

Abstract
Aurones, derivatives of 2-benylidenebenzofuran-3 (2H)-one, are natural products that serve as plant pigments. There have been reports that some of these substances fluoresce, but little information about their optical properties is in the literature. In this report, series of aurone derivatives were synthesized as possible fluorescent probes that can be excited by visible light. We found that an amine substituent shifted the lowest energy absorption band from the near-UV to the visible region of the electromagnetic spectrum. Four amine-substituted aurone derivatives were synthesized to explore the effect of this substituent on the absorption and emission properties of the aurone chromophore. The emission maxima and intensities of the molecules are strongly dependent on the nature of the substituent and the solvent polarity. Overall, the emission intensity increases and the maximum wavelength decreases in less polar solvents; thus, the aurones may be useful probes for hydrophobic sites on biological molecules. A limited investigation with model protein, nucleic acid and fixed cells supports this idea. It is known that the sulfur analog of aurone can undergo photoinduced E/Z isomerization. This possibility was investigated for one of the aminoaurones, which was observed to reversible photoisomerize. The two isomers have similar absorption spectra, but the emission properties are distinct. We conclude that appropriately substituted aurones are potentially useful as biological probes and photoswitches.
Keywords: Aurones, Fluorescent probes, Biological macromolecules
Introduction
Organic molecules that fluoresce in the visible region of the electromagnetic spectrum are frequently employed as probes for biological systems. Small organic molecules can cause minimal perturbation of the biological macromolecule, provide flexibility in spectral range selection, reduce background fluorescence and are easy to use. There are a variety of commercially available fluorophores that absorb and fluoresce in the visible region. These are based on three types of chromophores: xanthenes (fluorescein, rhodamine), boron dipyrromethenes and cyanines. However, most of these molecules are relatively bulky with small Stokes’ shift [1, 2]. We are interested in discovering fluorescent probes for biomolecules from new chromophores that can be observed with visible light. Our ideal probe is one that has a minimal effect on the structure of the biomolecular target; thus, one of our goals is to discover chromophores with small molecular footprints.
Molecules that possess a planar 1,2-diamino-1,2-diketoethylene functionality, such as the core structure of indigo, absorb visible light at energies that are unusually low for such a small molecular system [3]. The so-called hemiindigos, which possess an indolinone ring conjugated to an aromatic system, also have anomalously long wavelength absorption maxima. These molecules are synthetically more accessible than indigos, particularly when an asymmetrically substituted product is desired [4]. A number of hemi-indigo and hemi-thioindigo derivatives have been synthesized, and optical properties of many of these have been assessed. [5, 6]. The hemi-thioindigos have been particularly studied as light-activated molecular switches [7–9].

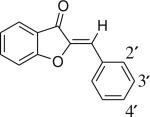
The oxygen-containing member of this molecular family, hemi-oxindigo, is the natural compound aurone. Aurones [2-benylidenebenzofuran-3(2H)-ones] are pigments found in vegetable, fruits and flowers and belong to the flavonoid class of natural products [10]. Compared to the natural abundance of flavones, aurones have a limited occurrence. Like all subclasses of flavonoids, the naturally occurring aurones are mostly found in the hydroxylated, methoxylated and glycosylated forms. The UV–vis absorption characteristics of naturally occurring aurones have been well documented. They absorb longer wavelength light than any of the other closely related flavonoid pigments. For example, the constitutional isomers flavone, 2-hydroxychalcone and aurone display absorption maxima at 297 nm, 312 nm and 379 nm, respectively [11]. The low energy absorption band of the aurone chromophore is attributed to the cross conjugation between ring B and coumaranone moeity [12]. Electron rich substituents on the B ring of the aurone produce bathochromic shifts in the aurone spectrum [13]. Fluorescence has been noted from some aurones [14, 15], but the fluorescence spectra of these molecules have not been reported to date.
In this work, we first synthesized a small series of aurone derivatives to explore the effect of the 4′-substituent on the low energy absorption band of the chromophore. We reasoned that a “push-pull” arrangement of functional groups in the chromophore would give the greatest red shift in the absorption spectrum relative to the parent molecule. We discovered that a 4′-amine produced a substantial red shift in the absorption maximum relative to the parent aurone, so additional fluorophores containing this substituent (1, 2, 3 and 4) were synthesized and studied. The aminoaurones absorb and fluoresce in the visible region of the electromagnetic spectrum. The spectral properties of these derivatives are dependent on the nature of the substituents on the B ring and solvent polarity. The molecules interact non-covalently with biological macromolecules and their binding results in change in the absorption and emission maximum of the probe. In some structures an enhancement in the quantum yield of the probe accompanies macromolecular binding. Photoisomerization was investigated for one of the molecules, which was found to be reversible. The aminoaurones therefore present a new class of visible range fluorescent molecules with potential utility as environmentally sensitive probes and as photoswitches.

Experimental Details
Materials and Methods
Proton (1H) and (13C) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker AM-360 MHz spectrometer as a solution in deuterated chloroform. Chemical shifts are reported in parts per million relative to tetramethylsilane. All solvents were reagent grade unless stated otherwise. Reactions were monitored by thin layer chromatography (TLC) with pre-coated silica gel plates. Silica gel (100–200 mesh) was used for all chromatographic purifications.
Synthesis of the Aurone Derivatives
Aurone derivatives were synthesized by acid-catalyzed condensation reactions between benzofuran-3(2H)-one and the respective aromatic aldehyde [12]. Benzofuran-3(2H)-one was synthesized in two steps starting from 1-(2-hydroxyphenyl) ethanone (Aldrich) [16, 17].

The known aurone derivatives were synthesized from the commercially available aromatic aldehydes: benzaldehyde, salicylaldehyde, p-bromobenzaldehyde, p-nitrobenzaldehyde, p-acetamidobenzaldehyde and 3-hydroxy-4-methoxybenzaldehyde [13]. The structures were confirmed by proton NMR. The aldehyde 2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinoline-9-carbaldehyde (julolidine-9-carbaldehyde), used for the synthesis of compound 3, was prepared in the laboratory starting from 2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinoline (julilodine) [18].
2-[1-(4-Acetamido-phenyl)-meth-(Z)-ylidene]-benzofuran-3-one (1)
A mixture of benzofuran-3(2H)-one (134 mg, 1 mmol), p-acetamidobenzaldehyde (163 mg, 1 mmol) and neutral alumina (5 g) were stirred at room temperature in 5 mL of methylene chloride [19]. Alumina was removed by filtration and washed with acetone. The product 2-[1-(4-acetamido-phenyl)-meth-(Z)-ylidene]-benzofuran-3-one was obtained as yellow solid (250 mg). The product was recrystallized from hot methanol and 100 mg (0.3 mmol, 36%) of the product 1 was recovered as yellow solid. 1H NMR (360 MHz, CO(CH3)2: δ 9.43 (s, 1H, NH), 7.98 (d, J=8.79 Hz, 1H), 7.82-7.77 (m, 4H), 7.51 (d, J=8.42 Hz, 1H), 7.33 (t, J=8.06 Hz, 1H), 6.84 (s, 1H), 2.12 (s, 3H).
2-[1-(4-Amino-phenyl)-meth-(Z)-ylidene]-benzofuran-3-one (2)
Compound 2 was prepared by hydrolysis of compound 1: Compound 1 (60 mg, 0.21 mmol) was dissolved in 4 mL of hot ethanol and a drop of concentrated hydrochloric acid was added. The solution was refluxed overnight. The solution was washed with saturated NaHCO3 solution and extracted in methylene chloride. Methylene chloride was removed in vacuo to obtain 50 mg (0.2 mmol, 95%) of 2 as a orange solid. 1H NMR (360 MHz, CDCl3): δ 7.80 – 7.76 (m, 3H), 7.62 (t, J=7.32 Hz, 1H), 7.31 (d, J= 7.33 Hz, 1H), 7.19 (t, J=7.69 Hz, 1H), 6.87 (s, 1H), 6.72 (d, J=8.78 Hz, 2H), 4.07 (s, 2H, NH); 13C NMR (90 MHz, CDCl3): δ 184.3, 165.5, 148.6, 145.2, 136.1, 133.7 (2C), 124.4, 123.0, 122.4, 122.2, 114.9 (2C), 114.6, 112.8.
2-[1-(2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij]quinolin-9-yl)-meth-(Z)-ylidene]-benzofuran-3-one (3)
Compound 3 was prepared from benzofuran-3(2H)-one (0.13 g, 1.01 mmol) and julolidine-9-carbaldehyde (0.2 g, 1.03 mmol) using the procedure described for synthesis of compound 4. The product was obtained as dark red crystals. The yield was 0.27 g (0.84 mmol, 83%). 1H NMR (360 MHz, CDCl3): δ 7.79 (d, J=8.06 Hz, 1H), 7.58 (t, J=7.7 Hz, 1H), 7.40 (s, 2H), 7.40 (s, 2H), 7.32 (d, J= 8.06 Hz, 1H), 7.17 (t, J=7.32 Hz, 1H), 6.84 (s, 1H), 3.30 – 3.26 (m, 4H), 2.83 – 2.78 (m, 4H), 2.02 – 1.94 (m, 4H); 13C NMR (90 MHz, CDCl3): δ 183.6, 164.9, 144.8, 144.5, 135.3, 131.3, 124 (2C), 122.6 (2C), 121.1, 118.8, 116.1, 112.7, 49.9 (2C), 27.7 (2C), 21.4 (2C).
2-[1-(8-Hydroxy-2,3,6,7-tetrahydro-1H,5H-pyrido[3,2,1-ij] quinolin-9-yl)-meth-(Z)-ylidene]-benzofuran-3-one (4)
A mixture of benzofuran-3(2H)-one (134 mg, 1 mmol), 8-hydroxy-julolidine-9-carbaldehyde (217 mg, 1 mmol) was refluxed overnight in ethanol containing 1 drop of concentrated hydrochloric acid. The solution was washed with saturated NaHCO3 solution and extracted to methylene chloride. The solvent was removed in vacuo. The product 4 was purified by flash chromatography using 1:1 hexanes: ethyl acetate. Rf=0.5 (60:40 hexanes: ethyl acetate). The product was obtained as bright purple crystals 40 mg (0.12 mmole, 12%). 1H NMR (360 MHz, CDCl3): δ 7.82 (d, J=8.42 Hz, 1H), 7.59 (t, J=8.06 Hz, 1H), 7.47 (s, 1H, OH), 7.34-7.31 (m, 3H), 7.20 (t, J= 7.32 Hz, 1H), 3.28-3.23 (m, 4H), 2.78-2.72 (m, 4H), 2.01-1.95 (m, 4H); 13C NMR (90 MHz, CDCl3): δ 182.2, 163.5, 153.5, 147.0, 142.4, 135.3, 131.7, 124.2, 123.1 (2C), 114.9, 113.8, 112.6, 107.1, 106.9, 50.2, 49.4, 27.3, 21.9, 21.1, 21.0.
Absorption and Emission Spectra of the Molecules
The absorption spectra of the molecules were recorded on a Hewlett Packard 8453 diode array spectrophotometer at room temperature. The fluorescence spectra were measured on a Jobin Yvon Fluoromax 3 spectrophotometer at 25°C in a thermostated cell holder. The solvents used for the spectroscopic studies were freshly distilled prior to spectroscopic measurements. The light-induced isomerization of these molecules was observed by irradiation of the sample at the desired wavelength in the spectrofluorimeter.
Determination of Molar Extinction Coefficient
The molar extinction coefficients for the compounds 2, 3 and 4 were determined by dissolving a known mass of the compound in ethanol to prepare a stock solution of 1 mM. The stock solution was subsequently diluted with ethanol to concentrations in the range of 0–60 μM. The absorbance at the maximum was recorded and plotted against the concentration to yield a straight line with slope equal to the extinction coefficient.
Determination of Quantum Yields
Fluorescence quantum yields (ϕF) were determined at 25°C in dilute solutions with an absorbance below 0.1 at the excitation wavelength. Quinine sulfate in 0.1 M H2SO4 (λex=347 nm) was used as a standard (ϕF=0.577) [20]. All solvents were dried before use. The slit width was kept as 2 nm for both excitation and emission. The quantum yields were determined using the following equation
where ϕF is the quantum yield of the molecule, A is the absorbance value, F is the area of the corrected emission spectrum after subtraction of the solvent background, and η is the refractive index of the solvent.
Binding of Aurone Derivatives to Biological Macromolecules
The binding of the aurone derivatives 2 and 3 to salmon sperm DNA and bovine serum albumin (in 0.05 M Tris HCl buffer, pH 7.4) at room temperature were checked using absorption spectrophotometry and spectrofluorimetry. Stock solutions of aurone derivatives were prepared in DMSO. The concentrations of BSA and DNA were determined based on the extinction coefficients: ε280 nm, water=0.677 (mg/mL)−1cm−1 and ε260 nm =13,800 Mbp−1cm−1 [21].
Confocal Microscopy
PC3 cells were grown on Lab-Tek II chambered coverglass for 24 h. Cells were washed three times with PBS (10 mM sodium phosphate, pH 7.2, 0.9% sodium chloride w/v) and then treated with methanol: acetone (1:1, v/v) for 10 min at 4°C. The cells were washed with PBS three times and then stained with the aurone derivatives 1, 2, 3 and 4 (50 μM) for 5 min. The coverglass was washed with PBS thrice to remove the excess stain. Excess buffer was gently removed and Gel/Mount (Biomedia Corp.) was added. Photomicrographs were obtained using a Zeiss LSM 510 META confocal scanning laser microscope. The excitation sources were 405 nm diode laser (for compounds 1 and 2), 488 nm Argon laser (for compound 3) and 543 nm HeNe laser (for compound 4). Emission was monitored using filters LP 475 nm (for compounds 1 and 2), LP 505 nm (for compound 3) and LP 560 nm (for compound 4).
Results
The UV–vis absorption spectra have been characterized for most of the naturally occurring aurones. To investigate the influence of electrochemically diverse substituents on the absorption spectrum of the aurone chromophore, a small series of B ring substituted derivatives were synthesized. Table 1 lists the absorption maxima of these derivatives in a polar and an apolar solvent. It is apparent from Table 1 that the presence of electron rich substituents on the B ring of the aurone shifts the absorption maximum to a longer wavelength; the most noteworthy derivative in the series is the 4′-aminoaurone (compound 2). The amine substituent extends the absorption maximum of the parent compound by about 50 nm into the visible region of the electromagnetic spectrum.
Table 1.
Effect of B ring substituents on the absorption maximum of aurone derivatives
|
Absorption (λmax, nm) | |
|---|---|---|
| Ethanol | Dioxane | |
| 4′-H (aurone) | 377 | 377 |
| 4′-Br | 381 | 383 |
| 4′-NO2 | 384 | 386 |
| 4′-NHCOCH3 (1) | 395 | 399 |
| 2′-OH | 402 | 398 |
| 4′-OCH3, 3′-OH | 414 | 412 |
| 4′-NH2 (2) | 441 | 426 |
Absorption Spectra of the 4′-Aminoaurone Derivatives
Structurally, the aminoaurones represent the classic D-π-A system with the electron deficient coumaranone moiety linked to the electron donating B ring. The accessibility of the lone pair of the 4′-substituent on ring B should affect the absorption maximum in predictable ways. Absorption spectra for compounds 1–4 in are illustrated in Fig. 1a. The most blue-shifted absorption spectrum of the four molecules is the amide 1, in which the nitrogen atom is bonded to the electron withdrawing carbonyl. Both compounds 2 and 3 have electron density readily available for donation to the aurone π-system, but the absorption maximum of 3 is considerably red shifted relative to the absorption maximum of 2 (61 nm in methanol; Table 2). The rigid ring structure in 3 eliminates free rotation of the N–C bond thereby enhancing the electron donating ability of the amine [22]. The additional electron donating group in compound 4 results in a further 15 nm red shift in the absorption maximum and an increase in molar absorptivity (Table 2).
Fig. 1.

a Normalized absorption spectra of 4′-aminoaurones in ethyl acetate: 1 (black), 2 (blue), 3 (green) and 4 (red). b Normalized emission spectra of 4′-aminoaurones in ethyl acetate: 1 (black), 2 (blue), 3 (green) and 4 (red). The excitation wavelengths were 399 nm (1), 426 nm (2), 483 nm (3), 490 nm (4)
Table 2.
Molar extinction coefficients of 4′-aminoaurone derivatives
| Compound | Absorption |
||
|---|---|---|---|
| Solvent | λmax (nm) | Log ελ | |
| 1 | Methanol | 395 | 4.09 |
| 2 | Methanol | 442 | 4.33 |
| 3 | Methanol | 503 | 4.15 |
| 4 | Methanol | 518 | 4.34 |
Fluorescence Spectra of the 4′-Aminoaurone Derivatives
The amine derivatives fluoresce in the visible region of the electromagnetic spectrum. The emission maximum shifts to longer wavelength when the 4′-substituent is an amine versus an amide (Fig. 1b), and the emission maxima observed for the amine derivatives are above 500 nm in nearly every solvent. Table 3 summarizes the absorption and emission energies of 1–4 in different solvents. Notable features include the large Stokes shift exhibited by all four molecules in all solvents. The data display a non-linear dependence of the Stokes shift on solvent polarity (data in Table 3, plot not shown), and the largest shifts are in the polar protic solvents. This observation points to a specific solvent interaction between the fluorophore and solvents containing hydrogen bond donors. Interestingly, the Stokes shift of 4 was smaller than that of 3 in all solvents, which is due to the consistently longer absorption maximum of 4 versus 3 without a corresponding increase in the wavelength of the emission maximum.
Table 3.
Absorption and emission maxima and stokes shift of aurones 1–4 in various solvents
|
|
Amide 1 |
Amine 2 |
Amine 3 |
Amine 4 |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Solvent | ET30 | Amax nm | Emmax nm | Δν cm−1 | Amax nm | Emmax nm | Δν cm−1 | Amax nm | Emmax nm | Δν cm−1 | Amax nm | Emmax nm | Δν cm−1 |
| Dioxane | 36 | 399 | 446 | 2640 | 426 | 489 | 3020 | 472 | 540 | 2670 | 486 | 546 | 2260 |
| Ethyl Acetate | 38.1 | 399 | 446 | 2640 | 426 | 501 | 3510 | 472 | 552 | 3070 | 483 | 552 | 2590 |
| Acetone | 42.2 | 398 | 460 | 3390 | 429 | 522 | 4150 | 481 | 575 | 3400 | 487 | 574 | 3110 |
| DMSO | 45.0 | 401 | 481 | 4150 | 452 | 548 | 3880 | 498 | 591 | 3160 | 510 | 595 | 2800 |
| Isopropanol | 48.6 | 395 | 468 | 3950 | 446 | 542 | 3970 | 497 | 579 | 2850 | 513 | 576 | 2130 |
| Ethanol | 51.9 | 395 | 482 | 4570 | 441 | 540 | 4160 | 501 | 580 | 2720 | 515 | 584 | 2290 |
| Methanol | 55.5 | 395 | 496 | 5160 | 442 | 542 | 4120 | 503 | 582 | 2740 | 518 | 588 | 2260 |
| Water | 63.1 | 407 | 521 | 5380 | 430 | 560 | 5400 | – a | – | – | 545 | 612 | 2010 |
Amine 3 was insoluble in water
The relative emission intensities of the aurones in a variety of solvents are collected in Table 4. The emission intensities of compounds 1 and 4 are relatively insensitive to solvent polarity, while those of compounds 2 and 3 increase dramatically with decreasing solvent polarity. Quantum yields for 1–4 were determined in two solvents (Table 5).
Table 4.
Relative fluorescence intensities of aurones 1–4 in various solvents
|
|
Relative fluorescence intensitya |
||||
|---|---|---|---|---|---|
| Solvent | ET30 | Amide 1 | Amine 2 | Amine 3 | Amine 4 |
| Dioxane | 36.0 | 0.7 | 18 | 47 | 8.9 |
| Ethyl acetate | 38.1 | 0.9 | 28 | 58 | 4.3 |
| Acetone | 42.2 | 2.4 | 23 | 3.0 | 8.3 |
| DMSO | 45.0 | 1.3 | 9.4 | 3.9 | 4.8 |
| Isopropanol | 48.6 | 1.0 | 4.9 | 3.2 | 4.5 |
| Ethanol | 51.9 | 0.8 | 2.2 | 2.9 | 2.0 |
| Methanol | 55.5 | 1.0 | 1.0 | 1.0 | 1.0 |
| Water | 63.1 | 0.4 | 0.2 | – | 0.2 |
Fluorescence intensity at emission maximum, relative to fluorescence intensity in methanol. Solutions were excited at the absorption maximum. The absorbance of each sample at the absorption maximum was 0.1
Table 5.
Quantum yields of selected 4′-aminoaurones
| Compound | Quantum yield (ϕ) |
|
|---|---|---|
| Methanol | Dioxane | |
| 1 | 0.011 | 0.004 |
| 2 | 0.002 | 0.035 |
| 3 | 0.002 | 0.282 |
| 4 | 0.005 | 0.025 |
Potential Probes for Biomacromolecules
The potential for the aminoaurones 2 and 3 to serve as probes for proteins and nucleic acids was assessed spectroscopically using model biomolecules. Figure 2 shows the effect of protein or DNA on the emission spectrum of 2. Compound 2 in presence of an equimolar concentration of BSA displayed 30-fold enhancement in the fluorescence intensity with a concomitant blue shift in the emission maximum (Fig. 2a). The derivative also exhibited change in fluorescence intensity in presence of salmon sperm DNA; an 8-fold increase in the emission intensity but no change in the emission maximum was observed in presence of equimolar DNA. This result indicates that binding of 2 to DNA does not result in change in the environment polarity of the fluorophore, which suggests that 2 may bind in a DNA groove. Compound 3 displayed 170-fold enhancement in the fluorescence intensity in presence of equimolar BSA, but did not show any change in its spectral properties in presence of DNA (Fig. 2b). The absence of an apparent interaction of aurone 3 with DNA may be due to the non-planarity of fused ring system.
Fig. 2.

a Fluorescence spectra of compound 2 (20 μM) in the presence of bovine serum albumin (20 μM) and salmon sperm DNA (20 μM) in 0.05 M Tris buffer, pH 7.2. Red: BSA+ 2 Green: DNA+3. Blue: 2 in buffer. Excitation wavelength=460 nm. b Fluorescence spectra of compound 3 (20 μM) in the presence of bovine serum albumin (20 μM) and salmon sperm DNA (20 μM) in 0.05 M Tris buffer, pH 7.2. Red: BSA+3 Green: DNA+3. Blue: 3 in buffer. Excitation wavelength=500 nm
A difference in cellular distribution of the probes was also noted. PC3 cells were grown on coverglass and fixed with methanol/acetone. The cells were exposed to 0.05 mM compound 1, 2, 3 or 4, washed with buffer, and then examined using confocal microscopy. Figure 3 shows that compound 1 and 2 uniformly labels the cell, labeling both the nucleic acid as well as the protein structure, whereas compound 3 does not label the nucleus. Compound 4 labels the cytoplasm more brightly than the nucleus.
Fig. 3.

Confocal microscopy images of fixed PC3 cells stained with compound 1 (a), 2 (B), 3 (c) and 4 (d). Scale bar=20 μm
Photoisomerization of the Aurones
Both hemi-indigos and hemi-thioindigos are known to undergo light induced isomerization [5, 6]. The possibility that aurones can also undergo photoisomerization was examined for one of the compounds (2) using NMR spectroscopy. The synthetic aurones are the thermodynamically more stable Z-isomer. This isomer can be distinguished from its E-counterpart from the distinct chemical shifts observed in 1H-NMR. In general, the olefinic proton of the Z-isomer is more shielded than that of the corresponding E-isomer [23]. The proton NMR of 2 in CDCl3 is shown in Fig. 4a. Figure 4b shows the sample after exposure to a 400 nm radiation for a period of 1 h. The decrease in the intensity of the 6.87 ppm singlet was accompanied by appearance of a new peak at 6.91 ppm, which corresponds to the olefinic proton of the E-isomer. Next, the NMR sample was exposed to lower energy radiation (480 nm) for a period of 1 h (Fig. 4c). A nearly quantitative conversion of the isomeric mixture to the Z-isomer was observed.
Fig. 4.

NMR spectrum of 2 in deuterated chloroform (CDCl3). a Z-isomer of 2 in CDCl3. The numbers 1–4 refer to the protons (or sets of equivalent protons) that are affected by the radiation of the Z-isomer to E-isomer. b after exposure to 400 nm radiation for 1 h. The olefinic peak (1) of the Z-isomer shifts from 6.87 to 6.91 ppm (3) (which is characteristic of the E-isomer). Note that the peak labeled (2) in the Z-isomer undergoes a significant downfield shift in the E-isomer (4). The ratio of E-isomer to Z-isomer is 60:40, based on the relative integration of the resolved peaks. C: Spectrum after exposure of the sample to 480 nm radiation for 1 h. About 95% of the sample is the Z-isomer occurs as is evident from the decrease in the 6.91 ppm peak

Absorption and emission spectra of the NMR sample were also collected. Figure 5a shows absorption spectra of samples corresponding to each spectrum from Fig. 4. The absorption maximum of the mixture of isomers was 433 nm, 9 nm greater than the absorption maximum of the single isomer. The absorption maximum reverted back to 426 nm after irradiation with 480 nm light. From these data we conclude that the absorption maximum of the Z-isomer is at higher energy than that of the E-isomer.
Fig. 5.

a Absorption spectra of compound 2 in chloroform before (black), after exposure to 1 h 400 nm (red) and after exposure to 1 h 480 nm radiation (green). The spectra are normalized to illustrate the spectral shift. b Fluorescence spectra of 2 in deuterated chloroform: before (black), after 400 nm (red) and after 480 nm (green) irradiation, respectively (λex=426 nm)
Emission intensity of the NMR samples was measured on undiluted solutions (Fig. 5b). The emission spectra are therefore asymmetric due to the inner filter effect. Since the shift in the absorption maximum is small, the inner filter effect is similar in all emission spectra and therefore their characteristics can be compared. The emission intensity is decreased by irradiation at 400 nm and is restored upon irradiation at 480 nm. The data suggests that the E-isomer is probably non-fluorescent.
We noted that the fluorescence was not completely restored after exposure to 480 nm light, and that the relative intensity of the short wavelength bands in the absorption spectrum was larger after photoreversion. It therefore appears that the isomerization may be accompanied by some photodecomposition. The photoreversion process was therefore investigated. The fluorescence of the CDCl3 sample was monitored while under continuously irradiation at 480 nm (Fig. 6). The fluorescence increased for about an hour, but then decreased with continuing irradiation. Therefore, the aminoaurone 2 is susceptible photodecomposition to as well as photoisomerization.
Fig. 6.

Fluorescence intensity of compound 2 decreases after exposure to 480 nm radiation after 55 min. Arrow indicates the time point of 3300 s. The emission wavelength was 535 nm
Discussion
Aurones have been known for over 70 years. However, very little is known about their fluorescence. In this study we have investigated the influence of various substituents on the absorption of the parent aurone and explored the use of aurone derivatives as potential fluorescent probes for biological macromolecules. Our study indicates that the aurone skeleton is suitable as a small molecule framework for fluorophores. The spectroscopic properties can be tuned by the nature of the substituent on the aromatic ring. Of the molecules examined, an amine substituent at the 4′-position produces the largest red shift in the absorption maximum compared to that of the parent molecule. Therefore, a small series of 4′-amine-containing molecules was synthesized. We found that the availability of the lone pair of electrons on the nitrogen atom strongly affects the absorption and fluorescence properties of the amine-substituted aurone. Acetylation of the amine (1) shifts the absorption and emission maxima to shorter wavelength, while restricting the rotation of the amine nitrogen (3) shifts the absorption and emission maxima to longer wavelength. There is a solvent effect on the Stokes shift of all amine-substituted aurones, but the relative emission intensity is strongly solvent dependent in just 2 and 3. Due to the large increase in emission intensity coupled with the wavelength shift in less polar solvent, both 2 and 3 are potential probes for hydrophobic sites on biological macromolecules. Interestingly, 2 associates with both protein and DNA, while 3 preferentially interacts with proteins, probably because 3 is not completely planar. Thus, the chemical substituents on the aurone not only define the spectral properties but also can be modified so as to make probes selective for DNA or protein labeling. The different substituents also seem to affect their interaction with components of fixed cells; specifically, 1 and 2 stain the entire cell, while 3 appears to be excluded from the nucleus of PC3 cells.
The thio homolog of the aurone, called hemi-thioindigo or thioaurone, is known to undergo photoinduced E,Z-isomerization. Therefore, the possibility of photochemical isomerization of the aurones was assessed with one of the aminoaurones. The proton NMR spectra of 2 clearly show reversible isomerization of 2 about the carbon-carbon double bond. The less stable E-isomer was not isolated in pure form, but the absorption and emission spectrum of the E-isomer-containing solutions indicate that this isomer is much less fluorescent than the Z-isomer. Further exploration of the aurones as photoswitches is in progress in our laboratory.
In summary, the aurone skeleton provides a small molecular framework on which a variety of novel fluorescent probes can be designed. These fluorescent probes have properties that are complementary to the commonly used dye classes. Even the largest molecule synthesized for this study is smaller than the xanthenes dyes such as fluorescein and rhodamine. The xanthenes have high quantum yields in polar environments, however, while the aminoaurones investigated so far need to be in a hydrophobic environment to be useful probes. Aurones are environmentally sensitive like cyanines, with lower quantum yields but significantly larger Stokes shifts. The aminoaurones can be observed using common microscopy excitation sources. The Z- and E-isomers of 2 can be interconverted photochemically and therefore may have applications as photoactivated switches. Finally, this family of molecules is relatively simple to synthesize. The absorption and emission maxima of new family members may be tuned to suit a particular application through functional group selection.
Acknowledgements
Funding from the National Institutes of Health (R15 GM093941 and R01 CA69571) is gratefully acknowledged.
Contributor Information
Ozlem Dilek, Department of Chemistry, Binghamton University, State University of New York, Binghamton, NY 13902, USA.
Kamalika Mukherjee, Department of Chemistry, Binghamton University, State University of New York, Binghamton, NY 13902, USA.
Dennis W. McGee, Department of Biological Sciences, Binghamton University, State University of New York, Binghamton, NY 13902, USA
Susan L. Bane, Department of Chemistry, Binghamton University, State University of New York, Binghamton, NY 13902, USA
References
- 1.Choi YH, Veal AD, Epicocconone KPA. New cell-permeable long Stokes’ shift fluorescent stain for live cell imaging and multiplexing. J Fluoresc. 2006;16:475–482. doi: 10.1007/s10895-005-0010-7. [DOI] [PubMed] [Google Scholar]
- 2.Haugland RP. Handbook of fluorescent probes and research chemicals. Molecular Probes, Inc.; Oregon: 2002. [Google Scholar]
- 3.Zollinger H. Color chemistry. 3rd edn. Wiley-VCH; Weisheim: 2003. [Google Scholar]
- 4.Konieczny MT, Konieczny W. Molecules. 2005;65:451–464. [Google Scholar]
- 5.Yamaguchi T, Seki T, Tamaki T, Ichimura K. Photochromism of hemithioindigo derivatives. I. Preparation and photo-chromic properties in organic solvents. Bull Chem Soc Japan. 1992;65:649–656. [Google Scholar]
- 6.Ikegami M, Aria T. Photoisomerization and fluorescence properties of hemiindigo compounds having intramolecular hydrogen bonding. Bull Chem Soc Japan. 2003;76:1783–1792. [Google Scholar]
- 7.Ichimura K, Seki T, Tamaki T, Yamaguchi T. Fatigue-resistant photochromic hemithioindigos. Chem Lett. 1990:1645–1646. [Google Scholar]
- 8.Eggers K, Fyles TM, Montoya-Palaez PJ. Synthesis and characterization of photo switchable lipids containing hemithioindigo chromophores. J Org Chem. 2001;66:2966–2977. doi: 10.1021/jo0056848. [DOI] [PubMed] [Google Scholar]
- 9.Steinle W, Ruck-Brown K. Synthesis and characterization of novel bifunctional hemithioindigo chromophores. Org Lett. 2003;5:141–144. doi: 10.1021/ol027102+. [DOI] [PubMed] [Google Scholar]
- 10.Boumendjel A. Aurones: a subclass of flavones with promising biological potential. Curr Med Chem. 2003;10:2621–30. doi: 10.2174/0929867033456468. [DOI] [PubMed] [Google Scholar]
- 11.Geissman TA, Harborne JB. Anthoclor pigments. X. Aureusin and cernuoside. J Am Chem Soc. 1955;77:4622–4624. [Google Scholar]
- 12.Geissman TA, Moje W. Anthochlor pigments. VIII. The pigments of Coreopsis grandiflora, Nutt. III. J Am Chem Soc. 1951;73:5765–5768. [Google Scholar]
- 13.Geissman TA. The chemistry of flavonoid compounds. The Macmillan Company; New York: 1962. [Google Scholar]
- 14.Asen S, Norris KH, Stewart RN. Copigmentation of aurone and flavone from petals of Antirrhinum majus. Phytochemistry. 1972;11:2739–2741. [Google Scholar]
- 15.Ono M, Maya Y, Haratake M, Ito K, Mori H, Nakayama M. Aurones serve as probes of β-amyloid plaques in Alzheimer's disease. Biochem Biophys Res Commun. 2007;361:116–121. doi: 10.1016/j.bbrc.2007.06.162. [DOI] [PubMed] [Google Scholar]
- 16.King LC, Ostrum KG. Selective bromination with copper (II) bromide. J Org Chem. 1964;29:3459–3461. [Google Scholar]
- 17.Shriner RL, Witte M. Derivatives of coumaran. V. Synthesis of 4-hydroxycoumaran-3-one. J Am Chem Soc. 1939;61:2328–2329. [Google Scholar]
- 18.Cai GL, Bozhkova N, Odingo J, Berova N, Nakanishi K. CD exciton chirality method—new red-Shifted chromophores for hydroxyl-groups. J Am Chem Soc. 1993;115:7192–7198. [Google Scholar]
- 19.Varma RS, Varma M. Alumina-mediated condensation—a simple synthesis of aurones. Tetrahedron Lett. 1992;33:5937–5940. [Google Scholar]
- 20.Lakowicz RJ. Principles of fluorescence spectroscopy. Springer Science+Business Media, LLC; 2006. [Google Scholar]
- 21.Völker J, Klump HH, Breslauer JK. Communication between noncontacting macromolecules. Proc Natl Acad Sci USA. 2001;98:7694–7699. doi: 10.1073/pnas.141221298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Jiang X, Zhao C, Chen R, Qina P, Suna L. Donor– acceptor molecules containing thiophene chromophore: synthesis, spectroscopic study and electrogenerated chemiluminescence. Tetrahedron Lett. 2006;47:4961–4964. [Google Scholar]
- 23.Hastings JS, Heller GH. The stereochemistry of aurones 2-substituted benzylidenebenzofuran-3(2H)-ones. J Chem Soc Perkins. 1972;I:2128–2132. [Google Scholar]