

Abstract
Genomic instability is a critical driver in the process of cancer formation. At the same time, inducing DNA damage by irradiation or genotoxic compounds constitutes a key therapeutic strategy to kill fast‐dividing cancer cells. Sensing of DNA lesions initiates a complex set of signalling pathways, collectively known as the DNA damage response (DDR). Deciphering DDR signalling pathways with high‐throughput technologies could provide insights into oncogenic transformation, metastasis formation and therapy responses, and could build a basis for better therapeutic interventions in cancer treatment. Mass spectrometry (MS)‐based proteomics emerged as a method of choice for global studies of proteins and their posttranslational modifications (PTMs). MS‐based studies of the DDR have aided in delineating DNA damage‐induced signalling responses. Those studies identified changes in abundance, interactions and modification of proteins in the context of genotoxic stress. Here we review ground‐breaking MS‐based proteomics studies, which analysed changes in protein abundance, protein‐protein and protein‐DNA interactions, phosphorylation, acetylation, ubiquitylation, SUMOylation and Poly(ADP‐ribose)ylation (PARylation) in the DDR. Finally, we provide an outlook on how proteomics studies of the DDR could aid clinical developments on multiple levels.
Keywords: Biomedicine, Cancer, DNA damage response, DNA–protein interaction, Mass Spectrometry, PTM analysis
Abbreviations
- ATM
ataxia‐telangiectasia mutated
- ATR
ATM and RAD3 related
- DDR
DNA damage response
- DSB
DNA double strand break
- ICL
interstrand crosslink
- IR
γ‐irradiation
- MMS
methyl methanesulfonate
- SUMO
small ubiquitin‐like modifier
- UV
ultraviolet
1. DNA damage response in cancer formation and treatment
Despite the great variety of endogenous and exogenous sources that threaten the integrity of the DNA, our genomes are remarkably stable. This is due to the action of the DNA damage response (DDR). DDR signalling processes comprise the recognition of sites of DNA damage and the recruitment of factors, which transmit and amplify the damage signal, and finally execute the adequate cellular responses 1. These responses to DNA damage include: chromatin rearrangements to allow access to the damaged DNA, DNA repair, cell cycle arrest, and alignment of cellular housekeeping functions, such as transcription, translation and cellular metabolism 2, 3. Damage beyond repair can lead to initiation of apoptosis (or other forms of programmed cell death), or senescence. Cells, which survive in the presence of unrepaired damage and re‐enter the cell cycle might ultimately become cancerous (Fig. 1) 1. This is reflected in hereditary cancer syndromes linked to dysfunctional DDR pathways 4 and the enhanced genomic instability in spontaneously arising, non‐hereditary types of cancers 5. Excessive DNA damage has further been associated with accelerated ageing 1, 6.
Figure 1.
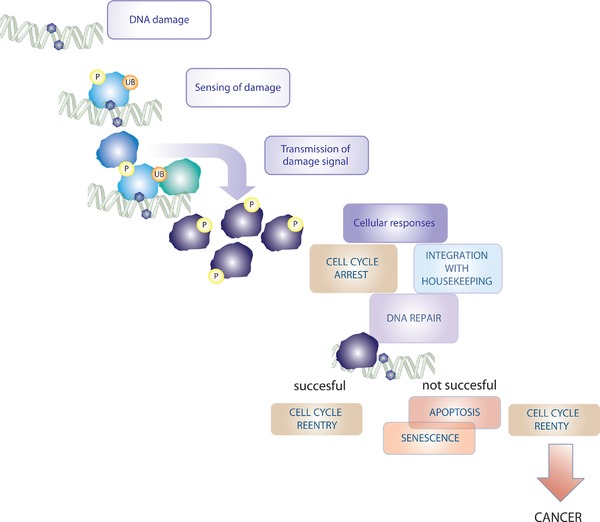
DNA damage signalling response. After sensing of DNA damage by proteins, which are either involved in DNA metabolism, or specifically recruited to aberrant DNA structures, a PTM‐based signalling cascade is set into motion. This cascade enhances the nuclear damage signal and leads the damage signal down to effector components, which are involved in DNA repair, cell cycle arrest, and the integration of DNA damage with on‐going cellular housekeeping processes. If DNA repair is successful cells can re‐enter the cell cycle. If repair is not successful, the initiation of apoptosis or terminal arrest (senescence) can ensue. If cells re‐enter the cell cycle in the presence of unrepaired DNA, this can lead to cancer formation.
While silencing of the proper response to DNA damage is seen as an enabling factor of cancer formation 7, on the other hand cancer treatment commonly relies on DNA damage induction by genotoxic drugs or irradiation 8. In recent years, the potential to specifically exploit DDR defects of tumour cells (e.g. deficiencies in homologous recombination repair) has emerged as a strategy for finding novel drugs and cancer biomarkers 4, 9. Utilising the concept of synthetic lethality in cancer cells is also emerging as a powerful strategy for anticancer therapy 10, 11.
The DDR comprises a complex signalling network in which proteins and their posttranslational modifications (PTMs) play crucial roles on a multitude of levels.
Proteins involved in DNA metabolism, as well as specialised DNA damage sensor proteins sense various DNA lesions. Often damage sensing proteins are intimately linked with the DNA repair pathways, which repair specific types of lesions 12.
Sensing of aberrant DNA structures generally sets in motion a signalling cascade in which PTMs are added to sensor proteins, chromatin proteins and signalling factors (Fig. 1) 13. PTM enrichment at sites of damage serves as a recruitment platform for further signalling factors involved in damage sensing, DNA repair, and transmission to downstream effector molecules.
Amongst the earliest activated sensors in the DDR are nuclear protein kinases and E3 ligases, which modify substrate proteins by site‐specific phosphorylation and ubiquitylation, respectively 14. Key upstream modifying enzymes include the PI3‐K‐related protein kinases ataxia‐telangiectasia mutated (ATM) and ATM and RAD3 related (ATR). While ATM reacts to the presence of DNA double strand breaks (DSBs), ATR activity is triggered by RPA‐coated single strand breaks 15, 16. In the response to DSBs also E3 ubiquitin ligases such as RNF8 and RNF168 are crucially important 17.
Enzymes involved in DDR‐PTM‐cascades, such as kinases and poly(ADP‐ribose) (PAR) polymerase (PARP) enzymes have been identified as promising cancer biomarkers and drug targets 4, 18, 19. The potential to exploit DDR factors for improving the success of cancer therapy makes a better understanding of DNA damage signalling cascades and their apical regulators an important task for researchers today.
A better understanding of the intricate signalling responses evoked by DNA damage requires high‐throughput technologies. Mass spectrometry (MS)‐based proteomics has emerged as a highly sensitive, high‐throughput, technique, which allows snapshots of cellular proteomes at a given cellular state 20, 21. Shotgun proteomics has tremendous discovery power on multiple levels. The technique allows studying the abundance of proteins 22, their interactions with other proteins or other cellular macromolecules such as DNA 23, 24, and their modification by PTMs 25, 26.
Different groups have attempted MS‐based analyses of the responses to different kinds of damage stimuli. Those included studies of PTM changes 27, 28, 29, 30, 31, 32, 33, 34, changes in interactions between proteins or between proteins and DNA 35, 36, 37, and changes in protein abundance 30, 31, 32 (Fig. 2).
Figure 2.
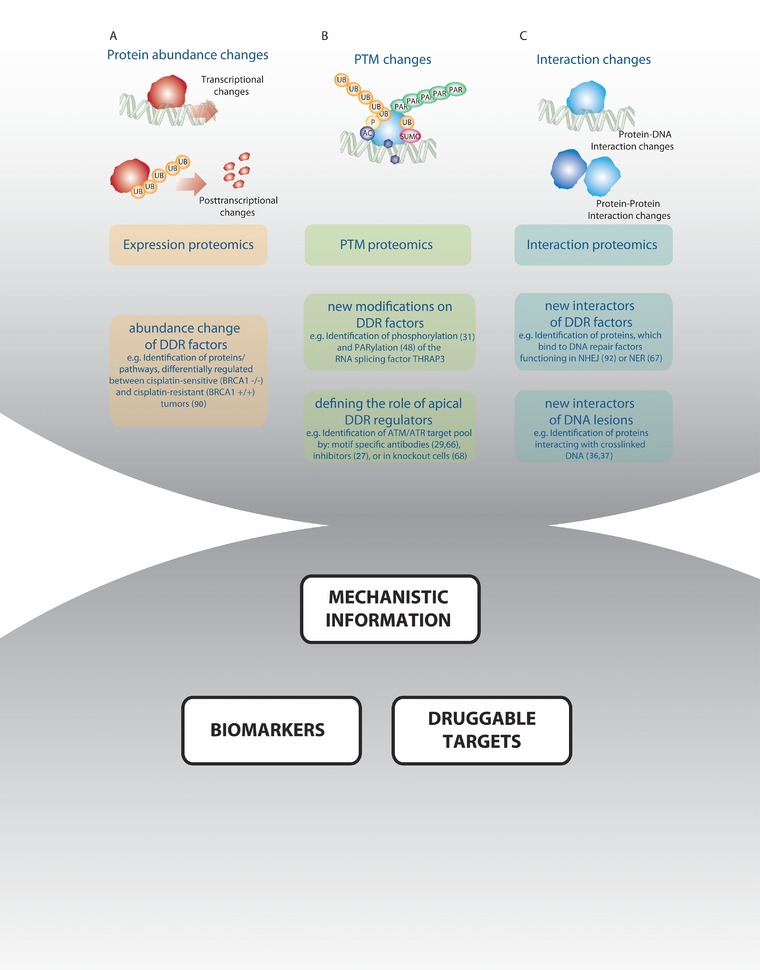
Proteomics techniques to study DNA damage‐induced changes in protein interactions, protein abundance and PTM modifications. (A) Expression proteomics can measure changes in protein abundance. Those can result from transcription changing mechanisms or from posttranscriptional mechanisms, which are induced by DNA damage. (B) PTM proteomics can measure PTM changes, which are induced by DNA damage. (C) Interaction proteomics can identify changes in protein‐protein and protein‐DNA interactions after DNA damage. Those data can help to clarify or corroborate drug mechanisms of action, and lead to identification of drug targets and biomarkers.
2. Exploring the DDR using MS
In contrast to techniques, which rely on antibodies and thus inherently preclude identification of new proteins and modifications, MS‐based proteomics has the power to identify novel players of DDR signalling processes 38.
In shotgun proteomics proteins are generally digested prior to analysis using proteases, in a so‐called bottom‐up approach. Often, sample complexity is reduced by on‐ or offline fractionation, or by the enrichment of proteins of peptides prior to MS analysis 39. Moreover, most workflows include an on‐line chromatographic separation step, before peptides are ionised and analysed by MS. Currently, mass spectrometers of the orbitrap type are the most commonly used 39.
The “bottom‐up” shotgun proteomics approach entails several limitations. Most shotgun proteomics experiments use trypsin as the exclusive protease, because it generates MS‐friendly peptides. This approach, however, neglects the proteome space, to which trypsin is blind 40. In the future top‐down analysis of individual proteins, i.e. direct MS analysis of intact proteins without a protease digestion step 41 and middle‐down proteomics using different proteases that generate longer polypeptides than trypsin could increase proteome coverage 42.
Another challenge of proteomics lies in the fact that the target database against which MS spectra are searched is a generic human database. This precludes the possibility to analyse specific mutation profiles of individual cell types, which might be highly relevant when studying cancer genomes that generally undergo massive rearrangements 43. Proteogenomics approaches, where genomic data from the cell line or tissue sample under investigation are used as reference database, could in the future serve as an elegant means to overcome this problem 44, 45.
The dynamic range of protein abundance within a cell is very large, spanning multiple orders of magnitude. Housekeeping proteins (such as ribosomal proteins), which generally remain steady over a great number of cellular conditions, are often highly abundant. In contrast, levels of signalling‐relevant proteins and signalling‐relevant PTMs are often low 20. To capture lowly abundant proteins and PTMs advances in fractionation and enrichment methods can help.
3. Analyzing PTMs in the DDR using MS
In DDR signalling processes, individual PTMs are covalently attached to signalling proteins with different kinetics. Multiple ways exist to dynamically regulate the process of PTM addition. The abundance, localisation and specificity of the enzymes, which add and remove PTMs (e.g. kinases and phosphatases) is regulated upon different stimuli. This regulation depends on transcriptional changes in the expression of these enzymes or alteration in their posttranslational modifications, e.g. by kinase auto‐phosphorylation. Moreover, cofactors can either bring enzyme and substrate together or sequester them. Cofactors themselves can also undergo transcriptional and posttranscriptional regulation. In the case of protein‐modifiers such as ubiquitin and small ubiquitin‐like modifier (SUMO) also the extent of the free pool of modifiers can influence the speed and efficiency of PTM attachment (Fig. 3).
Figure 3.
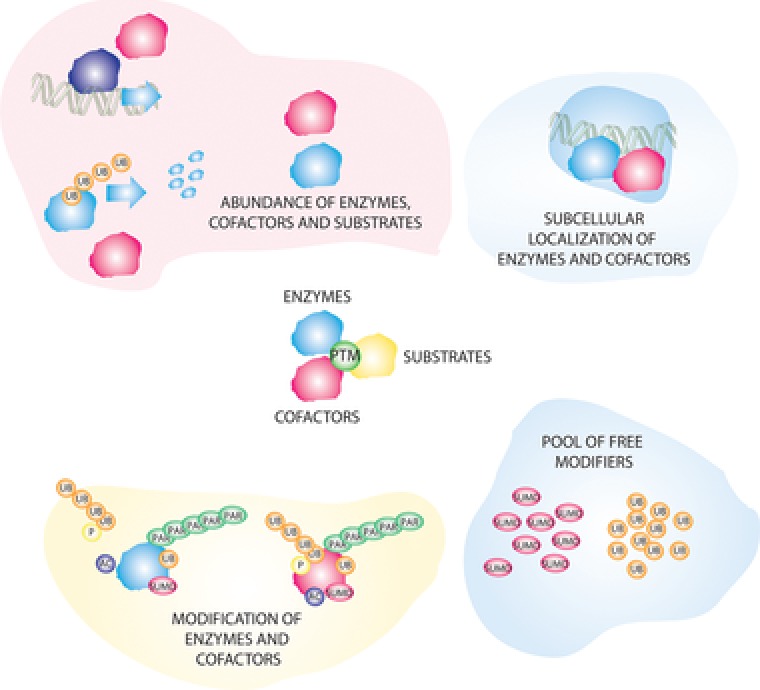
Modes of regulation of PTM responses after DNA damage. Different levels of integration exist for the dynamics and specificity of PTMs. Those include regulation of the (A) abundance (B) localisation (C) modification of enzymes and co‐factors. For small protein modifiers regulation can also occur on the level of the pool of free modifiers.
This highly dynamic nature of PTMs can make it difficult to choose the optimal timeframe for the analysis of PTM‐responses to DNA damage. Most PTM studies are limited to one or few timepoints, due to the often high requirement of input material in PTM proteomics. It is however, important to consider that the endpoint the researcher chooses will bias the scope of the identified results. While posttranslational responses can be very fast, transcriptional responses might take longer. Moreover, depending on the strength of the damage pulse, choosing a late analysis timepoint might mean that repair of the DNA lesion could already have occurred. Also, the massive contribution of cell cycle changes to the expression and modification of proteins should not be neglected when interpreting the results of DDR studies 46. It is important to consider the difference between signalling events and cellular responses, which are caused by a halt of cell cycle progression, especially at later timepoints after DNA damage 31.
When analysing PTMs by MS it is important to take into account any special requirements related to sample preparation workflows and MS instrumentation. For highly dynamic PTMs it is important to counteract their reversal by blocking the activity of the responsible enzymes, for example by use of phosphatase inhibitors to preserve phosphorylation, or of alkylating chemicals to inhibit deubiquitinating enzymes 20, 47. Also artificial addition of PTMs during sample preparation should be avoided, e.g. by inhibiting the activity of kinases or PARylating enzymes during sample preparation steps by addition of Mg2+ scavengers such as EDTA/EGTA or PARP inhibitors, respectively 20, 48. Modification usually changes the physiochemical properties of peptides, thus impacting their digestion efficiency and behaviour during chromatography and ionisation 49.
Since modified versions of peptides often exist in sub‐stoichiometric quantities, enrichment steps are generally required to analyse those by MS. Enrichment methods can rely on the physicochemical properties or structure/ sequence‐specific features of proteins and peptides. Phosphorylated peptides can for example be enriched by using metal ion‐based affinity capture (MOAC) or immobilised metal affinity chromatography (IMAC) 50. Titanium dioxide (TiO2) metal ion‐based affinity capture was used in different studies that provided global snapshots of phosphoproteomes after DNA damage 30, 31, 32, 51.
Other enrichment strategies target specific protein sequence, or structural features, using antibodies or binding domains. Antibody‐based enrichment is commonly used for enrichment of tyrosine phosphorylated peptides, and has also been successfully applied for enriching ubiquitylated and acetylated peptides in DDR studies 28, 31, 33. In their landmark study of the phosphorylation response to DNA damage, Matsuoka et al. used antibodies targeting the S/T‐Q motif, which is specific for the protein kinases ATM and ATR 29.
Also, binding domains of specific modifications, such as phospho‐binding domains can be used to “fish out” their interactors, as shown by Blasius et al. who detected interactors of 14‐3‐3 proteins in the context of UV treatment 35. Domains with a high affinity for PAR were further used to enrich PARylated proteins after DNA damage 48.
Nevertheless, for some PTM‐types good antibodies are not yet available. Moreover, sample preparation conditions used for IP‐based enrichment can interfere with stability of modifications. For enrichment of those proteins, researchers generally rely on the exogenous expression of tagged versions of proteins, as done for studies of SUMOylation responses 52.
While enrichment is still a prerequisite for analysing lowly abundant peptide species, enrichment strategies are generally accompanied by an increase in workload, instrument time and a decrease in reproducibility. Those limit the resolution at the levels of time, cell type and damage‐inducer studied.
Most changes in the abundance of proteins or in the occupancy of modified versions of a protein are no on/off situation 53. Full stoichiometry of PTMs is only reached in rare cases, such as phosphorylation changes during the mitotic phase of the cell cycle 46. To accurately identify the abundance of peptides, different strategies exist. Those include label‐free quantification, Stable isotope labeling with amino acids in cell culture (SILAC)‐based quantification and chemical labelling strategies 20. Most studies of PTM‐changes in the DDR used SILAC‐based quantitation as method of choice 28, 29, 31, 33.
Another specific challenge posed by PTM proteomics experiments is the downstream computational analysis and bioinformatic interpretation. Multiple modified variants can exist of the same protein, which might have different biological functions. Most DNA damage studies followed the strategy of treating PTM changes similar to changes in protein expression. Using pathway and network analyses DDR PTM studies pointed towards novel signalling routes, implicating RNA metabolism, in particular RNA splicing, in the response to DNA damage 27, 29, 31, 32. They moreover served to confirm ubiquitylation‐mediated regulation of nucleotide excision repair (NER) after ultraviolet light (UV)‐exposure 28, 33.
It is, however, important to note that the modification of a signalling molecule does not necessarily correlate with its activity. On the contrary, modification can target a protein for deactivation or even degradation 54. Moreover, not all modifications are biologically relevant. Indeed, many of them are considered part of the biological noise 55.
Only follow‐up studies, using targeted biological experiments can provide final certainty about the relevance of specific PTMs. Targeted validation led to identification of DNA damage‐mediated phosphorylation and PARylation of the RNA splicing factor THRAP3 31, 48, or the ubiquitylation‐mediated regulation of RPA 28, 56.
4. Phosphorylation in the DDR
Site‐specific protein phosphorylation is the best described PTM functioning in the DDR. Phospho‐signalling regulates all stages of the DDR (Fig. 1). Fast phospho‐responses lead to the recruitment of DNA repair factors and signalling molecules to damaged DNA 14, 30. Subsequently, phosphorylation can serve to retain those factors at sites of damage. The phosphorylation of downstream signalling molecules further regulates later cellular responses. Those can “take the long road” by phosphorylating and thus modulating the activity of transcription factors 57, 58. Among those transcription factors, is the key cellular hub protein p53, termed the guardian of the genome. Phosphorylation by DDR kinases can activate p53 by disrupting the regulatory loop between p53 and its negative regulator MDM2 59, 60, 61.
Next to regulating the functions of transcription factors, phosphorylation‐mediated signalling can also take a shortcut, by directly regulating downstream effector molecules 58. Those include for example the CDC25 family of phosphatases, which can remove the highly‐conserved inhibitory phosphorylation marks from the N‐terminal part of cyclin‐dependent kinases (CDKs). Checkpoint kinase‐mediated phosphorylation attenuates CDC25 protein stability by priming it for proteasomal degradation. It further induces interaction with 14‐3‐3, sequestering CDC25 proteins from CDK1. Both mechanisms result in an induction of cell cycle arrest 62, 63.
Given this highly complex phosphorylation‐mediated signalling network, phosphoproteomics can be a vital technique to discover new phosphorylation‐mediated phenotypes in the DDR. Those might ultimately be translated into new biomarkers and drug targets 64.
Special interest has been invested into studying the function of the PI3‐K‐related protein kinases ATM and ATR, which are the principal sensor kinases that are immediately activated after DNA damage. Together with their direct downstream targets Chk1 and Chk2, ATM and ATR regulate a pleiotropic array of processes after DNA damage 16, 51. Their substrate pool reflects the whole spectrum of the DDR and mediates cell cycle arrest, DNA repair and cell survival 15, 65. Phosphoproteomics studies have aimed to answer the following questions:
Which targets are comprised in the substrate pool of ATM and ATR?
Which processes do their targets likely mediate?
What is the distribution of nuclear and non‐nuclear phosphorylation events?
Which other kinases might be important for the DDR?
The boundaries of discovery within phosphoproteomics analyses of the DDR are defined by choice of enrichment method, MS instrumentation, quantification method, subcellular fractionation, timepoint and damage inducer (Table 1). Indeed, the number of quantified phosphosites within bulk‐phosphoproteomics studies increased from earlier studies, which identified around 3000–5000 phosphosites 27, 30 to over 10 000 phosphosites in more recent analyses 31, 32.
Table 1.
Proteomics studies of the DDR
| Study | Cell line | DNA damage‐inducer | Timing | Enrichment | Number of sites/proteins | Major affected pathways/factors |
|---|---|---|---|---|---|---|
| Matsuoka 2007 29 | 293T cells | γ−irradiation | 1 h after IR | S/T‐Q motif specific AB | 905 phosphosites on 700 proteins induced after IR |
|
| Stokes et al. 66 | M059K glioblastoma cells, GM18366 Seckel syndrome cells and GM00200‐matched control cells | UV 50 mJ/cm2 | 2 h after UV | S/T‐Q motif specific AB | 570 sites phosphorylated in UV‐damaged cells |
|
| Bensimon 2010 27 | G361 human melanoma cell line | Neocarzinostatin (NCS) | 10, 30, 120, and 360 min | TiO2 | 2871 phosphosites on 1099 proteins |
|
| Bennetzen 2010 30 | GM00130 | γ−irradiation | 5 timepoints: 0 min, 5 min, 20 min, 1h, 8 h | ERLIC and TiO2 | 5204 phosphosites, 594 regulated |
|
| Pines 2011 32 | Mouse ES cells | Cisplatin | 4 h | SCX and TiO2 |
|
|
| Beli 2012 31 | U2OS human sarcoma cells | γ−irradiation etoposide |
|
|
|
|
| Povlsen 2012 33 | U2OS human sarcoma cells | UV‐irradiation | 1 h after UV | Di‐Gly AB SCX | 6700 UB sites | Proteome‐wide analysis of ubiquitylation changes after UV. Identification of PAF15 mono‐ubiquitylation. |
| Elia et al. 2015 28, 56 | HeLa |
|
1 h after UV or IR |
|
|
Combination of global ubiquitin and acetyl proteomics. Global increase in K6‐ and K33‐linked polyubiquitination. Cullin‐RING ligases mediate 10% of DNA damage‐induced ubiquitination events. |
| Hendriks et al. 2015 80 | HeLa and U2OS | Methyl methanesulfonate (MMS) | 90 min |
|
755 SUMO‐2 sites, 362 regulated after MMS | SUMOylation of chromatin modifiers, transcription factors, DNA repair factors, and nuclear body components. |
| Xiao et al. 2015 52 | U2OS | Hydroxy Urea | 2 h, 24 h | His10‐SUMO‐2 pulldown | 566 SUMO target proteins | SUMO network including replication factors, transcriptional regulators, DNA damage response factors |
| Jungmichel et al. 2013 48 | U2OS cells | H2O2, MMs, UV, IR | 1 h for genotoxic stresses, 10 min for H2O2 | Af1521 domain pulldown | 165 proteins, which significantly increase in PARylation |
|
| Warmoes et al. 2013 90 | Murine BRCA1‐/‐, p53‐/‐ tumors; CDH1‐/‐, p53‐/‐ tumors | cisplatin | 24 h | Gel‐based proteomics |
|
|
| Mazouzi et al. 68 |
|
1 μM aphidicolin |
|
Fe(III)‐NTA‐based phosphoenrichment | 13 801 phosphosites on 4094 proteins |
|
| Boeing et al. 67 | HEK293 | UV 30 J/cm2 | 3 h after UV | Di‐Gly AB For ubiquitylated peptides |
|
|
Matsuoka et al. identified over 700 ATM/ ATR/ DNA‐PK substrates by combining a number of S/T‐Q motif antibodies 29. This study for the first time highlighted the breadth of the ATM/ATR target pool and revealed the intersection of the ATM‐mediated DDR with other cellular processes such as PI3K−AKT signalling 29.
Stokes et al. used an S/T‐Q antibody‐based approach to examine the effect of UV radiation on ATM/ ATR substrate phosphorylation. While they found extensive overlap to the substrates that had been identified by Matsuoka et al. they also found a number of UV‐specific substrates. UV radiation leads to a strong activation of ATR kinase. The authors aimed to decipher potentially ATR‐specific substrates by testing their phosphorylation in cells from Seckel‐syndrome patients, which have very low ATR levels and fail to activate UV‐induced ATR‐based responses 66.
Subsequent studies analysed nuclear 27, 30 or whole‐cell 31, 32 phosphoproteomes in the context of different DNA damage types. Those comprised ɣ‐irradiation 30, UV radiation 67, replication stress induced by aphidicolin 68 and stress evoked by various genotoxicants such as cisplatin, neocarzinostatin or etoposide 27, 31, 32.
All studies found an enrichment of the ATM/ATR‐substrate motif [S/T‐Q] among DNA damage‐induced phosphorylation sites [27, 30, 31, 32, 67, 68]. Bennetzen et al., who performed a time‐resolved analysis, found ATM‐dependent phosphorylation sites amongst the early responders, in line with ATM mediating fast responses to DNA damage 30. While Bensimon et al. found only 10% of the identified phosphosites carrying an S/T‐Q motif; addition of the ATM inhibitor KU55933 counteracted 60% of DNA damage‐modulated phosphosites 27. Similarly Mazouzi et al. found an enrichment of the S/T‐Q motif for around 50% of phosphosites induced by 4 h of aphidicolin treatment, over 70% of which were mediated by ATM. Their findings highlighted the role of ATM‐signalling in early replication stress. At the later replication stress timepoint, 24 h, the number of ATM‐regulated sites decreased to around 50% 68.
Different studies suggested ATM‐dependent and independent activation of the NFκ‐B signalling pathway 51. Interestingly, Choi et al. performed MS‐based analysis of ATM‐dependent protein composition of different cellular compartments. They found that the chromatin association of ANXA1, a protein that has been linked to NFκ‐B signalling, depended on ATM activity 69. Furthermore, also Beli et al. found DNA damage‐induced phospho‐regulation of members of the NFκ‐B pathway 31.
Studies, which analyzed whole‐cell phosphoproteome changes after DNA damage, allowed deciphering the different dynamics and biology of nuclear and non‐nuclear phosphorylation events 31, 32. Beli et al. found that DNA damage‐induced phosphorylation events were enriched in the nuclear compartment, which was particularly true for S/T‐Q phosphosites 31. While nuclear phosphorylation was mainly related to DNA metabolic processes, cytoplasmic events were enriched for proteins involved in cell cycle regulation 31. Pines et al. found processes related to cytoskeleton rearrangements changed after DNA damage in embryonic stem cells 32.
Despite the clear overrepresentation of S/T‐Q motif‐containing peptides after different types of DNA damage, phosphoproteomics studies of the DDR suggested the modulation of the activity of other kinases. Proline‐directed phosphorylation, which is common for both cell cycle kinases and stress kinase family members, was found enriched among peptides, whose phosphorylation decreased after DNA damage 27, 31. This might be due to the activation of phosphatases or decreased activity of kinases.
A number of kinases were phospho‐targets themselves, including cytoplasmic kinases involved in cytoskeleton rearrangements 32. Interestingly, phosphosites on p38, BUB1 and OXSR1 conformed to S/T‐Q motifs 31. Nevertheless, it is important to stress that phosphorylation of a protein is not the same as its activation.
Taken together, phosphoproteomics studies of the DDR indicated that next to the clearly vital and wide‐ranging effect of ATM and ATR, other kinases might be important in the DDR 51. Kinases, ordinarily involved in other cellular signalling events, such as stress kinases or cell cycle kinases, can be drawn into DDR signalling processes. Phosphorylation of those kinases on S/T‐Q motifs suggests extensive crosstalk with the “classical” DDR kinases 51. This complex phosphorylation network is likely destined to function as a cellular buffer, ensuring the faithful execution of the DDR, even if one of the players is missing. For instance, p53‐deficient cells have been shown to rely on a p38‐MAPK/MK2 signalling module for checkpoint activation 70, 71. Indeed, only a few factors within the intricate phospho‐signalling network are vital for cellular survival, including the kinase ATR, as well as its downstream target Chk1 15, 61.
4.1. Ubiquitylation and SUMOylation in the response to DNA damage
Ubiquitin, a small 76 amino acid long protein highly conserved in eukaryotes, is covalently attached onto target lysines involving a three‐enzyme process 47. The ubiquitin machinery classically includes an E1 activating enzyme, an E2 conjugating enzyme and an E3 ubiquitin ligase 72. The large number of E3 ubiquitin ligases confers specificity within the ubiquitin system 73. Removal of ubiquitin requires deubiquitinase (DUB) enzymes 74.
Importantly, ubiquitin can be added to a number of lysines of ubiquitin itself, resulting in a variety of ubiquitin chains, including K6, K11, K48 and K63 linkage 75, whereas, K11 and K48 ubiquitin chains generally target proteins for proteasomal degradation, other types of ubiquitin chains and mono‐ubiquitylation events can alter protein features.
Next to ubiquitin a number of other small ubiquitin‐like protein modifiers exist. Amongst them, SUMO is probably the best studied in the DDR 52, 76, 77. SUMO, similar to ubiquitin, is added in a 3‐step process. A major difference to the ubiquitin system lies in the fact that the number of SUMOylases is much smaller than that of kinases and ubiquitinases, comprising only one E2 and a limited number of E3 ligases 78.
Both substrate degradation and change of protein properties induced by the ubiquitin‐ and SUMO‐systems are highly relevant for DDR signalling processes 47, 73 (Fig. 2) 78. Ubiquitylation regulates the signalling response downstream of DNA double strand breaks, as well as DNA repair pathways such as NER or Fanconi Anemia 78. SUMOylation also regulates various nuclear processes, including transcription and cell cycle regulation. A number of DNA repair factors can be found SUMOylated after DNA damage 77.
However, the induction of degradation of protein substrates by the ubiquitin proteasome systems can pose a challenge in the interpretation of ubiquitin proteomics results 28, 33. The decrease in an ubiquitylated peptide species can result either from its deubiquitylation or from its degradation 33. To identify cases in which protein degradation is responsible for decrease in ubiquitylated peptides, a comparison to the un‐modified proteome is a possibility. Alternatively, including proteasome inhibitors can enrich for species, which are targeted for degradation by ubiquitin after DNA damage. Those include for example the cell cycle regulators CDC25A and CDC25B 28. However, proteasome inhibition massively boosts the overall abundance of ubiquitylated proteins and leads to a depletion of the pool of free ubiquitin. Thus, extra care has to be taken in the interpretation of proteomics data from such treatments.
Different enrichment strategies were employed to analyse the ubiquitin‐response to UV radiation or γ‐irradiation (IR) treatment, as well as SUMOylation responses to replication stress and methyl methanesulfonate (MMS) 28, 33, 34, 52, 67, 77. Schwertmann et al. used the FK2‐ubiquitin antibody to enrich ubiquitylated proteins after UV treatment 34. The studies by Povlsen et al., Elia et al. and Boeing et al. utilised antibodies that recognise the glycine dipeptide (di‐Gly), which results from tryptic cleavage of ubiquitylated peptides 28, 33, 67. While the use of di‐Gly antibodies allows bulk enrichment of great numbers of ubiquitin sites, and permits studying endogenous proteins, it entails certain limitations. Not only ubiquitin, but also the ubiquitin‐like modifiers NEDD8 or ISG15 leave a di‐Gly remnant on the acceptor lysine. The modifying molecule cannot be distinguished using this strategy 47. Nevertheless, the amount of ISG15 or NEDD8‐modified proteins was found to be negligible in comparison to ubiquitylated proteins 79. While di‐Gly antibodies cannot identify the ubiquitin chain linked to an individual peptide, the bulk changes in different types of linkages can be quantified. Interestingly, Elia et al. found a strong increase in K6‐ and a less pronounced increase in K33‐linked ubiquitin chains after UV but not IR exposure 28. K6‐chains have been related to BRCA1, a key DDR protein in vitro 75.
Xiao et al. and Hendriks et al. used expression of epitope‐tagged, exogenous SUMO molecules 52, 80. Since SUMO modifications are quickly removed by deSUMOylases during sample preparation, and to date no suitable deSUMOylase inhibitor exists, expression of tagged SUMO versions provides a viable alternative, allowing lysis under harsh conditions 80.
Upstream regulators of phosphorylation responses can be explored by identification of linear sequence motifs, which can help inferring upstream kinases (or kinase families). Also inhibitor studies can aid identifying kinases, which are responsible for phosphorylation responses 27, 30, 31. In contrast, linear target motifs are much less prevalent for the ubiquitin system and good inhibitors for E3 ligases are rare 81. Elia et al. made a first attempt to identify upstream enzymes, which might be relevant for bulk ubiquitin changes after DNA damage, by combining the Cullin‐Ring (CRL) ligase inhibitor MLN4924 with DNA damage induction. They found that CRLs mediated around 10% of UV‐induced ubiquitylation events 28.
Similar to phosphorylation, also ubiquitylation and SUMOylation were found to be enriched in the nuclear compartment after DNA damage 28, 33, 80. Moreover, ubiquitylation and SUMOylation events were particularly enriched on proteins, which are involved in the repair of DNA lesion caused by the damage inducers employed. Those included for example protein ubiquitylation of factors involved in the NER pathway, which is crucial for the repair of UV lesions 28, 33, 34, or SUMOylation of factors involved in the response to replication stress, which is caused by Hydroxyurea treatment 52. Moreover, processes related to the mitotic spindle, were found changed after UV and IR, with many of those proteins decreased in ubiquitylation 28.
Identification of the specific sites of SUMOylation has posed a considerable challenge to researchers. Hendriks et al. 80. combined SILAC‐based quantification of SUMOylated proteins with label free site‐identification. They were able to map 755 SUMO‐2 sites, of which 362 were regulated after MMS treatment. Interestingly, next to identifying proteins involved in DDR processes, Hendriks et al. found that MMS also induced SUMOylation of chromatin proteins. SUMOylation targets were moreover found to functionally interact, further establishing the concept of SUMO group modifications.
4.2. Other PTMs: acetylation and PARylation
Next to ubiquitylation and phosphorylation, a number of other modifications are emerging to be relevant in the DDR. Prevalent examples are two types of PTMs, acetylation and PARylation, which are of high clinical relevance. Inhibitors of PARP enzymes and histone deacetylases (HDACs) have entered clinical trials or clinical use in cancer treatment 19, 82.
Lysine acetylation impacts gene expression by modifying chromatin interacting proteins, including histone tails and non‐histone proteins. HDACs have been implicated as radiation sensitizers, and acetylation has been shown to regulate a number of DNA damage‐relevant processes 48, 83. Amongst the prominent DDR targets of acetylation is the transcription factor p53. Acetylation enhances p53 protein stability and transcriptional activity. The latter is further aided by acetylation‐dependent chromatin relaxation in p53 target genes 84.
A number of proteomics investigations set out to analyse acetylation responses to DNA damage 28, 31, 85. DNA damage‐induced acetylation changes were enriched in the nuclear compartment 28, 31, 85. However, both acetylation and deacetylation changes were found to be significantly less pronounced than changes in (de)ubiquitylation and (de)phosphorylation at 1 h after IR and UV damage 28, 31. Interestingly, Bennetzen et al. found an early wave of deacetylation 5 min after IR 85.
In a recent large‐scale proteomics screen, Elia et al. improved the enrichment protocol for acetylation sites, by combining it with deep SCX fractionation. This yielded an unprecedented depth in acetylation site identification, which equaled the range of ubiquitin‐ and phosphoproteomics studies 28. A number of known DDR factors, such as DNAPK and PARP1, showed dynamic acetylation after DNA damage 28.
Another PTM, whose relevance for the DDR is increasingly appreciated is PARylation. PARylation is a reversible posttranslational modification that is excessively added to proteins and other biomolecules after DNA damage. Inhibitors of PARP enzymes are in clinical studies for cancer treatment and have been shown to be especially successful in the context of HR‐deficiency 9. The PARP inhibitor Olaparib was recently approved by the FDA for treatment of cancer patients 19.
Despite the fact that PARylation cascades are highly relevant for the DDR, high‐throughput studies of PARylation were hampered by the difficulty of PAR enrichment. In 2013, Jungmichel et al. used an Af1521 domain, which showed strong ADP‐Ribose‐binding features, to fish out PARylated proteins from cellular lysates, which had been exposed to different DNA damage stimuli 48. Using as SILAC approach the authors compared the wild‐type PARP binding domain to a mutant version, which lacked affinity to PARylated proteins. Similar to other studied PTMs, PARylation events after DNA damage were enriched in the nucleus. The authors confirmed a large number of DNA repair factors as PARylation targets after DNA damage. Moreover, similar to studies of the phosphorylation machinery, the authors also found proteins involved in RNA metabolism to be PARylated in the context of (genotoxic) stress. Those included the RNA splicing factor THRAP3, whose localisation had been shown to also depend on phosphorylation in an earlier study 31.
5. Studying PTM crosstalk by MS
PTM crosstalk is a key means for signal integration in the DDR. Enzymes that function in specific PTM pathways are targets for modification by other PTMs, including for example the phosphorylation of ubiquitinases 30, 31. While crosstalk on the pathway level suggests reciprocal regulation of different PTM classes, functional validation is required to draw final conclusions about PTM‐based regulation of enzymes.
Next to regulating enzymatic activity of other PTM‐modifiers, different PTMs can also converge on the same protein, sometimes even on the same amino acid residue. The addition of multiple PTMs has great regulatory potential, including the modulation of positive and negative interactions 54. The combinatorial logic of different PTMs is large, considering the different types of PTMs, as well as their potentially different functional outcomes. This PTM integration is vital in regulating DDR signalling hubs, such as the DNA clamp loader PCNA or the transcription factor p53 78, 86.
Many different types of modifications target lysines. Among those are acetylation, methylation, ubiquitylation and SUMOylation. This phenomenon presents the idea of specific lysine residues in signalling proteins to function as cellular modification hubs that integrate different PTM pathways. Indeed, different studies suggested the potential for reciprocal regulation between ubiquitylation and acetylation 79, 85. The potential crosstalk between the acetylation and ubiquitylation system in response to DNA damage was tackled by Elia et al. on a global level. However, they only discovered a small proportion of reciprocal modification (increased ubiquitylation and decreased acetylation or vice versa) on the same lysine residue 28.
It is important to note that the peptide‐centric approach severely limits the ability of MS to analyse PTM crosstalk. PTMs, which are further apart than the typical length of a tryptic peptide cannot be analysed. Moreover, different PTMs might require different enrichment strategies and cannot be properly analysed within the same sample. Finally, determination of site occupancy would be required to truly assess crosstalk between PTMs on a global scale.
5.1. Changes in protein abundance after DNA damage
DNA damage can regulate protein expression on multiple levels. Bulkier DNA lesions can directly block transcription 87. Moreover, signalling downstream of DNA damage can lead to changes in gene expression and posttranscriptional mechanisms can change the stability of RNA transcripts or proteins 88. Due to those posttranscriptional mechanisms, gene expression levels are not always well correlated with protein abundance. Indeed, the correlation between gene transcripts and proteins seems to be highly context‐dependent 89. In line with this observation, Pines et al. found little direct correlation between regulation on RNA and protein level, when comparing the cisplatin response in embryonic stem cells. They confirmed this lack of correlation using targeted biology experiments 32.
Different studies analysed the regulation of protein abundance after DNA damage. Overall, changes in protein levels were much less pronounced than PTMs changes 31. Proteins, which did change after DNA damage were enriched for target genes of the transcription factor p53, which is regulating pleiotropic responses after different types of DNA damage 31, 32, 86.
In 2013, Warmoes et al. searched for protein biomarkers in cisplatin‐sensitive, BRCA1‐ and p53‐deficient and cisplatin‐resistant BRCA1‐proficient, p53‐deficient mouse tumors. They analysed proteome changes 24 h after cisplatin treatment, a timepoint were DNA damage induction in tumor cells was evident, yet no excessive amounts of apoptosis did occur. Enriching cisplatin‐regulated protein networks for functional information they found categories associated with “M‐phase” and “chromosome segregation” and “DNA metabolic process/deoxyribonucleotide metabolic process” enriched in BRCA1‐deficient tumors. They further discovered enrichment of proteins involved in fatty acid metabolism in cisplatin resistant tumors. Indeed, knockdown of one of the identified fatty acid metabolism factors, FASN, could sensitize resistant tumor cells to cisplatin treatment 90.
An interesting mechanism of transcriptional repression after DNA damage was proposed by Hendriks et al., who suggested that SUMOylation of chromatin modifiers could lead to transcriptional repression after DNA damage 77, 80. They found SUMOylation of various chromatin proteins. Those included the transcriptional co‐activators P300 and CBP, SUMOylation of which had been previously reported to suppress transcription. They moreover discovered SUMOylation‐mediated recruitment of the histone demethylase JARID1C, which led to demethylation of the transcriptionally‐activating histone marks H3K4me2 and H3K4me3. Those and other SUMO‐mediated changes in chromatin modifiers might act in concert to repress transcription after MMS treatment 80.
5.2. Changes in protein‐protein interaction and protein‐DNA interaction induced by DNA damage
Signalling in the DDR requires recognition of the presence of damaged DNA. The initial recognition of a DNA lesion induces a great number of rearrangements in the nuclear architecture. Those include the recruitment of DNA damage‐specific proteins to the DNA, alterations in chromatin e.g. to make the damage accessible, or form docking platforms for repair factors, and changes in proteins involved in DNA metabolism, which are already present at the DNA 14, 91. Altogether those rearrangements result from changes in protein‐protein and protein‐DNA interactions, which can be measured by MS‐based interaction proteomics (Fig. 2).
A number of studies analysed the proteins binding to a single DDR factor or a whole group of proteins, functioning in the same DDR pathway. Xing et al. used MS to analyse proteins binding to 19 factors functioning in the DSB repair pathway NHEJ 92, whereas Boeing et al. analysed the interactome of the NER factors CSB and RNAPII in the context of UV stress 67.
Next to studying the interactions of a single protein or a group of proteins another strategy lies in the pull‐down of a specific protein‐binding domain. Phospho‐binding domains were found to be crucial for early signalling processes in the DDR. Those include BRCT, FHA and 14‐3‐3 domains, to name a few 93. In 2014, Blasius et al. analysed the 14‐3‐3 interactome in the context of UV radiation and caffeine‐mediated PI3K‐kinase family inhibition 35. 14‐3‐3 proteins are highly conserved phospho‐binders, which regulate a number of cellular functions, such as cell cycle halt by binding to Chk and CDC25 proteins 58. Next to known damage‐induced binders, such as Chk1, the authors found proteins and protein complexes involved in RNA metabolism. Those included the nuclear exosome component Rbm7 35.
Two elegant studies recently combined novel MS‐technology with sophisticated follow‐up experiments to decipher the recruitment of proteins to cross‐linked DNA 36, 37. Interstrand crosslinks (ICLs) are extremely toxic lesions, which affect both transcription and replication by hindering the crucial separation of the DNA strands. ICLs are formed by exposure to chemotherapeutic drugs such as cisplatin or mitomycin C and their repair involves a complex mixture of repair pathways, including the Fanconi Anemia pathway 94.
Raeschle et al. developed a technique they termed chromatin mass spectrometry (CHROMASS) to decipher protein recruitment during ICL repair. They used cross‐linked and undamaged sperm chromatin, which underwent replication in Xenopus extracts, and analysed protein binding at different timepoints. The authors found DNA repair factors strongly enriched in the damaged chromatin, compared with the undamaged one. This enrichment depended on DNA replication, as the recruitment of those factors was inhibited by replication inhibitor geminin 37.
Liang et al. created a DNA structure that contained a single, well‐defined ICL. They incubated this DNA structure with nuclear extracts of HeLa cells, which had been exposed to mitomycin C. Using MS, they identified the protein UHFR1, which was enriched at ICL‐DNA compared with the control 36. UHFR1 was recruited to chromatin after treatment with crosslinking agents, and was required for recruitment of the Fanconi Anemia pathway component FANCD2 36. Those two initial analyses open up possibilities for the study of other DNA lesions, by pulling down damaged DNA and the binding components for other types of DNA lesions.
5.3. Outlook: clinical relevance of proteomics studies of the DDR
Proteomics can be relevant for multiple steps of drug discovery processes, including the identification of novel drug targets, highlighting drug mechanisms of action and biomarker discovery 95 (Fig. 2). In the future, proteomics might also become relevant as a diagnostic tool.
5.4. Proteomics as a tool to discover new drug targets, biomarkers and drug mechanisms of action in model systems
Most proteomics studies of the DDR to date focused on the description of global responses to a single or limited number of DNA damage inducers in model systems. Those studies could identify known and novel signalling routes and highlight their key players. Those are especially valuable for providing a better understanding of drug mechanisms of action, but can also help identifying potential new drug targets and biomarkers.
In the future, powerful proteomics technologies can be a valuable source for network medicine approaches, which base biomarkers and drug targets on a network of events (protein signature), rather than a single marker or target 96. Pioneering studies, such as mid‐level resolution phosphorylation analyses by the Yaffe lab, could predict sensitivity to DNA damage‐inducing drugs in breast cancer cells 97. Initial efforts have explored the predictive power of large‐scale phosphoproteomics datasets in the study of signalling pathways in model organisms and drug sensitivity in cancer cells 98, 99. Nevertheless, predictive modelling generally requires a high‐resolving power of time‐points, high reproducibility and high coverage, in order not to miss crucial data points. Proteomics analyses are now on a good way to attain the speed, sensitivity and reproducibility that will allow designing studies with high numbers of timepoints, replicates and different DNA damage‐inducers.
5.5. Diagnostic clinical application of proteomics
To take the next step into the clinic, proteomics will have to master the challenges posed by mass spectrometric analyses of tissues 53. This is relevant for biomarker discovery from tissue samples, and for proteomics becoming a true diagnostic tool.
Next to the aforementioned limitations of MS‐based proteomics analysis, specific challenges for tissue proteomics relate to tissue availability. Those are particularly critical for analysis of prognostic patient biopsy samples. Sample amounts are especially relevant for PTM studies, whereas for expression proteomics nowadays minute amounts of sample might suffice.
Next to the availability of samples also their quality is important. It is not always feasible to gain fresh samples. Formalin‐fixed, paraffin‐embedded (FFPE) samples pose a viable alternative for both proteomics and PTM proteomics 100. However, it is worth noting that ischemia‐induced artifacts can occur during the preparation of tissue samples. Those can drastically change patterns of dynamic PTMs such as phosphorylation 101, 102.
Another challenge lies in the inherent heterogeneity of tissue samples. On the quest for biomarkers, this can pose a distinct challenge to the researcher. Heterogeneity between samples requires an increase in the number of replicates in order to identify confident biomarkers 53. Heterogeneity amongst tissue cell types or amongst different tumor cell populations can potentially be addressed by tissue micro‐dissection prior to MS analysis 103. Targeted MS approaches could further serve as an alternative to biomarker discovery from patient samples 104.
The authors have declared no conflict of interest.
Acknowledgment
The authors thank all lab members for fruitful discussion. We especially thank Simon Bekker‐Jensen and Stephanie Munk for their valuable input on the manuscript. Work at The Novo Nordisk Foundation Center for Protein Research (CPR) is funded in part by a generous donation from the Novo Nordisk Foundation (Grant number NNF14CC0001). The proteomics technology developments applied was part of a project that has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No 686547 (J.V.O.). The work was supported by the Danish Research Council (research career program FSS Sapere Aude to L.V.S and the Danish Cancer Society R90‐A5844 KBVU project grant (J.V.O.).
Olsen J. V., Proteomics 2017, 1600018.
Colour Online: See the article online to view Figs. 1–3 in colour.
6 References
- 1. Jackson, S. P. , Bartek, J. , The DNA‐damage response in human biology and disease. Nature 2009, 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harper, J. W. , Elledge, S. J. , The DNA damage response: ten years after. Mol. Cell 2007, 28, 739–745. [DOI] [PubMed] [Google Scholar]
- 3. von Stechow, L. , van de Water, B. , Danen, E. H. J. , Unraveling DNA damage response‐signaling networks through systems approaches. Arch. Toxicol. 2013, 87, 1635–1648. [DOI] [PubMed] [Google Scholar]
- 4. Curtin, N. J. , DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [DOI] [PubMed] [Google Scholar]
- 5. Vollebergh, M. A. , Jonkers, J. , Linn, S. C. , Genomic instability in breast and ovarian cancers: translation into clinical predictive biomarkers. Cell. Mol. Life Sci. 2012, 69, 223–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoeijmakers, J. H. , DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- 7. Bartek, J. , Lukas, J. , Bartkova, J. , DNA damage response as an anti‐cancer barrier: damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle 2007, 6, 2344–2347. [DOI] [PubMed] [Google Scholar]
- 8. Helleday, T. , Petermann, E. , Lundin, C. , Hodgson, B. , Sharma, R. A. , DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [DOI] [PubMed] [Google Scholar]
- 9. Lord, C. J. , Ashworth, A. , The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [DOI] [PubMed] [Google Scholar]
- 10. Tutt, A. , Robson, M. , Garber, J. E. , Domchek, S. M. et al., Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancet 2010, 376, 235–244. [DOI] [PubMed] [Google Scholar]
- 11. Hopkins, A. L. , Network pharmacology: the next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [DOI] [PubMed] [Google Scholar]
- 12. Rouse, J. , Jackson, S. P. , Interfaces between the detection, signaling, and repair of DNA damage. Science 2002, 297, 547–551. [DOI] [PubMed] [Google Scholar]
- 13. Lukas, J. , Lukas, C. , Bartek, J. , More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell. Biol. 2011, 13, 1161–1169. [DOI] [PubMed] [Google Scholar]
- 14. Dantuma, N. P. , van Attikum, H. , Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016, 35, 6–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cimprich, K. A. , Cortez, D. , ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shiloh, Y. , Ziv, Y. , The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [PubMed] [Google Scholar]
- 17. Pellegrino, S. , Altmeyer, M. , Interplay between ubiquitin, SUMO, and Poly(ADP‐Ribose) in the cellular response to genotoxic stress. Front. Genet. 2016, 7, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen, P. , Alessi, D. R. , Kinase drug discovery–what's next in the field? ACS Chem. Biol. 2013, 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. O'Connor, M. J. , Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [DOI] [PubMed] [Google Scholar]
- 20. Olsen, J. V. , Mann, M. , Status of large‐scale analysis of post‐translational modifications by mass spectrometry. Mol. Cell. Proteomics 2013, 12, 3444–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Walther, T. C. , Mann, M. , Mass spectrometry‐based proteomics in cell biology. J. Cell. Biol. 2010, 190, 491–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Walther, T. C. , Olsen, J. V. , Mann, M. , Yeast expression proteomics by high‐resolution mass spectrometry. Methods Enzymol. 2010, 470, 259–280. [DOI] [PubMed] [Google Scholar]
- 23. Wepf, A. , Glatter, T. , Schmidt, A. , Aebersold, R. , Gstaiger, M. , Quantitative interaction proteomics using mass spectrometry. Nat. Methods 2009, 6, 203–205. [DOI] [PubMed] [Google Scholar]
- 24. Hein, M. Y. , Hubner, N. C. , Poser, I. , Cox, J. et al., A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [DOI] [PubMed] [Google Scholar]
- 25. Lundby, A. , Lage, K. , Weinert, B. T. , Bekker‐Jensen, D. B. et al., Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Olsen, J. V. , Blagoev, B. , Gnad, F. , Macek, B. et al., Global, in vivo, and site‐specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [DOI] [PubMed] [Google Scholar]
- 27. Bensimon, A. , Schmidt, A. , Ziv, Y. , Elkon, R. et al., ATM‐dependent and ‐independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal. 2010, 3, rs3. [DOI] [PubMed] [Google Scholar]
- 28. Elia, A. E. , Boardman, A. P. , Wang, D. C. , Huttlin, E. L. et al., Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol. Cell 2015, 59, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuoka, S. , Ballif, B. A. , Smogorzewska, A. , McDonald, E. R., 3rd et al., ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [DOI] [PubMed] [Google Scholar]
- 30. Bennetzen, M. V. , Larsen, D. H. , Bunkenborg, J. , Bartek, J. et al., Site‐specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteomics 2010, 9, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Beli, P. , Lukashchuk, N. , Wagner, S. A. , Weinert, B. T. et al., Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pines, A. , Kelstrup, C. D. , Vrouwe, M. G. , Puigvert, J. C. et al., Global phosphoproteome profiling reveals unanticipated networks responsive to cisplatin treatment of embryonic stem cells. Mol. Cell. Biol. 2011, 31, 4964–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Povlsen, L. K. , Beli, P. , Wagner, S. A. , Poulsen, S. L. et al., Systems‐wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA‐damage bypass. Nat. Cell Biol. 2012, 14, 1089–1098. [DOI] [PubMed] [Google Scholar]
- 34. Schwertman, P. , Lagarou, A. , Dekkers, D. H. , Raams, A. et al., UV‐sensitive syndrome protein UVSSA recruits USP7 to regulate transcription‐coupled repair. Nat Genet. 2012, 44, 598–602. [DOI] [PubMed] [Google Scholar]
- 35. Blasius, M. , Wagner, S. A. , Choudhary, C. , Bartek, J. , Jackson, S. P. , A quantitative 14‐3‐3 interaction screen connects the nuclear exosome targeting complex to the DNA damage response. Genes Dev. 2014, 28, 1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang, C. C. , Zhan, B. , Yoshikawa, Y. , Haas, W. et al., UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep. 2015, 10, 1947–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raschle, M. , Smeenk, G. , Hansen, R. K. , Temu, T. et al., DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross‐links. Science 2015, 348, 1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Picotti, P. , Bodenmiller, B. , Aebersold, R. , Proteomics meets the scientific method. Nat. Methods 2013, 10, 24–27. [DOI] [PubMed] [Google Scholar]
- 39. Cox, J. , Mann, M. , Quantitative, high‐resolution proteomics for data‐driven systems biology. Annu. Rev. Biochem. 2011, 80, 273–299. [DOI] [PubMed] [Google Scholar]
- 40. Solari, F. A. , Dell'Aica, M. , Sickmann, A. , Zahedi, R. P. , Why phosphoproteomics is still a challenge. Mol. BioSystems 2015, 1487–1493. [DOI] [PubMed] [Google Scholar]
- 41. Tran, J. C. , Zamdborg, L. , Ahlf, D. R. , Lee, J. E. et al., Mapping intact protein isoforms in discovery mode using top‐down proteomics. Nature 2011, 480, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Giansanti, P. , Aye, T. T. , van den Toorn, H. , Peng, M. et al., An augmented multiple‐protease‐based human phosphopeptide atlas. Cell Rep. 2015, 11, 1834–1843. [DOI] [PubMed] [Google Scholar]
- 43. Bouwman, P. , Jonkers, J. , The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat. Rev. Cancer 2012, 12, 587–598. [DOI] [PubMed] [Google Scholar]
- 44. Dimitrakopoulos, L. , Prassas, I. , Diamandis, E. P. , Nesvizhskii, A. et al., Proteogenomics: opportunities and caveats. Clin. Chem. 2016, 62, 551–557. [DOI] [PubMed] [Google Scholar]
- 45. Mertins, P. , Mani, D. R. , Ruggles, K. V. , Gillette, M. A. et al., Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olsen, J. V. , Vermeulen, M. , Santamaria, A. , Kumar, C. et al., Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [DOI] [PubMed] [Google Scholar]
- 47. Beaudette, P. , Popp, O. , Dittmar, G. , Proteomic techniques to probe the ubiquitin landscape. Proteomics 2016, 16, 273–287. [DOI] [PubMed] [Google Scholar]
- 48. Jungmichel, S. , Rosenthal, F. , Altmeyer, M. , Lukas, J. et al., Proteome‐wide identification of poly(ADP‐Ribosyl)ation targets in different genotoxic stress responses. Mol. Cell 2013, 52, 272–285. [DOI] [PubMed] [Google Scholar]
- 49. Switzar, L. , Giera, M. , Niessen, W. M. , Protein digestion: an overview of the available techniques and recent developments. J. Proteome Res. 2013, 12, 1067–1077. [DOI] [PubMed] [Google Scholar]
- 50. Beltran, L. , Cutillas, P. R. , Advances in phosphopeptide enrichment techniques for phosphoproteomics. Amino Acids 2012, 43, 1009–1024. [DOI] [PubMed] [Google Scholar]
- 51. Bensimon, A. , Aebersold, R. , Shiloh, Y. , Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [DOI] [PubMed] [Google Scholar]
- 52. Xiao, Z. , Chang, J. G. , Hendriks, I. A. , Sigurethsson, J. O. et al., System‐wide analysis of SUMOylation dynamics in response to replication stress reveals novel small ubiquitin‐like modified target proteins and acceptor lysines relevant for genome stability. Mol. Cell. Proteomics 2015, 14, 1419–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pagel, O. , Loroch, S. , Sickmann, A. , Zahedi, R. P. , Current strategies and findings in clinically relevant post‐translational modification‐specific proteomics. Exp. Rev. Proteomics 2015, 12, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hunter, T. , The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 2007, 28, 730–738. [DOI] [PubMed] [Google Scholar]
- 55. Beltrao, P. , Bork, P. , Krogan, N. J. , van Noort, V. , Evolution and functional cross‐talk of protein post‐translational modifications. Mol. Syst. Biol. 2013, 9, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Elia, A. E. , Wang, D. C. , Willis, N. A. , Boardman, A. P. et al., RFWD3‐dependent ubiquitination of rpa regulates repair at stalled replication forks. Mol. Cell 2015, 60, 280–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jaehnig, E. J. , Kuo, D. , Hombauer, H. , Ideker, T. G. , Kolodner, R. D. , Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep. 2013, 4, 174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Medema, R. H. , Macurek, L. , Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [DOI] [PubMed] [Google Scholar]
- 59. Banin, S. , Moyal, L. , Shieh, S. , Taya, Y. et al., Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- 60. Khosravi, R. , Maya, R. , Gottlieb, T. , Oren, M. et al., Rapid ATM‐dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 14973–14977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhou, B. B. , Bartek, J. , Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [DOI] [PubMed] [Google Scholar]
- 62. Kiyokawa, H. , Ray, D. , In vivo roles of CDC25 phosphatases: biological insight into the anti‐cancer therapeutic targets. Anticancer Agents Med. Chem. 2008, 8, 832–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bartek, J. , Lukas, J. , Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [DOI] [PubMed] [Google Scholar]
- 64. Cutillas, P. R. , Role of phosphoproteomics in the development of personalized cancer therapies. Proteomics Clin. Appl. 2015, 9, 383–395. [DOI] [PubMed] [Google Scholar]
- 65. Shiloh, Y. , ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [DOI] [PubMed] [Google Scholar]
- 66. Stokes, M. P. , Rush, J. , Macneill, J. , Ren, J. M. et al., Profiling of UV‐induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boeing, S. , Williamson, L. , Encheva, V. , Gori, I. et al., Multiomic analysis of the UV‐induced DNA damage response. Cell Rep. 2016, S2211–1247(16)30474–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mazouzi, A. , Stukalov, A. , Muller, A. C. , Chen, D. et al., A comprehensive analysis of the dynamic response to aphidicolin‐mediated replication stress uncovers targets for ATM and ATMIN. Cell Rep. 2016, pii:S2211–1247(16)30366–7. [DOI] [PubMed] [Google Scholar]
- 69. Choi, S. , Srivas, R. , Fu, K. Y. , Hood, B. L. et al., Quantitative proteomics reveal ATM kinase‐dependent exchange in DNA damage response complexes. J. Proteome Res. 2012, 11, 4983–4991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Reinhardt, H. C. , Aslanian, A. S. , Lees, J. A. , Yaffe, M. B. , p53‐deficient cells rely on ATM‐ and ATR‐mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reinhardt, H. C. , Yaffe, M. B. , Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 2009, 21, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kerscher, O. , Felberbaum, R. , Hochstrasser, M. , Modification of proteins by ubiquitin and ubiquitin‐like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [DOI] [PubMed] [Google Scholar]
- 73. Komander, D. , The emerging complexity of protein ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [DOI] [PubMed] [Google Scholar]
- 74. Hussain, S. , Zhang, Y. , Galardy, P. J. , DUBs and cancer: the role of deubiquitinating enzymes as oncogenes, non‐oncogenes and tumor suppressors. Cell Cycle 2009, 8, 1688–1697. [DOI] [PubMed] [Google Scholar]
- 75. Kulathu, Y. , Komander, D. , Atypical ubiquitylation ‐ the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat. Rev. Mol. Cell Biol. 2012, 13, 508–523. [DOI] [PubMed] [Google Scholar]
- 76. Morris, J. R. , More modifiers move on DNA damage. Cancer Res. 2010, 70, 3861–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Hendriks, I. A. , Vertegaal, A. C. , SUMO in the DNA damage response. Oncotarget 2015, 6, 15734–15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bergink, S. , Jentsch, S. , Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [DOI] [PubMed] [Google Scholar]
- 79. Kim, W. , Bennett, E. J. , Huttlin, E. L. , Guo, A. , Li, J. , Possemato, A. et al., Systematic and quantitative assessment of the ubiquitin‐modified proteome. Mol. Cell 2011, 44, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hendriks, I. A. , Treffers, L. W. , Verlaan‐de Vries, M. , Olsen, J. V. , Vertegaal, A. C. , SUMO‐2 orchestrates chromatin modifiers in response to DNA damage. Cell Rep. 2015, 1778–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Danielsen, J. M. , Sylvestersen, K. B. , Bekker‐Jensen, S. , Szklarczyk, D. et al., Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol. Cell. Proteomics 2011, 10, M110 003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lakshmaiah, K. C. , Jacob, L. A. , Aparna, S. , Lokanatha, D. , Saldanha, S. C. , Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [DOI] [PubMed] [Google Scholar]
- 83. Groselj, B. , Sharma, N. L. , Hamdy, F. C. , Kerr, M. , Kiltie, A. E. , Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br. J. Cancer 2013, 108, 748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Brooks, C. L. , Gu, W. , The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bennetzen, M. V. , Larsen, D. H. , Dinant, C. , Watanabe, S. et al., Acetylation dynamics of human nuclear proteins during the ionizing radiation‐induced DNA damage response. Cell Cycle 2013, 12, 1688–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Brooks, C. L. , Gu, W. , New insights into p53 activation. Cell Res. 2010, 20, 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ljungman, M. , The transcription stress response. Cell Cycle 2007, 6, 2252–2257. [DOI] [PubMed] [Google Scholar]
- 88. Derks, K. W. , Hoeijmakers, J. H. , Pothof, J. , The DNA damage response: the omics era and its impact. DNA Repair 2014, 19, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jovanovic, M. , Rooney, M. S. , Mertins, P. , Przybylski, D. et al., Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 2015, 347, 1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Warmoes, M. , Jaspers, J. E. , Xu, G. , Sampadi, B. K. et al., Proteomics of genetically engineered mouse mammary tumors identifies fatty acid metabolism members as potential predictive markers for cisplatin resistance. Mol. Cell. Proteomics 2013, 12, 1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Altmeyer, M. , Lukas, J. , To spread or not to spread–chromatin modifications in response to DNA damage. Curr. Opin. Genet. Dev. 2013, 23, 156–165. [DOI] [PubMed] [Google Scholar]
- 92. Xing, M. , Yang, M. , Huo, W. , Feng, F. et al., Interactome analysis identifies a new paralogue of XRCC4 in non‐homologous end joining DNA repair pathway. Nat. Commun. 2015, 6, 6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Reinhardt, H. C. , Yaffe, M. B. , Phospho‐Ser/Thr‐binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 2013, 14, 563–580. [DOI] [PubMed] [Google Scholar]
- 94. Deans, A. J. , West, S. C. , DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Morris, M. K. , Chi, A. , Melas, I. N. , Alexopoulos, L. G. , Phosphoproteomics in drug discovery. Drug Discov. Today 2014, 19, 425–432. [DOI] [PubMed] [Google Scholar]
- 96. Barabasi, A. L. , Gulbahce, N. , Loscalzo, J. , Network medicine: a network‐based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lee, M. J. , Ye, A. S. , Gardino, A. K. , Heijink, A. M. et al., Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks. Cell 2012, 149, 780–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Casado, P. , Rodriguez‐Prados, J. C. , Cosulich, S. C. , Guichard, S. et al., Kinase‐substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013, 6, rs6. [DOI] [PubMed] [Google Scholar]
- 99. Vaga, S. , Bernardo‐Faura, M. , Cokelaer, T. , Maiolica, A. et al., Phosphoproteomic analyses reveal novel cross‐modulation mechanisms between two signaling pathways in yeast. Mol. Syst. Biol. 2014, 10, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wakabayashi, M. , Yoshihara, H. , Masuda, T. , Tsukahara, M. et al., Phosphoproteome analysis of formalin‐fixed and paraffin‐embedded tissue sections mounted on microscope slides. J. Proteome Res. 2014, 13, 915–924. [DOI] [PubMed] [Google Scholar]
- 101. Mertins, P. , Yang, F. , Liu, T. , Mani, D. R. et al., Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteomics 2014, 13, 1690–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Gajadhar, A. S. , Johnson, H. , Slebos, R. J. , Shaddox, K. et al., Phosphotyrosine signaling analysis in human tumors is confounded by systemic ischemia‐driven artifacts and intra‐specimen heterogeneity. Cancer Res. 2015, 75, 1495–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liu, N. Q. , Braakman, R. B. , Stingl, C. , Luider, T. M. et al., Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J. Mammary Gland Biol. Neoplasia 2012, 17, 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Narumi, R. , Murakami, T. , Kuga, T. , Adachi, J. et al., A strategy for large‐scale phosphoproteomics and SRM‐based validation of human breast cancer tissue samples. J. Proteome Res. 2012, 11, 5311–5322. [DOI] [PubMed] [Google Scholar]