure of a trimeric variant of the Epstein‐Barr virus glycoprotein B. Proc Natl Acad Sci USA 106: 2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero E, Albertini AA, Gaudin Y (2015a) Recent mechanistic and structural insights on class III viral fusion glycoproteins. Curr Opin Struct Biol 33: 52–60 [DOI] [PubMed] [Google Scholar]
- Baquero E, Albertini AA, Raux H, Buonocore L, Rose JK, Bressanelli S, Gaudin Y (2015b) Structure of the low pH conformation of Chandipura virus G reveals important features in the evolution of the vesiculovirus glycoprotein. PLoS Pathog 11: e1004756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero E, Buonocore L, Rose JK, Bressanelli S, Gaudin Y, Albertini AA (2012) Crystallization and preliminary X‐ray analysis of Chandipura virus glycoprotein G. Acta Crystallogr Sect F Struct Biol Cryst Commun 68: 1094–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquero E, Albertini AA, Vachette P, Lepault J, Bressanelli S, Gaudin Y (2013) Intermediate conformations during viral fusion glycoprotein structural transition. Curr Opin Virol 3: 143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeri Erba E, Petosa C (2015) The emerging role of native mass spectrometry in characterizing the structure and dynamics of macromolecular complexes. Protein Sci 24: 1176–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke HG, Heldwein EE (2015) Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog 11: e1005227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouli S, Ciferri C, Nikitin PA, Calo S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, Carfi A (2015) Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat Commun 6: 8176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LH, Klein DE, Schmidt AG, Pena JM, Harrison SC (2014) Sequential conformational rearrangements in flavivirus membrane fusion. eLife 3: e04389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM (2008) Mechanics of membrane fusion. Nat Struct Mol Biol 15: 675–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessau M, Modis Y (2013) Crystal structure of glycoprotein C from Rift Valley fever virus. Proc Natl Acad Sci USA 110: 1696–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doms RW, Keller DS, Helenius A, Balch WE (1987) Role for adenosine triphosphate in regulating the assembly and transport of vesicular stomatitis virus G protein trimers. J Cell Biol 105: 1957–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrer P, Gaudin Y, Ruigrok RW, Graf R, Brunner J (1995) Photolabeling identifies a putative fusion domain in the envelope glycoprotein of rabies and vesicular stomatitis viruses. J Biol Chem 270: 17575–17581 [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlin A, Raux H, Baquero E, Lepault J, Gaudin Y (2014) Characterization of pH‐sensitive molecular switches that trigger the structural transition of vesicular stomatitis virus glycoprotein from the postfusion state toward the prefusion state. J Virol 88: 13396–13409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, Whitt MA (1995) Vesicular stomatitis virus glycoprotein mutations that affect membrane fusion activity and abolish virus infectivity. J Virol 69: 1435–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, Whitt MA (1998) Attenuation of recombinant vesicular stomatitis viruses encoding mutant glycoproteins demonstrate a critical role for maintaining a high pH threshold for membrane fusion in viral fitness. Virology 240: 349–358 [DOI] [PubMed] [Google Scholar]
- Gaudin Y, Ruigrok RW, Knossow M, Flamand A (1993) Low‐pH conformational changes of rabies virus glycoprotein and their role in membrane fusion. J Virol 67: 1365–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin Y, de Kinkelin P, Benmansour A (1999) Mutations in the glycoprotein of viral haemorrhagic septicaemia virus that affect virulence for fish and the pH threshold for membrane fusion. J Gen Virol 80: 1221–1229 [DOI] [PubMed] [Google Scholar]
- Gaudin Y (2000) Reversibility in fusion protein conformational changes. The intriguing case of rhabdovirus‐induced membrane fusion. Subcell Biochem 34: 379–408 [DOI] [PubMed] [Google Scholar]
- Hammond C, Helenius A (1995) Quality control in the secretory pathway. Curr Opin Cell Biol 7: 523–529 [DOI] [PubMed] [Google Scholar]
- Harrison SC (2015) Viral membrane fusion. Virology 479–480: 498–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M (1996) A new generation of the IMAGIC image processing system. J Struct Biol 116: 17–24 [DOI] [PubMed] [Google Scholar]
- Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC (2006) Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313: 217–220 [DOI] [PubMed] [Google Scholar]
- Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC (2013) Influenza‐virus membrane fusion by cooperative fold‐back of stochastically induced hemagglutinin intermediates. eLife 2: e00333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky TS, Lamb RA (2014) Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 5: 24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeetendra E, Ghosh K, Odell D, Li J, Ghosh HP, Whitt MA (2003) The membrane‐proximal region of vesicular stomatitis virus glycoprotein G ectodomain is critical for fusion and virus infectivity. J Virol 77: 12807–12818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010) Xds. Acta Crystallogr D Biol Crystallogr 66: 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM (2008) The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat Struct Mol Biol 15: 1024–1030 [DOI] [PubMed] [Google Scholar]
- Klein DE, Choi JL, Harrison SC (2013) Structure of a dengue virus envelope protein late‐stage fusion intermediate. J Virol 87: 2287–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson ND, Stillman EA, Whitt MA, Rose JK (1995) Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA 92: 4477–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Modis Y (2014) A novel membrane fusion protein family in Flaviviridae? Trends Microbiol 22: 176–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libersou S, Albertini AA, Ouldali M, Maury V, Maheu C, Raux H, de Haas F, Roche S, Gaudin Y, Lepault J (2010) Distinct structural rearrangements of the VSV glycoprotein drive membrane fusion. J Cell Biol 191: 199–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutteke T (2009) Analysis and validation of carbohydrate three‐dimensional structures. Acta Crystallogr D Biol Crystallogr 65: 156–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehmood S, Allison TM, Robinson CV (2015) Mass spectrometry of protein complexes: from origins to applications. Annu Rev Phys Chem 66: 453–474 [DOI] [PubMed] [Google Scholar]
- Navaza J, Lepault J, Rey FA, Alvarez‐Rua C, Borge J (2002) On the fitting of model electron densities into EM reconstructions: a reciprocal‐space formulation. Acta Crystallogr D Biol Crystallogr 58: 1820–1825 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Pierson TC, Kielian M (2013) Flaviviruses: braking the entering. Curr Opin Virol 3: 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao BL, Basu A, Wairagkar NS, Gore MM, Arankalle VA, Thakare JP, Jadi RS, Rao KA, Mishra AC (2004) A large outbreak of acute encephalitis with high fatality rate in children in Andhra Pradesh, India, in 2003, associated with Chandipura virus. Lancet 364: 869–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S, Gaudin Y (2002) Characterization of the equilibrium between the native and fusion‐inactive conformation of rabies virus glycoprotein indicates that the fusion complex is made of several trimers. Virology 297: 128–135 [DOI] [PubMed] [Google Scholar]
- Roche S, Bressanelli S, Rey FA, Gaudin Y (2006) Crystal structure of the low‐pH form of the vesicular stomatitis virus glycoprotein G. Science 313: 187–191 [DOI] [PubMed] [Google Scholar]
- Roche S, Rey FA, Gaudin Y, Bressanelli S (2007) Structure of the prefusion form of the vesicular stomatitis virus glycoprotein g. Science 315: 843–848 [DOI] [PubMed] [Google Scholar]
- Rose NF, Roberts A, Buonocore L, Rose JK (2000) Glycoprotein exchange vectors based on vesicular stomatitis virus allow effective boosting and generation of neutralizing antibodies to a primary isolate of human immunodeficiency virus type 1. J Virol 74: 10903–10910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JK, Whitt MA (2001) Rhabdoviridae: the viruses and their replication. In Fields Virology, Knipe DM, Howley PM. (eds). 4th edn, pp 1221–1224. Philadelphia, PA: Lippincott Williams & Wilkins; [Google Scholar]
- Sanchez‐San Martin C, Sosa H, Kielian M (2008) A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 4: 600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell MJ, Buonocore L, Whitt MA, Rose JK (1996) The minimal conserved transcription stop‐start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J Virol 70: 2318–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanifer ML, Cureton DK, Whelan SP (2011) A recombinant vesicular stomatitis virus bearing a lethal mutation in the glycoprotein gene uncovers a second site suppressor that restores fusion. J Virol 85: 8105–8115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr 66: 22–25 [DOI] [PubMed] [Google Scholar]
- Zeev‐Ben‐Mordehai T, Vasishtan D, Hernandez Duran A, Vollmer B, White P, Prasad Pandurangan A, Siebert CA, Topf M, Grunewald K (2016) Two distinct trimeric conformations of natively membrane‐anchored full‐length herpes simplex virus 1 glycoprotein B. Proc Natl Acad Sci USA 113: 4176–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ghosh HP (1994) Characterization of the putative fusogenic domain in vesicular stomatitis virus glycoprotein G. J Virol 68: 2186–2193 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Review Process File