

Abstract
The advent of deep sequencing technologies has resulted in the deciphering of tremendous amounts of genetic information. These data have led to major discoveries, and many anecdotes now exist of individual patients whose clinical outcomes have benefited from novel, genetically guided therapeutic strategies. However, the majority of genetic events in cancer are currently undrugged, leading to a biological gap between understanding of tumor genetic etiology and translation to improved clinical approaches. Functional screening has made tremendous strides in recent years with the development of new experimental approaches to studying ex vivo and in vivo drug sensitivity. Numerous discoveries and anecdotes also exist for translation of functional screening into novel clinical strategies; however, the current clinical application of functional screening remains largely confined to small clinical trials at specific academic centers. The intersection between genomic and functional approaches represents an ideal modality to accelerate our understanding of drug sensitivities as they relate to specific genetic events and further understand the full mechanisms underlying drug sensitivity patterns.
GENOMICS—STRENGTHS AND LIMITATIONS
Deep sequencing technology has changed the face of nearly every area of biological research and has led to a number of breakthroughs in clinical medicine (Koboldt et al. 2013). As a result, the concept of personalized medicine that is driven by genomic analysis has become pervasive. Although genomically guided precision medicine is a very powerful concept, its implementation is impeded by a lack of biological understanding of the pharmacological ramifications of most tumor-associated genetic lesions. In addition, the combinatorial complexity of tumor genetics further complicates the assignment of drugs to mutational events.
One illustration of this point comes from hematologic malignancies and the elegant work to decipher the genetic etiology of some of these diseases such as acute myeloid leukemia (AML) (The Cancer Genome Atlas Research Network 2013). A small number of the genetic lesions found in AML are targetable, including mutations or fusions involving signaling proteins (e.g., kinases such as FLT3, KIT) and metabolic pathways (e.g., IDH1/2). However, the majority of genetic events observed in AML (as well as most other malignancies) do not fall within this class of genes but in alternative gene families such as those that play a role in regulating epigenetic processes or transcriptional splicing. Some of these mutational events may eventually be directly targetable through drug discovery efforts; however, many of the mutations result in loss of function of the mutated protein. Consequently, drug targeting strategies must focus on biological events downstream from these loss-of-function mutations, and our understanding of most of these downstream events is not sufficiently mature to predict actionable drug targets.
This dilemma is not confined to AML or even to hematologic malignancies, but is a common issue in solid tumors as well, where currently targetable lesions are often confined to kinase point mutations (e.g., BRAF), kinase fusions (e.g., ALK fusions), and some of the very same metabolic pathway lesions (IDH1/2). In addition, many types of solid tumors exhibit significantly higher mutational burden rates, adding to the challenges of assigning the best drugs to patients on the basis of a complex genetic profile. In addition to the knowledge gap preventing easy clinical translation of many genetic events, there are also some technical limitations to current sequencing approaches. The short-read technology now favored by most laboratories results in limited capacity to detect chromosomal breaks and rearrangements as well as copy-number variation, both issues that may ultimately be ameliorated with long-read sequencing technology. In addition, although sequencing is broadly applicable to many tumors and tumor types, there is a subset of cases where sequencing is infeasible because of low quantity/quality of tissue (e.g., formalin-fixed, paraffin-embedded), low tumor burden, and extreme heterogeneity of the tumor cells.
In recent years, many new functional screening strategies have emerged that aim to assess the impact of exogenous interventions such as drug exposure on tumor cell phenotype. These functional screening platforms deliver information regarding the capacity of drugs to elicit apoptotic responses of cancer cells without a priori knowledge of the mechanistic underpinnings of such responses. As such, these tools represent a tremendous opportunity to synergize with genomic data, where functional drug sensitivity can offer alignment of actionable strategies with mutational status and genomic data can help elucidate the etiologies of observed drug sensitivity patterns (Fig. 1).
Figure 1.
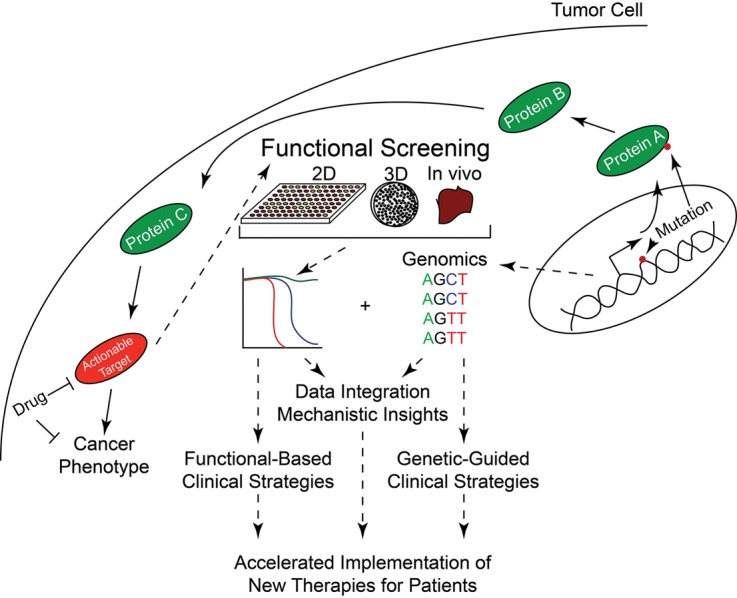
Functional genomics can accelerate elucidation of the genetic etiology of pharmacologic sensitivity patterns. Mutant genes in tumors often produce proteins that are not druggable either because of dearth of known agents that interact with the protein or the loss-of-function nature of the genetic event. However, the mutated protein may set off a cascade of intracellular signals that eventually culminates in a druggable protein being involved in the oncogenic process. Genomics can tell us the location of the mutant gene, and functional screens of a variety of formats can identify potentially effective drugs. Both functional and genomic tools can be used in isolation to drive clinical trials and decision-making; however, the intersection of genomic and functional data represents the most promising strategy for accelerated understanding of the mechanistic etiology of drug sensitivity patterns as well as the rapid deployment of gene-targeted therapies for cancer.
FUNCTIONAL APPROACHES—STRENGTHS AND LIMITATIONS
Recent advances in functional screening technology have yielded many variations in assay platforms for understanding responses of tumor cells to exogenous perturbations. Functional screening efforts in hematologic malignancies have often involved the culture of patient cells in conventional two-dimensional tissue culture platforms, sometimes with conventional culture conditions (Tyner et al. 2009, 2013; Pemovska et al. 2013) and other times using additives or feeder cell coculture that promote certain phenotypic aspects of the cells, such as preservation of primitive cell differentiation state (Pabst et al. 2014) and cell proliferation (Klco et al. 2013). Although this review focuses mainly on the application of libraries of small molecules to these functional screening platforms, as detailed below this strategy has also been used extensively with RNA interference (RNAi) libraries and more recently with clustered regularly interspersed short palindromic repeat (CRISPR)–Cas technology for mechanistically focused projects.
For solid tumors, two-dimensional culture that retains fidelity to characteristics of the original tumor has proven more challenging and has required the development of a technique called conditional reprogramming, which involves specific media conditions and protocols using feeder cell layers (Liu et al. 2012; Suprynowicz et al. 2012). Analysis of rare circulating tumor cells from solid tumor patients has been another successful avenue for functional screening of solid tumors in two dimensions (Brouzes et al. 2009; Yu et al. 2014). In other cases, solid tumor cells have been grown in three-dimensional organoids (Sato et al. 2009) or organotypic cultures comprised of multiple cell types rederived into a three-dimensional structure (Ridky et al. 2010) or live three-dimensional tissue slices (Vaira et al. 2010), which retain important geographical positioning properties and cell interactions of the original tumor. In many instances, these two- and three-dimensional approaches are used to assess the ability of drugs to halt growth or induce death of tumor cells; however, other approaches use readouts with shorter turnaround time such as target/pathway engagement (Schayowitz et al. 2012; Hilhorst et al. 2013; Tahiri et al. 2013), activation (Irish et al. 2004), or induction of apoptotic responses (Ni Chonghaile et al. 2011) as useful surrogates for eventual phenotypic impacts on tumor cells.
In vivo approaches are also commonly explored. Many investigators make use of xenograft animal models for propagation of tumor cells and measurement of the impact of drugs on tumor growth in vivo. In addition, recent approaches have been developed to implant drug libraries into tumors in situ with subsequent resection and imaging to determine each drug's impact on the local environment into which the drug was microinjected (Jonas et al. 2015; Klinghoffer et al. 2015). A more comprehensive survey and description of the functional screening landscape can be found in Friedman et al. (2015b).
Finally, the recent advances in immunotherapies for oncology have opened the door to functional testing of these agents. Although this is a very new area, investigators are beginning to examine the capacity of immune checkpoint inhibitors to elicit T-cell activation responses when cultured ex vivo in a milieu of tumor cells (Lamble et al. 2016). It is also conceivable that the ex vivo and in vivo platforms described above could be useful for understanding activity profiles of antibody–drug conjugates or chimeric antigen receptor T cells. Indeed, xenograft models have already been routinely used for validation of in vivo activity of these classes of agents.
Although functional screening is clearly a rapidly expanding field offering many different modalities and opportunities to evaluate candidate drug responses ex vivo and in vivo, there are also some current limitations to these approaches. The start-up costs and expertise to obtain specialized instrumentation and personnel for execution of these screens can be substantial, which may represent an activation barrier for some institutions. In addition, the tissue requirements to perform these screens are often larger than the requirements to perform genomic studies, making the subset of cases for which the technology is not feasible larger for some functional approaches relative to genomics. Finally, although many of the ex vivo approaches, particularly for the hematologic malignancies, can actually deliver data faster than most current genomic facilities, there can be a significant delay in performing the experiments for approaches that require expansion of tumor cells in vivo or ex vivo.
Hence, like the genomic approaches, these functional screening strategies have strengths as well as limitations and the relative strengths and weaknesses of functional and genomic approaches are both ideally suited to complementing the other. As noted above, deep sequencing has revolutionized our ability to efficiently decipher somatic genetic lesions associated with individual patient tumors, yet most are not currently actionable. Functional strategies have the benefit of offering results that can be immediately acted upon in a clinical setting (with proper assay setup and oversight, as noted below); however, the widespread application of findings to patients can be limited without substantial mechanistic exploration. By integrating functional and genomic data sets, it is possible to generate understanding of complex genotype-to-phenotype correlations and, thereby, accelerate discovery of the biology of tumorigenesis as well as the clinical implementation of novel therapies.
CLINICAL IMPLEMENTATION OF FUNCTIONAL OR GENOMIC STRATEGIES
Both functional and genomic approaches to personalized medicine face hurdles of implementation, although often for different reasons. Although notable clinical trials incorporate the use of genetic markers for stratification of patients onto specific therapies, these trials are impeded by the paucity of solid hypotheses that enable assignment of specific drugs to specific genetic events. Conversely, functional approaches have the capacity to make predictions of effective therapies for large proportions of patients; however, the validation of these hypotheses in a clinical setting has been impeded by less than widespread acceptance of functional assays for clinical decision-making as well as the other limitations around start-up cost, tissue requirements, and timing noted above. Indeed, the integration of both genomic and functional approaches in a clinical setting holds great promise where patients with immediately targetable genetic lesions could be treated on the basis of genetic findings, whereas functional assays could be used to generate candidate drugs for patients not exhibiting targetable genetic lesions. In general, there are at least three criteria that are important for implementation of these tools in a clinical setting, all of which are relevant to both functional and genomic approaches.
-
Precision. It is vital that any assay or device that is used for clinical decision-making be performed in a stable manner that offers reproducible results. At a minimum, this reproducibility should be validated by internal controls demonstrating consistency of results over time and, ideally, the results should be cross validated with external laboratories performing the same or similar technique. External validation is currently much easier for genomic strategies where nucleic acids can be stably stored for long periods of time and sent to any of a multitude of other laboratories capable of running the same test(s). For functional assays, this manner of external validation has been more difficult because many of these assays require freshly harvested, viable cells and the number of laboratories offering functional tests with regulatory approval has been small. However, in recent years more and more centers and companies have emerged with functional screening capabilities, and even though these tests are not performed in identical manner, it is still becoming possible to orthogonally validate results between platforms at multiple centers.
Demonstrating the precision and reproducibility of functional testing results is especially important in light of recent analyses of the Cancer Cell Line Encyclopedia (Barretina et al. 2012) and the Cancer Genome Project (Garnett et al. 2012) suggesting inconsistent drug sensitivity results between these two collections (Haibe-Kains et al. 2013). Although follow-up analyses showed a higher level of concordance when controlling for genomic variation (The Cancer Cell Line Encyclopedia and Genomics of Drug Sensitivity in Cancer Investigators 2015), the thresholds for precision by which functional screening studies should strive and be evaluated is still a point of discussion (Safikhani et al. 2016). Clearly, for translating these assays into a clinical setting, the highest standards for rigor must be demonstrated.
Oversight. As both functional and genomic tools are explored for clinical use in prospective trials, regulatory approval is important. Investigators using such tools for cutting edge trials must consider steps such as certification of assays with the College of American Pathologists and/or the Food and Drug Administration. Putting in place controls as noted above to ensure precision and reproducibility of the assay will be a component of assay oversight as will other common steps such as clear delineation of specimen/protocol tracking and chain of custody. In addition, as new informatics tools are developed that can integrate functional and genomic data (as well as other data sources) to generate informed treatment predictions, these computational tools will also be subject to regulatory oversight at the point of clinical implementation.
Accuracy. Although it is clearly important that genomic and functional techniques are performed in a reproducible manner with proper regulatory oversight, it is also clearly desirable that the assay results are reproducibly accurate in predicting clinical responses. This is the most challenging criterion to satisfy, especially as it is somewhat circular in the setting of testing these tools in clinical trials—the point of such trials is to demonstrate assay accuracy. As a result, it will be important that oversight bodies bear in mind that much work remains to be done in demonstrating accuracy and clinical relevance of both functional and genomic tools in reviewing proposed trials that aim to demonstrate assay/biomarker accuracy.
MECHANISTIC INSIGHTS AIDED BY FUNCTIONAL SCREENING
Even as genomic and functional strategies are being advanced separately for clinical purposes, the research world is already integrating these tools to accelerate discovery. Although there is far too much of this work ongoing for a comprehensive review here, the following represent some examples of functional genomic data integration that has led to novel insights into genotype/phenotype correlations in a few distinct categories.
Linkage of specific genotype to gene target. Some of the earliest examples of successfully combining functional with genetic events occurred with the use of RNAi screens (Paddison et al. 2004; Silva et al. 2004), and this work continues with more modern RNAi and/or CRISPR–Cas technologies. Some examples of important findings in this category include the identification of the transcriptional repressor, REST, as a tumor suppressor in colorectal cancer associated with loss-of-function chromosomal deletions or frameshift mutations that lead to increased signaling of progrowth signaling pathways (Westbrook et al. 2005); the ribosomal subunit protein, RPS14, as a critical region of the 5q-deletions observed in myelodysplasia (Ebert et al. 2008); the cell surface receptor, ROR1, as a functional target in acute lymphoblastic leukemia cases harboring a 1;19 chromosomal translocation (Bicocca et al. 2012); and identification of dependence on several kinase targets such as WEE1 in head and neck squamous cell carcinoma with TP53 mutation (Moser et al. 2014). In some instances, these approaches have used primary patient cells to conduct the functional genomic techniques, whereas others have used cell line models (Schlabach et al. 2008).
Linkage of specific genotype to drug activity/pathway dependence. Similar to the use of RNAi to elucidate functional gene targets that are associated with specific genetic disease subsets, small-molecule assays have similarly been used to directly identify candidate drugs that may be useful in association with specific genetic markers. Some examples include the use of JAK inhibitors for Philadelphia-negative neutrophilic leukemia cases harboring mutation of CSF3R (Maxson et al. 2013); the identification of axitinib as a selective inhibitor of BCR-ABL harboring a T315I mutation (Pemovska et al. 2015); and discovery of JAK pathway activation and sensitivity to JAK inhibitors in AML cases with biallelic CEBPA mutations (Lavallee et al. 2016).
Enrichment of drug efficacy in defined diagnostic subsets. Although the identification of a genetic marker that mechanistically explains drug sensitivity represents a clear and desirable objective of functional genomics, there are also many findings of functional targets (both targetable and nontargetable with current agents) that associate with broader diagnostic categories. Some examples include identification of the molecular scaffold, CARD11, as an activator of NF-κB signaling in activated B-cell-like diffuse large B-cell lymphoma (Ngo et al. 2006); activity of BH3 mimetics such as venetoclax for a variety of hematologic malignancy subsets including chronic lymphocytic leukemia, multiple myeloma, and acute myeloid leukemia (Davids et al. 2013; Pan et al. 2014; Anderson et al. 2016; Touzeau et al. 2016); and distinct activity of BH3 or SMAC mimetics for distinct subsets of acute lymphoblastic leukemia (Fischer et al. 2015; McComb et al. 2016). The predicted clinical activity of some of these agents, such as venetoclax, has been validated in recent clinical trials (Roberts et al. 2016).
Understanding drug resistance, predicting effective drug combinations. Finally, a number of studies have used functional genomic approaches to understand drug resistance, particularly for targeted agents in the setting of specific genetic events. Examples include strategies to tackle resistance to MAP/ERK kinase (MEK)/phosphoinositide 3-kinase (PI3K) inhibitor combinations in lung cancer with RAS mutations (Hata et al. 2014); fibroblast growth factor 2 (FGF2) as an autocrine/paracrine mechanism of resistance in gastrointestinal stromal tumors (Javidi-Sharifi et al. 2015), chronic myeloid leukemia (Traer et al. 2014), and RAS mutant solid tumors (Manchado et al. 2016); resistance to ALK and/or epidermal growth factor receptor (EGFR) inhibitors in lung cancer with fusions or mutations in these genes (Wilson et al. 2015); and modalities to overcome BRAF inhibitor resistance in melanoma (Friedman et al. 2015a). Finally, although many of these approaches have been carried out using in vitro strategies, there have also been related studies in vivo, such as the identification of strategies to overcome BRAF resistance in melanoma in vivo (Jonas et al. 2016).
THE PATH FORWARD
Although tremendous work has already been done and major discoveries have resulted from functional genomics, there is clearly much work still to do. In the research world, it is critical to continue performing these integrated analyses with emerging drugs, combinations of drugs, and technologies such as CRISPR–Cas as well as cutting-edge genomic tools such as single-cell analytics and long-read technology. As technologies evolve and data sets become more complex and larger, it will also be important to refine and develop new computational tools to integrate data sets. Many technologies for this purpose have already been developed involving predictors of functional relevance from sequence/structure data (discussed here in Gonzalez-Perez et al. 2013), development of large databases of gene/protein interaction networks (e.g., STRING) (Szklarczyk et al. 2015), and tools to calculate potentially relevant connections between functionally implicated and mutant genes/proteins (e.g., HitWalker) (Bottomly et al. 2013, 2016). Although this subject is too large to extensively discuss in this commentary, each approach has strengths and limitations; hence, more work is needed to develop best practices for this computational problem.
In the clinical realm, there is a great deal of work that must be done as well. In addition to the steps outlined above for advancing functional and genomic approaches separately, it will also be important to utilize integrated data in a clinical setting. This will require that the computational approaches noted above are designed in a manner that is compatible with patient care. In addition, new computational visualization tools are needed such that these large functional genomic data sets can be rapidly recalled, displayed, and digested for individual patients by clinical personnel working independently and/or working in tandem with researchers in a tumor board-like setting.
In sum, functional genomics has already delivered tremendous insights into the pathogenesis, diagnosis, possible therapeutics, and resistance mechanisms for a wide diversity of cancer subtypes, and new applications of this strategy will continue to impact on cancer research and the deployment of new therapeutic strategies for patients well into the future.
Competing Interest Statement
J.W.T. receives research support from Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Seattle Genetics, Syros, and Takeda and is on the Scientific Advisory Board for Leap Oncology.
References
- Anderson MA, Deng J, Seymour JF, Tam C, Kim SY, Fein J, Yu L, Brown JR, Westerman D, Si EG, et al. 2016. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 127: 3215–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicocca VT, Chang BH, Masouleh BK, Muschen M, Loriaux MM, Druker BJ, Tyner JW. 2012. Crosstalk between ROR1 and the pre-B-cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell 22: 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomly D, Wilmot B, Tyner JW, Eide CA, Loriaux MM, Druker BJ, McWeeney SK. 2013. HitWalker: variant prioritization for personalized functional cancer genomics. Bioinformatics 29: 509–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomly D, McWeeney SK, Wilmot B. 2016. HitWalker2: visual analytics for precision medicine and beyond. Bioinformatics 32: 1253–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouzes E, Medkova M, Savenelli N, Marran D, Twardowski M, Hutchison JB, Rothberg JM, Link DR, Perrimon N, Samuels ML. 2009. Droplet microfluidic technology for single-cell high-throughput screening. Proc Natl Acad Sci 106: 14195–14200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davids MS, Letai A, Brown JR. 2013. Overcoming stroma-mediated treatment resistance in chronic lymphocytic leukemia through BCL-2 inhibition. Leuk Lymphoma 54: 1823–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, et al. 2008. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451: 335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Forster M, Rinaldi A, Risch T, Sungalee S, Warnatz HJ, Bornhauser B, Gombert M, Kratsch C, Stutz AM, et al. 2015. Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet 47: 1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Amzallag A, Pruteanu-Malinici I, Baniya S, Cooper ZA, Piris A, Hargreaves L, Igras V, Frederick DT, Lawrence DP, et al. 2015a. Landscape of targeted anti-cancer drug synergies in melanoma identifies a novel BRAF-VEGFR/PDGFR combination treatment. PLoS One 10: e0140310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Letai A, Fisher DE, Flaherty KT. 2015b. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer 15: 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. 2012. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483: 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Mustonen V, Reva B, Ritchie GR, Creixell P, Karchin R, Vazquez M, Fink JL, Kassahn KS, Pearson JV, et al. 2013. Computational approaches to identify functional genetic variants in cancer genomes. Nat Methods 10: 723–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJ, Quackenbush J. 2013. Inconsistency in large pharmacogenomic studies. Nature 504: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA, Walton Z, Sarosiek KA, Letai A, Heist RS, et al. 2014. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Res 74: 3146–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilhorst R, Houkes L, Mommersteeg M, Musch J, van den Berg A, Ruijtenbeek R. 2013. Peptide microarrays for profiling of serine/threonine kinase activity of recombinant kinases and lysates of cells and tissue samples. Methods Mol Biol 977: 259–271. [DOI] [PubMed] [Google Scholar]
- Irish JM, Hovland R, Krutzik PO, Perez OD, Bruserud O, Gjertsen BT, Nolan GP. 2004. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 118: 217–228. [DOI] [PubMed] [Google Scholar]
- Javidi-Sharifi N, Traer E, Martinez J, Gupta A, Taguchi T, Dunlap J, Heinrich MC, Corless CL, Rubin BP, Druker BJ, et al. 2015. Crosstalk between KIT and FGFR3 promotes gastrointestinal stromal tumor cell growth and drug resistance. Cancer Res 75: 880–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas O, Landry HM, Fuller JE, Santini JT Jr, Baselga J, Tepper RI, Cima MJ, Langer R. 2015. An implantable microdevice to perform high-throughput in vivo drug sensitivity testing in tumors. Sci Transl Med 7: 284ra257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas O, Oudin MJ, Kosciuk T, Whitman M, Gertler FB, Cima MJ, Flaherty KT, Langer R. 2016. Parallel in-vivo assessment of drug phenotypes at various time points during systemic BRAF inhibition reveals tumor adaptation and altered treatment vulnerabilities. Clin Cancer Res 22: 6031–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klco JM, Spencer DH, Lamprecht TL, Sarkaria SM, Wylie T, Magrini V, Hundal J, Walker J, Varghese N, Erdmann-Gilmore P, et al. 2013. Genomic impact of transient low-dose decitabine treatment on primary AML cells. Blood 121: 1633–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinghoffer RA, Bahrami SB, Hatton BA, Frazier JP, Moreno-Gonzalez A, Strand AD, Kerwin WS, Casalini JR, Thirstrup DJ, You S, et al. 2015. A technology platform to assess multiple cancer agents simultaneously within a patient's tumor. Sci Transl Med 7: 284ra258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER. 2013. The next-generation sequencing revolution and its impact on genomics. Cell 155: 27–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamble A, Kosaka Y, Huang F, Sasser AK, Adams H, Tognon C, Laderas T, McWeeney SK, Loriaux MM, Tyner JW, et al. 2016. Mass cytometry as a modality to identify candidates for immune checkpoint inhibitor therapy within acute myeloid leukemia. Blood 128: 2829. [Google Scholar]
- Lavallee V-P, Krosl J, Lemieux S, Boucher G, Gendron P, Pabst C, Boivin I, Marinier A, Guidos CJ, Meloche S, et al. 2016. Chemo-genomic interrogation of CEBPA mutated AML reveals recurrent CSF3R mutations and subgroup sensitivity to JAK inhibitors. Blood 127: 3054–3061. [DOI] [PubMed] [Google Scholar]
- Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, et al. 2012. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol 180: 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchado E, Weissmueller S, Morris JPt, Chen CC, Wullenkord R, Lujambio A, de Stanchina E, Poirier JT, Gainor JF, Corcoran RB, et al. 2016. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 534: 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, Bottomly D, Wilmot B, McWeeney SK, Tognon CE, et al. 2013. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med 368: 1781–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McComb S, Aguade-Gorgorio J, Harder L, Marovca B, Cario G, Eckert C, Schrappe M, Stanulla M, von Stackelberg A, Bourquin JP, et al. 2016. Activation of concurrent apoptosis and necroptosis by SMAC mimetics for the treatment of refractory and relapsed ALL. Sci Transl Med 8: 339ra370. [DOI] [PubMed] [Google Scholar]
- Moser R, Xu C, Kao M, Annis J, Lerma LA, Schaupp CM, Gurley KE, Jang IS, Biktasova A, Yarbrough WG, et al. 2014. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res 20: 4274–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo VN, Davis RE, Lamy L, Yu X, Zhao H, Lenz G, Lam LT, Dave S, Yang L, Powell J, et al. 2006. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 441: 106–110. [DOI] [PubMed] [Google Scholar]
- Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, Deng J, Anderson KC, Richardson P, Tai YT, et al. 2011. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 334: 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabst C, Krosl J, Fares I, Boucher G, Ruel R, Marinier A, Lemieux S, Hebert J, Sauvageau G. 2014. Identification of small molecules that support human leukemia stem cell activity ex vivo. Nat Methods 11: 436–442. [DOI] [PubMed] [Google Scholar]
- Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O'Shaughnessy A, Gnoj L, Scobie K, et al. 2004. A resource for large-scale RNA-interference-based screens in mammals. Nature 428: 427–431. [DOI] [PubMed] [Google Scholar]
- Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, et al. 2014. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 4: 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemovska T, Kontro M, Yadav B, Edgren H, Eldfors S, Szwajda A, Almusa H, Bespalov MM, Ellonen P, Elonen E, et al. 2013. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov 3: 1416–1429. [DOI] [PubMed] [Google Scholar]
- Pemovska T, Johnson E, Kontro M, Repasky GA, Chen J, Wells P, Cronin CN, McTigue M, Kallioniemi O, Porkka K, et al. 2015. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nature 519: 102–105. [DOI] [PubMed] [Google Scholar]
- Ridky TW, Chow JM, Wong DJ, Khavari PA. 2010. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat Med 16: 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, et al. 2016. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 374: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safikhani Z, El-Hachem N, Quevedo R, Smirnov P, Goldenberg A, Juul Birkbak N, Mason C, Hatzis C, Shi L, Aerts HJ, et al. 2016. Assessment of pharmacogenomic agreement. F1000Res 5: 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. 2009. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459: 262–265. [DOI] [PubMed] [Google Scholar]
- Schayowitz A, Bertenshaw G, Jeffries E, Schatz T, Cotton J, Villanueva J, Herlyn M, Krepler C, Vultur A, Xu W, et al. 2012. Functional profiling of live melanoma samples using a novel automated platform. PLoS One 7: e52760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z, Smogorzewska A, Sowa ME, Ang XL, et al. 2008. Cancer proliferation gene discovery through functional genomics. Science 319: 620–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JM, Mizuno H, Brady A, Lucito R, Hannon GJ. 2004. RNA interference microarrays: high-throughput loss-of-function genetics in mammalian cells. Proc Natl Acad Sci 101: 6548–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suprynowicz FA, Upadhyay G, Krawczyk E, Kramer SC, Hebert JD, Liu X, Yuan H, Cheluvaraju C, Clapp PW, Boucher RC Jr, et al. 2012. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci 109: 20035–20040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. 2015. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiri A, Roe K, Ree AH, de Wijn R, Risberg K, Busch C, Lonning PE, Kristensen V, Geisler J. 2013. Differential inhibition of ex-vivo tumor kinase activity by vemurafenib in BRAF(V600E) and BRAF wild-type metastatic malignant melanoma. PLoS One 8: e72692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Cell Line Encyclopedia and Genomics of Drug Sensitivity in Cancer Investigators. 2015. Pharmacogenomic agreement between two cancer cell line data sets. Nature 528: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touzeau C, Ryan J, Guerriero J, Moreau P, Chonghaile TN, Le Gouill S, Richardson P, Anderson K, Amiot M, Letai A. 2016. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 30: 761–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traer E, Javidi-Sharifi N, Agarwal A, Dunlap J, English I, Martinez J, Tyner JW, Wong M, Druker BJ. 2014. Ponatinib overcomes FGF2-mediated resistance in CML patients without kinase domain mutations. Blood 123: 1516–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, Willis SG, Erickson H, Kovacsovics T, O'Hare T, Heinrich MC, et al. 2009. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc Natl Acad Sci 106: 8695–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner JW, Yang WF, Bankhead A, Fan G, Fletcher LB, Bryant J, Glover JM, Chang BH, Spurgeon SE, Fleming WH, et al. 2013. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res 73: 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaira V, Fedele G, Pyne S, Fasoli E, Zadra G, Bailey D, Snyder E, Faversani A, Coggi G, Flavin R, et al. 2010. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proc Natl Acad Sci 107: 8352–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, Zhao JJ, Roberts TM, Mandel G, Hannon GJ, et al. 2005. A genetic screen for candidate tumor suppressors identifies REST. Cell 121: 837–848. [DOI] [PubMed] [Google Scholar]
- Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, Van Allen EM, Corsello SM, Capelletti M, Calles A, Butaney M, et al. 2015. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell 27: 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, et al. 2014. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science 345: 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]