

Abstract
Despite preventive human papilloma virus (HPV) vaccination efforts, cervical cancer remains a leading cause of death in women worldwide. Development of therapeutic approaches for cervical cancer are hampered by a lack of mechanistic insight during tumorigenesis. The cytoskeletal protein Keratin 17 (KRT17;K17) is robustly expressed in a broad array of carcinomas, including in cervical tumors, where it has both diagnostic and prognostic value. In this study, we have established multiple functional roles for K17 in the promotion of cervical tumorigenesis in vivo using the established HPV16tg mouse model for cervical squamous cell carcinoma. In HPV16tg/+;Krt17−/−relative to HPV16tg/+ reference female mice, onset of cervical lesions is delayed and closely paralleled by marked reductions in hyperplasia, dysplasia and vascularization. In addition, loss of Krt17 is associated with a cytokine polarization and recruitment of effector immune cells to lesion-prone cervical epithelia. Further, we observed marked enhancement of terminal differentiation in HPV16tg/+;Krt17−/−cervical epithelium accompanied by a stimulation and expansion in the expression of p63, a known basal/reserve cell marker in this tissue. Altogether, the data suggest that the loss of Krt17 may foster an overall protective environment for lesion-prone cervical tissue. In addition to providing new insights into the immunomodulatory and cellular mechanisms of cervical tumorigenesis, these findings may help guide the development of future therapies including vaccines.
INTRODUCTION
Cervical cancer is the second leading cause of death for women worldwide. In total, 12 900 new cases and 4100 deaths were estimated to occur in the United States in 2015.1 The development of cervical cancer is a multi-step biological process thought to be initiated by infection of cervical epithelia with high risk, oncogenic strains of the human papilloma virus (HPV; type 16 or 18). When targeted to the proliferative basal cells of the cervical epithelium, integration of HPV16 genes into the host genome causes chronic infection.2,3 The HPV16 E6 and E7 oncoproteins interfere with host DNA synthesis and apoptosis, as well as cell cycle control, leading to aberrant epithelia growth and appearance of cervical intraepithelial lesions (CIN). If left uncontrolled, CINs can progress through increasing stages of dysplasia from LSIL (low-grade squamous intraepithelial lesion) to HSIL (high-grade squamous intraepithelial lesions), which can further develop into squamous cell carcinomas (SCC).4 Many of these key attributes are reproduced in the HPV16tg/+ mouse model,5 which has been extensively used to study various aspects of the cellular and molecular pathophysiology of cervical cancer.
Despite longstanding knowledge regarding how high-risk HPV strains can infect and hijack host cell processes to promote cervical tumorigenesis,6–8 why selected patients exhibit more aggressive cancer growth or respond differently to chemotherapeutic treatments largely remains a mystery. What is clear, however, is that HPV infection itself is a poor indicator of clinical outcome, as many HPV infections are transient and poorly correlate with progression from CIN to HSIL and SCC.9,10 Moreover, extrinsic factors such as increased estrogen levels or a compromised immune system can also promote cervical tumorigenesis.11,12 Although biomarkers such as p16INK4a and Ki67 have been used extensively in the clinic to identify CIN and LSIL precursor lesions, the type I intermediate filament protein keratin 17 (gene, KRT17; protein, K17) has emerged as a powerful diagnostic and prognostic biomarker with preferential expression in more advanced HSIL and SCC cervical lesions.13–15
In both humans and mice, K17 exhibits an expression pattern normally restricted to specific compartments within ectoderm-derived epithelial appendages (for example, hair, nail, tooth, thymus, various glands). However, KRT17/K17 can be rapidly induced in response to mechanical insult (wounding), chemical stimulation (phorbol ester) or environmental challenge (UV irradiation) in all complex epithelia. In addition, upregulation of K17 expression is correlated with lesion progression and poor prognosis in several epithelial cancers, including those occurring in the cervix (see below), skin, lung, ovary, larynx, pancreas, breast and gut.16–23 In mouse models for SCC or BCC (basal cell carcinoma), the genetic loss of Krt17 attenuates tumorigenesis, in part through a dampening of a tumor-promoting cytokine profile that results in reduced recruitment of inflammatory and effector immune cell to sites of tumor formation.23 A similar phenomenon occurs in tumor cell models in culture,22–24 adding to the evidence that K17 contributes to regulating cytokine expression in a skin keratinocyte-autonomous fashion. From this progress, one infers that K17’s status must be upgraded from biomarker to effector in a variety of chronic inflammatory epithelial disorders.
In cervical epithelia, expression of K17 is highly correlated with more advanced stages of cervical cancer growth and, moreover, higher K17 expression is strongly associated with a poorer prognosis for the patient.15 Further, K17 is expressed in the reserve cells of the cervix, which are stem-like and give rise to both the columnar endocervical and the squamnocolumnar ectocervical epithelia.25 When targeted by HPV, these reserve cells are thought to function as precursors of malignant lesions.26 Keratin 17 has already been shown to exert an impact on a growing number of tumor paradigms.22–24,27 Given all this, a functional role for K17 in vivo in normal cervical epithelia and during cervical tumorigenesis seems likely, but has not been examined to date. In this study, we have exploited a well-established animal model, the HPV16tg/+ mouse,5 to test whether and how the loss of Krt17/K17 impacts cervical tumorigenesis in vivo.
RESULTS
Keratin 17 expression in cervical epithelia correlates with lesion progression
In the HPV16tg/+ mouse model, expression of the early genes E1–E7 for HPV type 16 is driven by the K14 gene promoter. With no additional treatment in the FVB strain background, HPV16tg/+ mice spontaneously develop SCC-like lesions in the skin with complete penetrance on the ears, between 60 and 120 days of age, but show no sign of cervical lesions.5 HPV16tg/+ female mice subcutaneously implanted with slow release 17β-estradiol pellets at 1 month of age develop cervical lesions that mimic the transformation from normal epithelia to dysplastic lesions over the course of 180 days.5 This transformation is most prominent in the endocervical cervico-uterine transformation zone, though lesions may also arise near the ectocervical epithelia5 (Supplementary Figure S1A). This progression phenocopies the transition of normal epithelia to CIN-LSIL-HSIL-SCC in human cervical cancer, and accounts for the popularity of this model for examining the molecular mechanisms governing cervical tumorigenesis.
Relative to wild-type (WT) mice, estrogen implantation in HPV16tg/+ mice results in the onset of epithelial hyperplasia, with some papillation, at 3 months post implantation (Supplementary Figure S1B). Dysplasia has set in by 6 months post implantation as determined by marked disorganization of the basal epithelium and increased nuclear staining throughout suprabasal epithelium, in addition to more pronounced papillation (Figure 1a, Supplementary Figure S1B–C). Further, HPV16tg/+ mice exhibit significantly increased expression of p16INK4a (also called Cdkn2a), a known marker of cervical dysplasia,28 over time post implantation (Figure 1b), with a pronounced upregulation throughout the transformation zone epithelium (Figure 1c). By 6 months post implantation, none of the mice exhibited lesions that had progressed to in situ or invasive carcinomas. Consistent with previous reports, estrogen-implanted WT mice show no sign of basal epithelial layer disorganization, papillation or elevated p16INK4a expression over the same time frame.5
Figure 1.
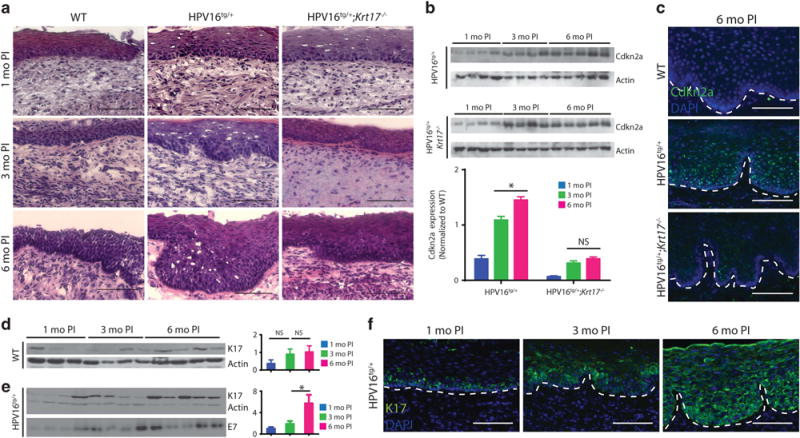
Increased K17 expression correlates with cervical lesion progression. (a) H&E staining of transformation zone epithelium for wild-type (WT), HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 1-, 3- and 6 months post implantation. Bar = 100 μm. (b) Immunoblots for Cdkn2a in cervical tissue of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 1-, 3- and 6 months post implantation. Each lane represents a distinct biological replicate. Actin serves as loading control. Densitometric quantification shown below. (c) Immunostaining for Cdkn2a (green) in transformation zone epithelium of WT, HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 6 months post implantation. Nuclei are stained with DAPI (blue). Dotted line separates epithelium (above line) from underlying stroma (below line). Bar = 100 μm. (d, e) Immunoblots for K17 in cervical tissue of (d) WT and (e) HPV16tg/+ mice at 1-, 3- and 6 months post implantation. Each lane represents a distinct biological replicate. Actin serves as loading control. E7 indicates transgene expression. Densitometric quantification of K17 expression normalized to WT shown at right. (f) Immunostaining for K17 (green) in transformation zone epithelium of HPV16tg/+ mice at 1-, 3- and 6 months post implantation. Nuclei are stained with DAPI (blue). Dotted line separates epithelium (above line) from underlying stroma (below line). Bar = 100 μm. All error bars are s.e.m. *P<0.05. NS; no significance.
Expression of K17 in cervical epithelia remains relatively low and constant in WT mice (Figure 1d), but increases in HPV16tg/+ mice over time post implantation (Figure 1e). In estrogen-implanted WT mice, K17 immunoreactivity is restricted to basal cells in a patchy pattern in the transformation zone and does not expand into the suprabasal layers over time post implantation (Supplementary Figure S1D). In estrogen-implanted HPV16tg/+ mice, by contrast, K17 is not only initially restricted to the basal layer at 1 month post implantation but has markedly increased and expanded throughout the suprabasal layers of the cervical epithelium at 3- and 6 months post implantation (Figure 1f). There were no obvious differences between genotypes with regards to the expression of the related type I keratin 14 (K14) (Supplementary Figure S1E). Consistent with findings in human cervical carcinoma (Smedts et al.,13 Escobar-Hoyos et al.15,29), therefore, a clear induction of K17 and expansion into the suprabasal layers coincides with lesion progression in the HPV16tg/+ cervical epithelium. Based on these observations, we next assessed the functional roles for K17 toward the early stages of cervical tumorigenesis in estrogen-implanted HPV16tg/+ mice.
Loss of Keratin 17 attenuates cervical lesion progression
Relative to the HPV16tg/+ reference model, several morphological and molecular readouts for cervical tumorigenesis were attenuated in the HPV16tg/+;Krt17−/− mice. The hyperplasia and dysplasia observed in transformation zones of HPV16tg/+ mice over time post implantation were reduced with the loss of K17 (Figure 1a). At 3 months post implantation, immunostaining for phosphor-histone H3 reflecting mitotic activity was significantly upregulated in HPV16tg/+ relative to WT controls, but blunted with the loss of K17 (Figure 2a). In addition, vascularization in the surrounding stromal tissue, as determined by PECAM-1 immunostaining, was increased in cervical tissue from HPV16tg/+ mice but reduced as well in HPV16tg/+;Krt17−/− mice (Figure 2b). Consistent with the latter, a reduction in mast cell density was observed in the cervical stromal compartment of HPV16tg/+;Krt17−/− relative to HPV16tg/+ mice (Figure 2c). The cervical epithelium in the transformation zone, but not ectocervical epithelium, was significantly thickened in HPV16tg/+ mice relative to WT controls, and reduced in HPV16tg/+;Krt17−/− mice (Figure 2d). Further, the significant induction of p16INK4a over time in cervical epithelia of HPV16tg/+ mice is blunted in HPV16tg/+;Krt17−/− mice (Figures 1b and c). There was no obvious difference between genotypes in the expression of the transgene driver K14 (Supplementary Figure S1E), HPV E7 transgene (Supplementary Figure S1F) or apoptosis (data not shown). Altogether, the data point to the occurrence of a delay in HPV16-induced cervical tumorigenesis when Krt17 is ablated.
Figure 2.
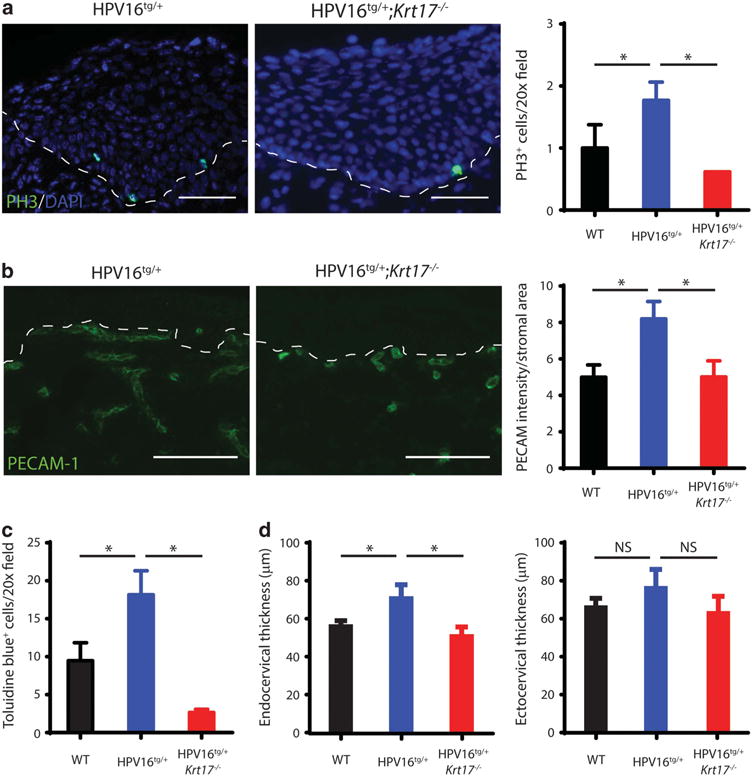
Markers of cervical lesion progression are attenuated with the loss of K17. (a) Immunostaining for phospho-histone H3 (PH3; green) in transformation zone epithelium of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 3 months post implantation. Nuclei are stained with DAPI (blue). Dotted line separates epithelium (above line) from underlying stroma (below line). Bar = 100 μm. Quantification of PH3+ cells per field at right. (b) Immunostaining for PECAM-1 (green) in underlying stroma of transformation zone epithelium of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 3 months post implantation. Dotted line separates epithelium (above line) from underlying stroma (below line). Bar = 100 μm. Quantification of PECAM-1 pixel intensity per stromal area at right. (c) Quantification of toluidine blue-positive cells per field (images not shown) of transformation zone epithelium of WT, HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 3 months post implantation. (d) Thickness (in μm) of transformation zone epithelium (left) and ectocervical epithelium (right) of WT, HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 3 months post implantation. All error bars are s.e.m. *P<0.05.
Inflammation and inflammatory gene expression in cervical epithelia are elevated with the loss of keratin 17
Our previous work on the impact of Krt17 deficiency on Gli2tg/+-driven23 and HPV16tg/+-driven22 tumorigenesis in skin prompted us to examine the molecular changes that parallel the delay in cervical tumorigenesis in HPV16tg/+;Krt17−/− mice relative to HPV16tg/+ controls. We assessed the levels of several gene transcripts of interest using quantitative reverse transcriptase–PCR in cervical tissue of HPV16tg/+ or HPV16tg/+;Krt17−/− relative to WT mice at 1 month post implantation, which precedes lesion onset based on histological observations. Although several transcripts associated with various signaling and growth pathways changed at least twofold in the absence of Krt17 (Supplementary Figure S2A), the most striking finding was the marked elevation in transcript levels for numerous Th1-associated pro-inflammatory cytokines including Ifng, Cxcl9, Cxcl10, Cxcl11, Ido1, Mmp13, Tnfa, Il1b, Mmp9, Tgfb and Cxcl5 (Figure 3a). No upregulation was observed for Il17a (Th17-associated cytokine) or Il10 and Il22 (Th2-associated cytokines) (Figure 3a). Strikingly, at 3 months post implantation these same inflammatory gene transcripts exhibit a near opposite expression pattern with the Th1-associated cytokines being relatively unchanged and Th17 and Th2 cytokines being robustly upregulated in cervical tissue in the absence of Krt17 (Figure 3b). Altogether, the data indicate Krt17 may regulate cytokine polarization during cervical tumorigenesis, a role it is known to have during ear skin tumorigenesis.23
Figure 3.
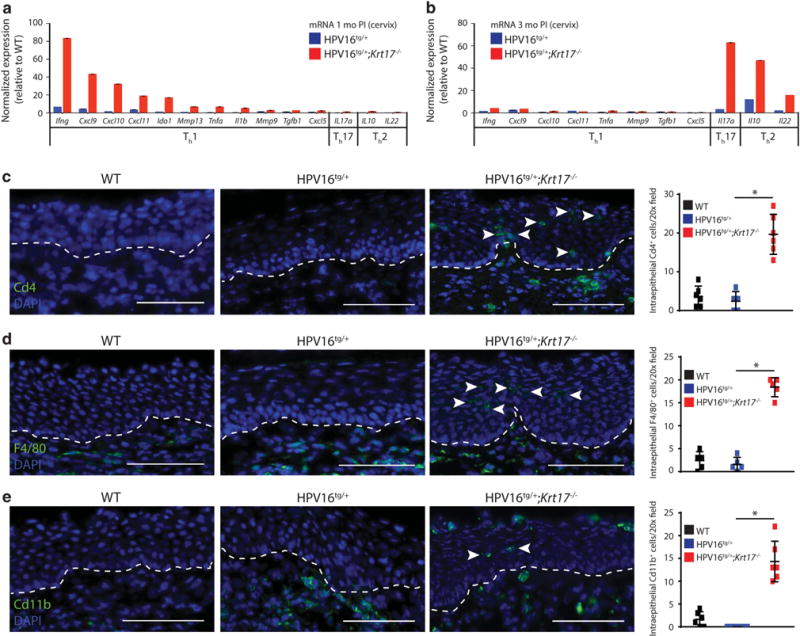
Inflammation is increased in cervical tissue in the absence of K17. (a, b) Normalized expression for gene transcripts in cervical tissue of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at (a) 1 month post implantation and at (b) 3 months post implantation. (c–e) Immunostaining for (c) Cd4, (d) F4/80 and (e) Cd11b in transformation zone epithelium of WT, HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 3 months post implantation. Nuclei are stained with DAPI (blue). Dotted line separates epithelium (above line) from underlying stroma (below line). Arrowheads denote intraepithelial immunostained cells. Bar = 100 μm. Graphs at right depict the number of intraepithelial immune cells per field across genotypes. All error bars are s.e.m. *P<0.05.
Correlating with these molecular changes in inflammatory cytokine expression was an increased recruitment of numerous types of immune cell effectors to the transformation zone of HPV16tg/+;Krt17−/− cervical epithelia at 3 months post implantation including CD4+ T cells (Figure 3c), F4/80+ macrophages (Figure 3d) and CD11b+ leukocytes (Figure 3e), typically in an intraepithelial pattern. Cervical epithelia from WT or HPV16tg/+ mice exhibit little to no recruitment of these immune cell effectors. Intraepithelial immune cells were not observed at 1 month post implantation in any of these genotypes (data not shown).
We recently reported that genetic ablation of Krt17 attenuates pro-inflammatory gene expression in tumor-prone ear skin tissue from HPV16tg/+ mice,22 a finding that seems at odds with the role K17 appears to have in cervical tissue of the same mouse model. The only technical differences between these two studies are the tissue type being examined and the use of estrogen implantation in the current study. We confirmed that ear skin tissue of estrogen-implanted HPV16tg/+ mice still exhibit elevated expression of a number of pro-inflammatory gene transcripts (comparable to unimplanted mice22), and that this increase is largely blunted in ear skin of HPV16tg/+;Krt17−/− mice (Supplementary Figure S2C). Thus, although an overall outcome of attenuated HPV16-induced tumorigenesis is observed for both skin and cervical tissues of the same set of mice when Krt17 expression is lost, the mechanisms governing the expression of select pro-inflammatory cytokines and the induction and site (for example, intraepithelial in cervix versus stromal in skin) of an inflammatory response during tumorigenesis appear to be at odds in these two tissue types.
In an attempt to define what may be distinctly different between cervical and skin tumorigenesis in the same set of HPV16tg/+;Krt17−/− mice, we next examined the status of Aire and hnRNP K in cervical epithelia. These two proteins act as regulators of gene expression and physically interact with K17 in skin keratinocytes, and have been previously linked to the promotion of tumor growth in a K17-dependent fashion in the skin.22,24 No substantial change could be detected in the level or localization of Aire mRNA transcripts in cervical epithelia from HPV16tg/+;Krt17−/− mice relative to HPV16tg/+ mice at 1 month post implantation, even though Aire expression remains inducible and Krt17-dependent in ear skin tissue of the same set of mice (Supplementary Figure S2D, S2F). Though Hnrnpk transcript levels are not Krt17-dependent in cervical tissue (Supplementary Figure S2E), expression of hnRNP K protein is substantially reduced in cervical epithelium lacking Krt17 (Supplementary Figure S2G), an outcome that differs from previous observations in skin tumor keratinocytes.24 Although these findings do not rule out a function for Aire or hnRNP K in cervical epithelia, they suggest that neither Aire nor hnRNP K influences cervical tumorigenesis in a K17-dependent fashion as they do in the skin, thus substantiating the existence of fundamental differences regarding the role of K17 in these two distinct tissue settings.
Differentiation of cervical epithelia robustly increases in the absence of keratin 17
From the quantitative reverse transcriptase–PCR screening, S100A8 and S100A9 were upregulated in cervical epithelia of HPV16tg/+;Krt17−/−, but not HPV16tg/+, mice (Figure 4a). Intriguingly, yet again, these findings are opposite between ear and cervical tissue samples harvested from the same set of mice (Figure 4a). We confirmed that S100A8 was upregulated at the protein level in the cervical but not in the ear skin tissue of HPV16tg/+;Krt17−/− mice relative to HPV16tg/+ controls (Figure 4b). Several S100 proteins are viewed as markers of differentiation,30 which prompted us to assess the differentiation status of these two epithelia in HPV16tg/+;Krt17−/− mice. We found that several differentiation markers, including keratin 4 (Krt4), keratin 1 (Krt1), keratin 10 (Krt10), involucrin (Ivl) and filaggrin (Flg) were robustly upregulated at the transcript level in cervical epithelia from HPV16tg/+;Krt17−/− relative to HPV16tg/+ control mice at both 1- and 3 months post implantation (Figure 4c). Keratin 1 and filaggrin protein levels were confirmed to be increased in cervical epithelia from HPV16tg/+;Krt17−/− mice (Figure 4d), and K1, specifically, was increased in transformation zone epithelia, but not in the ear skin, of HPV16tg/+;Krt17−/− mice relative to HPV16tg/+ controls (Figures 4e and f). Finally, the increase in epithelial differentiation markers in the cervix is related to the Krt17 expression status and is not specific to the HPV16tg/+ transgene status, estrogen implantation or age. Indeed, unimplanted, non-transgenic, aged Krt17−/− mice also exhibited an increased expression of markers of cervical epithelial differentiation markers (at the transcript level) relative to WT littermates (Supplementary Figure S3A). This is also the case for S100A8 protein which, when assessed via indirect immunofluorescence, was markedly increased in the outermost suprabasal layer in the cervico-uterine transformation zone of aged Krt17−/− mice (Supplementary Figure S3B). Collectively, these findings point to a fundamental difference in the manner with which K17 impacts tumorigenesis in cervical versus skin epithelia, and suggest that the protective effect of the loss of Krt17 in the cervical epithelium may be in part linked to a stimulation of terminal differentiation of tumor-prone cells.
Figure 4.
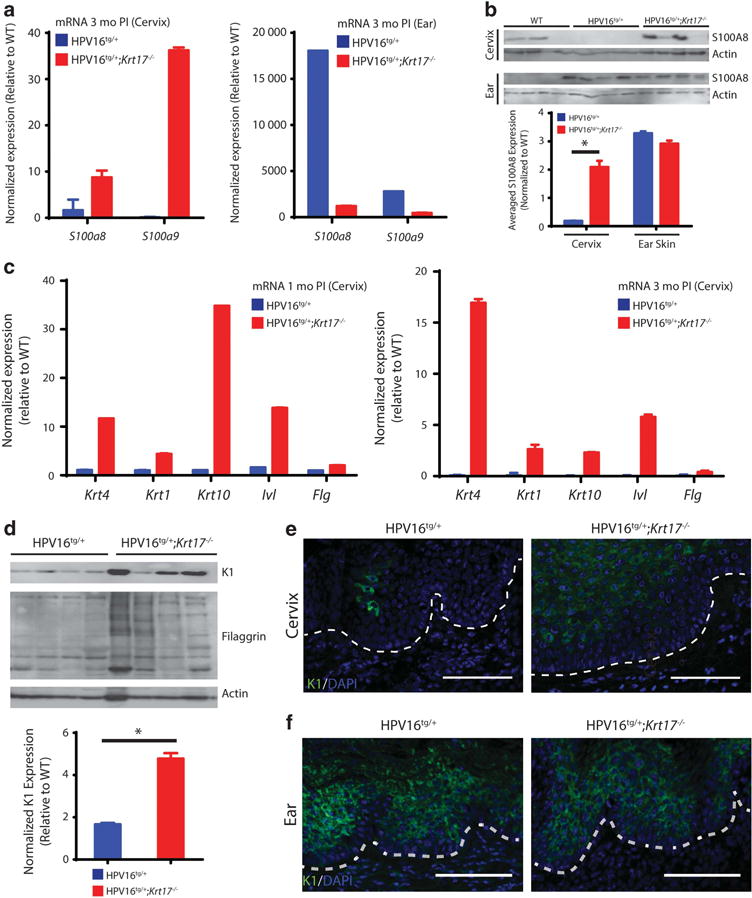
Differentiation of cervical epithelia is induced in the absence of K17. (a) Normalized expression for S100a8 and S100a9 gene transcripts in cervical (left) and ear (right) tissue of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 3 months post implantation. (b) Immunoblots for S100a8 in cervical (top) and ear (bottom) tissues of WT, HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 3 months post implantation. Each lane represents a distinct biological replicate. Actin serves as loading control. Densitometric quantification below. (c) Normalized expression for differentiation markers in cervical tissue of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 1 month (left) and 3 months (right) post implantation. (d) Immunoblots for K1 and filaggrin in cervical tissue of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 1 month post implantation. Each lane represents a distinct biological replicate. Actin serves as loading control. Densitometric quantification below. (e, f) Immunostaining for K1 in (e) transformation zone epithelium and (f) ear skin epidermis of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 1 month post implantation. Nuclei are stained with DAPI (blue). Dotted line separates epithelium (above line) from underlying stroma (below line). Bar = 100 μm. All error bars are s.e.m. *P<0.05. NS; no significance.
Keratin 17 is required for restricting p63 expression to a basal cell compartment within cervical epithelium
With the goal of identifying a tissue-specific regulatory function for K17, we next examined whether K17 may influence the status of 14-3-3σ (also called Stratifin) or p63, as these are known to be key regulators of epithelial proliferation, differentiation and tumorigenesis often in a tissue- or context-dependent manner.31,32 Further, K17 interacts with 14-3-3σ in growth-stimulated skin keratinocytes.27,33 Similar to our findings for multiple markers of differentiation, the transcript levels of 14-3-3σ in cervical tissue is robustly increased in HPV16tg/+;Krt17−/− mice at 1 month post implantation relative to HPV16tg/+ controls (Figure 5a). However, transcript levels of 14-3-3σ are similarly elevated in ear skin of HPV16tg/+;Krt17−/− mice relative to HPV16tg/+ controls (Figure 5b). Along with K17, p63 is a marker of reserve cells in cervical epithelia.25 Similar to that observed for K17 immunostaining (Supplementary Figure S1C), p63 exhibits a patchy immunostaining pattern throughout the basal layer of the transformation zone in cervical epithelia of either WT or HPV16tg/+ mice at 1 month post implantation, (Figure 5c). In cervical epithelia of HPV16tg/+;Krt17−/− mice, however, p63 expression is markedly expanded into and throughout the suprabasal epithelial layers of the transformation zone, as visualized at 1 month post implantation and more strikingly at 3 months post implantation (Figure 5c).
Figure 5.
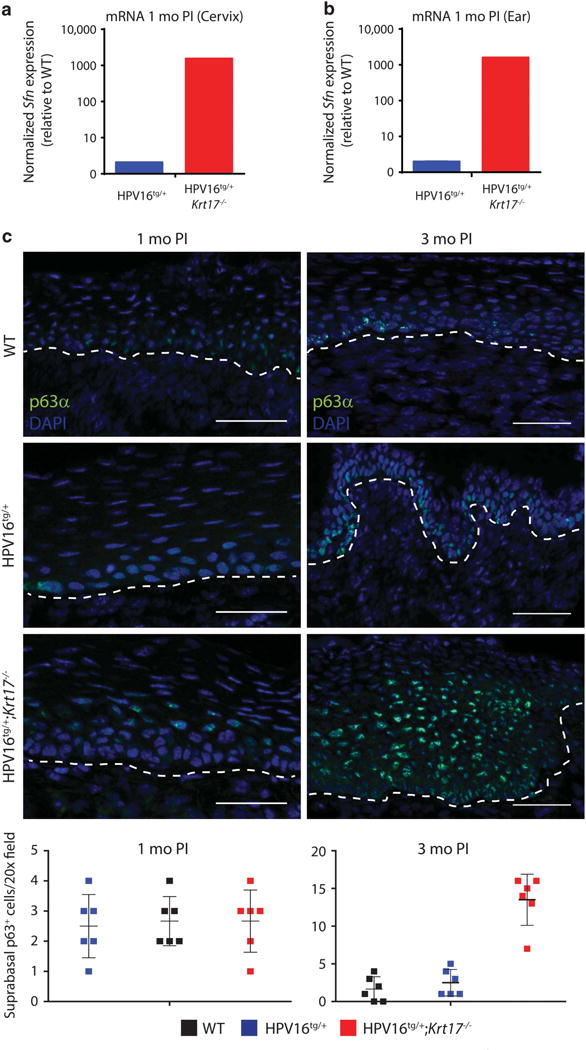
p63 expression is expanded throughout suprabasal cervical epithelium in the absence of K17. (a, b) Normalized expression for Sfn gene transcript in (a) cervical and (b) ear tissue of HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 1 month post implantation. (c) Immunostaining for p63α in transformation zone epithelium of WT, HPV16tg/+ and HPV16tg/+;Krt17−/− mice at 1 month (left) and 3 months (right) post implantation. Nuclei are stained with DAPI (blue). Dotted line separates epithelium (above line) from underlying stroma (below line). Bar = 100 μm. Graphs below depict the number of suprabasal cells immunopositive for p63 per field across genotypes. All error bars are s.e.m. *P<0.05.
DISCUSSION
With this study herein, we have determined that K17 has a functional role in promoting cervical tumorigenesis in vivo using the well-established HPV16tg mouse model coupled to estrogen implantation. Several metrics of tumor growth including hyper-proliferation (hyperplasia), increased vascularization and dysplasia that steadily develop in an estrogen-dependent fashion in female HPV16tg/+ mice are significantly attenuated in mice genetically ablated for Krt17. These findings extend the notion that keratins have a substantial role as effectors of disease progression and further highlight the molecular contributions of K17 as a tumor promoter.
Cancer-associated immunology is complex and poorly understood, reflected in part by the diversity of the immune response and by spatial and temporal heterogeneity of a developing tumor. Accordingly, the design and implementation of therapeutic cancer vaccines has been challenging. FDA-approved therapeutic vaccines targeting inflammatory molecules are currently being utilized to treat various cancers (excluding cervical tumors so far), with many more currently in phase III clinical trials.34 Consistent with complexity, our study shows that intra-tumor expression of several inflammatory and immune cytokines during cervical tumorigenesis significantly depends on a cytoskeletal protein, namely K17, albeit in a manner that fluctuates along time. At 1 month post implantation, cervical tissue from HPV16tg/+; Krt17−/− mice exhibit what can be approximated to be a Th1high/Th17low/Th2low profile compared with HPV16tg/+ mice. By 3 months post implantation, however, this profile has switched to a Th1low/Th17high/Th2high profile. K17-dependent cytokines such as IFNγ, TNFα and IL10, for instance, can activate distinct populations and subtypes of macrophages (for example, M1 and M2) in addition to T cells, and these effectors can each have profound influence on the proliferation-differentiation balance of a developing tumor.35 Regardless of whether T-cell or macrophage polarization is key for the progression of cervical tumors, our findings suggest that the timing of any immunotherapy approach will likely be crucial in the treatment’s success. The utility of K17 expression as a diagnostic/prognostic biomarker combined with immunotherapy approaches targeting specific K17-dependent cytokines may prove to be a powerful therapeutic tool for the treatment of existing cervical cancers.
The primary target cell for HPV infection is thought to be the reserve cell, a stem-like cell that resides in the basal cervical epithelium and gives rise to the columnar endocervical and squamocolumnar ectocervical epithelia in humans.26 This population of cells has also been described in the cervico-uterine transformation zone and ectocervical epithelium of mice.36 Interestingly, both K17 and p63 have been identified as markers of the reserve cell in normal cervical epithelia,25,37 and have also been identified as a marker of cancer stem-like cells derived from cervical carcinomas.38 Although the loss of K17 has no obvious bearing on p63 expression and differentiation in skin epidermis, we observed an expansion of p63 throughout the suprabasal cervical epithelium correlating with marked upregulation of differentiation markers only in the absence of Krt17. Moreover, we identified Lgr5, a known marker of stemness39 and prognostic indicator for cervical carcinoma,40 to be reduced in cervical tissue in the absence of K17 (see Supplementary Figure S2A). Although the full properties and roles of K17 in reserve and/or progenitor basal cells of the cervical epithelium have yet to be deciphered, our finding of K17 influencing p63 and Lgr5 expression, as well as the balance between proliferation and differentiation, suggests that K17 may have a key role in the regulation of cell fate decisions within the cervical epithelium. Whether this is specific to terminal differentiation, reprogramming, cellular fitness and/or transdifferentiation requires future studies.
Our attempt to identify a molecular mechanism for K17 in the regulation of cervical tumorigenesis was initially guided by a recent study using the same HPV16tg mouse model, which demonstrated K17 to be required for the timely onset and early progression of tumorigenesis in ear skin.22 Although the loss of K17 attenuates tumorigenesis in both ear and cervical epithelia, the current study conveys that the underlying mechanisms are most likely distinct in these two tissue types. The ribonucleoprotein hnRNP K, in conjunction with K17, regulates many Th1 effector cytokines in skin tumor keratinocytes.24 In cervical tissue, however, these cytokine transcripts remain elevated even though hnRNP K protein expression is substantially decreased. We infer that the massive upregulation of Ifng, a major stimulator of the Th1-type immune response, is able to override the impact of a hnRNP K deficiency in cervical tissue (but not in skin) lacking K17. In addition, we recently reported Aire (autoimmune regulator) to be inducibly expressed in a K17-dependent manner in skin keratinocytes.22 That Aire is absent or expressed at below-detectable levels in HPV16tg/+ cervical epithelium, coupled to the observed increase in intraepithelial immune cells only in cervical epithelium, raises the possibility that Aire exerts a tissue-specific function as a regulator of local immune tolerance during tumor growth.
We also identified other gene transcripts to be expressed in a K17-dependent manner in cervical tissue. These include genes associated with adhesion (Epcam1 was down, whereas Lamb1 was up), Notch signaling (Hes1 was reduced), Gli signaling (Hip1 was elevated but Ptch1 was decreased), Wnt signaling (Lgr5 was reduced) and mTOR signaling (Mtor, Rptor and Rictor were down; however, a downstream indicator of mTOR activation (S6 phosphorylation) was elevated; see Supplementary Figure S2B). Whether there is a functional role for these molecules in estrogen-driven HPV16-induced cervical tumorigenesis remains to be determined.
At last, the ability of K17 to regulate gene expression in skin tumor keratinocytes involves, in part, a nuclear-localized form of K17. Nuclear-localized K17 also occurs in biopsies from cases of basal cell carcinoma in human skin22 and human cervical carcinoma, and in human cervical carcinoma-derived cell lines in culture.29 K17 has been reported to be critical for the nuclear-to-cytoplasmic translocation of the cell cycle inhibitor p27KIP1 in human cervical epithelial cells in culture,29 an influence that may have a direct bearing on the proliferation-differentiation balance in the cervix. Whether a nuclear-localized form of K17 and a primary impact on cell cycle regulation have a role in the setting of HPV16-induced and estrogen-promoted tumorigenesis in the mouse cervical epithelium represent open issues worth examining in the future.
Altogether, our data support a model whereby K17 promotes tumorigenesis in multiple tissue types of the same mouse, but with divergent, tissue-dependent mechanisms. A likely contributing factor to our tissue-dependent cellular and molecular findings is that K17 is naturally expressed in a patchy pattern in the basal layer of cervical epithelium in WT mice, whereas it is absent in normal interfollicular epidermis. Beyond this, it appears very likely that K17 is being regulated and specified differently, via interaction with distinct and powerful regulators, to control inflammation, proliferation and differentiation in the setting of HPV16-induced tumorigenesis.
MATERIALS AND METHODS
Transgenic mice and estrogen treatment
All procedures involving mice were performed using protocols reviewed and approved by the Johns Hopkins University Animal Care and Use Committee. Generation of K14-HPV16 (HPV16tg) mice (FVB/N strain; obtained from the National Cancer Institute Mouse Repository, strain 01XT3) and HPV16tg/+;Krt17−/− mice has been described,5,22 as were relevant genotyping protocols.22 All mice were fed rodent chow and water ad libitum upon weaning. One-month-old virgin female mice were anesthetized using short exposure to 20% isoflurane (v/v in propylene glycol) and implanted subcutaneously in the dorsal region of the neck via surgical trocar with 90-day release, 0.05 mg 17β-estradiol pellets (Innovative Research of America, Sarasota, FL, USA). Animals were euthanized and tissue samples were harvested for histology, RNA and protein at 1-, 3-and 6 months post implantation. Male mice were excluded from this study. Mice deemed sickly by veterinary staff were not utilized. The sample size of mice required for this study was empirically determined from previous experience.22,23 Investigator blinding and randomization were not conducted.
Tissue collection, histology and RNA in situ hybridization
Tissues were harvested from three to five mice for each genotype (WT, HPV16tg/+; HPV16tg/+;Krt17−/−) at each time point (1-, 3- and 6 months) post implantation. For each mouse, both ears and the cervix were removed, quickly embedded in Tissue-Tec OCT compound (Sakura Finetek, Torrance, CA, USA) and stored at − 20 °C prior to sectioning. In total, 5–7 μM-thick tissue sections were subjected to histological stains as described.22 Tissues used for RNA in situ hybridization were immediately fixed in freshly prepared 4% paraformaldehyde prior to embedding and processed for Aire in situ hybridization as described.22
RNA and protein isolation
Tissues of three to five mice for each genotype (WT, HPV16tg/+; HPV16tg/+; Krt17−/−) at each time point (1-, 3- and 6 months) post implantation were prepared for RNA and protein isolation using Trizol (Invitrogen, Carlsbad, CA, USA). Tissue was homogenized and directly underwent phase separation. Protein and RNA were isolated for each sample as directed by the manufacturer’s protocol. All protein lysates were prepared for immunoblotting in urea sample buffer as described.22 All RNA samples were treated with RNAse-free DNAseI (Qiagen, Valencia, CA, USA) and cleaned on RNEasy mini columns (Qiagen). Concentration and purity for all RNA samples were determined by spectrophotometry. quantitative reverse transcriptase–PCR was performed and analyzed as described.22 A list of all target-specific oligonucleotide primers used for quantitative reverse transcriptase–PCR is provided in Supplementary Table S1.
Antibodies
Primary antibodies used included rabbit polyclonal antisera directed against K17,41 K14 (Covance #AF64, Princeton, NJ, USA), K1 (Biolegend #AF109, San Diego, CA, USA), phospho-histone H3 (Cell Signaling #9701, Danvers, MA, USA), CDKN2A (Thermo Scientific #MA1-16664, Waltham, MA, USA) and filaggrin (Biolegend #PRB-417P); goat polyclonal antisera against S100A8 (Santa Cruz #SC-8113, Dallas, TX, USA); rat monoclonal antibodies against CD11b (eBiosciences #11-0112-81, San Diego, CA, USA), F4/80 (AbD Serotec #MCA497, Raleigh, NC, USA) and CD4 (BD Biosciences #550280, San Jose, CA, USA); and mouse monoclonal antisera against HPV E7 (Invitrogen #28-0006), PECAM-1 (Chemicon #CBL1337, Billerica, MA, USA), p63α (Genetex #GTX23239, Irvine, CA, USA) and β-actin (Sigma #A5441, St Louis, MO, USA). Secondary antibodies used included Alexa 488 (Invitrogen) for indirect immunofluorescence and goat anti-rabbit HRP (Sigma) and goat anti-mouse HRP (Sigma) for indirect chemiluminescence.
Western blotting, indirect immunofluorescence and microscopy
For immunoblotting, all samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), and subjected to immunoblot analysis using relevant antibodies. Blots were developed using ECL Select developing solution (GE Healthcare, Pittsburgh, PA, USA). For immunofluorescence analysis, all frozen tissue sections were blocked for 1-h at room temperature in 5% normal goat serum (in 1 × PBS) prior to application of primary antibody. Species-relevant immunoglobulin G controls (Santa Cruz) were used as negative controls. All tissue sections were imaged using a Zeiss fluorescence microscope with Apotome attachment. Images for the same marker were acquired at equal exposure, pixel range and gamma values. All images of a similar type were equally brightened, contrasted and cropped using ImageJ software for optimal presentation.
Image quantification and description of statistical methods
Signal quantification for western blots and K17, PECAM-1, K1 and S100A8 immunofluorescence images was conducted using ImageJ software. A manual cell count was performed for the quantification of phospho-histone H3+, CD4+, F4/80+, CD11b+, p63+ and toluidine blue+ cells. Counts were conducted from 10–15 distinct images (20 × magnification) from three to five biological replicates per genotype. The number of cells stained positive relative to negative controls was manually counted and averaged first per mouse, and then per genotype. All densitometry quantifications were calculated by dividing the band intensity for the target of interest by the loading control, and then averaging all biological replicates across a similar trait (for example, time point, genotype). The papillation index was determined from 4–10 hematoxylin and eosin images (20 ×) of two mice with each genotype (WT, HPV16tg/+ and HPV16tg/+;Krt17−/−) at each time point (1, 3 and 6 months post implantation). Length measurements for the epithelial-stromal interface and the epithelial-lumen interface were determined using the freehand line selection and measure features in ImageJ software. All graphs were generated in either Microsoft Excel or Prism (GraphPad) software. All error bars represent the standard error of the mean (s.e.m.). All P-values were generated by simple t-test with two-sided distribution and equal variance (Microsoft Excel).
Supplementary Material
Acknowledgments
We thank members of the Coulombe laboratory for support, Dr Michelle Kerns for critical reading of the manuscript and Drs Janice Evans, Hyo Lee and TC Wu for advice and assistance. These studies were supported by NIH research grants CA160255 and AR44232 (to PAC) and NIH training grant CA009110 (to RH) from the National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Hopman AH, Smedts F, Dignef W, Ummelen M, Sonke G, Mravunac M, et al. Transition of high-grade cervical intraepithelial neoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. J Pathol. 2004;202:23–33. doi: 10.1002/path.1490. [DOI] [PubMed] [Google Scholar]
- 3.Hopman AH, Theelen W, Hommelberg PP, Kamps MA, Herrington CS, Morrison LE, et al. Genomic integration of oncogenic HPV and gain of the human telomerase gene TERC at 3q26 are strongly associated events in the progression of uterine cervical dysplasia to invasive cancer. J Pathol. 2006;210:412–419. doi: 10.1002/path.2070. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell MF, Hittelman WN, Hong WK, Lotan R, Schottenfeld D. The natural history of cervical intraepithelial neoplasia: an argument for intermediate endpoint biomarkers. Cancer Epidemiol Biomarkers Prev. 1994;3:619–626. [PubMed] [Google Scholar]
- 5.Arbeit JM, Howley PM, Hanahan D. Chronic estrogen-induced cervical and vaginal squamous carcinogenesis in human papillomavirus type 16 transgenic mice. Proc Natl Acad Sci USA. 1996;93:2930–2935. doi: 10.1073/pnas.93.7.2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Helt AM, Funk JO, Galloway DA. Inactivation of both the retinoblastoma tumor suppressor and p21 by the human papillomavirus type 16 E7 oncoprotein is necessary to inhibit cell cycle arrest in human epithelial cells. J Virol. 2002;76:10559–10568. doi: 10.1128/JVI.76.20.10559-10568.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mighty KK, Laimins LA. The role of human papillomaviruses in oncogenesis. Recent Results Cancer Res. 2014;193:135–148. doi: 10.1007/978-3-642-38965-8_8. [DOI] [PubMed] [Google Scholar]
- 8.Thomas M, Pim D, Banks L. The role of the E6–p53 interaction in the molecular pathogenesis of HPV. Oncogene. 1999;18:7690–7700. doi: 10.1038/sj.onc.1202953. [DOI] [PubMed] [Google Scholar]
- 9.Plummer M, Schiffman M, Castle PE, Maucort-Boulch D, Wheeler CM, ALTS Group A 2-year prospective study of human papillomavirus persistence among women with a cytological diagnosis of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesion. J Infect Dis. 2007;195:1582–1589. doi: 10.1086/516784. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez R, Fadare O. Longitudinal cytological follow-up of patients with a papanicolaou test interpretation of “atypical squamous cells of undetermined significance” that was followed by a negative reflex test for high-risk human papillomavirus types. Int J Gynecol Pathol. 2008;27:108–112. doi: 10.1097/pgp.0b013e31804216b8. [DOI] [PubMed] [Google Scholar]
- 11.Chung SH, Franceschi S, Lambert PF. Estrogen and ERalpha: culprits in cervical cancer? Trends Endocrinol Metab. 2010;21:504–511. doi: 10.1016/j.tem.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee BN, Follen M, Shen DY, Malpica A, Adler-Storthz K, Shearer WT, et al. Depressed type 1 cytokine synthesis by superantigen-activated CD4+ T cells of women with human papillomavirus-related high-grade squamous intraepithelial lesions. Clin Diagn Lab Immunol. 2004;11:239–244. doi: 10.1128/CDLI.11.2.239-244.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smedts F, Ramaekers F, Troyanovsky S, Pruszczynski M, Robben H, Lane B, et al. Basal-cell keratins in cervical reserve cells and a comparison to their expression in cervical intraepithelial neoplasia. Am J Pathol. 1992;140:601–612. [PMC free article] [PubMed] [Google Scholar]
- 14.Martens J, Baars J, Smedts F, Holterheus M, Kok MJ, Vooijs P, et al. Can keratin 8 and 17 immunohistochemistry be of diagnostic value in cervical cytology? A feasibility study. Cancer. 1999;87:87–92. doi: 10.1002/(sici)1097-0142(19990425)87:2<87::aid-cncr8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 15.Escobar-Hoyos LF, Yang J, Zhu J, Cavallo JA, Zhai H, Burke S, et al. Keratin 17 in premalignant and malignant squamous lesions of the cervix: proteomic discovery and immunohistochemical validation as a diagnostic and prognostic biomarker. Mod Pathol. 2014;27:621–630. doi: 10.1038/modpathol.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van de Rijn M, Perou CM, Tibshirani R, Haas P, Kallioniemi O, Kononen J, et al. Expression of cytokeratins 17 and 5 identifies a group of breast carcinomas with poor clinical outcome. Am J Pathol. 2002;161:1991–1996. doi: 10.1016/S0002-9440(10)64476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang YF, Lang HY, Yuan J, Wang J, Wang R, Zhang XH, et al. Overexpression of keratin 17 is associated with poor prognosis in epithelial ovarian cancer. Tumour Biol. 2013;34:1685–1689. doi: 10.1007/s13277-013-0703-5. [DOI] [PubMed] [Google Scholar]
- 18.Cohen-Kerem R, Madah W, Sabo E, Rahat MA, Greenberg E, Elmalah I. Cytokeratin-17 as a potential marker for squamous cell carcinoma of the larynx. Ann Otol Rhinol Laryngol. 2004;113:821–827. doi: 10.1177/000348940411301008. [DOI] [PubMed] [Google Scholar]
- 19.Ide M, Kato T, Ogata K, Mochiki E, Kuwano H, Oyama T. Keratin 17 expression correlates with tumor progression and poor prognosis in gastric adenocarcinoma. Ann Surg Oncol. 2012;19:3506–3514. doi: 10.1245/s10434-012-2437-9. [DOI] [PubMed] [Google Scholar]
- 20.Toyoshima T, Vairaktaris E, Nkenke E, Schlegel KA, Neukam FW, Ries J. Cytokeratin 17 mRNA expression has potential for diagnostic marker of oral squamous cell carcinoma. J Cancer Res Clin Oncol. 2008;134:515–521. doi: 10.1007/s00432-007-0308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Y, Cui T, Yang L, Mireskandari M, Knoesel T, Zhang Q, et al. The diagnostic value of cytokeratin 5/6, 14, 17, and 18 expression in human non-small cell lung cancer. Oncology. 2011;80:333–340. doi: 10.1159/000329098. [DOI] [PubMed] [Google Scholar]
- 22.Hobbs RP, DePianto DJ, Jacob JT, Han MC, Chung BM, Batazzi AS, et al. Keratin-dependent regulation of Aire and gene expression in skin tumor keratinocytes. Nat Genet. 2015;47:933–938. doi: 10.1038/ng.3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Depianto D, Kerns ML, Dlugosz AA, Coulombe PA. Keratin 17 promotes epithelial proliferation and tumor growth by polarizing the immune response in skin. Nat Genet. 2010;42:910–914. doi: 10.1038/ng.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chung BM, Arutyunov A, Ilagan E, Yao N, Wills-Karp M, Coulombe PA. Regulation of C-X-C chemokine gene expression by keratin 17 and hnRNP K in skin tumor keratinocytes. J Cell Biol. 2015;208:613–627. doi: 10.1083/jcb.201408026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martens JE, Arends J, Van der Linden PJ, De Boer BA, Helmerhorst TJ. Cytokeratin 17 and p63 are markers of the HPV target cell, the cervical stem cell. Anticancer Res. 2004;24:771–775. [PubMed] [Google Scholar]
- 26.Herfs M, Vargas SO, Yamamoto Y, Howitt BE, Nucci MR, Hornick JL, et al. A novel blueprint for ‘top down’ differentiation defines the cervical squamocolumnar junction during development, reproductive life, and neoplasia. J Pathol. 2013;229:460–468. doi: 10.1002/path.4110. [DOI] [PubMed] [Google Scholar]
- 27.Sankar S, Tanner JM, Bell R, Chaturvedi A, Randall RL, Beckerle MC, et al. A novel role for keratin 17 in coordinating oncogenic transformation and cellular adhesion in Ewing sarcoma. Mol Cell Biol. 2013;33:4448–4460. doi: 10.1128/MCB.00241-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van de Putte G, Holm R, Lie AK, Trope CG, Kristensen GB. Expression of p27, p21, and p16 protein in early squamous cervical cancer and its relation to prognosis. Gynecol Oncol. 2003;89:140–147. doi: 10.1016/s0090-8258(03)00010-6. [DOI] [PubMed] [Google Scholar]
- 29.Escobar-Hoyos LF, Shah R, Roa-Pena L, Vanner EA, Najafian N, Banach A, et al. Keratin-17 Promotes p27KIP1 Nuclear Export and Degradation and Offers Potential Prognostic Utility. Cancer Res. 2015;75:3650–3662. doi: 10.1158/0008-5472.CAN-15-0293. [DOI] [PubMed] [Google Scholar]
- 30.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, et al. Functions of S100 proteins. Curr Mol Med. 2013;13:24–57. [PMC free article] [PubMed] [Google Scholar]
- 31.Romano RA, Smalley K, Magraw C, Serna VA, Kurita T, Raghavan S, et al. DeltaNp63 knockout mice reveal its indispensable role as a master regulator of epithelial development and differentiation. Development. 2012;139:772–782. doi: 10.1242/dev.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freeman AK, Morrison DK. 14-3-3 Proteins: diverse functions in cell proliferation and cancer progression. Semin Cell Dev Biol. 2011;22:681–687. doi: 10.1016/j.semcdb.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–365. doi: 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- 34.Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014;11:509–524. doi: 10.1038/nrclinonc.2014.111. [DOI] [PubMed] [Google Scholar]
- 35.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patterson AL, Pru JK. Long-term label retaining cells localize to distinct regions within the female reproductive epithelium. Cell Cycle. 2013;12:2888–2898. doi: 10.4161/cc.25917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martens JE, Smedts FM, Ploeger D, Helmerhorst TJ, Ramaekers FC, Arends JW, et al. Distribution pattern and marker profile show two subpopulations of reserve cells in the endocervical canal. Int J Gynecol Pathol. 2009;28:381–388. doi: 10.1097/PGP.0b013e31819932f8. [DOI] [PubMed] [Google Scholar]
- 38.Feng D, Peng C, Li C, Zhou Y, Li M, Ling B, et al. Identification and characterization of cancer stem-like cells from primary carcinoma of the cervix uteri. Oncol Rep. 2009;22:1129–1134. doi: 10.3892/or_00000545. [DOI] [PubMed] [Google Scholar]
- 39.Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- 40.Sun B, Ye X, Li Y, Zhang W. Lgr5 is a potential prognostic marker in patients with cervical carcinoma. Int J Clin Exp Pathol. 2015;8:1783–1789. [PMC free article] [PubMed] [Google Scholar]
- 41.McGowan KM, Coulombe PA. Onset of keratin 17 expression coincides with the definition of major epithelial lineages during skin development. J Cell Biol. 1998;143:469–486. doi: 10.1083/jcb.143.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.