

Abstract
Interest in amylose and its hybrids has grown over many decades, and a great deal of work has been devoted to developing methods for designing functional amylose hybrids. In this context, phosphorylase‐catalyzed polymerization shows considerable promise as a tool for preparing diverse amylose hybrids. Recently, advances have been made in the chemoenzymatic synthesis and characterization of amylose‐block‐polymers, amylose‐graft‐polymers, amylose‐modified surfaces, hetero‐oligosaccharides, and cellodextrin hybrids. Many of these saccharides provide clear opportunities for advances in biomaterials because of their biocompatibility and biodegradability. Important developments in bioapplications of amylose hybrids have also been made, and such newly developed amylose hybrids will help promote the development of new generations of glyco materials. WIREs Nanomed Nanobiotechnol 2017, 9:e1423. doi: 10.1002/wnan.1423
For further resources related to this article, please visit the WIREs website.
INTRODUCTION
Amylose is a linear polysaccharide in which a large number of glucose units are linked via α(1,4) glycosidic bonds1 (Figure 1(a)). Amylose is present in almost every plant system as an energy source2 and is therefore one of the most abundant raw materials. It is biodegradable, biocompatible, and renewable. Amylose adopts a left‐handed helical form in aqueous solution3 and can entrap hydrophobic molecules in the helix, which acts as a one‐dimensional supramolecular host molecule.4, 5, 6 The interest in amylose and their hybrid molecules therefore has been growing because of their potential applications.
Figure 1.
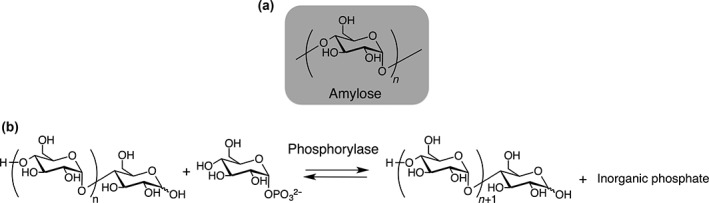
Chemical structure of amylose (a) and phosphorylase‐catalyzed synthesis of amylose (b).
Polysaccharides are structurally well‐defined molecules. This makes it difficult to synthesize polysaccharides containing amylose. In conventional chemical synthesis, the choice of substrates with appropriate leaving groups and protecting groups, catalysts, and experimental conditions (e.g., temperature and atmosphere) are crucial for obtaining stereo‐ and regiocontrolled oligosaccharides.7, 8 Such methods also require multiple synthetic steps and are time consuming. Chemical synthetic approaches are therefore unsuitable for obtaining structurally defined amylose.
Biocatalytic synthesis is a promising method and has several advantages over chemical synthetic approaches, such as mild reaction conditions (e.g., room temperature and aqueous solution) and high stereo‐ and regioselectivity of glycosidic bonds.9, 10, 11 In plants, amylose is synthesized from adenosine diphosphate (ADP)–glucose by granule‐bound starch synthase. Subsequently, a starch‐branching enzyme cleaves the resulting amylose and transfers the cleaved glucan in an α‐1,6 position. Pure amylose, therefore, can be obtained by the suppression of gene‐encoding starch‐branching enzymes.12 On the other hand, linear malto‐oligosaccharide is synthesized from ADP–glucose by glycogen synthase (GS) in mammalian cells.13 However, the substrate is fragile, and cumbersome procedures are needed. Therefore, these methods are not suitable for preparing amylose in practice. Alternatively, α‐glucan phosphorylases can be used for amylose preparation. α‐Glucan phosphorylases naturally catalyze the hydrolysis of glycosidic bonds in α(1,4)‐glucan in the presence of inorganic phosphate and the formation of glycosidic bonds in α(1,4)‐glucan in the absence of inorganic phosphate14 (Figure 1(b)). In glycosidic bond formation, phosphorylase catalyzes the addition of a glucose unit from glucose monophosphate to the nonreducing end of an amylose primer, with a minimum degree of polymerization (DP) of 3.15 The reducing ends of amylose primers can therefore be freely modified. The DP of enzymatically synthesized amylose can be controlled by changing the glucose monophosphate/amylose primer ratio. The reaction is analogous to living polymerization; therefore, the molecular weight distribution of the amylose is narrow.
This method has been used to prepare amylose and amylose hybrids, including block and graft copolymers, amylose‐grafted surfaces, and hetero‐oligosaccharides, as shown in Figure 2. We have coined the term ‘amylose engineering’ to describe the generation of amylose‐functionalized materials from amylose primers. An appropriate choice of amylose and other materials enables the addition of further functionalities and tuning and control of the materials for specific applications. Another aspect of this enzymatic polymerization is the entrapment of hydrophobic polymers in the amylose helix during polymerization (known as vine‐twining polymerization).16, 17, 18 This topic, however, has been reviewed recently19, 20, 21; therefore, we will not discuss it in detail.
Figure 2.
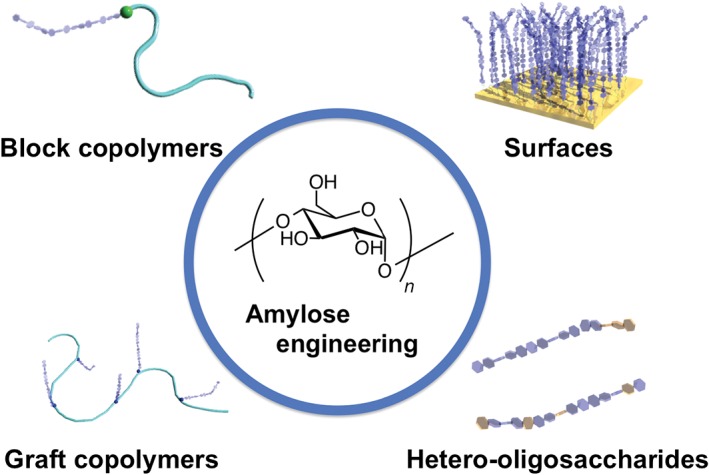
Schematic diagram of amylose‐based materials prepared by the phosphorylase‐catalyzed reaction of amylose primers.
In this review, we provide an overview of the chemoenzymatic synthetic routes for the preparation of amylose and its hybrids using phosphorylase. We focus on the synthesis and characterization of linear and grafted architectures in which amylose units are attached as side chains to synthetic and natural polymer backbones and surfaces (see section Enzymatic Synthesis of Amylose Hybrids by Phosphorylases). In the section Phosphorylase‐Catalyzed Synthesis of Hetero‐Oligosaccharides and Cellodextrins, we focus on the synthesis of hetero‐oligosaccharides and cellodextrin by phosphorylase‐catalyzed polymerization. Finally, some of the challenges in the application of these new amylose hybrid materials in biotechnology and therapeutics are highlighted (see section Bioapplications of Enzymatically Synthesized Amylose Hybrids).
ENZYMATIC SYNTHESIS OF AMYLOSE HYBRIDS BY PHOSPHORYLASES
Block Copolymers
As mentioned in the Introduction, phosphorylase catalyzes the addition of glucose units from glucose monophosphate to the nonreducing end of an amylose primer (e.g., maltopentaose) in the absence of inorganic phosphate. The reducing end of the primer can be functionalized, and the resulting primer can be recognized for phosphorylase. In 1987, Ziegast and Pfannemüller first used this method for the enzymatic synthesis of amylose hybrids.22 In their seminal work, they prepared two amylose‐chain‐functionalized poly(ethylene glycol)s (PEG); these were the first amylose‐based copolymers. It was found that a bifunctional primer gave amylose block copolymers with a uniform molecular weight distribution of amylose. This was ascribed by the authors to equal recognition of phosphorylases by both ends of the primer.
We also synthesized amylose‐b‐mPEG using phosphorylase‐catalyzed polymerization23, 24 (Figure 3(a)). The double helix formation is a well‐known property of amylose chains, and it leads to the formation of large aggregates and precipitation (retrogradation). Such low colloidal stability is a drawback in practical applications. However, this polymer (the amylose DP was 73) did not form a precipitate even after 1 day in aqueous solution. Amylose aggregation was effectively reduced by modification with water‐soluble PEG. Interestingly, the amylose‐b‐mPEG copolymer formed reverse micelles in chloroform solution, and a hydrophobic dye (methyl orange) was entrapped in the amylose helix in the reverse micelles.
Figure 3.
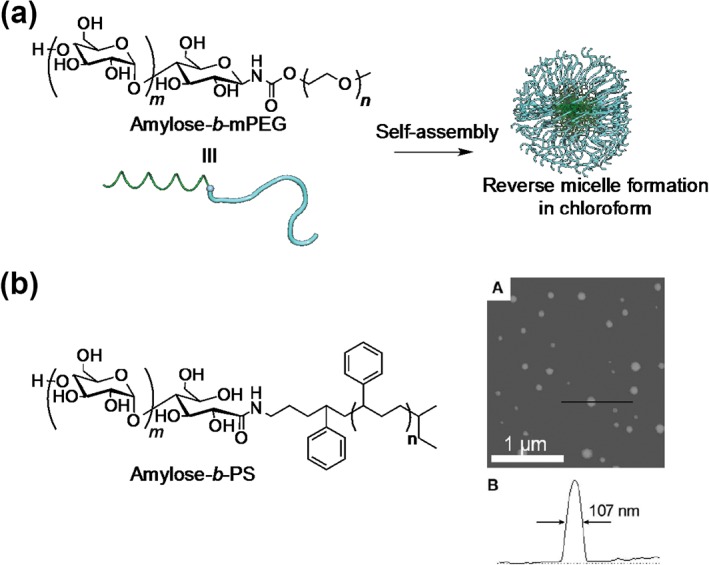
Chemical structure of amylose‐b‐mPEG and formation of reverse micelles in chloroform (a), and chemical structure of amylose‐b‐poly(styrene) and scanning force microscopy image of micellar aggregates of amylose‐b‐poly(styrene) in aqueous solution (b). (Reprinted with permission from Ref 26. Copyright 2005 American Chemical Society)
Loos and Stadler reported amphiphilic amylose‐b‐poly(styrene) polymers25 (Figure 3(b)), which were prepared by coupling maltoheptanolactone with amine‐functionalized poly(styrene) by reductive amination, followed by potato phosphorylase‐catalyzed polymerization. Although the primers were insoluble in aqueous solution, the enzymatic polymerization proceeded. This can be attributed to the partial formation of micellar aggregates.26, 27 Recently, the same research group used amphiphilic maltoheptaose‐b‐poly(2‐vinyl pyridine) as a primer.28
Phosphorylase‐catalyzed polymerization can be used not only with bifunctional primers but also with trifunctional primers. Rachmawati et al. prepared three‐armed poly(tetrahydrofuran) (PTHF)‐b‐maltoheptaose using a combination of cationic ring‐opening polymerization and reductive amination.29 The resulting polymers can be used as primers for the enzymatic synthesis of three‐armed PTHF‐b‐amylose.
More recently, we developed a series of amylose‐based star polymers (with one, two, four, and eight arms) using a combination of click chemistry and phosphorylase‐catalyzed polymerization30 (Figure 4(a)). The uniform length of the elongated amylose chains was confirmed using iodine encapsulation experiments. The resulting polymers acted as multivalent host molecules for hydrophobic molecules such as oligo(phenylene vinylene) (OPV), curcumin, bispyrenylpropane (BPP), and lipids. The encapsulation experiments showed that large elongated molecules (more than ca 2 nm) were essential for inclusion because various shorter, nonelongated molecules, e.g., pyrene, 1‐anilinonaphthalene‐8‐sulfonic acid (1,8‐ANS) and 2‐anilinonaphthalene‐6‐sulfonic acid (2,6‐ANS), did not form complexes (Figure 4(b)). It is worth noting that the Hill coefficients and binding constants increased with an increasing number of arms when the polymer encapsulated a cationic lipid (C16SP). We attribute these phenomena to neighboring helix formation in highly branched polymers.
Figure 4.
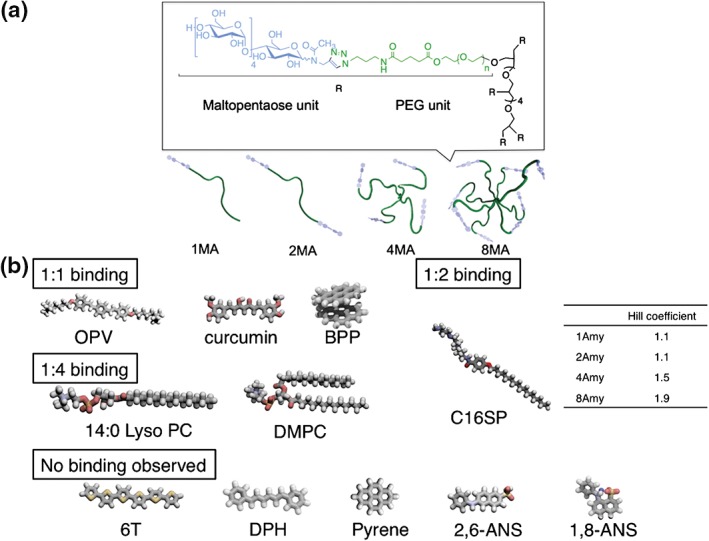
Chemical structures and diagrams of amylose‐primer‐modified poly(ethylene glycol)s (a) and chemical structures of hydrophobic guest molecules (b). (Reprinted with permission from Ref 29. Copyright 2015 American Chemical Society)
As already mentioned, amylose can wrap various hydrophobic polymers, e.g., PTHF, poly(caprolactone), poly(tetramethylene carbonate), and poly(lactide)s, during polymerization (vine‐twining polymerization). Tanaka et al. prepared maltoheptaose‐b‐poly(l‐lactide) (PLLA), which is a primer–guest molecule hybrid, via a copper‐catalyzed 1,3‐dipolar cycloaddition.31 The elongated amylose chains entrap other PLLA chains in their helical cavities during enzymatic polymerization, resulting in the formation of linear supramolecular polymers. More recently, a three‐armed Glc7–PLLA2 conjugate was used as a primer for the construction of supramolecular polymers.32 After enzymatic polymerization, the resulting amylose–PLLA2 formed an ionic gel in 1‐butyl‐3‐methylimidazolium chloride. The gelation was probably caused by the formation of inclusion complexes between the branched polymers. A similar approach was used to show inclusion complex formation by an amylose–PTHF conjugate.33
Branched Polysaccharides
Glycogen is a highly branched molecule consisting of α(1,4) glucan units and is synthesized by the action of glycogenin (primer), GS, and glycogen‐branching enzymes (GBEs). Glycogenin catalyzes the addition of several glucose units from uridine diphosphate‐glucose. After the addition of 8–12 glucose residues, GS catalyzes the further addition of glucose units, and then, GBE cleaves the saccharide chains and transfers them from α(1,4) positions to α(1,6) positions. Loos et al. have developed an enzymatic synthesis of branched polysaccharides based on this biologically inspired synthesis. They used glucan phosphorylase and GBE, which introduces branching points. This tandem enzymatic polymerization successfully produced highly branched polysaccharides.34, 35 They also used this strategy to obtain three‐arm branched polysaccharides.36
Amylose‐Grafted Polymers
Phosphorylase‐catalyzed polymerization is most frequently used for the synthesis of amylose‐grafted polymers. To the best of our best knowledge, the first enzymatically synthesized amylose‐g‐polymer was reported by Husemann and Reinhardt in 1962.37 They synthesized acetobromo oligosaccharide‐grafted 6‐trityl‐2,3‐dicarbanilylamylose. After removal of the protecting group, the resulting polymer served as a primer for producing amylose‐grafted amylose using potato phosphorylase‐catalyzed polymerization.
Amylose‐Grafted Synthetic Polymers
This method has been used for various primer‐graft‐polymers (naturally occurring polymers and synthetic polymers). The first amylose‐graft‐synthetic polymer was reported by Stadler et al.38 They prepared two macro primers, namely, maltoheptaose‐graft‐poly(dimethylsiloxane)s, via either acetal or amide linkages. Although both macro primers were recognized by potato phosphorylase, their reaction rates were much lower than that of maltopentaose. The lower reaction rates were attributed to the low solubilities of the macro primers in aqueous solutions.
Kobayashi et al. synthesized two different types of amylose‐graft‐poly(styrene) via homopolymerization of an amylose‐substituted styrene monomer (VAA) and enzymatic synthesis from maltopentaose‐carrying poly(styrene) [poly(VM5A); Figure 5], and investigated the structures of these polymers based on amylose–iodine complexations.39 The DP can be estimated from the maximum absorption wavelength (λ max) of an amylose–iodine complex.40, 41 The λ max value of the poly(VAA)–iodine complex was comparable to that of the polyiodide ion–amylose complex. This result suggests that the DP of poly(VAA) was nearly equal to that of amylose, and most of the primers were accessible to phosphorylase, enabling the production of VAA with a uniform amylose length. In contrast, the λ max of a poly(VAA‐co‐VM5A)–iodine complex indicated an amylose chain length much greater than that estimated using a phosphate assay. The authors assumed that the amylose chains were elongated from a few primers because of the steric configuration of the polymer. As a result, the iodine complexes showed much higher λ max values than expected.
Figure 5.
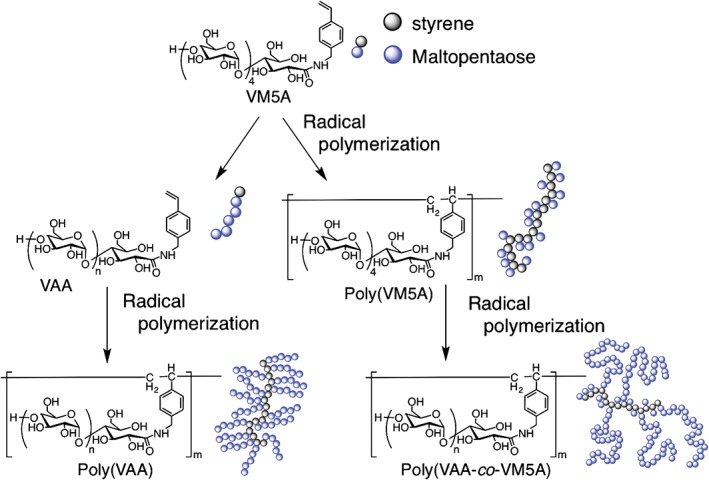
Schematic diagrams of structures of two different types of amylose‐graft‐polystyrene.
Amylose‐graft‐poly(acetylene),42 amylose‐graft‐poly(vinyl alcohol),43 and poly[styrene‐block‐(styrene‐graft‐amylose)]44 were also synthesized by phosphorylase‐catalyzed polymerization.
Amylose‐Graft‐Naturally Occurring Polymers
Saccharide‐conjugated peptides occur widely in living systems; for example, glycoproteins, peptidoglycans, and proteoglycans are found in plasma membranes, intercellular matrixes, and connective tissue, and they play important roles in biological activities. Inspired by such biological systems, Kamiya and Kobayashi reported amylose‐graft‐poly(l‐glutamic acid) conjugates prepared by phosphorylase‐catalyzed polymerization.45 This polymer formed helical structures in acidic aqueous solutions because poly(l‐glutamic acid) is a helix‐forming polymer. The helix‐to‐coil transition occurred at pH 6.1, which is higher than that of native poly(l‐glutamic acid) (pH 4.5). This was ascribed by the authors to electrostatic repulsion between neighboring carboxylate side chains because of the surrounding amylose chains.
We synthesized a series of amylose‐primer‐modified cholesteryl poly(l‐lysine) materials, which were prepared by coupling maltoheptaose and cholesterol‐bearing poly(l‐lysine)46 via reductive amination47 (Figure 6(a)). The resulting polymer self‐assembled into nanometer‐sized gel particles (i.e., a nanogel) of an average diameter of 50 nm and with a positive charge (19.6 mV). The nanogel showed a structural transition from a random coil to an α‐helical structure, induced by an increase in the pH. As expected, the nanogel acted as a primer for phosphorylase. The average diameter of the nanogel after enzymatic polymerization decreased to 30 nm. Interestingly, the zeta potential of the enzymatically polymerized nanogel (DP = 23) showed a weakly negative charge (−2.1 mV). These phenomena are attributed to the shielding of the cationic‐charged surfaces by the elongated amylose chains (Figure 6(b)).
Figure 6.
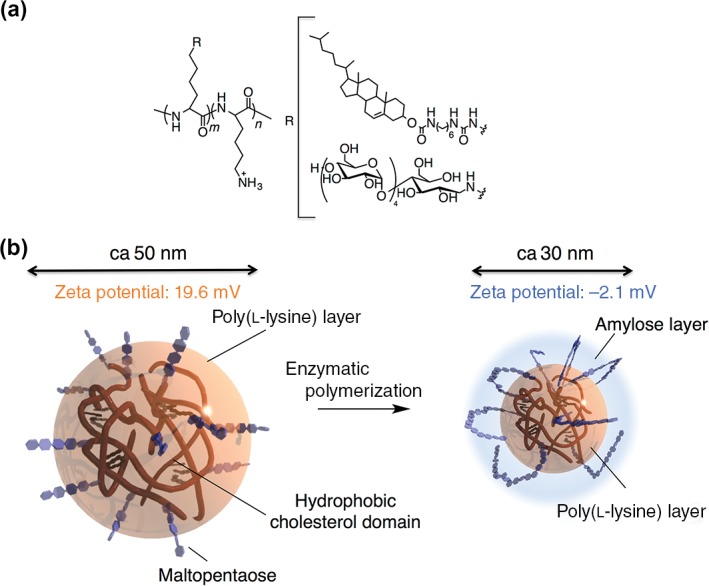
Chemical structure of maltopentaose‐modified cholesteryl poly(l‐lysine) (ChMaPLL) (a) and schematic diagram of ChMaPLL nanogel and amylose‐modified cholesteryl poly(l‐lysine) nanogel (b).
Natural polysaccharides are well‐known structurally diverse molecules and include linear polymers and branched hetero‐oligosaccharides (e.g., gum Arabic). Researchers have developed different types of branched hetero‐oligosaccharides based on the structures of natural polysaccharides. The first example of the chemoenzymatic synthesis of a branched hetero‐oligosaccharide was reported by Kadokawa et al. in 2007.48 They synthesized amylose‐graft‐chitosan, which is a linear polysaccharide consisting of α(1,4)‐linked 2‐amino‐2‐deoxy‐β‐D‐glucan units, by coupling maltoheptaose with chitosan, followed by phosphorylase‐catalyzed polymerization. Thus far, the synthesis of starch–chitosan hybrids by direct coupling of these polysaccharides is inefficient because of steric hindrance.49 These results suggest that the chemoenzymatic method is an efficient tool for the preparation of branched polysaccharides. The obtained polysaccharide hybrids were hardly soluble in any solvents because of helix formation by the amylose chains. The authors exploited this characteristic to prepare thin films and hydrogels consisting of branched polysaccharides. A more recent example, reported by the same group, is based on amylose‐graft‐cellulose.50 Amino‐functionalized polysaccharides are easily accessible starting materials for producing amylose‐graft‐polysaccharides using reductive amination. To obtain amylose‐graft‐cellulose, amine‐functionalized cellulose was prepared by treating tosylated cellulose with sodium azide, followed by reduction with sodium borohydride. The amylose‐graft‐cellulose was synthesized using the chemoenzymatic method described above. The polymers also formed thin films and hydrogels by simply drying. The gel was tougher than that formed from amylose‐graft‐chitosan. This chemoenzymatic method was recently extended to the preparation of amylose‐graft‐alginate, which is a linear polysaccharide consisting of (1,4)‐linked β‐d‐mannuronic acid and α‐l‐glucuronic acid units.51
Glycogen consists of linear chains of α(1,4)‐linked glucose residues, which are interlinked by α(1,6) glycosidic bonds. The resulting structure is highly branched and multivalent. Because a number of nonreducing ends of α(1,4)‐glucan are located on the surface, glycogen can act as a naturally occurring primer for phosphorylase. Kadokawa et al. performed α‐glucan phosphorylase‐catalyzed polymerization using glycogen as a primer and confirmed the elongation of amylose chains from glycogen.52 It was noted that the elongated amylose chains from the nonreducing ends of glycogen formed double helices among intermolecular glycogens, which acted as cross‐linking points to produce a hydrogel.
Amylose‐Grafted Surfaces
Because of its versatility, the chemoenzymatic approach is also suited for the modification of spherical and planar surfaces. The first example of an amylose‐modified silica gel was reported by Okamoto et al. in 1996.53 The modified silica gels were prepared using two different approaches (methods 1 and 2). In method 1, 3‐aminopropyltriethoxysilane‐modified maltopentaose was immobilized by reaction with silica gel. In method 2, maltoheptanolactone was treated with 3‐aminopropyl‐silanized silica gel. The two amylose‐modified silica gels were reacted with a large excess of 3,5‐dimethylphenyl isocyanate to convert the amylose hydroxy groups to the corresponding carbamate residues. The difference between the amounts of amylose immobilized on the silica surfaces using the two methods was negligible. These modified silica can be used as chiral stationary phases in high‐performance liquid chromatography because of their intrinsic chirality. They displayed excellent enantioseparation of 10 different racemates. Such systems are now widely used to separate racemic compounds into their enantiomers.54 Stadler et al. used a similar strategy to produce an amylose‐grafted silica gel and investigated its chiral discrimination.55
Another application of the chemoenzymatic method is the modification of two‐dimensional metal and inorganic planar surfaces. Vlist et al. synthesized amylose and branched polysaccharide brushes on silica surfaces via enzymatic polymerization.56 More recently, the same research group used this strategy to modify a gold surface.57 Such polysaccharide‐grafted surfaces have potential applications as antifouling materials for ship hulls, underwater pipes, and biomedical implants because of the highly hydrophilic nature of the polysaccharide.
PHOSPHORYLASE‐CATALYZED SYNTHESIS OF HETERO‐OLIGOSACCHARIDES AND CELLODEXTRINS
Hetero‐Oligosaccharides
Enzymes usually show strict specificities for substrate recognition in catalytic reactions. However, some enzymes have loose substrate specificities. This enables the synthesis of unique hetero‐oligosaccharide chains. Phosphorylases frequently have this property. Evers et al. reported that potato phosphorylase catalyzes the addition of d‐glycal to maltotetraose and degrades the resulting 2‐deoxy‐maltooligosaccharide to 2‐deoxy‐α‐d‐glucopyranosyl phosphate in the presence of inorganic phosphate.58 Since this discovery, various novel polysaccharide chains have been synthesized using phosphorylases (Figure 7).
Figure 7.
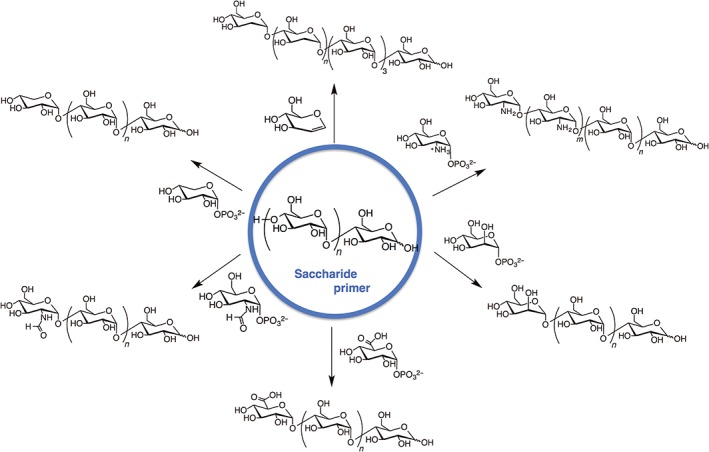
Phosphorylase‐catalyzed synthesis of various hetero‐oligosaccharides.
Kadokawa et al. first established this method for the preparation of hetero‐oligosaccharides.59 They used xylose monophosphate as a substrate and maltotetraose as a primer. Matrix‐assisted laser desorption/ionization time‐of‐flight mass spectrometry confirmed the formation of tetrasaccharides to octasaccharides containing one xylose unit and oligosaccharides consisting of two xylose units. Glucoamylase hydrolytic studies showed that xylose was added to the nonreducing end of the obtained hetero‐oligosaccharide. The authors further extended this method to the preparation of N‐formyl‐α‐glucosaminylated maltooligosaccharide,60 α‐glucuronylated maltooligosaccharide,61 α‐mannosylated maltooligosaccharide,62 chitin and chitosan stereoisomers,63 and non‐natural heteroaminopolysaccharides.64
A further example of enzymatic synthesis of a hetero‐oligosaccharide was reported by the same research group.65 In this case, dendritic amphoteric α‐glucans were prepared by sequential glucuronylation and glucosaminylation of highly branched cyclic dextrin (a glucan dendrimer). The zeta potentials of the polymers changed from positive to negative when the pH changed from acidic to basic. Such amphoteric polysaccharides could have useful bioapplications, such as carriers in drug‐delivery systems and in regenerative tissue scaffolds.
Cellodextrins
Cellulose is a linear polysaccharide consisting of β(1,4)‐linked glucose units and is the most abundant natural resource on Earth. Cellulose is synthesized by cellulose synthases, which are β‐glycosyltransferase‐type enzymes, at the plasma membrane. Low‐molecular‐weight cellulose (cellodextrin) can be prepared using the enzyme cellodextrin phosphorylase. Cellodextrin phosphorylase also catalyzes the addition of glucose units from glucose monophosphate to the nonreducing ends of cellobiose acceptors.11
Serizawa et al. used this method to prepare post‐functionalizable two‐dimensional crystalline cellulose nanosheets66 (Figure 8). An azide‐containing cellulose oligomer was synthesized by reacting β‐glycosyl azide as a primer and α‐glucose monophosphate as a substrate in the presence of cellodextrin phosphorylase. The resulting cellulose oligomers (average DP 10) aligned and formed nanosheets of an average thickness of 5.5 nm. Wide‐angle X‐ray scattering and attenuated total reflectance Fourier‐transform infrared spectroscopy studies showed that the nanosheet formed a cellulose II allomorph, which had an antiparallel saccharide arrangement against the sheet. The azide groups of cellodextrin were therefore located on the sheet surface. Such structures offer advantages such as the possibility of nanosheet post‐functionalization. As proof of concept, the nanosheet was incubated with 1‐ethynylpyrene as a model substrate in the presence of CuSO4 and sodium ascorbate; a pyrene‐conjugated nanosheet was successfully obtained. This method enables the fabrication of advanced functional cellulose sheets.
Figure 8.
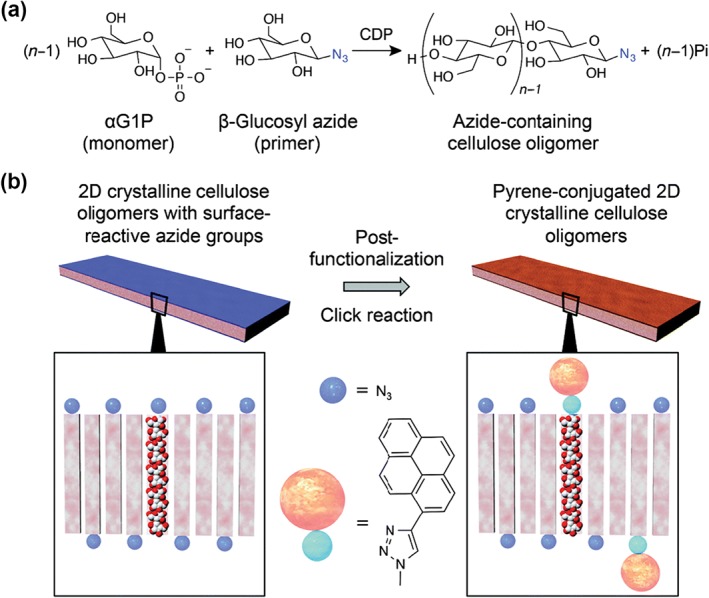
(a) Synthetic scheme for cellodextrin with azide groups at reducing end by cellulose phosphorylase‐catalyzed polymerization (a) and schematic illustration of post‐functionalization of azide groups to give cellulose nanosheets with 1‐ethynylpyrene groups through copper‐catalyzed click reactions (b). (Reproduced with permission from Ref 65. Copyright 2015 The Royal Society of Chemistry)
More recently, the same research group reported that a cellulose nanosheet acted as an artificial hydrolytic enzyme.67 The nanosheet was prepared by cellodextrin phosphorylase‐catalyzed polymerization, as described above. When the resulting nanosheet was incubated with p‐nitrophenyl acetate (PNP) as a model substrate, there was no significant difference in the presence and absence of the nanosheet. Previously, they showed that hydrolytic activities were derived from hydroxyl groups on nanosheets.68, 69 They therefore prepared a mechanically broken nanosheet with a large surface area by sonication to improve the activity. The activity of the resulting nanosheet was 500 times greater than that of the untreated nanosheet. They also showed that the efficiency of PNP hydrolysis depended on the nanosheet concentration. These studies will open up new potential applications of cellulose materials.
BIOAPPLICATIONS OF ENZYMATICALLY SYNTHESIZED AMYLOSE HYBRIDS
Considering the biodegradability and biocompatibility of amylose, it is not surprising that amylose hybrids have found use in numerous biological and medical applications. In the sections below, we discuss applications of amylose hybrids in protein science, DNA manipulation, and as carriers in drug‐delivery systems.
Reconstitution of Membrane Proteins
Membrane proteins participate in pivotal biological processes such as cell signaling, transportation, and adhesion. However, there have been fewer studies of membrane proteins than of water‐soluble proteins. This is because of their extremely low solubility in aqueous solution. We therefore need to embed membrane proteins in hydrophobic spaces such as lipid bilayers to study their use as functional materials. A typical method for the reconstitution of membrane proteins in liposomes is to solubilize the target proteins using a surfactant–phospholipid mixture and then remove the surfactant using dialysis or chromatography. This method, however, is generally time consuming and gives low yields of reconstituted membrane proteins. Alternative methods are therefore needed.
An example of a novel membrane protein reconstitution method based on enzymatically triggered micelle–vesicle transitions was reported by us in 2006.70 We synthesized a series of amylose primer‐modified surfactants, which were prepared by the treatment of maltopentaose lactone and alkylamines (octyl, dodecyl, and hexadecyl). The surfactants self‐assembled into micelles and acted as primers for phosphorylase. On enzymatic polymerization, the micelles collapsed (Figure 9(a)). The surface tension of the reaction solution increased and reached a plateau. The surface tension value after 150 min was comparable to that of the buffer solution. This was ascribed to the increased hydrophilicity of the surfactant caused by the elongation of saccharide chains, eventually leading to micelle dissociation. We further investigated enzymatically induced micelle‐to‐vesicle transitions in phospholipid–primer surfactant systems. A mixture of l‐α‐dipalmitoyl phosphatidylcholine and a primer surfactant formed micelles of a mean hydrodynamic diameter of 21 nm. Enzymatic polymerization caused an increase in the sizes of the assemblies. The resulting reaction mixture contained liposomes (123 nm), as confirmed by transmission electron microscopy. Liposome formation was attributed to the release of the surfactant from the mixed micelles as a result of an increase in the hydrophilicity of the surfactant. We used this system to reconstitute bacteriorhodopsin (BR), which is a model membrane protein, into liposomes. First, BR was solubilized in micelles consisting of a primer‐modified surfactant. Second, the resulting micelles were mixed with the phospholipid–primer surfactant micelle solution. Finally, the mixture was incubated with phosphorylase for 6 h (Figure 9(b)). BR‐reconstituted liposomes were successfully formed. The liposomes showed greater light‐induced H+ influx and light responsiveness than did the mixed‐micelle system.
Figure 9.
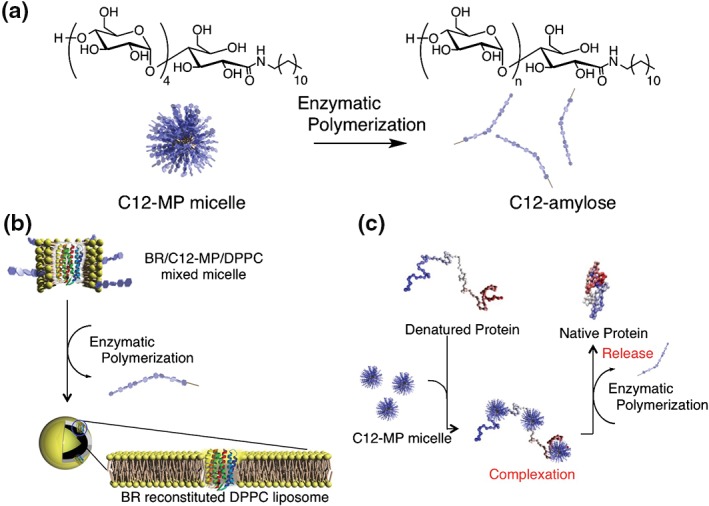
Schematic diagram of phosphorylase‐catalyzed reaction of amylose‐primer surfactant (a), membrane protein reconstitution on l‐α‐dipalmitoyl phosphatidylcholine (DPPC) liposome by enzyme‐responsive micelle–vesicle transition system (b), and enzyme‐responsive artificial chaperone system based on amylose‐primer surfactant (c).
In addition to the reconstitution method, effective folding of recombinant proteins is an important tool in protein science. We also used this enzyme‐responsive molecular assembly system to develop a novel protein‐folding method71 (Figure 9(c)). Specifically, micelles based on the surfactant described above suppressed precipitation of carbonic anhydrase B (CAB) on heating. On addition of phosphorylase, its substrate, and an activator, polymerization was accompanied by the recovery of enzymatic activity. This is attributed to a decrease in the surfactant hydrophobicity. Polymerization increased the surfactant hydrophilicity, and the resulting surfactant was removed from the CAB/surfactant complex. As a result, the CAB gradually refolded and recovered its activity. This system was also used to fold chemically denatured proteins and showed a higher efficiency than cyclodextrin as a surfactant‐stripping agent.
More recently, other polysaccharide materials based on enzymatically synthesized glycogen (ESG)‐bearing hydrophobic groups have been developed for artificial chaperone systems.72, 73 The chaperone‐like activity was greater than that of a previously reported system based on cholesteryl—group‐modified pullulan. This is attributed to the unique architecture of hyperbranched ESGs, which provide sufficient space for protein refolding but not for irreversible aggregation of denatured proteins.
DNA Chaperone System
DNA strand exchange between double‐stranded DNA (dsDNA) and single‐stranded DNA (ssDNA) is an important biological process for genetic maintenance and DNA repair. Such processes are catalyzed by recombinases (e.g., Eukaryotic Rad 51, E. coli RecA proteins). These enzymes, called DNA chaperones, bind with DNAs and form intermediate complexes with both dsDNA and ssDNA, resulting in the exchange of DNA strands. Artificial compounds can also catalyze DNA strand exchange. Maruyama et al. reported that poly‐l‐lysine‐graft‐dextran significantly accelerates strand exchange reactions when used as an artificial DNA chaperone.74 The DNA chaperone activities of self‐assembled cationic nanogels were also reported by our research group.75 These polymers have highly localized positive charges, which allow them to bind electrostatically with DNA strands, resulting in DNA strand concentration. Based on this work, we prepared a series of spermine‐functionalized amylose star copolymers and investigated their artificial DNA chaperone activities.30 Strand exchange was accelerated in the presence of the polymers, and the rate of strand exchange clearly increased as the number of amylose arms increased. The increased rate is attributed to a decrease in the dissociation constant of DNA. This decrease is probably caused by an increase in the apparent cation concentration, resulting in enhanced DNA binding.
Nucleic Acid‐Delivery Systems
Research on nucleic acid delivery has recently expanded because of its potential applications in next‐generation therapeutic strategies for treating inheritable or acquired diseases. However, nucleic acids are easily digested by nucleases. In addition, nucleic acids are large anionic molecules, and they do not cross cell membrane. Consequently, the development of safe and efficient gene carriers is needed. Among the materials under development for use as gene carriers, polysaccharides are one of the most promising because of their low toxicity, biocompatibility, and biodegradability.76, 77, 78, 79, 80, 81 Our group has developed a gene‐delivery system based on enzymatically synthesized polysaccharides. The system consists of cycloamylose (CA) functionalized with cationic groups. CA, which is a macrocyclic polysaccharide consisting of α(1, 4)‐glucose units, is produced through the treatment of linear amylose with 4‐α‐glucanotransferase. We reported a series of cycloamylose derivatives that effectively delivered plasmid DNA (pDNA),82 siRNA,83 and CpG DNA84 in vitro and in vivo.85 We also developed a novel gene‐delivery system using hexadecyl‐group‐bearing cationic CA and a phospholipid‐degrading enzyme.86
More recent examples of enzymatically synthesized polysaccharide‐based gene carriers are amylose star copolymers.87 The molecular weight of siRNA is much smaller than that of pDNA, and a strong interaction is needed to form stable nanoparticle complexes. We therefore took advantage of the multivalency of star polymers. We prepared spermine‐functionalized single‐ and eight‐armed amylose‐based star polymers (Figure 10(a)) and compared their siRNA carriers’ activities. As expected, the eight‐armed polymer formed more compact complexes with siRNA than did the single‐armed polymer. The siRNA/eight‐armed polymer complexes were effectively internalized into Renca cells and suppressed mRNA levels by 65% (Figure 10(b)). This is probably because of the formation of smaller complexes between siRNA and the eight‐armed polymer, which might enhance cell internalization, leading to higher gene‐silencing efficiency.
Figure 10.
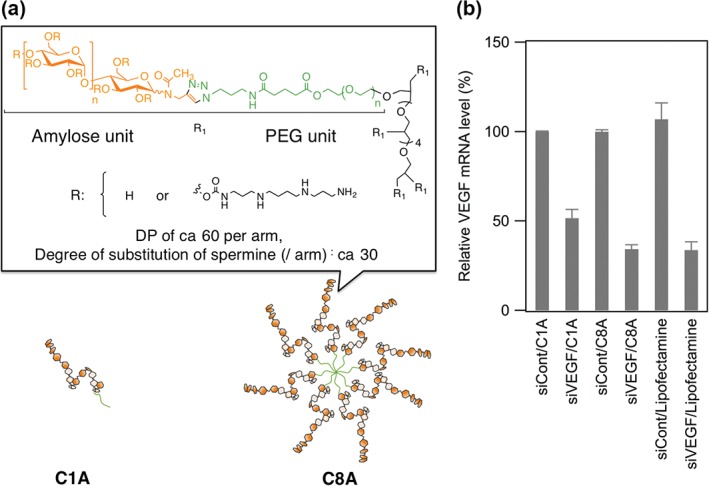
Chemical structures and diagrams of spermine‐modified amylose star polymers (a) and gene‐silencing effect of siRNA/cationic polymer complexes (b).
CONCLUSION
Amylose synthesis by phosphorylase‐catalyzed polymerization has been studied for more than three decades. Over the last 10 years, significant improvements not only in phosphorylase‐catalyzed polymerization but also in coupling methods for primers have enabled the synthesis of a variety of amylose hybrids with different compositions, structures, dimensions, and chemical functionalization. Amylose hybrids with unique properties have been developed. However, there are few reports of bioapplications of amylose hybrids. This means that amylase‐based materials are still a developing field. However, given the biocompatibility and biodegradability of amylose hybrids, and the possibility of additional functionalization, it is anticipated that amylose hybrids will find multiple applications in biomedicine in the near future.
Conflict of interest: The authors have declared no conflicts of interest for this article.
REFERENCES
- 1. Damager I, Engelsen SB, Blennow A, Lindberg Møller B, Motawia MS. First principles insight into the α‐glucan structures of starch: their synthesis, conformation, and hydration. Chem Rev 2010, 110:2049–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schnepp Z. Biopolymers as a flexible resource for nanochemistry. Angew Chem Int Ed 2013, 52:1096–1108. [DOI] [PubMed] [Google Scholar]
- 3. Roger P, Axelos MAV, Colonna P. SEC − MALLS and SANS studies applied to solution behavior of linear α‐glucans. Macromolecules 2000, 33:2446–2455. [Google Scholar]
- 4. Putseys JA, Lamberts L, Delcour JA. Amylose‐inclusion complexes: formation, identity and physico‐chemical properties. J Cereal Sci 2010, 51:238–247. [Google Scholar]
- 5. Star A, Steuerman DW, Heath JR, Stoddart JF. Starched carbon nanotubes. Angew Chem Int Ed 2002, 41:2508–2512. [DOI] [PubMed] [Google Scholar]
- 6. Sanji T, Kato N, Kato M, Tanaka M. Helical folding in a helical channel: chiroptical transcription of helical information through chiral wrapping. Angew Chem Int Ed 2005, 44:7301–7304. [DOI] [PubMed] [Google Scholar]
- 7. Boltje TJ, Buskas T, Boons G‐J. Opportunities and challenges in synthetic oligosaccharide and glycoconjugate research. Nat Chem 2009, 1:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schuerch C. Systematic approaches to the chemical synthesis of polysaccharides. Acc Chem Res 1973, 6:184–191. [Google Scholar]
- 9. Kobayashi S, Makino A. Enzymatic polymer synthesis: an opportunity for green polymer chemistry. Chem Rev 2009, 109:5288–5353. [DOI] [PubMed] [Google Scholar]
- 10. S‐i S, Uyama H, J‐i K, Kimura S, Kobayashi S. Enzymes as green catalysts for precision macromolecular synthesis. Chem Rev 2016, 116:2307–2413. [DOI] [PubMed] [Google Scholar]
- 11. J‐i K. Precision polysaccharide synthesis catalyzed by enzymes. Chem Rev 2011, 111:4308–4345. [DOI] [PubMed] [Google Scholar]
- 12. Carciofi M, Blennow A, Jensen SL, Shaik SS, Henriksen A, Buléon A, Holm PB, Hebelstrup KH. Concerted suppression of all starch branching enzyme genes in barley produces amylose‐only starch granules. BMC Plant Biol 2012, 12:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ugalde JE, Parodi AJ, Ugalde RA. De novo synthesis of bacterial glycogen: agrobacterium tumefaciens glycogen synthase is involved in glucan initiation and elongation. Proc Natl Acad Sci 2003, 100:10659–10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Illingworth B, Brown DH, Cori CF. The de novo synthesis of polysaccharide by phosphorylase. Proc Natl Acad Sci U S A 1961, 47:469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Whelan WJ, Bailey JM. The action pattern of potato phosphorylase. Biochem J 1954, 58:560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. J‐i K, Kaneko Y, Tagaya H, Chiba K. Synthesis of an amylose‐polymer inclusion complex by enzymatic polymerization of glucose 1‐phosphate catalyzed by phosphorylase enzyme in the presence of polyTHF: a new method for synthesis of polymer‐polymer inclusion complexes. Chem commun 2001, 5:449–450. [Google Scholar]
- 17. Yang L, Zhang B, Liang Y, Yang B, Kong T, Zhang L‐M. In situ synthesis of amylose/single‐walled carbon nanotubes supramolecular assembly. Carbohydr Res 2008, 343:2463–2467. [DOI] [PubMed] [Google Scholar]
- 18. Rachmawati R, Woortman AJJ, Loos K. Facile preparation method for inclusion complexes between amylose and polytetrahydrofurans. Biomacromolecules 2013, 14:575–583. [DOI] [PubMed] [Google Scholar]
- 19. J‐i K. Preparation and applications of amylose supramolecules by means of phosphorylase‐catalyzed enzymatic polymerization. Polymers 2012, 4:116–133. [Google Scholar]
- 20. J‐i K. Architecture of amylose supramolecules in form of inclusion complexes by phosphorylase‐catalyzed enzymatic polymerization. Biomolecules 2013, 3:369–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaneko Y, J‐i K. Vine‐twining polymerization: a new preparation method for well‐defined supramolecules composed of amylose and synthetic polymers. Chem Rec 2005, 5:36–46. [DOI] [PubMed] [Google Scholar]
- 22. Ziegast G, Pfannemüller B. Phosphorolytic syntheses with di‐, oligo‐ and multi‐ functional primers linear and star‐shaped hybrid polymers. Carbohydr Res 1987, 160:185–204. [Google Scholar]
- 23. Akiyoshi K, Kohara M, Ito K, Kitamura S, Sunamoto J. Enzymatic synthesis and characterization of amphiphilic block copolymers of poly(ethylene oxide) and amylose. Macromol Rapid Commun 1999, 20:112–115. [Google Scholar]
- 24. Akiyoshi K, Maruichi N, Kohara M, Kitamura S. Amphiphilic block copolymer with a molecular recognition site: induction of a novel binding characteristic of amylose by self‐assembly of poly(ethylene oxide)‐block‐amylose in chloroform. Biomacromolecules 2002, 3:280–283. [DOI] [PubMed] [Google Scholar]
- 25. Loos K, Stadler R. Synthesis of amylose‐block‐polystyrene rod − coil block copolymers. Macromolecules 1997, 30:7641–7643. [Google Scholar]
- 26. Loos K, Müller AHE. New routes to the synthesis of amylose‐block‐polystyrene rod − coil block copolymers. Biomacromolecules 2002, 3:368–373. [DOI] [PubMed] [Google Scholar]
- 27. Loos K, Böker A, Zettl H, Zhang M, Krausch G, Müller AHE. Micellar aggregates of amylose‐block‐polystyrene rod − coil block copolymers in water and THF. Macromolecules 2005, 38:873–879. [Google Scholar]
- 28. Kumar K, Woortman AJJ, Loos K. Synthesis of amylose‐b‐P2VP block copolymers. Macromol Rapid Commun 2015, 36:2097–2101. [DOI] [PubMed] [Google Scholar]
- 29. Rachmawati R, de Gier HD, Woortman AJJ, Loos K. Synthesis of telechelic and three‐arm polytetrahydrofuran‐block‐amylose. Macromol Chem Phys 2015, 216:1091–1102. [Google Scholar]
- 30. Nishimura T, S‐a M, S‐i S, Akiyoshi K. Glyco star polymers as helical multivalent host and biofunctional nano‐platform. ACS Macro Lett 2015, 4:367–371. [DOI] [PubMed] [Google Scholar]
- 31. Tanaka T, Sasayama S, Nomura S, Yamamoto K, Kimura Y, J‐i K. An amylose‐poly(l‐lactide) inclusion supramolecular polymer: enzymatic synthesis by means of vine‐twining polymerization using a primer–guest conjugate. Macromol Chem Phys 2013, 214:2829–2834. [Google Scholar]
- 32. Tanaka T, Gotanda R, Tsutsui A, Sasayama S, Yamamoto K, Kimura Y, J‐i K. Synthesis and gel formation of hyperbranched supramolecular polymer by vine‐twining polymerization using branched primer–guest conjugate. Polymer 2015, 73:9–16. [Google Scholar]
- 33. Rachmawati R, Woortman AJJ, Kumar K, Loos K. Inclusion complexes between polytetrahydrofuran‐b‐amylose block copolymers and polytetrahydrofuran chains. Macromol Biosci 2015, 15:812–828. [DOI] [PubMed] [Google Scholar]
- 34. van der Vlist J, Palomo Reixach M, van der Maarel M, Dijkhuizen L, Schouten AJ, Loos K. Synthesis of branched polyglucans by the tandem action of potato phosphorylase and Deinococcus geothermalis glycogen branching enzyme. Macromol Rapid Commun 2008, 29:1293–1297. [Google Scholar]
- 35. Ciric J, Loos K. Synthesis of branched polysaccharides with tunable degree of branching. Carbohydr Polym 2013, 93:31–37. [DOI] [PubMed] [Google Scholar]
- 36. Jvd V, Faber M, Loen L, Dijkman TJ, Asri LATW, Loos K. Synthesis of hyperbranched glycoconjugates by the combined action of potato phosphorylase and glycogen branching enzyme from Deinococcus geothermalis . Polymers 2012, 4:674–690. [Google Scholar]
- 37. Husemann VE, Reinhardt M. Über die darstellung definiert verzweigter polysaccharide. II. Einführung von oligosacchariden als seitenketten in amylose und deren verlängerung durch enzymatische synthese. Die Makromol Chem 1962, 57:129–149. [Google Scholar]
- 38. Vv B, Jonas G, Stadler R. Enzymic grafting of amylose from poly(dimethylsiloxanes). Macromolecules 1995, 28:17–24. [Google Scholar]
- 39. Kobayashi K, Kamiya S, Enomoto N. Amylose‐carrying styrene macromonomer and its homo‐ and copolymers: synthesis via enzyme‐catalyzed polymerization and complex formation with iodine. Macromolecules 1996, 29:8670–8676. [Google Scholar]
- 40. Fales FW. The linear relationship between iodine staining and average chain length of the unbranched amyloglucans. Biopolymers 1980, 19:1535–1542. [Google Scholar]
- 41. Banks W, Greenwood CT, Khan KM. The interaction of linear, amylose oligomers with iodine. Carbohydr Res 1971, 17:25–33. [Google Scholar]
- 42. Sasaki Y, Kaneko Y, J‐i K. Chemoenzymatic synthesis of amylose‐grafted polyacetylene by polymer reaction manner and its conversion into organogel with DMSO by cross‐linking. Polym Bull 2008, 62:291–303. [Google Scholar]
- 43. Kaneko Y, S‐i M, J‐i K. Chemoenzymatic synthesis of amylose‐grafted poly(vinyl alcohol). Polym Chem 2010, 1:193–197. [Google Scholar]
- 44. Narumi A, Kawasaki K, Kaga H, Satoh T, Sugimoto N, Kakuchi T. Glycoconjugated polymer 6. Synthesis of poly[styrene‐block‐(styrene‐graft‐amylose)] via potato phosphorylase‐catalyzed polymerization. Polym Bull 2003, 49:405–410. [Google Scholar]
- 45. Kamiya S, Kobayashi K. Synthesis and helix formation of saccharide‐poly(L‐glutamic acid) conjugates. Macromol Chem Phys 1998, 199:1589–1596. [Google Scholar]
- 46. Akiyoshi K, Ueminami A, Kurumada S, Nomura Y. Self‐association of cholesteryl‐bearing poly(l‐lysine) in water and control of its secondary structure by host − guest interaction with cyclodextrin. Macromolecules 2000, 33:6752–6756. [Google Scholar]
- 47. Morimoto N, Yamazaki M, Tamada J, Akiyoshi K. Polysaccharide‐hair cationic polypeptide nanogels: self‐assembly and enzymatic polymerization of amylose primer‐modified cholesteryl poly(l‐lysine). Langmuir 2013, 29:7509–7514. [DOI] [PubMed] [Google Scholar]
- 48. S‐i M, Kaneko Y, J‐i K. Chemoenzymatic synthesis of amylose‐grafted chitosan. Macromol Rapid Commun 2007, 28:863–867. [Google Scholar]
- 49. Tang R, Du Y, Fan L. Dialdehyde starch‐crosslinked chitosan films and their antimicrobial effects. J Polym Sci Part B Polym Phys 2003, 41:993–997. [Google Scholar]
- 50. Omagari Y, S‐i M, Kaneko Y, J‐i K. Chemoenzymatic synthesis of amylose‐grafted cellulose. Macromol Biosci 2009, 9:450–455. [DOI] [PubMed] [Google Scholar]
- 51. Omagari Y, Kaneko Y, J‐i K. Chemoenzymatic synthesis of amylose‐grafted alginate and its formation of enzymatic disintegratable beads. Carbohydr Polym 2010, 82:394–400. [Google Scholar]
- 52. Izawa H, Nawaji M, Kaneko Y, J‐i K. Preparation of glycogen‐based polysaccharide materials by phosphorylase‐catalyzed chain elongation of glycogen. Macromol Biosci 2009, 9:1098–1104. [DOI] [PubMed] [Google Scholar]
- 53. Enomoto N, Furukawa S, Ogasawara Y, Akano H, Kawamura Y, Yashima E, Okamoto Y. Preparation of silica gel‐bonded amylose through enzyme‐catalyzed polymerization and chiral recognition ability of its phenylcarbamate derivative in HPLC. Anal Chem 1996, 68:2798–2804. [DOI] [PubMed] [Google Scholar]
- 54. Ikai T, Okamoto Y. Structure control of polysaccharide derivatives for efficient separation of enantiomers by chromatography. Chem Rev 2009, 109:6077–6101. [DOI] [PubMed] [Google Scholar]
- 55. Loos K, von Braunmühl V, Stadler R, Landfester K, Spiess HW. Saccharide modified silica particles by enzymatic grafting. Macromol Rapid Commun 1997, 18:927–938. [Google Scholar]
- 56. van der Vlist J, Schönen I, Loos K. Utilization of glycosyltransferases for the synthesis of a densely packed hyperbranched polysaccharide brush coating as artificial glycocalyx. Biomacromolecules 2011, 12:3728–3732. [DOI] [PubMed] [Google Scholar]
- 57. Mazzocchetti L, Tsoufis T, Rudolf P, Loos K. Enzymatic synthesis of amylose brushes revisited: details from X‐ray photoelectron spectroscopy and spectroscopic ellipsometry. Macromol Biosci 2014, 14:186–194. [DOI] [PubMed] [Google Scholar]
- 58. Evers B, Thiem J. Synthesis of 2‐deoxy‐maltooligosaccharides with phosphorylase and their degradation with amylases. Starch Stärke 1995, 47:434–439. [Google Scholar]
- 59. Nawaji M, Izawa H, Kaneko Y, Ji K. Enzymatic synthesis of α‐d‐xylosylated malto‐oligosaccharides by phosphorylase‐catalyzed xylosylation. J Carbohydr Chem 2008, 27:214–222. [Google Scholar]
- 60. Kawazoe S, Izawa H, Nawaji M, Kaneko Y, J‐i K. Phosphorylase‐catalyzed N‐formyl‐α‐glucosaminylation of maltooligosaccharides. Carbohydr Res 2010, 345:631–636. [DOI] [PubMed] [Google Scholar]
- 61. Umegatani Y, Izawa H, Nawaji M, Yamamoto K, Kubo A, Yanase M, Takaha T, J‐i K. Enzymatic α‐glucuronylation of maltooligosaccharides using α‐glucuronic acid 1‐phosphate as glycosyl donor catalyzed by a thermostable phosphorylase from Aquifex aeolicus VF5. Carbohydr Res 2012, 350:81–85. [DOI] [PubMed] [Google Scholar]
- 62. Shimohigoshi R, Takemoto Y, Yamamoto K, J‐i K. Thermostable α‐glucan phosphorylase‐catalyzed successive α‐mannosylations. Chem Lett 2013, 42:822–824. [Google Scholar]
- 63. J‐i K, Shimohigoshi R, Yamashita K, Yamamoto K. Synthesis of chitin and chitosan stereoisomers by thermostable [α]‐glucan phosphorylase‐catalyzed enzymatic polymerization of [α]‐d‐glucosamine 1‐phosphate. Org Biomol Chem 2015, 13:4336–4343. [DOI] [PubMed] [Google Scholar]
- 64. Yamashita K, Yamamoto K, J‐i K. Synthesis of non‐natural heteroaminopolysaccharides by α‐glucan phosphorylase‐catalyzed enzymatic copolymerization: α(1 → 4)‐linked glucosaminoglucans. Biomacromolecules 2015, 16:3989–3994. [DOI] [PubMed] [Google Scholar]
- 65. Takata Y, Shimohigoshi R, Yamamoto K, Kadokawa J‐I. Enzymatic synthesis of dendritic amphoteric α‐glucans by thermostable phosphorylase catalysis. Macromol Biosci 2014, 14:1437–1443. [DOI] [PubMed] [Google Scholar]
- 66. Yataka Y, Sawada T, Serizawa T. Enzymatic synthesis and post‐functionalization of two‐dimensional crystalline cellulose oligomers with surface‐reactive groups. Chem Commun 2015, 51:12525–12528. [DOI] [PubMed] [Google Scholar]
- 67. Serizawa T, Kato M, Okura H, Sawada T, Wada M. Hydrolytic activities of artificial nanocellulose synthesized via phosphorylase‐catalyzed enzymatic reactions. Polym J 2016, 48:539–544. [Google Scholar]
- 68. Serizawa T, Sawada T, Okura H, Wada M. Hydrolytic activities of crystalline cellulose nanofibers. Biomacromolecules 2013, 14:613–617. [DOI] [PubMed] [Google Scholar]
- 69. Serizawa T, Sawada T, Wada M. Chirality‐specific hydrolysis of amino acid substrates by cellulose nanofibers. Chem Commun 2013, 49:8827–8829. [DOI] [PubMed] [Google Scholar]
- 70. Morimoto N, Ogino N, Narita T, Kitamura S, Akiyoshi K. Enzyme‐responsive molecular assembly system with amylose‐primer surfactants. J Am Chem Soc 2007, 129:458–459. [DOI] [PubMed] [Google Scholar]
- 71. Morimoto N, Ogino N, Narita T, Akiyoshi K. Enzyme‐responsive artificial chaperone system with amphiphilic amylose primer. J Biotechnol 2009, 140:246–249. [DOI] [PubMed] [Google Scholar]
- 72. Takahashi H, S‐i S, Akiyoshi K. Amphiphilic polysaccharide nanoballs: a new building block for nanogel biomedical engineering and artificial chaperones. ACS Nano 2011, 5:337–345. [DOI] [PubMed] [Google Scholar]
- 73. Takeda S, Takahashi H, S‐i S, Sasaki Y, Akiyoshi K. Amphiphilic nanogel of enzymatically synthesized glycogen as an artificial molecular chaperone for effective protein refolding. RSC Adv 2013, 3:25716–25718. [Google Scholar]
- 74. Kim WJ, Ishihara T, Akaike T, Maruyama A. Comb‐type cationic copolymer expedites DNA strand exchange while stabilizing DNA duplex. Chem Eur J 2001, 7:176–180. [DOI] [PubMed] [Google Scholar]
- 75. Morimoto N, Tamada J, S‐i S, Shimada N, Kano A, Maruyama A, Akiyoshi K. Interaction of self‐assembled cationic nanogels with oligo‐DNA and function as artificial nucleic acid chaperone. Chem Lett 2009, 38:496–497. [Google Scholar]
- 76. Zhou Y, Yang B, Ren X, Liu Z, Deng Z, Chen L, Deng Y, Zhang L‐M, Yang L. Hyperbranched cationic amylopectin derivatives for gene delivery. Biomaterials 2012, 33:4731–4740. [DOI] [PubMed] [Google Scholar]
- 77. Deng J, Zhou Y, Xu B, Mai K, Deng Y, Zhang L‐M. Dendronized chitosan derivative as a biocompatible gene delivery carrier. Biomacromolecules 2011, 12:642–649. [DOI] [PubMed] [Google Scholar]
- 78. Raemdonck K, Naeye B, Buyens K, Vandenbroucke RE, Høgset A, Demeester J, De Smedt SC. Biodegradable dextran nanogels for RNA interference: focusing on endosomal escape and intracellular siRNA delivery. Adv Funct Mater 2009, 19:1406–1415. [Google Scholar]
- 79. Song Y, Sun Y, Zhang X, Zhou J, Zhang L. Homogeneous quaternization of cellulose in NaOH/urea aqueous solutions as gene carriers. Biomacromolecules 2008, 9:2259–2264. [DOI] [PubMed] [Google Scholar]
- 80. J‐i J, Ikai T, Okazaki A, Yamamoto M, Hirano Y, Tabata Y. Expression profile of plasmid DNA by spermine derivatives of pullulan with different extents of spermine introduced. J Control Release 2007, 118:389–398. [DOI] [PubMed] [Google Scholar]
- 81. Mochizuki S, Sakurai K. A novel polysaccharide/polynucleotide complex and its application to bio‐functional DNA delivery system. Polym J 2009, 41:343–353. [Google Scholar]
- 82. Toita S, Morimoto N, Akiyoshi K. Functional cycloamylose as a polysaccharide‐based biomaterial: application in a gene delivery system. Biomacromolecules 2010, 11:397–401. [DOI] [PubMed] [Google Scholar]
- 83. Toita S, Soma Y, Morimoto N, Akiyoshi K. Cycloamylose‐based biomaterial: nanogel of cholesterol‐bearing cationic cycloamylose for siRNA delivery. Chem Lett 2009, 38:1114–1115. [Google Scholar]
- 84. Tahara Y, Yasuoka J, Sawada S, Sasaki Y, Akiyoshi K. Effective CpG DNA delivery using amphiphilic cycloamylose nanogels. Biomater Sci 2015, 3:256–264. [DOI] [PubMed] [Google Scholar]
- 85. Fujii H, Shin‐Ya M, Takeda S, Hashimoto Y, S‐a M, S‐i S, Adachi T, Akiyoshi K, Miki T, Mazda O. Cycloamylose‐nanogel drug delivery system‐mediated intratumor silencing of the vascular endothelial growth factor regulates neovascularization in tumor microenvironment. Cancer Sci 2014, 105:1616–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Toita S, S‐i S, Akiyoshi K. Polysaccharide nanogel gene delivery system with endosome‐escaping function: co‐delivery of plasmid DNA and phospholipase A2. J Control Release 2011, 155:54–59. [DOI] [PubMed] [Google Scholar]
- 87. Nishimura T, Umezaki K, Mukai S‐a, Sawada S‐i, Akiyoshi K. Amylose‐based cationic star polymers for siRNA delivery. Biomed Res Int 2015, 2015:6. [DOI] [PMC free article] [PubMed] [Google Scholar]