

Abstract
Background
Mediterranean islands host a disproportionately high level of biodiversity and endemisms. Growing phylogeographic evidence on island endemics has unveiled unexpectedly complex patterns of intra-island diversification, which originated at diverse spatial and temporal scales. We investigated multilocus genetic variation of the Corsican-Sardinian endemic lizard Podarcis tiliguerta with the aim of shedding more light on the evolutionary processes underlying the origin of Mediterranean island biodiversity.
Results
We analysed DNA sequences of mitochondrial (12S and nd4) and nuclear (acm4 and mc1r) gene fragments in 174 individuals of P. tiliguerta from 81 localities across the full range of the species in a geographic and genealogical framework. We found surprisingly high genetic diversity both at mitochondrial and nuclear loci. Seventeen reciprocally monophyletic allopatric mitochondrial haplogroups were sharply divided into four main mitochondrial lineages (two in Corsica and two in Sardinia) of Miocene origin. In contrast, shallow divergence and shared diversity within and between islands was observed at the nuclear loci. We evaluated alternative biogeographic and evolutionary scenarios to explain such profound discordance in spatial and phylogenetic patterning between mitochondrial and nuclear genomes. While neutral models provided unparsimonious explanations for the observed pattern, the hypothesis of environmental selection driving mitochondrial divergence in the presence of nuclear gene flow is favoured.
Conclusions
Our study on the genetic variation of P. tiliguerta reveals surprising levels of diversity underlining a complex phylogeographic pattern with a striking example of mito-nuclear discordance. These findings have profound implications, not only for the taxonomy and conservation of P. tiliguerta. Growing evidence on deep mitochondrial breaks in absence of geographic barriers and of climatic factors associated to genetic variation of Corsican-Sardinian endemics warrants additional investigation on the potential role of environmental selection driving the evolution of diversity hotspots within Mediterranean islands.
Electronic supplementary material
The online version of this article (doi:10.1186/s12862-017-0899-x) contains supplementary material, which is available to authorized users.
Keywords: Cyto-nuclear discordance, Evolutionary history, Genetic diversity, Local adaptation, Mediterranean island, Phylogeography
Background
The Mediterranean Basin has long been recognised as one of the richest global biodiversity hotspots [1]. A large fraction of such diversity, and particularly endemic diversity, is hosted within the over 5,000 islands scattered throughout the Mediterranean Sea [2]. The level of diversity and endemism is especially high in large continental islands such as the Tyrrhenian islands Corsica and Sardinia, which are considered a regional biodiversity hotspot within the Mediterranean area [2–4].
These islands offer an ideal setting for investigating the evolutionary processes behind the origin and the structure of current patterns of insular biodiversity hotspots because: (i) they have a complex topography with a diversity of landscapes and microclimatic regions, spanning from Mediterranean to alpine climates [5, 6], which combined with the imprints of Plio-Pleistocene climatic oscillations [7–10] contribute to the diversification and persistence of old lineages [11–13]; (ii) the palaeogeographical evolution of the Corsican-Sardinian system within the Western Mediterranean is well established [14–18], thus providing a useful framework for biogeographic and molecular inferences [19]; and (iii) emerging phylogeographic and phylogenetic data on many endemics allow conclusions to be drawn within a comparative framework [see e.g. [19, 20].
In the last decade, Corsican-Sardinian species have been the subject of intensive phylogeographic surveys, especially regarding amphibians and reptiles [12, 13, 20–34], which have revealed how an essential component of the Tyrrhenian biodiversity hotspot is represented by the genetic variation held within and among populations of these endemic species. A significant realization of these studies is that the current patterns of genetic structure and diversity of these endemic species have been historically shaped by an unexpectedly diverse array of evolutionary and demographic processes acting across unrelated spatial and temporal scales [20]. In fact, essentially each phylogeographical reconstruction carried out so far on Corsican-Sardinian species suggested an idiosyncratic scenario for the evolution of the current geographical patterns of intraspecific genetic diversity [12, 19, 20, 30, 31]. This suggests that we are still far from either an exhaustive inventory or a deep understanding of the evolutionary processes underlying the origins and diversity of this biodiversity hotspot.
In regard to this, the Tyrrhenian wall lizard Podarcis tiliguerta offers an intriguing case study as previous genetic assessments uncovered extraordinarily high level of diversity with contrasting patterns between different genetic markers [21, 24]. This species is common and locally abundant across a variety of shrubby and open habitats [35] from the sea level (including tiny islets) up to 1800 m asl in the mountain regions [36]. Based on the current continuous distribution of P. tiliguerta within both Corsica and Sardinia [37, 38] and associated with the fact that these two islands were largely and persistently connected into a single landmass during the Pleistocene glaciations [18, 39], we may have expected low genetic differentiation between populations with most of the genetic diversity shared within and between the main islands. In contrast, preliminary mitochondrial datasets [24, 40, 41], based on a few individuals and short gene fragments, showed three highly divergent lineages, one in Corsica and two in Sardinia, with genetic distances between them exceeding those typically found between reptile species [24, 42]. On the other hand, allozyme data from 15 variable loci suggested a lower genetic distance between Corsican and Sardinian populations (with no alternatively fixed alleles) with a latitudinal clinal variation in allele frequencies at some loci and an overall isolation-by distance pattern suggesting reduced gene-flow between populations [21].
Therefore, while all previous studies found extraordinarily high genetic diversity and substantial differentiation between populations, mitochondrial data indicate that P. tiliguerta may be a species complex [24] whereas nuclear data depict P. tiliguerta as a single species geographically structured in local populations [21], suggesting a possible mito-nuclear discordance. Comparing the mitochondrial and nuclear patterns of diversity and levels of divergence found within P. tiliguerta by previous studies, and identifying the evolutionary processes underlying their formation, is difficult. Allozyme analyses lack a genealogical framework to understand the evolutionary relationships between alleles, and genetic diversity and divergence between populations may be underestimated due to the occurrence of iso-electrophoretic alleles (distinct alleles with equal electrophoretic mobility). On the other hand, mitochondrial assessments only account for a single genealogical realization and previous studies have likely underestimated the genetic diversity of P. tiliguerta due to the short gene fragment used, limited geographic sampling, and the low number of individuals analysed, as supported by the finding that new haplotypes and lineages were sampled as a few more individuals were added in each study [40, 41].
In this study, we investigated the phylogeographic structure and evolutionary history of P. tiliguerta based on mitochondrial and nuclear genealogies sampled across the entire geographic range of the species. We found an extraordinarily high level of genetic diversity which has deep roots in the mitochondrial genome whereas it shows minimal phylogenetic and geographic structure in the nuclear genome. We evaluated the role of biogeographic and evolutionary processes at different temporal and spatial scales to explain the origin of the striking pattern of genetic richness and mito-nuclear discordance found within P. tiliguerta and we discussed possible generalizations within the Tyrrhenian biodiversity hotspot.
Methods
Genetic data collection
We sampled 174 individuals of P. tiliguerta from 81 localities across the entire species range (Fig. 1). Sampling design was informed by genetic analyses on a preliminary set of samples and refined during a four-year collection period (2009–2012). This was necessary to cover the high level of diversity found in P. tiliguerta. Individuals from additional 10 Podarcis species from Portugal, Spain, Italy, Malta, Slovenia, and Greece were collected and used in phylogenetic analyses together with sequences obtained from GenBank. Detailed information regarding individual and locality codes, geographic coordinates of sampling locality and GenBank accession numbers for all sequences used in this study is reported in Table 1. The source of data retrieved from GenBank is reported in Tables 2 and 3.
Fig. 1.
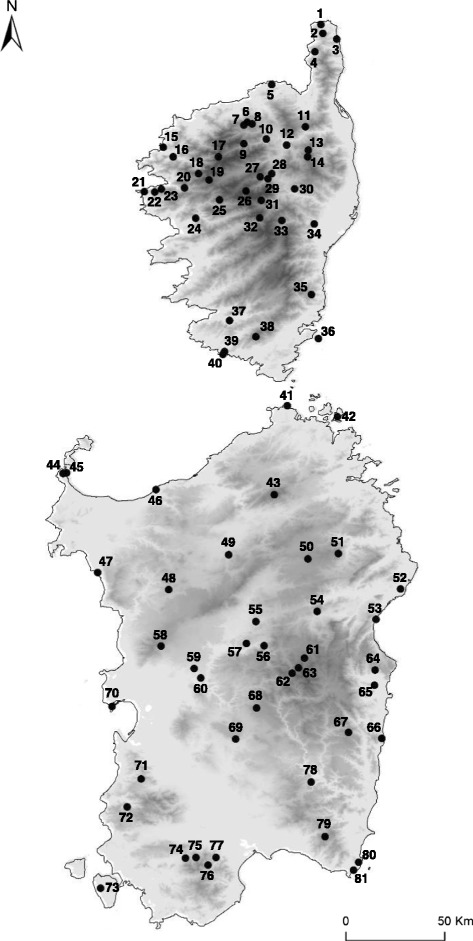
Map of the study area showing sampling locations of the Tyrrhenian wall lizard, Podarcis tiliguerta. Detailed information on sampling localities and specimens is found in Table 1
Table 1.
Geographical location and codes for the studied populations and individuals of Podarcis tiliguerta. Genbank accession numbers of the sequences are provided for each gene
Table 2.
GenBank accession numbers of the sequences used in this study to build the phylogeny of the genus Podarcis. The references for the sequences obtained from GenBank are reported
| Species | 12S | nd4 |
|---|---|---|
| Podarcis bocagei | DQ081064 [61] | DQ081153 [61] |
| Podarcis carbonelli | DQ081065 [61] | DQ081154 [61] |
| Podarcis cretensis | KY561996 a | KY562001 a |
| Podarcis filfolensis | KY561997 a | KJ027796 [58] |
| Podarcis gaigae | AF133444 [126] | KY562002 a |
| Podarcis hispanica | DQ081070 [61] | DQ081171 [61] |
| Podarcis guadarramae | AF469452 [61] | DQ081165 [61] |
| Podarcis lilfordi | KY561998 a | KY562003 a |
| Podarcis melisellensis | AF133448 [126] | KY562004 a |
| Podarcis milensis | AF133449 [126] | KY562005 a |
| Podarcis muralis | KX080575 [44] | KF372393 [60] |
| Podarcis peloponnesiaca | AF133451 [126] | KY562006 a |
| Podarcis pityusensis | KY561999 a | KY562007 a |
| Podarcis raffonei | KY562000 a | KJ027980 [58] |
| Podarcis sicula | KX080574 [44] | KF372035 [60] |
| Podarcis taurica | AF080279 [127] | KY562008 a |
| Podarcis tiliguerta lineage1 | KY562020 a | KY562427 a |
| Podarcis tiliguerta lineage2 | KY562081 a | KY562492 a |
| Podarcis tiliguerta lineage3 | KY562092 a | KY562503 a |
| Podarcis tiliguerta lineage4 | KY562146 a | KY562557 a |
| Podarcis vaucheri | HQ898229 [61] | HQ898028 [61] |
| Podarcis wagleriana | DQ017659 [40] | KJ027979 [58] |
| Scelarcis perspicillata | KX080591 [44] | KX081031 [44] |
| Teira dugesii | KX080595 [44] | KX081035 [44] |
Numbers in square brackets after GenBank accession numbers refer to publications that generated the cited GenBank data (asequences generated in this study)
Table 3.
Genetic diversity estimates at nd4 and mc1r loci in Podarcis lizards
| Species | Species range (km2) | nd4 | mc1r | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | ns | S | h | Hd (SD) | π (SD) | K | N | ns | S | h | Hd (SD) | π (SD) | K | ||
| P. bocagei [55–57] | 58387 | 22 | 661 | 18.0 | 18.0 | 0.978 (0.021) | 0.00321 (0.00034) | 2.1 | 12 | 583 | 5.0 | 3.0 | 0.621 (0.118) | 0.00377 (0.00075) | 2.2 |
| P. carbonelli [55, 56] | 4684 | 25 | 661 | 25.0 | 20.0 | 0.980 (0.017) | 0.00607 (0.00058) | 4.0 | - | - | - | - | - | - | - |
| 22 | 661 | 23.5 | 18.1 | 0.980 | 0.00610 | 4.0 | |||||||||
| P. filfolensis [58] | 338 | 94 | 661 | 32.0 | 27.0 | 0.900 (0.020) | 0.00471 (0.00040) | 3.1 | 130 | 583 | 20.0 | 27.0 | 0.904 (0.012) | 0.00535 (0.00024) | 3.1 |
| 22 | 661 | 17.4 | 12.3 | 0.904 | 0.00473 | 3.1 | 12 | 583 | 9.8 | 7.7 | 0.899 | 0.00537 | 3.1 | ||
| P. lilfordi [59] | <100 | 92 | 583 | 23.0 | 27.0 | 0.920 (0.015) | 0.00520 (0.00037) | 3.0 | |||||||
| 12 | 583 | 11.6 | 8.5 | 0.924 | 0.00532 | 3.1 | |||||||||
| P. muralis [60] | 1830883 | 77 | 661 | 141.0 | 55.0 | 0.987 (0.005) | 0.03089 (0.00112) | 20.4 | 162 | 583 | 35.0 | 31.0 | 0.835 (0.017) | 0.00308 (0.00025) | 1.8 |
| 22 | 661 | 97.5 | 19.3 | 0.985 | 0.03096 | 20.5 | 12 | 583 | 6.7 | 6.2 | 0.832 | 0.00304 | 1.8 | ||
| P. pityusensis [59] | 741 | 44 | 583 | 10.0 | 14.0 | 0.897 (0.022) | 0.00416 (0.00031) | 2.4 | |||||||
| 12 | 583 | 7.0 | 7.4 | 0.900 | 0.00423 | 2.5 | |||||||||
| P. tiliguerta a | 32898 | 148 | 661 | 220.0 | 104.0 | 0.9936 (0.0017) | 0.09994 (0.00191) | 65.8 | 250 | 583 | 92.0 | 148.0 | 0.9897 (0.0020) | 0.00691 (0.00024) | 4.0 |
| 22 | 661 | 190.2 | 20.7 | 0.994 | 0.10072 | 66.5 | 12 | 583 | 17.6 | 11.3 | 0.989 | 0.00685 | 4.0 | ||
| P. vaucheri species complex [55, 56, 61, 62] | 305794 | 87 | 661 | 135.0 | 68.0 | 0.995 (0.003) | 0.03492 (0.00166) | 23.1 | - | - | - | - | - | - | - |
| 22 | 661 | 94.5 | 20.9 | 0.995 | 0.03558 | 23.5 | |||||||||
| P. wagleriana b | 23914 | 101 | 661 | 30.0 | 28.0 | 0.828 (0.029) | 0.00396 (0.00023) | 2.6 | 186 | 583 | 13.0 | 16.0 | 0.790 (0.020) | 0.00289 (0.00021) | 1.7 |
| 22 | 661 | 12.8 | 9.9 | 0.833 | 0.00398 | 2.6 | 12 | 583 | 5.9 | 5.3 | 0.764 | 0.00276 | 1.6 | ||
N number of sampled gene copies, ns number of sites of the alignment used for the calculations, S number of segregating sites, h number of haplotypes, Hd haplotype diversity, π nucleotide diversity, K average number of pairwise differences, SD standard deviation. For the diversity measures S, h, Hd, π and K values calculated under a resampling approach are also reported below values calculated on the original datasets (see text for details). Numbers in square brackets after species names refer to publications that generated the sequence data (asequences generated in this study; bSalvi et al., unpublished sequence data). Species range (extent of occurrence) is based on spatial data from [IUCN, 2016]
Tissue samples were collected as tail tips and stored in ethanol; each individual was then released at the place of capture. Genomic DNA was extracted following standard high-salt protocols [43]. We amplified two mitochondrial gene fragments, 12S rRNA (12S) and NADH dehydrogenase subunit four with flanking tRNASer, tRNAHis, and tRNALeu (nd4), and two nuclear gene fragments, Melanocortin receptor 1 (mc1r) and acetylcholinergic receptor M4 (acm4), by polymerase chain reaction (PCR). Primers and PCR protocols used for the amplification of the molecular markers are described in [44].
Additionally, we cloned the mc1r PCR products of eight selected heterozygous individuals (showing 4–6 polymorphisms), because for this marker a pilot phasing analysis carried out on a preliminary subset of individuals (N = 35) showed high uncertainty of haplotypic phase estimate. PCR products were ligated into pGEM-T Easy Vector Systems kit (Promega) according to the manufacturer’s instructions. The output of the ligation reaction was then transformed into Escherichia coli competent cells and grown on standard LB medium with ampicillin/IPTG/X-Gal. For each sample we randomly selected six colonies for sequencing in order to account for PCR/cloning errors resulting from misincorporation of individual nucleotides [45]. We used a conventional blue/white screening to select the 48 positive colonies which were then amplified using universal primers pUC/M13F and pUC/M13R. PCR reactions were carried out in 25 μL volumes containing 1X PCR buffer, MgCl2; 1 mM each dNTP, 2U of GoTaq DNA polymerase (Promega), 0.4 μM each primer and 2 μL of colony DNA. After verification of successful amplification, the inserts were sequenced from both strands with the same primers used for amplification. Purification and sequencing of PCR products and plasmid DNA from positive clones were carried out by Macrogen Inc.
Data analysis
We used GENEIOUS 6.0 (www.geneious.com) to check electropherograms, calculate consensus sequences, perform multiple sequence alignment, and to annotate both tRNAs in the nd4 fragment and codon positions in coding regions. Sequence divergence (uncorrected p-distance) was assessed using MEGA 6 [46]. Heterozygous sequences within the acm4 and mc1r fragments were phased using PHASE 2.1.1. [47]. Three independent runs were conducted using a model with recombination (−MR0 option), 1000 initial iterations discarded as burn-in, one as thinning interval, and 1000 post-burnin iterations. After monitoring the goodness of fit for each run according to the program’s manual, we accepted haplotype reconstructions which yielded the same result in all of the three runs. Cloned mc1r sequences from each selected individual were used to determine the constituent alleles (haplotypes). Cloning-determined haplotypes of these individuals were first compared with corresponding haplotypes inferred by PHASE, in order to assess the inference accuracy, and then used as ‘known phase’ in a further PHASE analysis in order to improve haplotype estimation. For each nuclear gene dataset recombination detection was performed in RDP 4.77 [48] using five different algorithms: BootScan [49], GENECONV [50], MaxChi [51], RDP [52], and SiScan [53].
For each gene we estimated the following summary statistics of genetic diversity using DNASP 5 5.10.01 [54]: number of segregating sites (S), number of haplotypes (h), haplotype diversity (Hd), nucleotide diversity (π), and average number of pairwise differences (K) both overall and for clades defined within the species by phylogenetic analyses. Since DNASP removes entirely sites with missing data, in order to maximize the length of alignments used for diversity calculations we used fully phased haplotype datasets, we excluded sequences with more than 1.5% of missing data, and recoded the few remaining missing data with the most common nucleotide at that site (thus using the information of these sites while retaining the observed level of variation). In order to compare the degree of genetic diversity observed in P. tiliguerta with congeneric species, we compiled a synopsis of genetic diversity estimates for nd4 and mc1r loci for other Podarcis lizards based on data from previous phylogeographic studies which sampled fairly well each species range [55–62]. Haplotype phases for mc1r sequences downloaded from GenBank were resolved for each species separately, using PHASE (100 initial iterations). Retrieved datasets had different sizes, both in terms of number of sites and number of sequences, which may affect comparisons of diversity measures. Therefore, first we trimmed all alignments at the same number of sites, and then we also calculated genetic diversity statistics following the resampling approach described in [60]. Resampling was performed with the aid of a series of scripts written in Python 2.7.1 and taking advantage of DNASP “batch mode” calculations. Tajima’s D [63] test were used to assess whether mtDNA and nucDNA sequences of P. tiliguerta fitted a neutral model of evolution. D values were estimated in DNASP and their significance was assessed through 10,000 coalescent simulations under the hypothesis of population equilibrium and selective neutrality.
We performed a Bayesian evolutionary analysis in BEAST 1.8.0 [64] to estimate phylogenetic relationships among mitochondrial haplotypes and associate a time/age at each node of the phylogeny. BEAST allows the incorporation of the uncertainty associated with phylogenetic estimates, calibration dates and among-lineage variation of substitution rates in a single analysis [64]. In addition BEAST allows sampling the root of the tree by using the molecular clock method [65] without the need of using an outgroup taxon. This is particularly desirable when outgroup rooting is problematic as for P. tiliguerta which shows deep branching of mitochondrial lineages with unresolved position within the Podarcis phylogeny [24]. One the other hand, for our dataset the choice of a proper tree prior among those available in BEAST is not straightforward as we have an intraspecific dataset where we expect a deep geographic structure [24]. Thus, both a simple coalescent prior [e.g. [66]] which assumes that the individuals analysed are drawn from a single panmictic population – and a multi-species coalescent prior [64, 67] which allows accounting for multiple divergent populations each one following a coalescent process but assume reproductive isolation between them – are not fully appropriate for our data. Therefore, while we acknowledged that available priors are a rough simplification of real situations, we explored the sensitivity of our phylogenetic and dating results to different prior choices and we ran multiple analyses using both priors. Simple coalescent models were implemented in BEAST using the skyline coalescent prior. The multi-species coalescent model was implemented in the *BEAST extension. For both analyses, we implemented the HKY model for the nd4 coding region and the HKY + G + I for non-coding region (tRNAs +12S) according to the best partition schemes and substitution models selected by PARTITION FINDER 1.1.1 [68] under the Bayesian Information Criterion (BIC). We implemented a relaxed uncorrelated lognormal clock model with the normal distribution N (0.0115, 0.00075) for the nd4 substitution rate prior (parameter ucld.mean). This rate was estimated by [58] for the same nd4 region used in this study in a calibrated phylogeny of Sicilian Podarcis based on the palaeogeographic events associated to the splits between P. filfolensis and P. wagleriana and between the latter and P. raffonei (for a full account on the specific calibration points and methods used see [58]). Similar estimate of substitution rates were previously obtained for Podarcis lizards for the same nd4 gene fragment [55]. BEAST and *BEAST were run twice, with 100 million iterations per run, sampling every 10000 steps. Convergence diagnostic and summary of posterior samples of parameters were assessed in TRACER 1.6 (available at http://beast.bio.ed.ac.uk/Tracer); sampled trees from independent runs were combined in LogCombiner; and Maximum Clade Credibility Trees and Bayesian Posterior Probabilities associated to nodes (BPP) were calculated in TreeAnnotator (burn-in = 25%). We verified consistency of results with different models through additional analyses using unlinked substitution models for the nd4 codon positions and strict clock models.
In order to identify the main phylogenetic discontinuities along terminal branches of the mitochondrial phylogeny we used the statistical parsimony network approach [69] implemented in the software TCS 1.21 [70]. Subnetworks obtained under the 95% probability criterion for a parsimonious connection have been successfully used as an objective way to identify significant genetic discontinuities in mitochondrial DNA sequence dataset [71].
A preliminary mitochondrial dataset [24] of about 700 base pairs (12S and cytb) showed two highly divergent mitochondrial lineages in P. tiliguerta (maximum cytb divergence = 15%) which neither form a monophyletic group nor have a resolved position in the Podarcis phylogeny. Therefore we used one representative of each P. tiliguerta lineage recovered from our BEAST analyses to infer their phylogenetic relationships within Podarcis. In doing so we sequenced 10 Podarcis species from the same two fragment used in this study and retrieved Genbank sequences from nine additional Podarcis species (Table 2). We designated Scelarcis perspicillata and Teira dugesii as outgroups based on the mitochondrial phylogeny of Lacertini [44]. We performed a Bayesian analysis in BEAST, using the same settings as in the BEAST analysis of the P. tiliguerta dataset but using a Yule process of speciation as tree prior. A Maximum Likelihood (ML) analysis was also performed in Raxml GUI 1.1.3 [72], a graphical front-end for RaxML 7.4.2 [73]. ML searches included 100 random addition replicates and 1000 nonparametric bootstrap replicates, applying the general time-reversible model with gamma model of rate heterogeneity (GTRGAMMA) for both the nd4 and 12S partition. To determine if the monophyly of P. tiliguerta lineages could be rejected, we used Mesquite 3.03 [74] to generate a tree enforcing the monophyly of P. tiliguerta lineages; we then estimated per-site log likelihood values of the best ML tree and the constrained tree in RAxMLGUI and we compared these values using the Shimodaira–Hasegawa (SH) [75] and the approximately unbiased (AU) [76] tests as implemented in CONSEL [77].
We inferred the genealogical relationships between the acm4 and mc1r haplotypes detected in P. tiliguerta using the statistical parsimony network approach implemented in TCS. This method is particularly appropriate when few characters for phylogenetic analysis are available due to shallow levels of divergence [78] as we observed in the acm4 and mc1r datasets. Networks were constructed under the 95% probability criterion for a parsimonious connection and represented graphically using the tool tcsBU [79]. Additionally, since mc1r sequences are available in GenBank for other Podarcis species, we combined them with sequence data from the present study to examine the phylogenetic relationships of mc1r haplotypes from different populations and species. For this phylogenetic analysis we used the same mc1r datasets used for genetic diversity comparisons plus a few mc1r sequences available for two additional species, P. carbonelli and P. sicula [57, 80].
To investigate the hierarchical structure of the genetic diversity both at mitochondrial and nuclear loci we used the approach implemented in SAMOVA v.2 [81]. This method is based on the analysis of molecular variance (AMOVA) [82] through a simulated annealing procedure and allows defining groups of populations that are genetically homogeneous and maximally differentiated from each other, without the prior assumption of group composition [81]. Samples represented by less than four sequences were not included in the analysis. The 17 mitochondrial monophyletic sublineages previously identified by the phylogenetic analyses were treated as populations in our SAMOVA analyses; accordingly mtDNA sequences from neighbouring localities belonging to the same sublineage were pooled and geographic coordinates for each population were calculated as the geographic centroid of member localities. We ran the analysis both enforcing the geographical homogeneity of groups and without constraint for the geographic composition of the groups in order to investigate both spatial and non-spatial structure of the genetic dataset. SAMOVA was run three times for each value of predefined number of groups (K) using the TN + G substitution model, 100 random initial conditions and 10,000 iterations. For each gene we tested genetic structures from K = 2 until increasing K generated genetic structures with non-significant fixation indexes. Additionally, we explored patterns of genetic differentiation exhibited by nuclear genes by performing AMOVA using mtDNA-defined partitions. These analyses were conducted using ARLEQUIN 3.5.2.2 [83].
In order to explore whether nuclear genetic divergence of populations correlate with geographic distances we carried out Mantel tests and reduced major axis (RMA) regression analyses using IBDWS 3.23 [84]. Genetic differentiation was calculated both as simple FST (based on haplotype frequencies only) and PhiST (also accounting for haplotype divergence estimated as K2P distance) and significance was assessed through 1000 random permutations.
Finally, in order to assess whether past habitat suitability may have impacted the geographic distribution of P. tiliguerta and its genetic diversification, we performed species distribution modeling (SDM) under current and Last Glacial Maximum (LGM; ~21 thousands years ago, 21 kya) bioclimatic envelops using the maximum-entropy algorithm implemented in MaxEnt 3.3.3e [85]. We collected 1075 presence data of P. tiliguerta from personal observations (including sampled localities), previous literature and public databases. In order to correct for potential sampling biases in the distribution records [86], we selected 480 points of species occurrence with a minimum distance of 3 Km. The bioclimatic layers were downloaded from the WorldClim database website (www.worldclim.org). For the LGM prediction we used data from two different general circulation models (CCSM and MIROC). We built the models with a set of five variables that were not strongly correlated with each other (Pearson’s correlation coefficient, r2 < 0.80) and that we deemed as biologically significant for P. tiliguerta. Selected variables were: temperature seasonality (BIO4), temperature annual range (BIO7), mean temperature of the driest quarter (BIO9), annual precipitation (BIO12) and precipitation seasonality (BIO15). We ran Maxent with autofeatures, selecting at random 70% of the presence records as training data and 30% as test data for each species. We tested the quality of the models by calculating the area under the curve (AUC) of the receiver operated characteristics (ROC) plots [87]. We built models with an increasing regularization parameter β and between them we selected the model that best fit the data under the Akaike Information Criterion (AIC) with ENMTools 1.3 [88].
Results
The concatenated mitochondrial (mtDNA) dataset (12S + nd4) included 171 sequences and 1233 aligned positions (149 individuals sequenced for both fragments, 22 individuals sequenced only for the 12S). For three individuals we were only able to obtain nuclear sequences. The nuclear (nucDNA) datasets consisted of 250 (phased) sequences for mc1r (614 positions) and 216 for acm4 (401 positions) and had no missing data. Multiple sequence alignment did require four gap positions in the tRNAs and 12S regions; coding regions (nd4, acm4, and mc1r) did not require gap positions and their translation into amino acid sequences contained no stop codons. We obtained 43 mc1r sequences from the 48 positive colonies and within them we observed 57 instances of nucleotide misincorporation (0.21% of the total nucleotides) (Additional file 1: Table S1). When comparing mc1r haplotypes determined by cloning and inferred by PHASE they were identical at 33 of the 39 heterozygote sites (85%). The recombination tests applied in RDP did not find statistically significant evidence for recombination in any of the nuclear genes.
The overall level of sequence variation in the mitochondrial and nuclear loci was high (mtDNA: S = 285, h = 110, Hd = 0.995 ± 0.001, π = 0.07264 ± 0.00151, K = 82.442; acm4: S = 66, h = 105, Hd = 0.958 ± 0.008, π = 0.00818 ± 0.00037, K = 3.280; mc1r: S = 96, h = 157, Hd = 0.991 ± 0.002, π = 0.00688 ± 0.00023, K = 4.226). A summary of genetic diversity estimates and neutrality tests per locus for each lineage and island is given in the Additional file 2: Table S2. Genetic diversity estimated as S, H, and K, was higher in P. tiliguerta compared to congeneric species (Table 3). Significantly negative Tajima’s D values (excess of rare alleles), indicative of population expansion, purifying selection, or genetic hitchhiking, were found in acm4 (D = −2.121, P < 0.001) and mc1r (D = −2.222, P < 0.001). Non-significant D were found overall in mtDNA (D = 1.999, P = 0.979) and in all mtDNA lineages.
Bayesian phylogenetic trees inferred in BEAST (Fig. 2a) and *BEAST (not shown) based on mtDNA sequences showed four main lineages which are well supported (BPP = 1.00) and have geographic coherence: Lineage one including individuals from north and east Corsica, Lineage two from west Corsica, Lineage three from north Sardinia and Lineage four from south Sardinia. Relationships between the four main lineages are not statistically supported (BPP ≤ 0.88). Within these lineages, haplotypes clustered in 17 well supported sub-lineages (1A-1E, 2A-2C, 3A-3B, 4A-4G; all BPP = 1.00) with a strict phylogeographic association (i.e. formed by geographically proximate individuals). The same 17 sub-lineages were recovered as distinct sub-networks in the statistical parsimony analysis (Additional file 3: Figure S1).
Fig. 2.
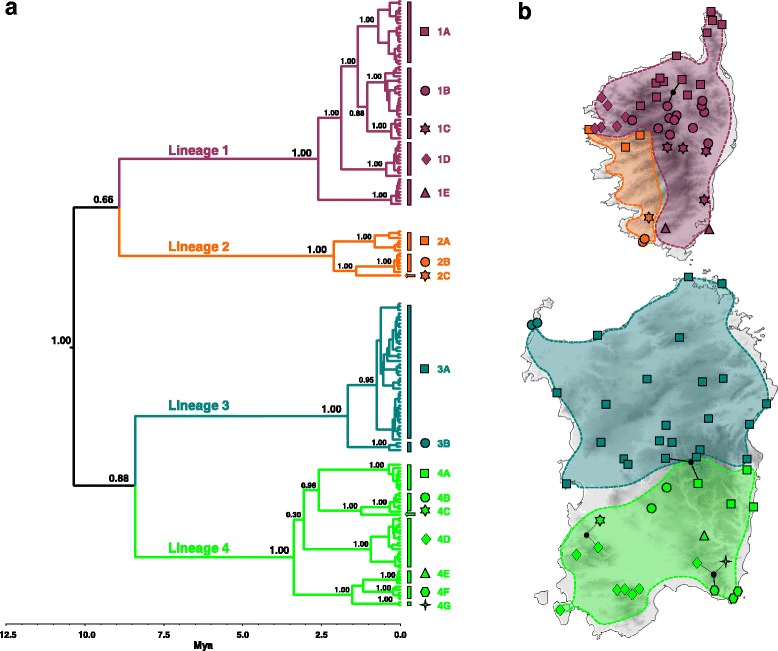
Bayesian time tree depicting the phylogenetic relationships among combined mitochondrial sequences (12S and nd4) of Podarcis tiliguerta (a). Bayesian Posterior Probabilities are reported in correspondence of the nodes of the main lineages (Lineage 1–4) and sublineages (1A-1E, 2A-2C, 3A-3B, 4A-4G) and the tree scale showed the estimated times to their most recent common ancestor (TMRCA) in million years (see text for further details and highest posterior density intervals). The map shows the geographic distribution of mitochondrial lineages and their putative ranges (b)
Average genetic distance between mtDNA lineages of P. tiliguerta was 11.65% at the nd4 and 5.23% at the 12S fragment (ranging from 11.1 to 12.4% for nd4 and from 3 to 6.5% for 12S). Within lineages average genetic distance was 2.65% at the nd4 and 1.07% at the 12S fragment (nd4: 1–4.2%; 12S: 0.6–1.3%).
The 12S substitution rate estimated by BEAST based on the nd4 rate prior implemented was 0.53% (0.37–0.69 95% HPD) per million years which is in agreement with substitution rates calculated for lacertid lizards for the same 12S gene region (0.55% ± 0.13% SD) in [89], based on a phylogeny calibrated using seven biogeographic calibration points. This correspondence provides further justification for the nd4 rate used in our molecular clock model.
According to our mitochondrial clock model the main cladogenetic events within P. tiliguerta occurred during the Miocene. The time to the most recent common ancestor (TMRCA) of the main lineages was estimated in BEAST at 10.3 million years ago (mya) (Fig. 2a) with an associated 95% highest posterior density (95% HPD) interval of 7.9–13.2 mya. For the Corsican lineages (Lineage 1 and 2) the TMRCA was estimated at 8.9 mya (95% HPD: 6.5–11.5) and for the Sardinian lineages (Lineage 3 and 4) at 8.4 mya (95% HPD: 6.1–11.1 mya), although these nodes received low posterior probability. The TMRCAs estimated for each main lineage are placed from the Early Pliocene to Early Pleistocene (3.4–1.7 mya; 95% HPD intervals: 4.3–0.6 mya) and their diversification in 17 sub-lineages bounds the period from the Late Pliocene to Early Pleistocene (3.1–1.3 mya; 95% HPD intervals: 3.9–1.0 mya). TMRCA estimates of the main lineages and the time of splits of the 17 sub-lineages calculated in *BEAST under the multi-species coalescent model (results not shown) are virtually identically to those calculated in BEAST. On the other hand, the TMRCA of the main lineages estimated in *BEAST (8.4 mya; 95% HPD: 6.9–10.2) is more recent compared to estimates from the BEAST analysis reported above.
The four mitochondrial lineages of P.tiliguerta represent deep branches in the mitochondrial phylogeny of Podarcis and are included in a basal polytomy (Additional file 4: Figure S2), although the hypothesis that they are monophyletic cannot be rejected (SH-test, P = 0.291; AU-test, P = 0.279).
Nuclear genealogies showed high haplotype diversity but shallow divergence and a lack of phylogeographic structure (Fig. 3). Both the mc1r and the acm4 networks showed a reticulated pattern of relationships between closely related haplotypes (maximum number of mutational steps: ten for mc1r and eight for the acm4). Nuclear haplotypes at both loci were broadly shared between individuals belonging to distinct mtDNA lineages and between Corsican and Sardinian individuals (Fig. 3).
Fig. 3.
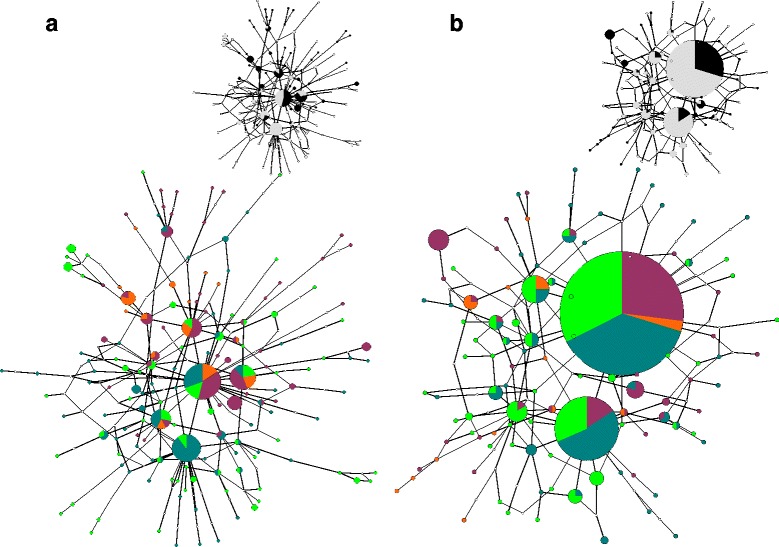
Statistical parsimony networks showing the genealogical relationships between the mc1r a and acm4 b haplotypes. Haplotypes are represented by circles with size proportional to their frequency; small white circles represent ‘missing’ or extinct haplotypes. Nuclear haplotype are coloured according to the mitochondrial lineage to which each individual belongs: the large networks map the four main lineages whereas the small networks in the top of panel a and b map Corsican (black) versus Sardinian (white) lineages
Phylogenetic relationships between mc1r haplotypes inferred from 946 phased sequences from nine Podarcis species show that, although separated by a few mutations, almost all haplotypes inferred (284, equal to 98.7% of the total) were species-specific (Additional file 5: Figure S3). Haplotypes carried by conspecific individuals tend to cluster together in the phylogenetic network, although a pattern of strict reciprocal monophyly between species was not observed. Four transpecific haplotypes (1.3% of the total) are shared between the species P. lilfordi, P. pityusensis and P. tiliguerta, and probably reflects a recent common history of these species.
The hierarchical genetic structure of P. tiliguerta inferred by SAMOVA analyses strictly correspond to the geographic partition of genetic lineages (or lack thereof) found by phylogenetic analyses (Fig. 4). Based on mtDNA data SAMOVA identified the four groups of populations corresponding to the main mtDNA lineages as the grouping option which best explains the among-group partitioning of molecular variance (K = 4; F CT = 0.67; P < 0.001). Consistent SAMOVA results were obtained either enforcing or not the spatial association between populations within groups. The subdivision into four main lineages explains 67% of the overall genetic variation compared with 26.8% due to differences between populations within lineages and 5.86% due to differences within populations. All variance components were highly significant providing evidence of a strong genetic structure (P < 0.001). SAMOVA analyses based on nucDNA data either enforcing or not the geographic homogeneity of groups showed a lack of genetic and geographic structure in nuclear genes. Partitioning populations in any number of groups explained less than 28 and 15% of the total amount of genetic variation for acm4 and mc1r respectively, whereas more than 65 and 71% of variation (for acm4 and mc1r respectively) was accounted by differences within populations. In most cases, and in particular for K = 2–4, all groups except one were defined by a single population. In all AMOVA analyses carried out on acm4 and mc1r, using mtDNA-defined partitions, 92–97% of the total genetic variation was explain by differences within populations whereas higher hierarchical level only explained 1–7% of the genetic variation (Additional file 6: Table S3).
Fig. 4.
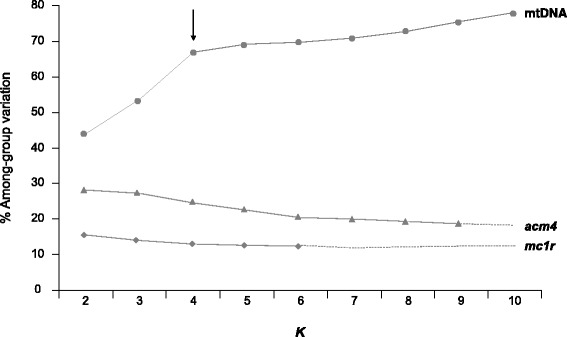
Summary of results of the spatial analysis of molecular variance (SAMOVA) analyses of the sampled populations of Podarcis tiliguerta for each locus. The percentage of variation explained by the among-group level of variation is reported for the best-clustering option at each pre-defined value of K (the number of groups). The arrow shows the best clustering option obtained for the mtDNA dataset, whereas SAMOVA analyses based on nuclear (mc1r and acm4) dataset do not allow identifying a population grouping best explaining the data. Dashed lines represent values of K for which fixation indexes were not significant
Mantel tests and scatterplots of pairwise genetic and geographic distances between populations showed no support for a pattern of isolation by distance at nuclear loci (Additional file 7: Figure S4). At the acm4 locus the association between either FST or on Phi-ST genetic distance and geographic distance was non-significant (FST: Z = 2525111.7735, r = 0.0733, one-sided P = 0.144; PhiST: Z = 6740977.5934, r = 0.0754, one-sided P = 0.130). At the mc1r locus the association between FST and geographic distance was not significant (Z = 3915308.0029, r = 0.0847, one-sided P = 0.124) whereas PhiST and geographic distance showed a weak but significant association (Z = 11681710.8232, r = 0.2539, one-sided P < 0.001) explaining less than 1% of the genetic variance at this locus (R2 = 0.064). However, like the previously generated scatter plots, the plot of the mc1r Phi-ST distance versus geographic distance failed to reveal a positive and monotonic relationship, showing instead a wide degree of scatter of genetic distance values over all geographic distances values (Additional file 7: Figure S4).
The AUC values of the SDM built using the five selected variable model were >0.7 for the training and the test data, indicating a good performance of the model. Projections of the model over LGM bioclimatic conditions using the MIROC and the CCSM databases produced comparable suitability areas, therefore only the MIROC model is shown (Fig. 5). Current species distribution model showed high bioclimatic suitability for P. tiliguerta across Corsica and Sardinia except for the Campidano plain in south Sardinia, which fits the known continuous distribution pattern of the species. Suitable bioclimatic conditions were widespread also during the LGM especially in the lowlands made available by marine regression such as in the wide expanse of land connecting Corsica and Sardinia. Lower suitability was apparent in mountainous regions of Corsica and western Sardinia.
Fig. 5.
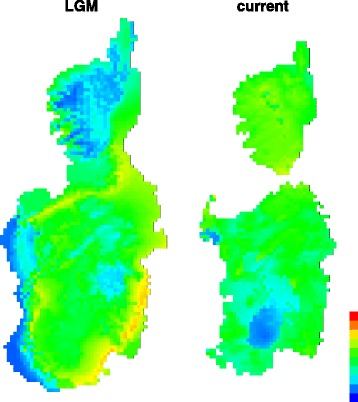
Species distribution models (SDM) based on bioclimatic variables for Podarcis tiliguerta under the present-day conditions (current) and the Last Glacial Maximum (LGM; ~21 thousands years ago, 21 kya). Warmer colours show areas with higher bioclimatic suitability
Discussion
An insular hotspot of genetic diversity within Podarcis tiliguerta
The Tyrrhenian wall lizard Podarcis tiliguerta shows unparalleled high genetic diversity compared either to island endemics or to continental species. Both at mitochondrial and nuclear loci, the amount of segregating sites, nucleotide diversity, and mean number of pairwise differences are many times greater in P. tiliguerta than in continental-island endemic species such as P. wagleriana or in species widespread across the Western Palaearctic such as P. muralis and the P. vaucheri species complex (Table 3). The values of mitochondrial nucleotide diversity found in P. tiliguerta are the highest observed in Podarcis lizards and among the highest observed in reptiles at the same loci. Such a vast nucleotide diversity is reflected in a complex phylogeographic pattern. Across the relatively small range of P. tiliguerta we found a geographic mosaic of 17 allopatric haplogroups sharply divided into four main mitochondrial lineages. The genetic divergence between these four lineages exceeds the mitochondrial differentiation usually observed between Podarcis species and in general between reptile species [24]. This high genetic diversity pattern is consistent with previous studies using mitochondrial [24, 40, 41] and nuclear [21] markers. In particular [21] found high level of heterozygosity within P. tiliguerta at allozyme loci with a value (H o = 0.066) that exceeds the average heterozygosity estimated in Podarcis species (H o = 0.053; [90]) and in reptile species (H o = 0.047; SD = 0.028 [91]).
The exceptional level of genetic diversity observed within P. tiliguerta is puzzling in view of its insular condition and continuous distribution across a restricted range (see Table 3 for species range size). Furthermore, the conditions for the long term persistence of such diversity have not been particularly more stable within the range of P. tiliguerta as it was for other Mediterranean islands compared to neighbouring continental areas [e.g. [58]]. Indeed, during the last glaciation northern Corsica was substantially influenced by polar air [92], and extensive glaciers formed on the central mountain chain during Pleistocene glacial phases [93]. Island endemics have long been considered as homogenous entities formed by a single panmictic population with size correlated to island size [94–96]. Likewise, P. tiliguerta is currently listed as Least Concern in the IUCN list, based on ‘its wide distribution and presumed large population’ [97]. The high genetic diversity, phylogeographic complexity and intra-island differentiation found within P. tiliguerta clearly challenges these simplified assumptions. Similar findings stand out from recent phylogeographic studies on Corsican Sardinian endemic vertebrates [13, 20, 32] as well as on other insular systems [e.g. [98, 99]] indicating that the complexity of evolutionary histories which took place in environmentally complex islands such as the Tyrrhenian islands engendered a wealth of diversity comparable to or even higher than that observed in continental settings.
Decoupled mito-nuclear histories at the root of the genetic diversity hotspot
Multilocus genetic variation in P. tiliguerta shows a profound discordance in spatial and phylogenetic patterning between mitochondrial and nuclear genomes. Two highly divergent mitochondrial lineages occur within each Corsica and Sardinia (Fig. 2), whereas nuclear variation is not phylogenetically (Fig. 3) or geographically (Fig. 4) structured. Under a neutral model of molecular evolution the mtDNA phylogeography of P. tiliguerta tells a very different story from nucDNA.
Divergence time estimates placed the coalescence of the Corsican and Sardinian mitochondrial lineages back to the Late Miocene (8–10 mya). Within-island divergence both in Corsica and Sardinia would have started accordingly somewhat later (7–9 mya), although the uncertainty over the monophyly of Corsican and Sardinian populations suggests that such postulation requires caution (Fig. 2; Additional file 4: Figure S2). The primary split between the biotas of Corsica and Sardinia occurred during the disjunction of the Corsica-Sardinia microplate which started 15 mya and was completed by 9 mya [14, 15, 100]. While such a palaeogeographic scenario may explain the onset of the between-islands divergence of mitochondrial lineages, on the other hand, Corsica and Sardinia have not remained geographically independent since their first separation. Further connections between them occurred during the Messinian Salinity Crisis (5.7–5.3 mya) [16, 101] and repeatedly in the Pleistocene at each ice age, from 2.58 mya up to 12 thousands years ago (12 kya) [18, 19]. Especially during the Pleistocene glaciations, faunal exchange between the two Islands has been intense, as documented in the other endemic lizards of Corsica and Sardinia, Archaeolacerta bedriagae [12, 27] and Algyroides fitzingeri [31], but also in other thermophilic endemic taxa (Hyla sarda [30]; Euleptes europaea (Salvi et al. unpublished data); see also preliminary data on Discoglossus sardus in [11]). Palaeogeographic data and results from SDM analyses (Fig. 5) suggest that also for P. tiliguerta the wide lowland area connecting Corsica and Sardinia during glaciation-induced marine regressions has offered suitable habitat and ample dispersal avenues between the two islands. If on one hand we have little evidence that the transitory separations between Corsica and Sardinian may have triggered the vicariance between the P. tiliguerta lineages of each island, on the other hand the geologic evolution of Corsica and Sardinia is clearly not associated to the ancient (Miocenic) intra-island divergence estimated between west and east Corsican (Lineage 1 and 2) lineages and between north and south Sardinian (Lineage 3 and 4) lineages. Deep phylogeographic partitions have been found in north Corsica, but not in Sardinia, for the endemic lizard A. bedriagae [12] and the Corsican newt Euproctus montanus [13] as well as in the snail Solatopupa guidoni [29]. While a possible association with an exceptionally dry period during the Pliocene may explain the allopatric divergence between the newt lineages [13], to date we still have no clues on possible historical (environmental) barriers which may have driven intra-island diversification in the former species.
Conversely, the pattern of genetic variation of nuclear markers is consistent with a more recent origin of the observed diversity and with a biogeographic scenario of high inter-island and intra-island connectivity. At both nuclear loci analysed we observed shallow divergence and extensive allele sharing between and within islands. This pattern may arise as a result of long-term demographic stability and absence of significant structuring processes in the recent evolutionary history, but this seems not to be the case for P. tiliguerta. Indeed, isolation by distance (IBD) analyses indicate a lack of regional equilibrium between genetic drift and gene flow at nuclear loci. The patterns of correlation between genetic and geographic distances (Additional file 4: Figure S4) strictly match the case III reported in [102], whereby genetic drift is more influential than gene flow. This pattern is expected if conditions conducive to dispersal have not been stable across the region for a long enough period of time for a migration–drift equilibrium to be established, such that at a regional level the species is fragmented (or has been recently fragmented) into small isolated populations [102]. Allozyme data on P. tiliguerta [21] also showed population fragmentation and extensive haplotype sharing, with a lack of alternatively fixed alleles between and within Corsica and Sardinia. Interestingly [21] found a latitudinal cline pattern of variation in the three loci displaying the larger allele frequency differences among populations (Idh-1, Gapd, and Gpi) and a significant IBD pattern in the overall allozyme frequencies. While it is difficult to disentangle IBD and clinal patterns of allozyme variation as the sampling of this study is structured along a north–south axis, such a latitudinal pattern lines up with changes in local bioclimatic regimes across the species range, leading to the hypothesis of a possible role for local adaptation in shaping allozyme frequencies and differentiation between populations [21].
Looking for the source of mito-nuclear discordance
Mito-nuclear discordance is a common phenomenon in animal systems [103], although the spatial and temporal degree of the discordance observed in P. tiliguerta is uncommon in literature (but see e.g. [104]]). In many cases it has been possible to conciliate the observed differences in spatial and phylogenetic patterns between mitochondrial and nuclear markers simply accounting for their different mode of inheritance and rates of evolution. Indeed, effective population size of the biparentally-inherited diploid nuclear genome is four times larger than the maternally-inherited haploid mitochondrial genome, which also displays higher mutation rates than the nuclear genome. Therefore, under neutral evolution, historical isolation between populations is readily imprinted in mitochondrial genomes in the form of allopatric genealogical clusters whereas more time is necessary for nuclear genealogies to be sorted (e.g. [105]). This scenario has been invoked to explain dissimilar mitochondrial and nuclear genealogies (including nd4 and mc1r respectively) in other Podarcis lizards such as P. fiflolensis [58], P. muralis [60] and the Iberian and North African species complex [106]. In these cases, further studies using nuclear markers with higher mutation rates (microsatellites) recovered the same groups identified by mitochondrial markers [107–109], corroborating the hypothesis of recent divergence with incomplete lineage sorting at some nuclear loci such as mc1r. On the other hand, in the case of P. tiliguerta the mitochondrial lineages have been independently evolving for as much as 7–13 million years and the genetic divergence observed between them is at the level of (or higher than) comparisons between distinct Podarcis species (see also Additional file 4: Figure S2). In these circumstances we may expect some level of sorting at nuclear loci between lineages as we actually observed at the mc1r when examining the phylogenetic relationships between haplotypes from different populations and species (Additional file 5: Figure S3).
If we assume that the deep phylogeographic breaks imprinted in the mitochondrial genome of P. tiliguerta are indicative of historical barriers to gene flow among populations, and based on previous studies on Podarcis [59, 80] we exclude a role of direct selection in shaping nuclear sequence variation, an alternative hypothesis to explain mito-nuclear discordance could be a scenario of allopatric divergence followed by secondary contact, with nuclear introgression mediated by male-biased asymmetries in dispersal or mating. Male-biased dispersal is unlikely to be the driver of such a pattern in P. tiliguerta as dispersal in Podarcis is not sex-biased and it would be in any case unrealistic to assume, under extensive gene flow at neutral nuclear loci, a complete lack of female dispersal over a few kilometres along phylogeographic breaks of >100 km during millions of years (see locations 21–22, 25–26 in Corsica or 62–63 and 64–65 in Sardinia, Figs. 1 and 2). On the other hand, strong asymmetries in male competitive ability and mating success, between distinct lizard lineages, may result in asymmetric hybridisation upon secondary contact [110]. While et al. [108] provided compelling evidence for such a scenario in the common wall lizard P. muralis and showed how allopatric divergence in sexually selected traits via male competition may determine asymmetric nuclear introgression following secondary contact, with replacement of nuclear characters of the sub-dominant lineage. In the case of P. muralis nuclear clines at neutral loci were shifted about 50 km compared to the mitochondrial break, and phenotypic clines (indicative of nuclear loci under sexual selection) were shifted even further (100–200 km). These kind of distances translated into the insular setting of Corsica and Sardinia may explain the replacement of sub-dominant lineages at nuclear loci and the maintenance of a sharp mitochondrial break within each island. However what makes this scenario less likely for P. tiliguerta is that these processes should have acted between islands as well, so that mitochondrial lineages should have been fixed at the zones of secondary contacts whereas one nuclear lineage would have replaced the others at the range-wide scale. An additional hypothesis to consider would be that of ghost mitochondrial DNA lineages corresponding to the remnants of former, now extinct species inhabiting the same regions which nuclear background was completely admixed. This scenario has been reported in other Podarcis species such as P. liolepis and P. hispanica [111]. However, this scenario seems highly farfetched for P. tiliguerta, since we would be in the presence of at least three such “ghosts”, which seems unlikely – particularly on an island setting. Moreover these scenarios are all hinged on allopatric divergence (e.g. a glacial refugia model in P. muralis [60]) and in the case of P. tiliguerta we have little biogeographic evidence for an ancient and prolonged vicariance of populations within and between islands, as discussed above.
Deep mitochondrial divergence can arise even in the absence of long-term barriers to gene flow as a result of stochastic and selective processes. Simulations show that strong phylogeographic structure at uniparental loci can form by chance in a continuous population, when dispersal and population size are low, due to the stochastic nature of the coalescent process [112–114]. While conditions for stochastic mtDNA lineage sorting are not met by P. tiliguerta (very low dispersal distances and population sizes), further studies have demonstrated that a small amount of selection for local adaptation dramatically increases the range of conditions – i.e. large population size and low to moderate dispersal - under which mitochondrial phylogeographic breaks can arise. Under this model a strong and stable mitochondrial structure can arise under rather weak selection and moderate dispersal in a continuously distributed species with gradual environmental variation [115]. These conditions and the expected pattern match rather well the case of P. tiliguerta. This scenario implies no long-term vicariance between populations, highly divergent mitochondrial clades that are geographically localized and unrelated to nuclear variation, and which have much longer coalescent time compared to neutral evolution [115]. This may explain why the origin and the persistence of mitochondrial partitions observed in P. tiliguerta do not fit the geological evolution of Corsica and Sardinia and in particular the historical geographic continuity and ecological connectivity of populations between and within islands. It may also account for the lack of association between mitochondrial and nuclear variation and the surprisingly deep divergence times estimated for the mitochondrial lineages under neutral expectations. Weak and/or not very recent selection may explain why selective neutrality tests failed to detect departure from neutrality in the mtDNA dataset, as these tests have been shown to have very low statistical power in such conditions [116].
There is increasing awareness and empirical evidence of selection on mtDNA due to the crucial role in metabolism of mtDNA encoded proteins and since they are part of a perfectly linked group that undergo a high mutation rate [reviewed in e.g. [115, 117, 118]]. A growing number of studies in animal systems showed that cases of striking mito-nuclear discordance can be explained by an association of mitochondrial variation with environmental variables such as climatic gradients [e.g. [119–122]]; see also [123]]. This indicates that geographically structured mtDNA diversity may reflect selective optima across environmental gradients rather than reliably track the species’ evolutionary history. The data collected in this study are not suitable to rigorously test the hypothesis of environmental selection on mtDNA and to understand the mechanisms through which putative female-linked selection operates. These may include selection on one or more mtDNA encoded proteins, W-linked genes, or at nuclear-encoded genes which are functionally related to mitochondrial (e.g. for proteins involved in the oxidative phosphorylation or related to mitochondrial metabolism). While much research will be needed to further investigate these aspects (including an exhaustive sampling across environmental gradients and a genomic-wide screening on mitochondrial, autosomal and sex-linked loci), preliminary evidence suggests that cyto-nuclear adaptation is a possible mechanism shaping genetic variation in Podarcis tiliguerta and in general in Corsican-Sardinian lizards. Indeed, [21] suggested that the latitudinal clinal pattern observed in P. tiliguerta at three highly heterogeneous allozyme loci (Idh-1, Gapd, and Gpi) was associated with local bioclimatic regimes. In the codistributed rock lizard A. bedriagae, [27] provided evidence for a significant association between the (non-clinal) variation at the Idh-1 locus and annual temperature and precipitation. Some of these enzymes such as the IDH have a close functional relationship with mitochondrial metabolism. Thus, these preliminary findings point to the interesting possibility that local adaptation leading to phylogeographic structure could influence both mitochondrial genes and nuclear genes functionally related to mitochondrial genes. This line of research appears particularly promising on Corsican Sardinian endemics for several reasons: (i) the well delimited and restricted spatial scale of this insular setting [19]; (ii) the availability of previous phylogeographic assessment and well known distribution of the species [12, 13, 20–34]; (iii) the growing number of cases of deep within-islands mitochondrial divergence that has been observed in the absence of a current or historical barrier to gene flow [12, 13, 29]; this study]; and (iv) preliminary evidence of association of allozyme genetic variation with climatic variables [21, 27]. Thus, Corsican-Sardinian endemic lizards appear a promising model for testing how and whether selection has been shaping patterns of within-species diversity in such a biodiversity hotspot.
Taxonomic and conservation implications
Based on the high value of genetic divergence observed between mitochondrial lineages of P. tiliguerta, it has been suggested that this species may represent a species complex [24]. Interpreting deep mitochondrial divergence as indicative of speciation would assume that mitochondrial lineages of P. tiliguerta will be the result of long-term barriers to gene flow under neutral evolution; an assumption which received little support in our study. Despite mitochondrial structure, both this study and [21] showed that nuclear variation is shared between and within Corsica and Sardinia with a lack of alternatively fixed alleles. Also morphological data on P. tiliguerta showed high variability with a north–south cline of variation across the main islands rather than a sharp transition between geographic units [124]. Bruschi and colleagues [125] based on 11 pholidotic characters failed to identify diagnostic characters between and within Corsican and Sardinian populations, although their sample was mainly composed by micro-insular populations (about 90% of the populations studied) from satellite islets surrounding Corsica and Sardinia, hence of little information as regards range-wide patterns. Therefore, at the moment we have no evidence of the existence of long-term barrier to gene flow within P. tiliguerta and convincing conclusions on the taxonomic value of the observed mitochondrial lineages can only be obtained after a fuller understanding of the evolutionary processes that led to the observed phylogeographic pattern.
On the other hand, whether the distinct phylogeographic groups observed within P. tiliguerta underline long-term isolation or local adaptation they indicate that this species does not represent in any case a single management and conservation unit. Therefore, each lineage should be individually targeted when setting strategies for the long-term conservation of P. tiliguerta and of its evolutionary potential.
Conclusions
Our study on the multilocus genetic variation of Podarcis tiliguerta reveals surprising levels of genetic diversity underlining a complex phylogeographic pattern with a striking example of mito-nuclear discordance. These findings have considerable implications for the taxonomy and conservation of P. tiliguerta as well as for our understanding of the processes involved in the evolution of biodiversity hotspots within Mediterranean islands. Neutral models based on long-term vicariance provide unparsimonious explanations for the deep phylogeographic breaks observed in mtDNA within and between islands, both in this species as well in other Corsican-Sardinian endemics. Growing empirical evidence suggests a possible role for local adaptation along a smooth environmental gradient underlying the origin and persistence of highly divergent, geographically localized, mitochondrial groups found in endemic amphibian and reptiles as well as of their geographic patterns of nuclear variation. These hypotheses could be thoroughly tested taking advantage of genomic resources. In this respect, this study represents one more step in our long-term aim to fully understand the evolutionary mechanisms underlying the rising of the Mediterranean diversity hotspot.
Acknowledgements
We are indebted with Miguel A. Carretero for his contribute to the conception of this study and his support in sample collection. We thank Pierluigi Bombi, Antigoni Kaliontzopoulou, João P.M.C. Maia, and Verónica Gomes for the help during fieldwork, Ana Perera for the help in laboratory work and Luis Machado for the help with species distribution modeling. Thanks to Daniele Canestrelli for fruitful discussion. Lizards were captured and handled under permits from the Italian Ministry of Environment (DPN-2009-0005106) and the Direction régionale de l’environnement, de l’aménagement et du logement (DREAL) de Corse.
Funding
This work was partially supported by FEDER through the COMPETE program, Portuguese national funds through the FCT (Fundação para a Ciência e a Tecnologia, Portugal) and by the project “Genomics and Evolutionary Biology” cofinanced by North Portugal Regional Operational Programme 2007/2013 (ON.2–O Novo Norte), under the National Strategic Reference Framework (NSRF-ERDF). DS, CP, and DJH, are supported by the FCT, Fundação para a Ciência e a Tecnologia (Portugal): DS, post-doctoral grant SFRH/BPD/105274/2014; CP, IF-contract IF/01597/2014; DJH, IF-contract IF/01627/2014. DS is currently supported by the program ‘Rita Levi Montalcini’ for the recruitment of young researchers at the University of L’Aquila.
Availability of data and materials
All sequence data generated in this study are publicly available in Genbank [Accession numbers KY561996-KY562564]. See also Tables 1, 2 and 3 for further details on sequence data used in this study.
Authors’ contributions
Conceived and designed the study: DS, DJH; Fieldwork sample collection: DS, DJH; Laboratory data collection: DS. Data analysis: DS, CP. Drafted the manuscript: DS. All authors reviewed and approved the manuscript. Provided funding: CP, DJH.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Lizards were captured and handled under permits from the Italian Ministry of Environment (DPN-2009-0005106) and the Direction régionale de l’environnement, de l’aménagement et du logement (DREAL) de Corse.
Abbreviations
- acm4
acetylcholinergic receptor M4
- BPP
Bayesian Posterior Probabilities
- BPP
Bootstrap Support
- mc1r
Melanocortin receptor 1
- ML
Maximum Likelihood
- nd4
NADH dehydrogenase subunit 4
Additional files
Summary results of haplotype phasing based on data from cloning. For each individual heterozygote sites are highlighted in green and the corresponding sequence of each replicated colony is reported. Nucleotide states in red font included in grey boxes indicate instances of nucleotide misincorporaitons (PCR/replication errors) in colonies and were solved following a parsimony approach. (PDF 103 kb)
Summary statistics of molecular diversity and neutrality tests for each locus and each mitochondrial clade within Podarcis tiliguerta. N: number of sampled gene copies; ns: number of sites of the alignment used for the calculations; S: number of segregating sites; h: number of haplotypes; Hd: haplotype diversity; π: nucleotide diversity; K: average number of pairwise differences; D: Tajima’s D values (1989). Significance values for the Tajima’s D tests based on 1000 coalescent simulations are shown next to the values (ns: P ≥ 0.05; *: 0.01 ≤ P < 0.05; **: 0.001 ≤ P < 0.01; ***: P < 0.001). (PDF 158 kb)
Results of statistical parsimony network analyses based on mtDNA data. Sub-networks recovered under the 95% limit for a parsimony connection. Haplotypes are represented by circles with size proportional to their frequencies, coloured according to the main mtDNA lineages and labelled according to the sublineages recovered in the phylogenetic analysis (see text for more details). Small white circles represent ‘unsampled or extinct haplotypes. (PDF 86 kb)
Bayesian phylogenetic tree of Podarcis based on mitochondrial sequences (12S and nd4). Bayesian Posterior Probabilities values > 0.90 are reported above the nodes; bootstrap values of the Maximum Likelihood analysis > 50 are reported below the nodes. (PDF 72 kb)
Statistical parsimony network showing the genealogical relationships between the mc1r haplotypes inferred for nine Podarcis species based on 946 sequences retrieved from Genbank [57–60, 80], generated in this study (P. tiliguerta) and unpublished (P. wagleriana; Salvi et al. in prep.). Haplotypes are represented by circles with size proportional to their frequency; small white circles represent ‘missing’ or extinct haplotypes. (PDF 104 kb)
Result of analyses of molecular variance (AMOVA) based on partitions defined by mtDNA variation of Podarcis tiliguerta. ns: P ≥ 0.05; *: 0.01 ≤ P < 0.05; **: 0.001 ≤ P < 0.01; ***: P < 0.001. (PDF 96 kb)
Patterns of correlation between genetic distances and geographic distance at nuclear loci. Scatterplots of the genetic distances (FST and PhiST) versus geographic distances (km) among population pairs of Podarcis tiliguerta at the loci acm4 and mc1r. The reduced major axis (RMA) regression lines are shown. (PDF 347 kb)
Contributor Information
Daniele Salvi, Email: danielesalvi.bio@gmail.com.
Catarina Pinho, Email: catarina@cibio.up.pt.
D. James Harris, Email: james@cibio.up.pt.
References
- 1.Médail F, Myers N. Mediterranean basin. In: Mittermeier RA, Gil PR, Hoffmann M, Pilgrim J, Brooks T, Mittermeier CG, Lamoreaux J, da Fonseca GAB, editors. Hotspots revisited: Earth’s biologically richest and most endangered terrestrial ecoregions. Mexico: CEMEX Monterrey, Conservation International, Washington and Agrupación Sierra Madre; 2004. pp. 144–147. [Google Scholar]
- 2.Médail F, Quézel P. Hot-spots analysis for conservation of plant biodiversity in the Mediterranean Basin. Ann Mo Bot Gard. 1997;84:112–127. doi: 10.2307/2399957. [DOI] [Google Scholar]
- 3.Médail F, Quézel P. Biodiversity hotspot in the mediterranean basin: setting global conservation priorities. Conserv Biol. 1999;13:1510–1513. doi: 10.1046/j.1523-1739.1999.98467.x. [DOI] [Google Scholar]
- 4.Thompson JD. Plant evolution in the mediterranean. New York: Oxford University Press; 2005. [Google Scholar]
- 5.Mouillot F, Paradis G, Andrei-Ruiz MC, Quilichini A. Corsica. In: Vogiatzakis IN, Pungetti G, Mannion AM, editors. Mediterranean island landscapes: natural and cultural approaches. New York: Springer Publishing; 2008. pp. 220–244. [Google Scholar]
- 6.Pungetti G, Marini A, Vogiatzakis IN. Sardinia. In: Vogiatzakis IN, Pungetti G, Mannion AM, editors. Mediterranean island landscapes: natural and cultural approaches. New York: Springer Publishing; 2008. pp. 146–169. [Google Scholar]
- 7.Kuhlemann J, Frisch W, Székely B, Dunkl I, Danišík M, Krumrei I. Würmian maximum glaciation in corsica. Austrian J Earth Sci. 2005;97:68–81. [Google Scholar]
- 8.Kuhlemann J, Rohling EJ, Krumrei I, Kubik P, IvyOchs S, Kucera M. Regional synthesis of mediterranean atmospheric circulation during the last glacial maximum. Science. 2008;321:1338–1340. doi: 10.1126/science.1157638. [DOI] [PubMed] [Google Scholar]
- 9.Fauquette S, Suc J-P, Guiot J, Diniz F, Feddi N, Zheng Z, Bessais E, Drivaliari A. Climate and biomes in the west mediterranean area during the pliocene. Palaeogeogr Palaeoclimatol Palaeoecol. 1999;152:15–36. doi: 10.1016/S0031-0182(99)00031-0. [DOI] [Google Scholar]
- 10.Médail F, Diadema K. Glacial refugia influence plant diversity patterns in the mediterranean basin. J Biogeogr. 2009;36:1333–1345. doi: 10.1111/j.1365-2699.2008.02051.x. [DOI] [Google Scholar]
- 11.Zangari F, Cimmaruta R, Nascetti G. Genetic relationships of the western mediterranean painted frogs based on allozymes and mitochondrial markers: evolutionary and taxonomic inferences (Amphibia, Anura, Discoglossidae) Biol J Linn Soc. 2006;87:515–536. doi: 10.1111/j.1095-8312.2006.00585.x. [DOI] [Google Scholar]
- 12.Salvi D, James Harris D, Bombi P, Carretero MA, Bologna MA. Mitochondrial phylogeography of the bedriaga’s rock lizard, Archaeolacerta bedriagae (Reptilia: Lacertidae) endemic to corsica and sardinia. Mol Phylogenet Evol. 2010;56:690–697. doi: 10.1016/j.ympev.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Bisconti R, Canestrelli D, Salvi D, Nascetti G. A geographic mosaic of evolutionary lineages within the insular endemic newt Euproctus montanus. Mol Ecol. 2013;22:143–156. doi: 10.1111/mec.12085. [DOI] [PubMed] [Google Scholar]
- 14.Alvarez W. Rotation of the corsica—sardinia microplate. Nature. 1972;235:103–105. [Google Scholar]
- 15.Cherchi A, Montadert L. Oligo—miocene rift of sardinia and the early history of the western mediterranean basin. Nature. 1982;298:736–739. doi: 10.1038/298736a0. [DOI] [Google Scholar]
- 16.Boccaletti M, Ciaranfi N, Cosentino D, Deiana G, Gelati R, Lentini F, et al. Palinspatic restoration and palaeogeographic reconstruction of the peri-Tyrrhenian area during the Neogene. Palaeogeogr Palaeoclimatol Palaeoecol. 1990;77:41–50. doi: 10.1016/0031-0182(90)90097-Q. [DOI] [Google Scholar]
- 17.Carmignani L, Decandia FA, Disperati L, Fantozzi PL, Lazzarotto A, Liotta D, Oggiano G. Relationships between the tertiary structural evolution of the sardinia-corsica-provencal domain and the northern apennines. Terra Nova. 1995;7:128–137. doi: 10.1111/j.1365-3121.1995.tb00681.x. [DOI] [Google Scholar]
- 18.Lambeck K, Antonioli F, Purcell A, Silenzi S. Sea-level change along the Italian coast for the past 10,000 yr. Quat Sci Rev. 2004;23:1567–1598. doi: 10.1016/j.quascirev.2004.02.009. [DOI] [Google Scholar]
- 19.Ketmaier V, Caccone A. Twenty years of molecular biogeography in the West Mediterranean islands of Corsica and Sardinia: lessons learnt and future prospects. In: Silva-Opps M, editor. Current Progress in Biological Research. 2013. InTech. doi:10.5772/55458. http://www.intechopen.com/books/current-progress-in-biological-research/twenty-years-of-molecular-biogeography-in-the-west-mediterranean-islands-of-corsica-and-sardinia-les.
- 20.Salvi D, Bisconti R, Canestrelli D. High phylogeographical complexity within mediterranean islands: insights from the corsican fire salamander. J Biogeogr. 2016;43:192–203. doi: 10.1111/jbi.12624. [DOI] [Google Scholar]
- 21.Capula M. Evolutionary genetics of the insular lacertid lizard Podarcis tiliguerta: genetic structure and population heterogeneity in a geographically fragmented species. Heredity. 1996;77:518–529. doi: 10.1038/hdy.1996.179. [DOI] [Google Scholar]
- 22.Ketmaier V, Argano R, Caccone A. Phylogeography and molecular rates of subterranean aquatic Stenasellid Isopod with a peri-Tyrrhenian distribution. Mol Ecol. 2003;12:547–555. doi: 10.1046/j.1365-294X.2003.01734.x. [DOI] [PubMed] [Google Scholar]
- 23.Ketmaier V, Casale A, Cobolli M, De Matthaeis E, Vigna TA. Biochemical systematics and phylogeography of the Percus strictus subspecies (Coleoptera, Carabidae), endemic to Sardinia. Ital J Zool. 2003;70:339–346. doi: 10.1080/11250000309356540. [DOI] [Google Scholar]
- 24.Harris DJ, Pinho C, Carretero MA, Corti C, Böhme W. Determination of genetic diversity within the insular lizard Podarcis tiliguerta using mtDNA sequence data, with a reassessment of the phylogeny of Podarcis. Amphib-reptil. 2005;26:401–407. doi: 10.1163/156853805774408676. [DOI] [Google Scholar]
- 25.Falchi A, Paolini J, Desjobert JM, Melis A, Costa J, Varesi L. Phylogeography of Cistus creticus L. on Corsica and Sardinia inferred by the TRNL-F and RPL32-TRNL sequences of cpDNA. Mol Phylogenet Evol. 2009;52:538–543. doi: 10.1016/j.ympev.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Salvi D, Capula M, Bombi P, Bologna MA. How many Archaeolacerta inhabit the corso-sardinian plate? allozyme variation and differentiation in Archaeolacerta bedriagae (Camerano, 1885) Amphib-reptil. 2009;30:463–470. doi: 10.1163/156853809789647077. [DOI] [Google Scholar]
- 27.Salvi D, Capula M, Bombi P, Bologna MA. Genetic variation and its evolutionary implications in a mediterranean island endemic lizard. Biol J Linn Soc. 2009;98:661–676. doi: 10.1111/j.1095-8312.2009.01313.x. [DOI] [Google Scholar]
- 28.Gentile G, Campanaro A, Carosi M, Sbordoni V, Argano R. Phylogeography of Helleria brevicornis Ebner 1868 (Crustacea, Oniscidea): old and recent differentiations of an ancient lineage. Mol Phylogenet Evol. 2010;54:640–646. doi: 10.1016/j.ympev.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Ketmaier V, Manganell G, Tiedemann R, Giusti F. Peri-Tyrrhenian phylogeography in the land snail Solatopupa guidoni (Pulmonata) Malacologia. 2010;52:81–96. doi: 10.4002/040.052.0106. [DOI] [Google Scholar]
- 30.Bisconti R, Canestrelli D, Colangelo P, Nascetti G. Multiple lines of evidence for demographic and range expansion of a temperate species (Hyla sarda) during the last glaciation. Mol Ecol. 2011;20:5313–5327. doi: 10.1111/j.1365-294X.2011.05363.x. [DOI] [PubMed] [Google Scholar]
- 31.Salvi D, Harris DJ, Perera A, Bologna MA, Carretero MA. Preliminary survey on genetic variation within the pygmy algyroides, Algyroides fitzingeri, across corsica and sardinia. Amphib-reptil. 2011;32:281–286. doi: 10.1163/017353711X556989. [DOI] [Google Scholar]
- 32.Bisconti R, Canestrelli D, Nascetti G. Has living on islands been so simple? Insights from the insular endemic frog Discoglossus montalentii. PLoS One. 2013;8:e55735. doi: 10.1371/journal.pone.0055735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dettori CA, Sergi S, Tamburini E, Bacchetta G. The genetic diversity and spatial genetic structure of the corso-sardinian endemic Ferula arrigonii Bocchieri (Apiaceae) Plant Biol. 2013;16:1005–1013. doi: 10.1111/plb.12145. [DOI] [PubMed] [Google Scholar]
- 34.Thibault J-C, Cibois A, Prodon R, Pasquet E. Quaternary history of an endemic passerine bird on corsica island: glacial refugium and impact of recent forest regression. Quatern Res. 2016;85:271–78. doi: 10.1016/j.yqres.2016.01.002. [DOI] [Google Scholar]
- 35.Bombi P, Salvi D, Luiselli L, Bologna MA. Modelling correlates of microhabitat use of two sympatric lizards: a model selection approach. Anim Biol. 2009;59:1–21. doi: 10.1163/157075609X417134. [DOI] [Google Scholar]
- 36.Bruschi S, Corti C, Capula M. Podarcis tiliguerta (Gmelin, 1789) In: Corti C, Capula M, Luiselli L, Razzetti E, Sindaco R, editors. Fauna d’Italia. Reptilia. Bologna: Edizioni Calderini de Il Sole 24 Ore Editoria Specializzata S.r.l; 2010. pp. 417–442. [Google Scholar]
- 37.Delaguerre M, Cheylan M. Atlas de répartition des batraciens et reptiles de corse. Ajaccio: Parc Naturel Régional de Corse; 1992. [Google Scholar]
- 38.Salvi D, Bombi P. Reptiles of Sardinia: updating the knowledge on their distribution. Acta Herpetol. 2010;5:161–177. [Google Scholar]
- 39.Thiede J. A glacial mediterranean. Nature. 1978;276:680–683. doi: 10.1038/276680a0. [DOI] [Google Scholar]
- 40.Podnar M, Mayer W. Can mitochondrial DNA draw the phylogenetic picture of central mediterranean island Podarcis? Herpetozoa. 2005;18:73–77. [Google Scholar]
- 41.Vasconcelos R, Harris DJ, Carretero MA, Pinho C, Corti C, Capula M, Bassu L, Spano G, Delauguerre M. Genetic diversity within corsican and sardinian specimens of the tyrrhenian wall lizard, Podarcis tiliguerta, estimated using mtDNA sequences. In: Corti C, Lo Cascio P, Biaggini M, editors. Mainland and insular lizards: a mediterranean perspective. Florence: Firenze University Press; 2006. [Google Scholar]
- 42.Harris DJ. Reassessment of comparative genetic distance in reptiles from the mitochondrial cytochrome b gene. Herp J. 2002;12:85–86. [Google Scholar]
- 43.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2. New York: Cold Spring Harbor Press; 1989. [Google Scholar]
- 44.Mendes J, Harris DJ, Carranza S, Salvi D. Evaluating the phylogenetic signal limit from mitogenomes, slow evolving nuclear genes, and the concatenated approach. new insights into the lacertini radiation using fast evolving nuclear genes and species trees. Mol Phylogenet Evol. 2016;100:254–267. doi: 10.1016/j.ympev.2016.04.016. [DOI] [PubMed] [Google Scholar]
- 45.Harrigan RJ, Mazza ME, Sorenson MD. Computation vs. cloning: evaluation of two methods for haplotype determination. Mol Ecol Resour. 2008;8:1239–1248. doi: 10.1111/j.1755-0998.2008.02241.x. [DOI] [PubMed] [Google Scholar]
- 46.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):vev003. doi: 10.1093/ve/vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin DP, Posada D, Crandall KA, Williamson C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res Hum Retroviruses. 2005;21:98–102. doi: 10.1089/aid.2005.21.98. [DOI] [PubMed] [Google Scholar]
- 50.Padidam M, Sawyer S, Fauquet CM. Possible emergence of new gemini viruses by frequent recombination. Virology. 1999;265:218–225. doi: 10.1006/viro.1999.0056. [DOI] [PubMed] [Google Scholar]
- 51.Smith JM. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34:126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 52.Martin D, Rybicki E. RDP: detection of recombination amongst aligned sequences. Bioinformatics. 2000;16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 53.Gibbs MJ, Armstrong JS, Gibbs AJ. Sister-scanning: a monte carlo procedure for assessing signals in recombinant sequences. Bioinformatics. 2000;16:573–582. doi: 10.1093/bioinformatics/16.7.573. [DOI] [PubMed] [Google Scholar]
- 54.Librado P, Rozas J. DnaSP v5. a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–1452. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 55.Pinho C, Harris DJ, Ferrand N. Contrasting patterns of population subdivision and historical demography in three western mediterranean lizard species inferred from mitochondrial DNA variation. Mol Ecol. 2007;16:1191–1205. doi: 10.1111/j.1365-294X.2007.03230.x. [DOI] [PubMed] [Google Scholar]
- 56.Pinho C, Ferrand N, Harris DJ. Reexamination of the iberian and north african Podarcis (squamata: lacertidae) phylogeny based on increased mitochondrial DNA sequencing. Mol Phylogenet Evol. 2006;38(1):266–273. doi: 10.1016/j.ympev.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 57.Pinho C, Rocha S, Carvalho BM, Lopes S, Mourão S, Vallinoto M, Brunes TO, Haddad CFB, Gonçalves H, Sequeira F, Ferrand N. New primers for the amplification and sequencing of nuclear loci in a taxonomically wide set of reptiles and amphibians. Conserv Genet Resour. 2010;2:181–185. doi: 10.1007/s12686-009-9126-4. [DOI] [Google Scholar]
- 58.Salvi D, Schembri PJ, Sciberras A, Harris DJ. Evolutionary history of the maltese wall lizard Podarcis filfolensis: insights on the ‘expansion-contraction’ model of the pleistocene biogeography. Mol Ecol. 2014;23:1167–1187. doi: 10.1111/mec.12668. [DOI] [PubMed] [Google Scholar]
- 59.Buades JM, Rodríguez V, Terrasa B, Pérez-Mellado V, Brown RP, Castro JA, Picornell A, Ramon MM. Variability of the mc1r gene in melanic and non-melanic Podarcis lilfordi and Podarcis pityusensis from the balearic archipelago. PLoS One. 2013;8(1):e53088. doi: 10.1371/journal.pone.0053088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salvi D, Harris DJ, Kaliontzopoulou A, Carretero MA, Pinho C. Persistence across Pleistocene ice ages in Mediterranean and extra-Mediterranean refugia: phylogeographic insights from the common wall lizard. BMC Evol Bio. 2013;13:147. doi: 10.1186/1471-2148-13-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaliontzopoulou A, Pinho C, Harris DJ, Carretero MA. When cryptic diversity blurs the picture: a cautionary tale from Iberian and North African Podarcis wall lizards. Biol J Linn Soc Lond. 2011;103:779–800. doi: 10.1111/j.1095-8312.2011.01703.x. [DOI] [Google Scholar]
- 62.Lima A, Pinho C, Larbes S, Carretero MA, Brito JC, Harris DJ. Relationships of Podarcis wall lizards from Algeria based on mtDNA data. Amphib-reptil. 2009;30(4):483–492. doi: 10.1163/156853809789647103. [DOI] [Google Scholar]
- 63.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 67.Heled J, Drummond AJ. Bayesian inference of species trees from multilocus data. Mol Biol Evol. 2010;27(3):570–580. doi: 10.1093/molbev/msp274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lanfear R, Calcott B, Ho SYW, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- 69.Templeton AR, Crandall KA, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132:619–633. doi: 10.1093/genetics/132.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1660. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 71.Hart MW, Sunday J. Things fall apart: biological species form unconnected parsimony networks. Biol Lett. 2007;3:509–512. doi: 10.1098/rsbl.2007.0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Silvestro D, Michalak I. RaxmlGUI: a graphical front-end for RAxML. Org Divers Evol. 2012;12:335–337. doi: 10.1007/s13127-011-0056-0. [DOI] [Google Scholar]
- 73.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 74.Maddison WP, Maddison DR. Mesquite: a modular system for evolutionary analysis. Version 3.04. 2015. http://mesquiteproject.wikispaces.com/.
- 75.Shimodaira H, Hasegawa M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol Biol Evol. 1999;16:1114–1116. doi: 10.1093/oxfordjournals.molbev.a026201. [DOI] [Google Scholar]
- 76.Shimodaira H. An approximately unbiased test of phylogenetic tree selection. Syst Biol. 2002;51:492–508. doi: 10.1080/10635150290069913. [DOI] [PubMed] [Google Scholar]
- 77.Shimodaira H, Hasegawa M. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics. 2001;17:1246–1247. doi: 10.1093/bioinformatics/17.12.1246. [DOI] [PubMed] [Google Scholar]
- 78.Posada D, Crandall KA. Intraspecific phylogenetics: Trees grafting into networks. Trends Ecol Evol. 2001;16:37–45. doi: 10.1016/S0169-5347(00)02026-7. [DOI] [PubMed] [Google Scholar]
- 79.Santos AM, Cabezas MP, Tavares AI, Xavier R, Branco M. tcsBU: a tool to extend TCS network layout and visualization. Bioinformatics. 2015;32:627–628. doi: 10.1093/bioinformatics/btv636. [DOI] [PubMed] [Google Scholar]
- 80.Fulgione D, Lega C, Trapanese M, Buglione M. Genetic factors implied in melanin-based coloration of the Italian wall lizard. J Zool. 2015;296:278–285. doi: 10.1111/jzo.12242. [DOI] [Google Scholar]
- 81.Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11(12):2571–2581. doi: 10.1046/j.1365-294X.2002.01650.x. [DOI] [PubMed] [Google Scholar]
- 82.Excoffier L, Smouse P, Quattro JM. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics. 1992;131:479–491. doi: 10.1093/genetics/131.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Excoffier L, Lischer HEL. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res. 2010;10:64–567. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 84.Jensen JL, Bohonak AJ, Kelley ST. Isolation by distance, web service. BMC Genet. 2005;6:13. doi: 10.1186/1471-2156-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Phillips SJ, Anderson RP, Schapire RE. Maximum entropy modelling of species’ geographic distributions. Ecol Model. 2006;190:231–259. doi: 10.1016/j.ecolmodel.2005.03.026. [DOI] [Google Scholar]
- 86.Fourcade Y, Engler JO, Rodder D, Secondi J. Mapping species distributions with Maxent using a geographically biased sample of presence data: a performance assessment of methods for correcting sampling bias. PLoS One. 2014;9:e97122. doi: 10.1371/journal.pone.0097122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu C, Berry PM, Dawson TP, Pearson RG. Selecting thresholds of occurrence in the prediction of species distribution. Ecography. 2005;28:385–393. doi: 10.1111/j.0906-7590.2005.03957.x. [DOI] [Google Scholar]
- 88.Warren DL, Seifert SN. Ecological niche modeling in Maxent: the importance of model complexity and the performance of model selection criteria. Ecol Appl. 2011;21:335–342. doi: 10.1890/10-1171.1. [DOI] [PubMed] [Google Scholar]
- 89.Carranza S, Arnold EN. A review of the geckos of the genus Hemidactylus (Squamata: Gekkonidae) from Oman based on morphology, mitochondrial and nuclear data, with descriptions of eight new species. Zootaxa. 2012;95:1–95. [Google Scholar]
- 90.Capula M. Ricerche sulla struttura genetica di Podarcis sicula, P. wagleriana e P. filfolensis (Reptilia: Lacertidae): aspetti tassonomici ed evolutivi. Bologna: Università degli studi di Bologna, PhD Thesis. 1990.
- 91.Nevo E. Genetic variation in natural populations: patterns and theory. Theor Pop Biol. 1978;13:1231–177. doi: 10.1016/0040-5809(78)90039-4. [DOI] [PubMed] [Google Scholar]
- 92.Kuhlemann J, Rohling EJ, Krumrei I, Kubik P, IvyOchs S, Kucera M. Regional synthesis of Mediterranean atmospheric circulation during the Last Glacial Maximum. Science. 2008;321:1338–1340. doi: 10.1126/science.1157638. [DOI] [PubMed] [Google Scholar]
- 93.Kuhlemann J, Frisch W, Székely B, Dunkl I, Kázmér M. Würmian maximum glaciation in Corsica. Austrian J Earth Sci. 2005;97:68–81. [Google Scholar]
- 94.Frankham R. Do island populations have less genetic variation than mainland populations? Heredity. 1996;78:311–327. doi: 10.1038/hdy.1997.46. [DOI] [PubMed] [Google Scholar]
- 95.Woolfit M, Bromham L. Population size and molecular evolution on islands. Proc R Soc Lond B. 2005;272:2277–2282. doi: 10.1098/rspb.2005.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Whittaker RJ, Fernández-Palacios JM. Island Biogeography: ecology, evolution, and conservation. Oxford: Oxford University Press; 2007. [Google Scholar]
- 97.Cheylan M, Corti C, Sindaco R, Romano R. Podarcis tiliguerta. In: The IUCN Red List of Threatened Species. 2009:e.T157293A5072953. http://dx.doi.org/10.2305/IUCN.UK.2009.RLTS.T157293A5072953.en.
- 98.Holland BS, Hadfield MG. Islands within an island: phylogeography and conservation genetics of the endangered Hawaiian tree snail Achatinella mustelina. Mol Ecol. 2002;11:365–376. doi: 10.1046/j.1365-294X.2002.01464.x. [DOI] [PubMed] [Google Scholar]
- 99.Barker BS, Waide RB, Cook JA. Deep intra-island divergence of a montane forest endemic: phylogeography of the Puerto Rican frog Eleutherodactylus portoricensis (Anura: Eleutherodactylidae) J Biogeogr. 2011;38:2311–2325. doi: 10.1111/j.1365-2699.2011.02578.x. [DOI] [Google Scholar]
- 100.Caccone A, Milinkovitch MC, Sbordoni V, Powell JR. Molecular biogeography: using the Corsica-Sardinia microplate disjunction to calibrate mitochondrial rDNA evolutionary rates in mountain newts (Euproctus) J Evol Biol. 1994;7:227–245. doi: 10.1046/j.1420-9101.1994.7020227.x. [DOI] [Google Scholar]
- 101.Krijgsman W, Hilgen F, Raffi I, et al. Chronology, causes and progression of the Messinian salinity crisis. Nature. 1999;400:652–654. doi: 10.1038/23231. [DOI] [Google Scholar]
- 102.Hutchinson DW, Templeton AR. Correlation of pairwise genetic and geographic distance measures: inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution. 1999;53:1898–1914. doi: 10.2307/2640449. [DOI] [PubMed] [Google Scholar]
- 103.Toews DPL, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol. 2012;21:3907–3930. doi: 10.1111/j.1365-294X.2012.05664.x. [DOI] [PubMed] [Google Scholar]
- 104.Giska I, Sechi P, Babik W. Deeply divergent sympatric mitochondrial lineages of the earthworm Lumbricus rubellus are not reproductively isolated. BMC Evol Biol. 2015;15:217. doi: 10.1186/s12862-015-0488-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Palumbi SR, Cipriano F, Hare MP. Predicting nuclear gene coalescence from mitochondrial data: the three time-rule. Evolution. 2001;55:859–868. doi: 10.1554/0014-3820(2001)055[0859:PNGCFM]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 106.Pinho C, Harris DJ, Ferrand N. Non-equilibrium estimates of gene flow inferred from nuclear genealogies suggest that Iberian and North African wall lizards are an assemblage of incipient species. BMC Evol Biol. 2008;8:63. doi: 10.1186/1471-2148-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rodríguez V, Brown RP, Terrasa B, Pérez-Mellado V, Picornell A, Castro JA, Ramon C. Genetic diversity and historical biogeography of the Maltese wall lizard, Podarcis filfolensis (Squamata: Lacertidae) Conserv Genet. 2014;15:295–304. doi: 10.1007/s10592-013-0538-z. [DOI] [Google Scholar]
- 108.While GM, Michaelides S, Heathcote RJP, MacGregor HEA, Zajac N, Beninde J, et al. Sexual selection drives asymmetric introgression in wall lizards. Ecol Lett. 2015;18:1366–1375. doi: 10.1111/ele.12531. [DOI] [PubMed] [Google Scholar]
- 109.Pinho C, Harris DJ, Ferrand N. Comparing patterns of nuclear and mitochondrial divergence in a cryptic species complex: the case of Iberian and North African wall lizards (Podarcis, Lacertidae) Biol J Linn Soc. 2007;91:121–133. doi: 10.1111/j.1095-8312.2007.00774.x. [DOI] [Google Scholar]
- 110.Canestrelli D, Porretta D, Lowe DW, Bisconti R, Carere C, Nascetti G. The tangled evolutionary legacies of range expansion and hybridization. Trends Ecol Evol. 2016;31:677–688. doi: 10.1016/j.tree.2016.06.010. [DOI] [PubMed] [Google Scholar]
- 111.Renoult JP, Geniez P, Bacquet P, Benoit L, Crochet PA. Morphology and nuclear markers reveal extensive mitochondrial introgressions in the Iberian Wall Lizard species complex. Mol Ecol. 2009;18:4298–4315. doi: 10.1111/j.1365-294X.2009.04351.x. [DOI] [PubMed] [Google Scholar]
- 112.Irwin DE. Phylogeographic breaks without geographic barriers to gene flow. Evolution. 2002;56:2383–2394. doi: 10.1111/j.0014-3820.2002.tb00164.x. [DOI] [PubMed] [Google Scholar]
- 113.Rauch EM, Bar-Yam Y. Theory predicts the uneven distribution of genetic diversity within species. Nature. 2004;431:449–452. doi: 10.1038/nature02745. [DOI] [PubMed] [Google Scholar]
- 114.Kuo C-H, Avise JC. Phylogeographic breaks in lowdispersal species: the emergence of concordance across gene trees. Genetica. 2005;124:179–186. doi: 10.1007/s10709-005-2095-y. [DOI] [PubMed] [Google Scholar]
- 115.Irwin DE. Local adaptation along smooth ecological gradients causes phylogeographic breaks and phenotypic clustering. Am Nat. 2012;180:35–49. doi: 10.1086/666002. [DOI] [PubMed] [Google Scholar]
- 116.Simonsen KL, Churchill GA, Aquadro CF. Properties of statistical tests of neutrality. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ballard JWO, Whitlock MC. The incomplete natural history of mitochondria. Mol Ecol. 2004;13:729–744. doi: 10.1046/j.1365-294X.2003.02063.x. [DOI] [PubMed] [Google Scholar]
- 118.Hudson RR, Turelli M. Stochasticity overrules the ‘three-time rule’: genetic drift, genetic draft and coalescence times for nuclear loci vs. mtDNA. Evolution. 2003;57:182–190. doi: 10.1111/j.0014-3820.2003.tb00229.x. [DOI] [PubMed] [Google Scholar]
- 119.Cheviron ZA, Brumfield RT. Migration-selection balance and local adaptation of mitochondrial haplotypes in rufouscollared sparrows (Zonotrichia capensis) along an elevational gradient. Evolution. 2009;63:1593–1605. doi: 10.1111/j.1558-5646.2009.00644.x. [DOI] [PubMed] [Google Scholar]
- 120.Dhillon RS, Schulte PM. Intraspecific variation in the thermal plasticity of mitochondria in killifish. J Exp Biol. 2011;214:3639–3648. doi: 10.1242/jeb.057737. [DOI] [PubMed] [Google Scholar]
- 121.Ribeiro ÂM, Lloyd P, Bowie RCK. A tight balance between natural selection and gene flow in a southern African arid-zone endemic bird. Evolution. 2011;65:3499–3514. doi: 10.1111/j.1558-5646.2011.01397.x. [DOI] [PubMed] [Google Scholar]
- 122.Pavlova A, Amos JN, Joseph L, Loynes K, Austin JJ, Keogh JS, Stone GN, Nicholls JA, Sunnucks P. Perched at the mito-nuclear crossroads: divergent mitochondrial lineages correlate with environment in the face of ongoing nuclear gene flow in an Australian bird. Evolution. 2013;67:3412–3428. doi: 10.1111/evo.12107. [DOI] [PubMed] [Google Scholar]
- 123.Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD. Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci. 2003;100:171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schneider B. Lacerta tiliguerta von Korsosardinien. Variabilitätsanalyse metrischer Merkmale. Das Aquar. 1973;7(48):246–247. [Google Scholar]
- 125.Bruschi S, Corti C, Carretero MA, Harris DJ, Lanza B, Leviton A. Comments on the status of the Sardinian-Corsican lacertid lizard Podarcis tiliguerta. Proc Calif Acad Sci. 2006;57:225–245. [Google Scholar]
- 126.Harris DJ, Arnold EN. Relationships and evolution of wall lizards, Podarcis (Reptilia, Lacertidae) based on partial mitochondrial DNA sequences. Copeia. 1999;3:740–754. [Google Scholar]
- 127.Harris DJ, Arnold EN, Thomas RH. Relationships of lacertid lizards (Reptilia: Lacertidae) estimated from mitochondrial DNA sequences and morphology. Proc R Soc Lond B Biol Sci. 1998;265(1409):1939–1948. doi: 10.1098/rspb.1998.0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All sequence data generated in this study are publicly available in Genbank [Accession numbers KY561996-KY562564]. See also Tables 1, 2 and 3 for further details on sequence data used in this study.