

Abstract
Aims:
Assessing whether epigenetic alterations mediate associations between environmental exposures and health outcomes is increasingly popular. We investigate the impact of exposure misclassification in such investigations.
Materials & methods:
We quantify bias and false-positive rates due to exposure misclassification in mediation analysis and assess the performance of the simulation extrapolation method (SIMEX). We evaluate whether DNA-methylation mediates smoking–birth weight relationship in the Norwegian Mother and Child Study birth cohort.
Results:
Ignoring exposure misclassification increases type I error in mediation analysis. The direct effect is underestimated and, when the mediator is a biomarker of the exposure, as is true for smoking, the indirect effect is overestimated.
Conclusion:
Misclassification correction plus cautious interpretation are recommended for mediation analyses in the presence of exposure misclassification.
Keywords: : DNA methylation, mediation analysis, misclassification
Evidence is accumulating that environmental exposures modify the epigenome. In humans, the best-studied epigenetic modification is methylation and the best-studied exposure is smoking. Smoking in adults has been reproducibly associated with alterations in methylation at specific loci [1]. Similar effects have been seen in newborns whose mothers smoked during pregnancy [2]. These smoking-methylation signals have been used to develop novel biomarkers of exposure [3–6]. In addition to its value as an exposure biomarker, there is great interest in the possibility that differential methylation at relevant loci mediates well-established associations between smoking and disease, both for adult [7,8] and in utero exposures [9,10].
It is widely acknowledged that measurement of human environmental exposures, including smoking, is prone to error [11]. Random error exists for all exposures. However, for smoking in particular, differential biased reporting occurs whereby some proportions of smokers falsely claim, on surveys, to be nonsmokers [12]. In addition, because of the well-publicized negative health impacts of maternal smoking during pregnancy on the developing fetus, pregnant women under-report smoking more than nonpregnant smokers of reproductive age [13]. Nonetheless, studies that address whether methylation signatures from smoking mediate its health outcomes have ignored the potential role of measurement error in assessment of smoking [7–10,14]. Given this measurement error, evaluation of mediation may be complicated by the fact that the proposed mediators, DNA sites differentially methylated by smoking, are excellent biomarkers that may better capture the exposure than self-reported smoking [3–6].
In the field of mediation analysis, bias introduced by measurement error in the mediator variable has been investigated [15–17]. However, misclassification of the exposure variable has not been well evaluated. Reduced birth weight is a well-established sequelae of maternal smoking during pregnancy [18]. Given strong evidence of differential methylation in newborns in relation to smoking by the mother [2], it has been of interest to consider whether these signals mediate the effects of maternal smoking on birth weight. It has recently been reported that differential DNA methylation of a single CpG site in placenta mediates up to 36% of the effect of smoking on lower birth weight [9]. In another study, differential methylation in newborn's blood at a single CpG site in a different gene was reported to mediate 19–46% of the relationship between smoking and birthweight [10]. Because self-reported smoking status during pregnancy is prone to misclassification, we were interested in evaluating the sensitivity of mediation analysis with methylation data to exposure misclassification bias. For this purpose, we considered a published scenario in perinatal epidemiology, for which we have relevant data [10], as an example.
Our study makes several contributions. First, we study the impact of exposure misclassification on the estimation of direct and indirect causal effects and testing of the indirect effect in methylation studies analytically. Second, we assess the impact of misclassification on estimation and testing via a simulation study. Third, we evaluate the ability of the SIMEX approach to adjust for exposure misclassification in this setting. Finally we use data from the Norwegian Mother and Child Cohort Study (MoBA) [19,20] to conduct a mediation analysis accounting for misclassification of self-reported smoking status using SIMEX. Our study provides evidence that ignoring misclassification can bias results of mediation analyses and shows the value of incorporating misclassification correction in mediation analysis in the context of environmental epigenetic studies.
Methods
Mediation analysis in the absence of exposure misclassification
With reference to our example of mediation of the effect of maternal smoking during pregnancy on newborn birth weight by smoking-related differential methylation, let A denote the exposure, maternal smoking and M denote the mediator, DNA methylation. Let Y denote the outcome, birth weight and C denote a vector of covariates representing potential confounders. The directed acyclic graph in Figure 1 describes the setting of mediation analysis. Mediation analysis can be employed to quantify how much of the total effect of maternal smoking on birth weight (Figure 1A) is explained by the indirect effect of smoking on birth weight that is mediated by the DNA-methylation level, relative to the direct effect of smoking on birthweight through pathways independent of DNA methylation (Figure 1B). Under the counterfactual framework for causal inference direct and indirect causal effects have been rigorously defined [21,22] (Section A1 of the Supplementary Material).
Figure 1. .

(A) Directed acyclic graph for average causal effect of sustained smoking during pregnancy (A) on birth weight (Y) (TE = 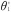 from Equation 1); (B) Directed acyclic graph for direct of sustained smoking during pregnancy (A) on birth weight (Y) and indirect effect of sustained smoking during pregnancy (A) on birth weight (Y) through DNA-methylation (M) (NDE = θ1 from Equation 3, NIE =
from Equation 1); (B) Directed acyclic graph for direct of sustained smoking during pregnancy (A) on birth weight (Y) and indirect effect of sustained smoking during pregnancy (A) on birth weight (Y) through DNA-methylation (M) (NDE = θ1 from Equation 3, NIE = 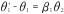 ) (C) denotes a vector of confounders.
) (C) denotes a vector of confounders.
To validly estimate direct and indirect effects, the following four confounding assumptions need to be satisfied. Conditioning on covariates C, there is no unmeasured confounding of the exposure–outcome relationship, the mediator–outcome relationship, the exposure–mediator relationship and there are no mediator–outcome confounders affected by the exposure. See [22,23] for further discussion of these assumptions. Furthermore, models for the outcome and mediator need to be correctly specified. For continuous outcome and mediator (as in the current setting of outcome birth weight and mediator methylation), under the assumption of no exposure–mediator interaction in the outcome model, typically made by published applications of mediation analysis in environmental epigenetics, if we specify three linear regression models
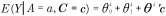 |
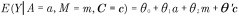 |
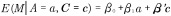 |
then the estimators of total effect (TE), direct effect (NDE) and indirect effect (NIE) take the form [24,25]:
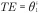 |
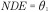 |
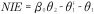 |
Under the assumption of no unmeasured confounding and that models 1–3 are correctly specified, these estimators (which are equivalent to the ones proposed by [24] in the psychology literature) can be interpreted as causal direct and indirect causal effects [22]. Estimators for direct and indirect effects in the presence of exposure-mediator interaction are given in Section A1 of the Supplementary Material. A discussion on the comparison between traditional and causal inference approaches to mediation analysis is given in [26].
The most popular test for indirect effects is based on the product method, also known as the Sobel test [27]. This is a Wald test for the null hypothesis H0: β1 θ2 = 0 based on the delta method standard error 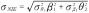 , where
, where 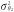 and
and 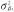 are the variances of the maximum likelihood estimates of θ2 and β1, respectively.
are the variances of the maximum likelihood estimates of θ2 and β1, respectively.
Mediation analysis in the presence of exposure misclassification
Let A* denote a binary exposure, self-reported smoking status during pregnancy in our example, potentially misclassified and A the true smoking status. We express, without loss of generality, measurement error in an additive form, A* = A + U. For a binary exposure, A* and A take values in {0, 1}, while the misclassification error U takes values in {-1,0, 1}. Assume that U is independent of the outcome, the mediator and the covariates, given true maternal smoking status A (i.e., misclassification error is nondifferential with respect to outcome, mediator and covariates). In this case the misclassification probabilities are characterized by sensitivity SN = P(A* = 1|A = 1) and specificity SP = P(A* = 0|A = 0) of the potentially misclassified exposure A*, yielding P(A*|A,M,Y,C) = P(A*|A). Under these assumptions misclassification is dependent on the true latent exposure because Cov(U,A) ≠ 0 [28]. This misclassification mechanism is realistic for self-reported maternal smoking during pregnancy and the results presented here can be easily extended to the case in which the error U is dependent on covariates as well. We expect perfect specificity (SP = 1) because it is reasonable to assume that if the mother is a nonsmoker (A = 0), she will report correctly to be a nonsmoker (A* = 0). However, we expect that some smoking mothers (A = 1) might incorrectly report being nonsmokers (A* = 0), leading to imperfect sensitivity (SN ≠ 1). Let, 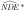 and
and 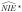 denote the naive direct and indirect effect estimators, respectively, when we fit the regression models in Equations 1–3 replacing the true exposure (A) with the self-reported exposure (A*). Let
denote the naive direct and indirect effect estimators, respectively, when we fit the regression models in Equations 1–3 replacing the true exposure (A) with the self-reported exposure (A*). Let 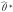 denote the naive outcome regression parameter estimators when the true exposure (A) is replaced with the self-reported exposure (A*) in Equation 2. Let
denote the naive outcome regression parameter estimators when the true exposure (A) is replaced with the self-reported exposure (A*) in Equation 2. Let 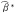 denote the naive mediator regression parameter estimators when the true exposure (A) is replaced with the self-reported exposure (A*) in Equation 3. In the results section we will assess analytically the bias of naive estimators of the natural direct and indirect effects and the type I error of the Sobel test when the assumptions given above hold and when exposure misclassification is ignored in the analysis.
denote the naive mediator regression parameter estimators when the true exposure (A) is replaced with the self-reported exposure (A*) in Equation 3. In the results section we will assess analytically the bias of naive estimators of the natural direct and indirect effects and the type I error of the Sobel test when the assumptions given above hold and when exposure misclassification is ignored in the analysis.
Correction approach
To correct for misclassification and obtain valid estimates of natural direct and indirect effects, we use a two-stage approach introduced in [15,16]. In the first stage, assuming plausible values for SP and SN, mediator and outcome regression coefficients are estimated using the SIMEX (simulation and extrapolation) approach to correct for misclassification of exposure [29,30]. In the second stage the SIMEX coefficient estimates are plugged into the formulas of NDE and NIE to obtain misclassification corrected estimates of the causal contrasts of interest with standard errors obtained via the bootstrap [29]. SIMEX has been shown to perform well in mediation analysis when the mediator is measured with error both in linear and nonlinear models [15]. For an assumed amount of measurement error (or misclassification error in this case), SIMEX simulates new datasets by additional error and calculates estimates for each of these new datasets, yielding data on the expected coefficient estimates as a function of the amount of measurement error. The procedure then fits a parametric model to this function, and then extrapolates this function back to the no-measurement error case. We used a quadratic model for the measurement error – coefficient estimate relationship because of its flexibility. Additional information on SIMEX and on its implementation, using the R package Simex can be found in [31]. Our code can be found in the Supplementary Material Section A8. When the amount of misclassification is not known from external validation data, as in our situation, we obtain SIMEX estimates under a range of specificity and sensitivity values.
Simulation study
We conducted a simulation study to assess the performances of the naive mediation analysis that ignores exposure misclassification and the SIMEX correction approach. In particular, we investigated the bias 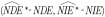 , relative bias
, relative bias 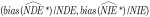 and variance
and variance 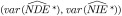 in the estimates of direct and indirect effects. Further, we studied Type I error rates of the Sobel test for H0: NIE = β1θ2 = 0 Note that the NIE will be zero if either (a) there is no effect of exposure on the mediator and no effect of the mediator on the outcome (θ2 = 0 from Equation 2 and β1 = 0 from Equation 3); or if (b) there is no effect of exposure on the mediator, but there is an effect of the mediator on the outcome (i.e., β1 = 0, θ2 ≠ 0); or if (c) there is an effect of exposure on the mediator, but there is no effect of the mediator on the outcome (i.e., β1 ≠ 0, θ2 = 0). In considering the indirect effect of smoking on birth weight through DNA methylation, we are particularly concerned about falsely rejecting the null hypothesis under this last scenario (β1 ≠ 0, θ2 = 0). The data generating process for these simulations, including sample size (n = 500) and distributions of exposure, mediator, covariates and outcome, was designed to mimic the recent study reporting that smoking-related differential methylation at specific CpG sites mediates part of the effect of maternal smoking exposure on birth weight [10]. Further, we specified mediator and outcome regression parameters according to the reported findings of this study. We assessed bias under the null hypothesis of no indirect effect and under the alternative hypothesis, assuming DNA methylation is a strong biomarker of the exposure. Therefore, under the alternative hypothesis we assumed β1 ≠ 0 in Equation 3 and θ2 ≠ 0 in Equation 2. Under the null hypothesis of no indirect effect we assumed β1 ≠ 0 in Equation 3 and θ2 = 0 in Equation 2. For simulations of type I error rates under the null hypothesis of no indirect effect, we considered the case in which the indirect effect is null because of no effect of methylation on birth weight (θ2 = 0) and no effect of smoking status on methylation (β1 = 0), but where smoking affects methylation (θ2 = 0, β1 ≠ 0). Full description of the simulation scenarios is given in Section A3 of Supplementary Material.
in the estimates of direct and indirect effects. Further, we studied Type I error rates of the Sobel test for H0: NIE = β1θ2 = 0 Note that the NIE will be zero if either (a) there is no effect of exposure on the mediator and no effect of the mediator on the outcome (θ2 = 0 from Equation 2 and β1 = 0 from Equation 3); or if (b) there is no effect of exposure on the mediator, but there is an effect of the mediator on the outcome (i.e., β1 = 0, θ2 ≠ 0); or if (c) there is an effect of exposure on the mediator, but there is no effect of the mediator on the outcome (i.e., β1 ≠ 0, θ2 = 0). In considering the indirect effect of smoking on birth weight through DNA methylation, we are particularly concerned about falsely rejecting the null hypothesis under this last scenario (β1 ≠ 0, θ2 = 0). The data generating process for these simulations, including sample size (n = 500) and distributions of exposure, mediator, covariates and outcome, was designed to mimic the recent study reporting that smoking-related differential methylation at specific CpG sites mediates part of the effect of maternal smoking exposure on birth weight [10]. Further, we specified mediator and outcome regression parameters according to the reported findings of this study. We assessed bias under the null hypothesis of no indirect effect and under the alternative hypothesis, assuming DNA methylation is a strong biomarker of the exposure. Therefore, under the alternative hypothesis we assumed β1 ≠ 0 in Equation 3 and θ2 ≠ 0 in Equation 2. Under the null hypothesis of no indirect effect we assumed β1 ≠ 0 in Equation 3 and θ2 = 0 in Equation 2. For simulations of type I error rates under the null hypothesis of no indirect effect, we considered the case in which the indirect effect is null because of no effect of methylation on birth weight (θ2 = 0) and no effect of smoking status on methylation (β1 = 0), but where smoking affects methylation (θ2 = 0, β1 ≠ 0). Full description of the simulation scenarios is given in Section A3 of Supplementary Material.
Analysis of Norwegian Mother & Child Study data
Finally, to assess the impact of misclassification of self-reported smoking status on estimates of mediation of the effect of maternal smoking on birthweight by specific methylation signals, we analyzed the same exposure–mediator–outcome scenario presented in [10] using data from the Norwegian Mother and Child Study (MoBa) pregnancy cohort. Naive analyses that ignore misclassification were conducted using the same approach employed in [10] and described in the previous sections. We then applied the SIMEX correction approach to assess the sensitivity to the results to exposure misclassification. MoBa is a large population-based pregnancy study targeting all women in Norway who gave birth between 1999 and 2008 [19,20]. Illumina HumanMethylation450K data from cord blood were measured on a subcohort of MoBa participants born between 2002 and 2004 (n = 1068), along with questionnaire data on smoking and potential confounders at about weeks 17 and 30 of pregnancy and cotinine measured in maternal plasma collected at about gestational week 18 of pregnancy [32]. Data on birthweight and gestational age were obtained from the Medical Birth Registry of Norway. Gestational age at birth was based mainly on routine fetal ultrasonographic examination at week 17–19, administered to more than 98% of Norwegian women. When ultrasound data were missing, gestational age was calculated using last menstrual period [33].
The Regional Committee for Medical Research Ethics, Norway and the NIEHS Institutional Review Board approved the study.
We first performed a naive mediation analysis in MoBa, ignoring exposure misclassification, to quantify the amount of mediation of the smoking–birth weight association due to methylation at each of three GFI1 CpG sites that were replicated in the published mediation analysis [10]. Gestational age was included as a linear variable as in the published mediation analysis [10] as well as in recent epigenome wide methylation analyses [34,35]. Women who reported smoking early in pregnancy but who quit early on were not considered sustained smokers. We used this exposure variable because the smoking methylation associations observed in previous studies are not seen for smoking that ends early in pregnancy but rather require more sustained exposure across the pregnancy [2,36].
To better evaluate the impact of misclassification we considered two definitions of sustained smoking. One is based on self-report alone. We then enhanced self-report by using cotinine measurements done at about 18 weeks so mothers who reported being nonsmokers but had cotinine values compatible with current smoking status were reclassified as smokers. We fitted the naive outcome and mediator regressions of the form of Equations 1–3 adjusting for sex, maternal age, maternal education, gestational age, parity and maternal prepregnancy BMI as potential confounders for comparability with the analysis presented in [10]. There were 1022 individuals with data on maternal smoking, the CpGs and all covariates available for these analyses. We then ran mediation analysis taking misclassification into account using the SIMEX method [30] for binary smoking status (sustained smoking across the pregnancy, yes or no). Applying SIMEX to outcome and mediator regressions, we obtained corrected estimates of the regression parameters and then used those estimates in the equations for the direct and indirect effects. Because SIMEX requires specifying sensitivity and specificity, and a wide range of sensitivities has been reported for self-reported smoking status [13,37], we assessed the robustness of the naive results to a wide range of plausible sensitivity (SN) parameter values (between 0.6 and 0.9). We assumed perfect specificity (SP = 1) because we do not expect pregnant women to falsely report smoking if they are nonsmokers. We used the bootstrap to estimate standard errors of the direct and indirect effects.
In addition to sustained smoking, we evaluated exposure to any smoking during the pregnancy. Women who reported smoking on either pregnancy questionnaire were coded as yes for any smoking without regard to whether they reported quitting early in pregnancy. This variable was used to classify maternal smoking in the discovery cohort in the published mediation analysis [10].
Results
Asymptotic bias & type I error
In the absence of exposure-mediator interaction, 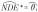 and by directly applying results on the impact of exposure misclassification in linear regression [28], the naive estimator of the natural direct effect is shown to be biased toward the null
and by directly applying results on the impact of exposure misclassification in linear regression [28], the naive estimator of the natural direct effect is shown to be biased toward the null 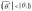 . The bias of
. The bias of 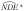 depends on the magnitude of the misclassification error and the true parameter θ1. We show in the Supplementary Material sections A5–A7 that under the special case of no direct effect, the naive estimator of the direct effect is unbiased.
depends on the magnitude of the misclassification error and the true parameter θ1. We show in the Supplementary Material sections A5–A7 that under the special case of no direct effect, the naive estimator of the direct effect is unbiased.
Exposure misclassification will bias the estimator of the exposure coefficient in the mediator model (β1) downward but will bias the estimator of the coefficient for the mediator in the outcome model (θ2) upward. The indirect effect, in the absence of exposure–mediator interaction, is the product of the coefficient for the exposure in the mediator model and the coefficient of the mediator in the outcome model (Equation 6 & Figure 1). Therefore, in theory, the bias of the naive indirect effect estimator can be in either direction. However, when the mediator is a strong biomarker for the exposure (i.e., β1 ≠ 0), as is the case for smoking methylation signals, our analytic results and simulation studies below show that the bias of the total effect estimator is larger than the bias of the natural direct effect estimator, leading to overestimation of the indirect effect. In other words, when the biomarker mediator captures the variability of true latent smoking exposure better than the self-reported measure of smoking, some of the direct effect is incorrectly attributed to the mediator (the indirect effect). Results on asymptotic bias in the presence of exposure-mediator interaction are less intuitive and for a full description of the results and proofs, the reader can refer to sections A4–A7 of the Supplementary Material.
Another important issue when studying a potential mediator that is a strong biomarker for the exposure is that under the null hypothesis of no indirect effect, 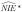 will be biased. Under this setting, one of the necessary conditions for the validity of type I error of the Sobel test for an indirect effect is not met. Therefore, under the null hypothesis of no indirect effect, if the exposure is misclassified and the mediator is a biomarker for the exposure, the Type I error rate will not be preserved. In the scenario we consider [10], the exposure is related to the mediator. Therefore, in reasonable scenarios of mediation analysis in environmental epigenetic studies, the naive mediation analysis is likely biased and there is risk of reporting false-positive findings of mediated effects through DNA methylation whenever the exposure is imperfectly measured and DNA methylation is a biomarker of the exposure.
will be biased. Under this setting, one of the necessary conditions for the validity of type I error of the Sobel test for an indirect effect is not met. Therefore, under the null hypothesis of no indirect effect, if the exposure is misclassified and the mediator is a biomarker for the exposure, the Type I error rate will not be preserved. In the scenario we consider [10], the exposure is related to the mediator. Therefore, in reasonable scenarios of mediation analysis in environmental epigenetic studies, the naive mediation analysis is likely biased and there is risk of reporting false-positive findings of mediated effects through DNA methylation whenever the exposure is imperfectly measured and DNA methylation is a biomarker of the exposure.
Simulation study
We now illustrate the bias of estimates of natural direct and indirect effects and type I error of tests for mediation in the presence of exposure misclassification. Under the simulation settings described in the previous section, in the presence of misclassification of a binary exposure due to misreport (Table 1 & Supplementary Figure 1), the direct effect is underestimated and the indirect effect is overestimated under all simulation scenarios (i.e., 1 the alternative hypothesis [β1 ≠ 0, θ2 ≠ 0] and 2 the null hypothesis of no indirect effect [β1 ≠ 0, θ2 = 0]). The exposure–mediator association (β1) and the exposure–outcome association (θ1) are underestimated and the mediator–outcome association (θ2) is overestimated (Supplementary Figure 2). Application of the SIMEX approach significantly reduces the bias of direct and indirect effect estimators, resulting in approximately unbiased estimates of direct and indirect effects (Supplementary Table 1).
Table 1. . Bias, Relative bias and variance of naive estimators of total effect, natural direct effect, natural indirect effect and proportion mediated for simulation scenario I assuming sensitivity = (0.70, 0.80, 0.90, 0.95), and specificity = 1.
| H1(β1a ≠ 0, ϑ2b ≠ 0) | True |
SN = 0.70 |
SN = 0.80 |
SN = 0.90 |
SN = 0.95 |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bias | Rel. bias | Var | Bias | Rel. bias | Var | Bias | Rel. bias | Var | Bias | Rel. bias | Var | ||
| TE |
-194 |
77.6 |
-0.40 |
990 |
58.2 |
-0.30 |
1013 |
32.98 |
-0.17 |
1305 |
19.4 |
-0.10 |
1397 |
| NDE |
-149 |
74.5 |
-0.50 |
1090 |
59.6 |
-0.40 |
1176 |
37.25 |
-0.25 |
1942 |
20.86 |
-0.14 |
1952 |
| NIE |
-45 |
-1.80 |
0.04 |
149 |
-0.9 |
0.02 |
193 |
-2.7 |
0.06 |
270 |
-2.25 |
0.05 |
330 |
| PM |
0.24 |
0.16 |
0.67 |
0.04 |
0.12 |
0.50 |
0.02 |
0.072 |
0.30 |
0.02 |
0.04 |
0.17 |
0.01 |
|
H0(β1a ≠ 0, ϑ2b = 0) | |||||||||||||
| TE |
-150 |
60.0 |
-0.40 |
990 |
45.0 |
-0.30 |
998 |
25.5 |
-0.17 |
1019 |
15.0 |
-0.10 |
1045 |
| NDE |
-150 |
75.0 |
-0.50 |
1090 |
59.6 |
-0.40 |
1177 |
37.5 |
-0.25 |
1305 |
20.86 |
-0.14 |
1398 |
| NIE |
0 |
-16 |
-16/0 |
127 |
-14 |
-14/0 |
174 |
-11 |
-11/0 |
225 |
-6 |
-6/0 |
319 |
| PM | 0 | 0.21 | 0.21/0 | 0.02 | 0.15 | 0.15/0 | 0.03 | 0.10 | 0.10/0 | 0.03 | 0.05 | 0.05/0 | 0.37 |
†Exposure-mediator association from Equation 3.
‡Mediator-outcome association from Equation 2.
NDE: Natural direct effect; NIE: Natural indirect effect; PM: Proportion mediated; SN: Sensitivity; SP: Specificity; TE: total effect; var: Variance.
We also note that the type I error of the Sobel test of the indirect effect is conservative in the absence of misclassification [38]. However, in the presence of exposure misclassification, the Type I error of the Sobel test of the indirect effect is elevated above the nominal 5% when the mediator is a strong biomarker of the exposure (Table 2).
Table 2. . Type I error of Sobel test for indirect effect for simulation scenario I assuming sensitivity = (0.70, 0.80, 0.90, 0.95), and specificity = 1.
| |
True† |
SN = 0.70 |
SN = 0.8 |
SN = 0.9 |
SN = 0.95 |
|
H0 (β1b = 0, ϑ2c = 0) |
0% |
0% |
0% |
0% |
0% |
| H0 (β1b = 0, ϑ2c = 0) | 4.9% | 27% | 18% | 10% | 6.5% |
†Type I error of Sobel test when the true exposure is used in the regression analyses.
‡Exposure–mediator association from Equation 3.
§Mediator–outcome association from Equation 2.
SN: Sensitivity.
Mediation analysis in the MoBa study
Küpers et al. [10] performed an epigenome-wide association study of the association between dichotomous maternal smoking (129 exposed, 129 unexposed) and DNA methylation data in the Groningen Expert Center for Kids with Obesity (GECKO) cohort and then analyzed the 35 top CpGs (those epigenome-wide significant at FDR < 0.05) to assess whether methylation at these CpGs mediates the effect of maternal smoking on birth weight. Among eight CpG sites in the GFI1 gene that showed the most robust mediation in the GECKO cohort, three gave significant Sobel p-values both in meta-analysis of two additional birth cohorts (Avon Longitudinal Study of Parents and Children (ALSPAC) and Generation R) [39,40] and in meta-analysis across all three cohorts. The authors reported a significant indirect effect whereby differential methylation of each of these three GFI1 CpGs mediated 19–46% of the decrease in birth weight in the GECKO discovery cohort and a smaller 12–19% in the three cohort meta-analysis.
Analyses for the MoBa cohort were conducted for the same three CpG loci in GFI1 using R software version 3.1.3. Commented code of the analyses can be found in the Supplementary Material Section A8.
Table 3 contains naive regression analyses of the MoBa cohort, using self-reported sustained smoking as the exposure, birth weight as the outcome and the three GFI1 CpGs (cg09935388, cg12876356, cg14179389) that were found to significantly mediate the smoking-birth weight association in [10]. Our study population of 1022 individuals with nonmissing data for all covariates included 117 women classified as sustained smokers by self-report.
Table 3. . Linear regression of self-reported sustained maternal smoking during pregnancy in relation to infant birth weight before and after adjustment for effects of maternal smoking on methylation at three CpGs in the GFI1 gene in the Norwegian Mother and Child Cohort Study.
| Regression model specification | Coeff† | SE | p-value |
|---|---|---|---|
| No mediator (CpG) adjustment |
-93.22 |
43.55 |
0.03 |
| Adjusting for cg09935388 |
-63.99 |
46.21 |
0.17 |
| Adjusting for cg12876356 |
-77.01 |
45.11 |
0.09 |
| Adjusting for cg14179389 | -82.48 | 45.42 | 0.07 |
†Regression coefficient interpretable as difference in birth weight, in grams between offspring of smoking mothers relative to nonsmokers.
Each separate linear regression model (only the specified CpG included) includes the following covariates: gestational age, child gender, maternal age, maternal education, parity, selection group and maternal prepregnancy BMI.
SE: Standard error.
In naive linear regression analyses, without any potential CpG mediator in the model, (Table 3) maternal smoking is significantly related to birthweight; birth weight was 93 g lower in newborns of smoking mothers. However, controlling for the CpG mediators attenuates the association between smoking and birth weight and it ceases to be statistically significant (Table 3). Among the three CpGs, all previously reported at epigenome wide Bonferroni significance in relation to maternal smoking in MoBa [32], adjustment for cg09935388 leads to the greatest reduction in the effect estimate for smoking on birthweight (Table 3). Among the three CpGs, cg09935388 also had the strongest association with smoking (β = 0.12 vs β = 0.07 for the other two).
In our MoBa data, naive mediation analysis implicates methylation at CpG cg09935388 as a potential mediator of the smoking–birth weight relationship (Table 4). We estimate a nonsignificant natural direct effect of smoking on birth weight (NDE = -64.1; 95% CI: -148.1–30.6), an indirect effect (via methylation) that is marginally statistically significant (NIE = -30.3; 95% CI: -60.5–0.0), based on the bootstrap CIs, which are recommended when sample size is small to moderate. The Sobel test yields stronger evidence of mediation (p = 0.021). The naive analyses estimate that 32% of the total effect of smoking on birthweight is mediated by this CpG. For the other two CpGs, which are less strongly associated with smoking and birthweight, the naive analyses provide weaker evidence of mediation: the indirect effects are nonsignificant and the proportions mediated are much lower (Table 4).
Table 4. . Estimates of natural direct and natural indirect effects of sustained maternal smoking, assessed by self-report, on birth weight and proportion mediated by three methylation cites (CpGs) in GFI1 in naive analyses and after SIMEX correction for measurement error in the Norwegian Mother and Child Study study.
| CpG | SN | NDE (95% CI) | NIE (95% CI) | PM |
|---|---|---|---|---|
| cg09935388 | Naive | -64.1 (-148.1 to 30.6) | -30.3 (-60.5 to 0.0) | 0.32 |
| 0.6 | -76.3 (-183.5 to 37.3) | -27.8 (-60.4 to 9.2) | 0.26 | |
| 0.7 | -73.8 (-171.3 to 35.9) | -28.4 (-59.0 to 6.0) | 0.27 | |
| 0.8 | -70.4 (-161.6 to 38.8) | -29.6 (-59.8 to 3.1) | 0.29 | |
| |
0.9 |
-66.3 (-153.5 to 34.4) |
-30.0 (-60.7 to 1.1) |
0.31 |
| cg12876356 | Naive | -78.0 (-158.9 to 11.7) | -16.3 (-38.0 to 6.8) | 0.17 |
| 0.6 | -86.5 (-183.8 to 17.3) | -13.9 (-39.6 to 12.5) | 0.14 | |
| 0.7 | -85.5 (-178.9 to 22.1) | -14.5 (-37.9 to 10.9) | 0.15 | |
| 0.8 | -82.1 (-170.6 to 15.5) | -15.3 (-38.7 to 9.6) | 0.16 | |
| |
0.9 |
-79.5 (-164.7 to 15.1) |
-16.0 (-38.4 to 7.9) |
0.17 |
| cg14179389 | Naive | -81.4 (-168.0 to 12.4) | -11.7 (-35.0 to 11.4) | 0.13 |
| 0.6 | -93.8 (-194.2 to 14.6) | -8.0 (-36.9 to 20.6) | 0.08 | |
| 0.7 | -91.3 (-184.2 to 14.0) | -9.4 (-36.3 to 18.0) | 0.10 | |
| 0.8 | -86.0 (-181.8 to 15.9) | -9.9 (-36.2 to 15.8) | 0.10 | |
| 0.9 | -85.0 (-173.7 to 15.4) | -10.9 (-35.3 to 13.1) | 0.11 |
The SIMEX corrected values are presented for four different values for sensitivity of the self-reported maternal smoking exposure variable: 0.6, 0.70, 0.80, 0.90 where specificity = 1. Median and 95% percentile CIs for the bootstrap estimates are in units of grams of birth weight.
NDE: Natural direct effect; NIE: Natural indirect effect; SN: Sensitivity; PM: Proportion mediated.
Correcting for potential misreporting of smoking during pregnancy using the SIMEX approach weakens the evidence for mediation at cg09935388 (Table 4). Under the assumption of fairly severe, but realistic [13] misclassification of smoking based on self-report in pregnant women (SN = 0.70), comparison of results after application of the SIMEX approach suggests that the direct effect of smoking on birth weight, not through methylation of CpG cg09935388, is underestimated (NDESN = 0.7 = -73.8; 95% CI: -171.3–35.9), and the indirect effect is overestimated (NIESN=0.7 = -28.4; 95% CI: -59.0–6.0; proportion mediated = 27%) by the naive analyses. Under all sensitivity values considered, SIMEX corrected analyses for CpGs cg12876356 and cg14179389 indicate severe under-estimation of the direct effect of smoking in the naive analysis and weaker evidence of mediation by the CpGs.
We repeated the mediation analyses (naive and SIMEX corrected) after enhancing the exposure variable by incorporating cotinine, a short-term biomarker of smoking, measured in mid-pregnancy. We reclassified as smokers 18 mothers who reported being nonsmokers, but had a cotinine level consistent with smoking resulting in 135 sustained smokers. This enhanced exposure variable should have less measurement error than smoking assessed by self-report alone. The reduction in birth weight for infants of smoking mothers is greater for this enhanced variable (-116 g; 95% CI: -195 to -36) than for the self-report alone variable (-93 g; 95% CI: -180 to -8) (Supplementary Table 2 includes results for the other two CpGs). In naive analyses (Table 5) for CpG cg0993538 when using this enhanced smoking variable, the direct effects are larger (-89 vs -64 g) compared with the mediation analysis using self-reported smoking in Table 4 (results for the other two CpGs in Supplementary Table 3). The indirect effects are smaller, have wider CIs and the proportion mediated is correspondingly much smaller (0.24 compared with 0.32) than in the analysis of self-reported smoking. Moreover, the Sobel test is marginally statistically significant (p-value = 0.051). Application of the SIMEX correction approach yields a more substantial reduction in the proportion mediated by CpG cg0993538 (from about 0.24 to 0.15) than in the analysis with self-reported smoking (reduced from about 0.32 to 0.26). SIMEX also increased the size and the precision of the estimated natural direct effect of smoking more than in the analysis of the self-reported variable (at sensitivity of 0.6, NDE was -89.4 [95% CI: -179.5–2.6] in the naive and -106.9 [95% CI: -216.4–8.1] in the SIMEX for the enhanced exposure variable compared with -64.1 [95% CI: -148.1–30.6] naive and -76.3 [95% CI: -183.5–37.3] SIMEX for the self-reported variable).
Table 5. . Estimates of natural direct and natural indirect effects of sustained maternal smoking, assessed by cotinine-enhanced self-report, on birth weight and proportion mediated by CpGs cg09935388 in GFI1 in both naive analyses and after SIMEX correction for measurement error in the Norwegian Mother and Child Study.
| CpG | SN | NDE (95% CI) | NIE (95% CI) | PM |
|---|---|---|---|---|
| cg09935388 | Naive | -89.4 (-179.5 to 2.6) | -27.8 (-59.7 to 5.1) | 0.24 |
| 0.6 | -106.9 (-216.4 to 8.1) | -18.7 (-60.4 to 21.7) | 0.15 | |
| 0.7 | -102.1 (-205.9 to 16.2) | -21.5 (-60.8 to 17.6) | 0.17 | |
| 0.8 | -97.9 (-195.5 to 4.2) | -23.9 (-59.6 to 11.9) | 0.20 | |
| 0.9 | -92.2 (-184.0 to -0.9) | -25.7 (-59.1 to 8.3) | 0.22 |
The SIMEX corrected values are presented for four different values for sensitivity of the self-reported maternal smoking exposure variable: 0.6, 0.70, 0.80, 0.90 where specificity = 1. Median and 95% percentile CIs for the bootstrap estimates are in units of grams of birth weight
NDE: Natural direct effect; NIE: Natural indirect effect; SN: Sensitivity; PM: Proportion mediated.
To evaluate in our MoBa data, the worst-case scenario for misclassification of exposure to maternal smoking, we also repeated the analysis using any smoking during pregnancy as the exposure variable. Women coded as yes to any smoking (N = 288) include the more than 50% of women who quit early in pregnancy. In linear regression, the coefficient for any smoking during pregnancy was - 40.0 g birthweight (SE = 30.9; p = 0.20) and was greatly reduced after adjustment for cg0993538 to -17.5 g (SE = 32.5; p = 0.59) with smaller reductions after adjustment for each of the other two CpGs (Supplementary Table 4). In naive mediation analyses there was a significant indirect effect -23.3 (95% CI: -41.4 to -2.9, Sobel test p = 0.011) of smoking on birthweight through this CpG but no significant direct effect of smoking on birthweight and the proportion mediated was much larger than in the sustained smoking analyses at 58%, even higher than that the 46% observed in the GECKO study of the same exposure variable [10] (Table 6). After SIMEX measurement error correction the proportion mediated was reduced, although the reduction was proportionally smaller than in the analyses of the two sustained smoking variables (Table 6). Results for the other two CpGs are reported in Supplementary Table 5.
Table 6. . Estimates of natural direct and natural indirect effects of any maternal smoking during pregnancy, assessed by self-report, on birth weight and proportion mediated by CpGs cg09935388 in GFI1 in both naive analyses and after SIMEX correction for measurement error in the Norwegian Mother and Child Study.
| CpG | SN | NDE (95% CI) | NIE (95% CI) | PM |
|---|---|---|---|---|
| cg09935388 | Naive | -16.7 (-84.6 to 52.9) | -23.3 (-41.4 to -2.9) | 0.58 |
| 0.6 | -21.1 (-112.1 to 64.6) | -26.8 (-51.6 to -0.9) | 0.55 | |
| 0.7 | -20.7 (-103.8 to 65.4) | -25.8 (-48.0 to -1.7) | 0.55 | |
| 0.8 | -18.7 (-96.0 to 63.5) | -25.0 (-46.1 to -2.1) | 0.57 | |
| 0.9 | -18.3 (-90.0 to 54.3) | -24.1 (-43.4 to -2.8) | 0.57 |
The SIMEX corrected values are presented for four different values for sensitivity of the self-reported maternal smoking exposure variable: 0.6, 0.70, 0.80, 0.90 where specificity = 1. Median and 95% percentile CIs for the bootstrap estimates are in units of grams of birth weight.
NDE: Natural direct effect; NIE: Natural indirect effect; PM: Proportion mediated; SN: Sensitivity.
Discussion
Mediation analysis is the primary tool for investigating the role of epigenenetic mechanisms in health effects of environmental exposures. Its use is increasing along with evidence for epigenetic impacts of smoking and other environmental exposures and the desire to identify biologic and public health implications of these epigenetic signals. However, mediation analysis is subject to various biases. It relies on stringent and untestable assumptions of no-unmeasured confounding and correct model specification. In observational studies, biases due to selection, missing data and measurement error further challenge the validity of mediation analysis [41]. Sensitivity analyses for violation of the assumptions of no-unmeasured confounding and no selection bias have been proposed [42,43]. Recently, Mendelian randomization (MR) estimation strategies have been suggested as an option to evaluate the reduced effect of measurement error in mediation analyses of methylation signals [44,45] in epigenetic studies. In MR, if there are genetic variants robustly associated with the exposure of interest, these can be used to help infer causality by serving as correctly measured instrumental variables, which are not associated with various confounders and are not directly influenced by the outcome of interest. Richmond et al. recently suggested that an earlier report that 30% of the association between adult smoking and lung cancer can be explained by methylation at a single smoking related CpG might be spurious and reflect measurement error in self-reported smoking [44]. They suggested that MR, using genetic variants related to smoking, might be a way to evaluate that possibility. Assumptions and application of MR analysis for the estimation of direct and indirect effects are further outlined in [45].
Here we examine the potential impact of exposure misclassification using the example of maternal smoking during pregnancy as the exposure and smoking-related methylation signals in the newborn as the putative mediators of the well-established relationship between smoking and reduced birth weight.
Our simulation studies and analyses of the MoBa data show that in environmental epigenetic studies on the mediating role of DNA methylation, when the methylation signal is a good biomarker of an exposure that is measured with error, there is a substantial risk of false positives and overestimation of the proportion mediated. This is the case for maternal smoking during pregnancy and newborn methylation where the smoking-related methylation signals are excellent biomarkers of exposure compared with self-report [3–6]. It is known that some smokers self-report as nonsmokers and this misreporting is more prominent during pregnancy because of the well-publicized health effects of smoking on the newborn and the attendant stigma to acknowledging this behavior [13]. The methylation signals detect smoking across pregnancy that is not reported by mothers. In addition, the degree of methylation difference at a site captures information about the amount and duration of smoking across the pregnancy which influences birth weight (or other health effects of maternal smoking) but is not captured by yes or no self-report. As a result, the indirect effect captures part of the direct effect because DNA methylation is less subject to measurement error than self-reported smoking, identifies falsely reported nonsmokers and captures quantitative information on duration and amount relevant to the outcome thus supplementing the measured exposure (self-reported smoking) itself. Thus mediation by the smoking methylation biomarker is overestimated.
We based our analyses on a recently published scenario whereby three CpGs in GFI1, differentially methylated in newborns in relation to self-reported maternal smoking, were reported to mediate 19–46% of the effect this in utero exposure on offspring birth weight [10]. These three CpGs are robust sites of differential methylation from maternal smoking reported in various studies including the MoBa dataset used for the current analysis [32]. The Küpers et al. [10] study was well conducted and included replication of the mediation finding in two additional high-quality birth cohorts [10]. Nonetheless, similar to other studies of smoking or other exposure-related methylation signals as mediators of exposure-related health effects, the potential contribution of misclassification was not estimated. We would expect misclassification to act the same way in the discovery and replication cohorts and thus replication of the apparent mediation does not reduce possibility that exposure misclassification contributes to the finding of mediation. Of interest, the proportion mediated by methylation at GFI1 cg09935388 was much higher in the GECKO discovery cohort (0.46), where the smoking variable was any smoking during pregnancy, than in the combined two replication cohorts (0.16) where the smoking variable was sustained smoking across the pregnancy [10]. The methylation signals in GFI1, like other top sites in epigenome wide analyses, reflect sustained smoking during the pregnancy as opposed to smoking that ends early in pregnancy that is captured by the any smoking variable [36]. Reduced birthweight is also more strongly associated with sustained smoking than any smoking [3]. The much stronger proportion mediated observed in the GECKO discovery cohort than for the combined replication cohorts may reflect the greater misclassification associated with any compared with sustained smoking during pregnancy. Accordingly, when we analyzed the any smoking during pregnancy variable in MoBa, we estimated a similarly greater proportion of mediation by CpG methylation than obtained with sustained smoking.
When we analyzed the cotinine-enhanced exposure variable, we found that the direct effect of smoking was larger and more precisely estimated and the proportion mediated was much lower than in the analysis of sustained smoking based on self-report alone. This finding has two implications. First, an enhanced measure of exposure, with less measurement error, leads to lower estimates of the proportion of mediation by the smoking CpG signal, a biomarker of the exposure. The proportion mediated was reduced more and the direct effect was both larger and more precisely estimated after SIMEX correction. Given that misclassification correction may be less widely used than it might be because of the perception that is generally leads to less precise effect estimates, this is an important result.
These results complement a body of literature on the impact of misclassification in mediation analysis under a regression framework. This previous work has focused on instances where the mediator, rather than the exposure, is measured with error or misclassified [15,16]. In our study methylation might be measured with some error, however, the three smoking associated CpGs have been reported at genome wide significance in at multiple individual studies of varying sizes [32,10] and are among the top eight findings in a meta-analysis of 13 cohorts [2]. Thus they probably have larger effect sizes and are likely measured with lower error than less robust signals.
All studies of exposure and methylation in blood are potentially confounded by effects of the exposure on cell composition. Thus this is a limitation in the interpretation of the study of [10], our analyses of the MoBa data and any other analyses examining potential mediation of health effects by exposure related methylation differences. A reference panel based on cord blood has become available to estimate cell type proportions for analyses of DNA extracted from whole cord blood [46]. When we add these estimated cell types to the model shown in Table 3, there is no further attenuation of the coefficient for birthweight in relation to sustained maternal smoking beyond that from adding cg09935388 to the model (β = -63.99 g before cell type adjustment vs -65.99 g after cell type adjustment). While these cell-type correction methods have limitations, this result provides some reassurance that the associations between maternal smoking, cg09935388 methylation and birthweight evaluated by [10] and followed up in this paper are not simply due to confounding by cell type. However, it should be noted that neither mediation analysis nor measurement error correction address the potential influence of exposure related difference in cell composition on reported findings regarding effects of exposure on methylation.
Integration of measurement error strategies within standard mediation analyses can reduce measurement error bias. It confers the important added benefit of realistically quantifying uncertainty around the mediation estimates. We illustrate a sensitivity analysis for misclassification bias employing the SIMEX correction approach [30], which can be implemented with easy-to-use software. The SIMEX approach for misclassified categorical variables allows the misclassification mechanism to be dependent on the true exposure status, as is typically the case for smoking where some smokers report themselves as nonsmokers on surveys. SIMEX can also be adopted for continuous variables and in nonlinear models as well (e.g., in the presence of exposure–mediator interaction) and if a categorical outcome, mediator or covariates are misclassified as well.
We included gestational age as a linear term for comparability with the mediation analysis of Kupers et al. [10]. Of note, in our MoBa data, methylation at cg09935388 was not significantly related to gestational age (p = 0.79). It is possible that in a study of birthweight a different form of gestational age, such as Z scores, might have been a better adjustment variable [47]. Nonetheless gestational age as a simple linear term has been robustly associated in recent genome wide analyses with methylation at many individual CpG sites [34,35]. Likewise, in both papers, there is likely measurement error in gestational lag. However, possible misspecification or misclassification of gestational age as an adjustment term in either study would not change our conclusion regarding measurement error in the self-reported smoking variable leading to potential overestimation of mediation of the smoking birthweight association by methylation at cg09935388, a strong biomarker of smoking.
There are limitations to this study. The SIMEX approach yields only approximately consistent estimates in small samples and therefore residual measurement error bias is possible despite the seemingly large sample size in our study. We, and no doubt others, plan to address these issues in future work. In the meanwhile, residual effects of misclassification on estimates of mediation are likely, given currently available exposure measures and measurement error correction methods. Thus, even when employing measurement error correction, great caution is warranted in the interpretation of mediation analysis involving a potential exposure biomarker. While our example focuses on methylation, the results are applicable to other types of mediators that may also capture some of the exposure under study. In this work we focused on evaluating the impact of misclassification on quantification of the mediating role of DNA methylation CpG sites at GFI1 reported in the literature. Application of misclassification correction approaches in epigenome-wide analysis is an important future direction.
Future perspective
Appreciation of the potential overestimation of mediation by epigenetic signals of exposure disease relationships will lead to both adoption of measurement error correction approaches and greater caution in interpreting apparent mediation in environmental epigenetics. Because current misclassification correction methods are even more effective when better exposure measures are used, both improved exposure assessment and novel statistical developments in the field of mediation analysis currently underway will improve both the validity and power of mediation analyses to quantify the role of DNA methylation or other epigenetic signals in mediating the effects of environmental exposures effects on human health across the life course.
Executive summary.
Analytic results, an extensive simulation study, and analysis of real data show that ignoring exposure misclassification when evaluating mediation of exposure disease relationships, or similar biomarkers of the exposure, can lead to false or exaggerated conclusions regarding mediation.
Measurement error correction approaches that acknowledge potential exposure misclassification can improve validity of findings on potential epigenetic targets on the pathway between environmental exposures and health outcomes. However, even when using these correction approaches caution is warranted in the interpretation of apparent mediation by epigenetic signals are good exposure biomarkers.
Supplementary Material
Acknowledgements
The authors are grateful to all the participating families in Norway who take part in the MoBa study. The authors thank F Day of NIEHS and for expert computing assistance.
Footnotes
Financial & competing interests disclosure
This work was supported in part by the Intramural Research Program of NIH, National Institute of Environmental Health Sciences (NIEHS). The Norwegian Mother and Child Cohort Study is supported by the Norwegian Ministry of Health and the Ministry of Education and Research, NIH/NIEHS (contract number N01-ES-75558 and ZO1 ES-49019), NIH/NINDS (grant number 1 UO1 NS 047537-01) and the Norwegian Research Council/FUGE (grant number 151918/S10), and the present project by the Norwegian Research Council/BIOBANK (grant no 221097). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 2016;9(5):436–447. doi: 10.1161/CIRCGENETICS.116.001506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am. J. Hum Genet. 2016;98(4):680–696. doi: 10.1016/j.ajhg.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]; • The large-scale meta-analysis across many birth cohorts identifies numerous replicable loci involved in response to maternal smoking in pregnancy with persistence into later childhood.
- 3.Reese SE, Zhao S, Wu MC, et al. DNA methylation score as a biomarker in newborns for sustained maternal smoking during pregnancy. Environ. Health Perspect. 2016 doi: 10.1289/EHP333. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Develops a biomarker in newborns of sustained maternal smoking during pregnancy using CpGs differentially methylated in relation to this exposure in the Norwegian Mother and Child Cohort Study.
- 4.Zhang Y, Yang R, Burwinkel B, Breitling LP, Brenner H. F2RL3 methylation as a biomarker of current and lifetime smoking exposures. Environ. Health Perspect. 2014;122(2):131. doi: 10.1289/ehp.1306937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Schottker B, Florath I, et al. Smoking-associated DNA methylation biomarkers and their predictive value for all-cause and cardiovascular mortality. Environ. Health Perspect. 2016;124(1):67–74. doi: 10.1289/ehp.1409020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shenker NS, Ueland PM, Polidoro S, et al. DNA methylation as a long-term biomarker of exposure to tobacco smoke. Epidemiology. 2013;24(5):712–716. doi: 10.1097/EDE.0b013e31829d5cb3. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H, Zheng Y, Zhang Z, et al. Estimating and testing high-dimensional mediation effects in epigenetic studies. Bioinformatics. 2016;32(20):3150–3154. doi: 10.1093/bioinformatics/btw351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds LM, Wan M, Ding J, et al. DNA methylation of the aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis. Circ. Cardiovasc. Genet. 2015;8(5):707–716. doi: 10.1161/CIRCGENETICS.115.001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morales E, Vilahur N, Salas LA, et al. Genome-wide DNA methylation study in human placenta identifies novel loci associated with maternal smoking during pregnancy. Int. J. Epidemiol. 2016;45(5):1644–1655. doi: 10.1093/ije/dyw196. [DOI] [PubMed] [Google Scholar]; • Identifies CpGs differentially methylated in placenta in relation to maternal smoking in a birth cohort and applies mediation analysis to assess the proportion of the effect of this exposure on birthweight mediated by one of these CpG.
- 10.Küpers LK, Xiaojing X, Soesma AJ, et al. DNA methylation mediates the effect of maternal smoking during pregnancy on birth weight of the offspring. Int. J. Epidemiol. 2015;44(4):1224–1237. doi: 10.1093/ije/dyv048. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Applies mediation analysis to assess the proportion of the effect of exposure to maternal smoking during pregnancy on birthweight explained by differential methylation in relation to this exposure at CpGs in the GFI1 gene. In the current paper, we evaluate this same exposure-methylation-birthweight scenario in data from the Norwegian Mother and Child Cohort and assess effects of measurement error correction.
- 11.Rhomberg LR, Chandalia JK, Long CM, Goodman JE. Measurement error in environmental epidemiology and the shape of exposure-response curves. Crit. Rev. Toxicol. 2011;41(8):651–671. doi: 10.3109/10408444.2011.563420. [DOI] [PubMed] [Google Scholar]
- 12.Rebagliato M. Validation of self reported smoking. J. Epidemiol. Commun. Health. 2002;56(3):163–164. doi: 10.1136/jech.56.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dietz PM, Homa D, England LJ. Estimates of nondisclosure of cigarette smoking among pregnant and nonpregnant women of reproductive age in the United States. Am. J. Epidemiolog. 2011;173(3):355–359. doi: 10.1093/aje/kwq381. [DOI] [PubMed] [Google Scholar]
- 14.Timms JA, Relton CL, Rankin J, Strathdee G, McKay JA. DNA methylation as a potential mediator of environmental risks in the development of childhood acute lymphoblastic leukemia. Epigenomics. 2016;8(4):519–536. doi: 10.2217/epi-2015-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valeri L, Lin X, VanderWeele TJ. Mediation analysis when a continuous mediator is measured with error and the outcome follows a generalized linear model. Stat. Med. 2014;33(28):4875–4890. doi: 10.1002/sim.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Studies bias of measurement error on a continuous mediator in mediation analysis and develops correction approaches using regression calibration, and SIMEX.
- 16.Valeri L, Vanderweele TJ. The estimation of direct and indirect causal effects in the presence of misclassified binary mediator. Biostatistics. 2014;15(3):498–512. doi: 10.1093/biostatistics/kxu007. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Studies bias of misclassification on a binary mediator in mediation analysis and develops correction approaches.
- 17.VanderWeele TJ, Valeri L, Ogburn EL. The role of measurement error and misclassification in mediation analysis. Epidemiology. 2012;23(4):561. doi: 10.1097/EDE.0b013e318258f5e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.General S. US Department of Health and Human Services; 2014. The health consequences of smoking—50 years of progress: a report of the surgeon general.www.surgeongeneral.gov/library/reports/50-years-of-progress/full-report.pdf [Google Scholar]
- 19.Magnus P, Birke C, Vejrup K, et al. Cohort Profile Update: The Norwegian Mother and Child Cohort Study (MoBa) Int. J. Epidemiol. 2016;45(2):382–388. doi: 10.1093/ije/dyw029. [DOI] [PubMed] [Google Scholar]
- 20.Rønningen KS, Paltiel L, Meltzer HM, et al. The Biobank of the Norwegian Mother and Child Cohort Study: a resource for the next 100 years. Eur. J. Epidemiol. 2006;21(8):619–625. doi: 10.1007/s10654-006-9041-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3(2):143–155. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- 22.Pearl J. Proceedings of the Seventeenth Conference on Uncertainty in Artificial Intelligence. Morgan Kaufmann Publishers Inc.; CA, USA: 2001. Direct and indirect effects; pp. 411–420. [Google Scholar]
- 23.Joffe M, Small D, Hsu CY. Defining and estimating intervention effects for groups that will develop an auxiliary outcome. Stat. Sci. 2007;22:74–97. [Google Scholar]
- 24.Baron RM, Kenny DA. The moderator–mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 1986;51(6):1173–1182. doi: 10.1037//0022-3514.51.6.1173. [DOI] [PubMed] [Google Scholar]
- 25.VanderWeele TJ, Vansteelandt S. Conceptual issues concerning mediation, interventions and composition. Stat. Interface. 2009;2:457–468. [Google Scholar]
- 26.Valeri L, VanderWeele TJ. Mediation analysis allowing for exposure–mediator interactions and causal interpretation: theoretical assumptions and implementation with SAS and SPSS macros. Psychol. Methods. 2013;18(2):137. doi: 10.1037/a0031034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobel ME. Asymptotic confidence intervals for indirect effects in structural equation models. Sociol. Methodol. 1982;13:290–312. [Google Scholar]
- 28.Aigner DJ. Regression with a binary independent variable subject to errors of observation. J. Econometrics. 1973;1:49–60. [Google Scholar]
- 29.Carroll RJ, Ruppert D, Stefanski LA, Crainiceanu CM. Measurement Error In Nonlinear Models: A Modern Perspective. CRC Press; FL, USA: 2006. [Google Scholar]
- 30.Küchenhoff H, Mwalili SM, Lesaffre E. A general method for dealing with misclassification in regression: the misclassification SIMEX. Biometrics. 2006;62(1):85–96. doi: 10.1111/j.1541-0420.2005.00396.x. [DOI] [PubMed] [Google Scholar]
- 31.Lederer W, Küchenhoff H. A short introduction to the SIMEX and MCSIMEX. The Newsletter of the R Project. 2006;6(4):26. [Google Scholar]; •• Provides theoretical explanation and practical description of the SIMEX approach for measurement error and misclassification correction using the R package ‘simex.’
- 32.Joubert BR, Håberg SE, Nilsen RM, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 2012;120(10):1425–1431. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Backe B. [Routine ultrasonography in obstetric care in Norway, 1994] Tidsskr. Nor Laegeforen. 1997;117(16):2314–2315. [PubMed] [Google Scholar]
- 34.Bohlin J, Håberg SE, Magnus P, et al. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 2016;17(1):207. doi: 10.1186/s13059-016-1063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knight AK, Craig JM, Theda C, et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 2016;17(1):206. doi: 10.1186/s13059-016-1068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joubert BR, Håberg SE, Bell DA, et al. Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol. Biomarkers Prev. 2014;23(6):1007–1017. doi: 10.1158/1055-9965.EPI-13-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patrick DL, Cheadle A, Thompson DC, Diehr P, Koepsell T, Kinne S. The validity of self-reported smoking: a review and meta-analysis. Am. J. Public Health. 1994;84(7):1086–1093. doi: 10.2105/ajph.84.7.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.MacKinnon DP, Warsi G, Dwyer JH. A simulation study of mediated effect measures. Multivar. Behav. Res. 1995;30(1):41–62. doi: 10.1207/s15327906mbr3001_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraser A, Macdonald-Wallis C, Tilling K, et al. Cohort profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int. J. Epidemiol. 2013;42(1):97–110. doi: 10.1093/ije/dys066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jaddoe VW, van Duijn CM, Franco OH, et al. The Generation R Study: design and cohort update 2012. Eur. J. Epidemiol. 2012;27(9):739–756. doi: 10.1007/s10654-012-9735-1. [DOI] [PubMed] [Google Scholar]
- 41.VanderWeele TJ. Explanation In Causal Inference: Methods For Mediation And Interaction. Oxford University Press; UK: 2015. [Google Scholar]
- 42.VanderWeele TJ. Bias formulas for sensitivity analysis for direct and indirect effects. Epidemiology. 2010;21(4):540. doi: 10.1097/EDE.0b013e3181df191c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valeri L, Coull BA. Estimating causal contrasts involving intermediate variables in the presence of selection bias. Stat. Med. 2016;35(26):4779–4793. doi: 10.1002/sim.7025. [DOI] [PubMed] [Google Scholar]
- 44.Richmond RC, Hemani G, Tilling K, Smith GD, Relton CL. Challenges and novel approaches for investigating molecular mediation. Hum. Mol. Gen. 2016;25(R2):R149–R156. doi: 10.1093/hmg/ddw197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burgess S, Daniel RM, Butterworth AS, Thompson SG EPIC-InterAct Consortium. Network Mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int. J. Epidemiol. 2015;44(2):484–495. doi: 10.1093/ije/dyu176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bakulski KM, Feinberg JI, Andrews SV, et al. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11(5):354–356. doi: 10.1080/15592294.2016.1161875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr. 2013;13(1):1. doi: 10.1186/1471-2431-13-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.