

Abstract
Bacterial biocatalysts play a key role in our transition to a bio-based, post-petroleum economy. However, the discovery of new biocatalysts is currently limited by our ability to analyze genomic information and our capacity of functionally screening for desired activities. Here, we present a simple workflow that combines functional metaproteomics and metagenomics, which facilitates the unmediated and direct discovery of biocatalysts in environmental samples. To identify the entirety of lipolytic biocatalysts in a soil sample contaminated with used cooking oil, we detected all proteins active against a fluorogenic substrate in sample’s metaproteome using a 2D-gel zymogram. Enzymes’ primary structures were then deduced by tryptic in-gel digest and mass spectrometry of the active protein spots, searching against a metagenome database created from the same contaminated soil sample. We then expressed one of the novel biocatalysts heterologously in Escherichia coli and obtained proof of lipolytic activity.
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-017-0247-9) contains supplementary material, which is available to authorized users.
Keywords: Zymogram, Lipase, Biocatalyst, Metagenomics, Metaproteomics
Main text
A conceptually straightforward way to identify new microbial biocatalysts is the screening of a multitude of organisms isolated from an environmental sample for a desired enzymatic activity [1]. However, due to our inability to cultivate the vast majority of microorganisms in the lab, such a screening will miss potentially more than 99% of organisms present in a given environmental sample [2]. To counter this problem, DNA-based, culture-independent approaches have now become the state-of-the-art in biocatalyst discovery. These methods rely on library-based screening efforts, where an expression library from environmental DNA is screened for a certain activity. Coining the term metagenome, this concept was introduced by Handelsmann and co-workers [3] and has been used e.g., in large-scale projects to identify lipolytic enzymes from soil metagenomes [4]. This approach typically involves screening hundreds of thousands of clones, and the number of biocatalytically active proteins discovered is dependent on the library size. An alternative is the in silico search for homologs of known biocatalysts in metagenomic datasets, a method we have recently employed ourselves [5], and which is comprehensively reviewed in [6]. This method uses known structural motifs to find novel enzymes in sequence databases. Rapid advances in sequence-based metagenomics and a plethora of publicly available DNA data have led to a widespread adoption in the scientific community. However, it can be argued that in silico screening loses the immediacy of an activity-based, i.e., structurally unbiased, discovery by adding an additional layer of abstraction in the form of DNA-sequence data.
Here, we present a functional metaproteomic approach as a method for rapid enzyme discovery. This method combines the immediacy of an activity-based screening with the independence from lab-cultivability of “meta-omic” approaches. This approach is conceptually comprehensive as it has the potential to discover all enzymes that exhibit an activity that can be screened for in an environmental sample, in principle facilitating the discovery of novel structure-function pairs. The method does not rely on a comprehensive evaluation of the metagenome and metaproteome data but rather utilizes both to simplify the discovery of proteins exhibiting a desired enzyme activity.
Metaproteomics is quickly becoming a well-established high-throughput “meta-omic” approach to study microbial ecology, as recently reviewed in [7] and [8]. Metaproteomics was developed by Bond and Wilmes to mine microbiomes for novel proteins from previously uncultured organisms [9], and one of its earliest applications was the functional study of biocatalysts that degrade organochloride pollutants [10].
We also used a functional metaproteomic approach to identify lipolytic enzymes from environmental sources. Thus, we collected samples from a site where we expected microorganisms harboring these activities to dwell in large numbers. We harvested one oil-contaminated soil sample from a restaurant’s used cooking oil disposal site and used it for enriching microorganisms with lipolytic activity. Proteins and DNA were isolated from the same sample (see Additional file 1 for more details). The proteins (600 μg of protein) were separated by two-dimensional (2D) polyacrylamide gel electrophoresis. After separation, proteins were refolded in the gel and an in-gel activity assay based on the fluorogenic lipase substrate para-methylumbelliferyl butyrate (pMUB) was performed. pMUB is a substrate that can be used to detect a wide variety of lipolytic and hydrolytic enzymes with high sensitivity [11]. Lipolytic enzymes present in the gel hydrolyzed pMUB and released butyric acid and p-methylumbelliferone, which is a fluorescent dye that can be detected under ultraviolet light. The intensity of the spot is dependent both on the quantity of the protein and its activity. With this method, we identified 14 lipolytically active spots in our protein sample (Fig. 1). These experiments were performed in duplicates. The fluorescing protein spots were then excised from the gels, tryptically digested and analyzed by mass spectrometry. Mass spectrometry-based protein identification is facilitated by searchable databases of predicted masses that arise from the fragmentation of tryptic peptides. We therefore created such a database from the metagenomics sequences we obtained from the DNA isolated from the sample. Through next-generation sequencing, we obtained a high quality DNA dataset with most sequences showing a Phred score higher than 35 indicating a base call accuracy close to 99.99% [12]. The sequenced raw data was assembled to recover original genome information and to predict proteins using the assembly software SPAdes v3.1.1 [13] and annotated using PROKKA v1.10 [14]. Prokka uses Prodigal to identify coding sequences in the assembled metagenome [15] and then transfers the annotation of the most significant match from a hierarchy of data sources to these sequences. Coding sequences that do not match are labeled as hypothetical protein. The obtained database contained approximately 161,000 proteins, of which 37.6% were annotated as hypothetical proteins with unknown function.
Fig. 1.
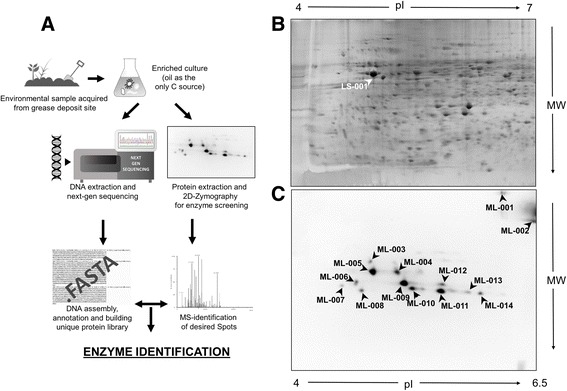
Functional metaproteomics as a tool to discover biocatalysts. a Schematic representation of the functional metaproteomics workflow. Metagenomics and functional metaproteomics combine the immediacy of an activity-based approach, while still retaining the comprehensive information of the metagenome. Optimization of DNA and protein extraction protocols can be found in Additional file 1: Figure S2 b 2D gel electrophoresis of the enriched sample stained with RuBPS Protein Gel Stain. LS-001 was excised as landmark spot. c In-gel activity assay for identifying lipolytic enzymes in the metaproteomic sample. Methylumbelliferyl-butyrate was hydrolyzed by lipolytic enzymes present in the gel. Resulting methylumbelliferone was detected under ultraviolet light. Fourteen spots (ML-001–ML-014) were manually excised for subsequent mass spectrometric analysis. Representative results shown, results of both technical replicates can be found in Additional file 1: Figure S3
This customized database was then used to analyze the mass spectra obtained from the 14 lipolytically active protein spots (Additional file 1: Table S1). Among these protein spots, we identified 2 serine-hydrolases, homologous to known lysophospholipase TesA from Pseudomonas species (ML-009 and ML-010). Additionally, 6 uncharacterized proteins, all homologous to a thioesterase from Pseudomonas species were found in the gel (ML-002, ML-003, ML-007, ML-008, ML-010, and ML-014). In total, 9 distinct primary structures of thioesterases, which matched the mass spectra generated from these spots, were present in our annotated metagenome database. These were all highly similar with minute differences (Additional file 1: Figure S1) and could not be unequivocally matched to the protein digests. All of these proteins are members of the family of SGNH hydrolases (named for their conserved and characteristic serine, glycine, asparagine, and histidine residues [16]), which are known to show lipolytic activity towards ester substrates [17]. Especially, TesA from Pseudomonas is known to have a preference for ester substrates with short- and mid-range carbon chain length [18]. In addition, we identified hydrolase ML-005, which is distantly homologous (35% identity) to the as of yet uncharacterized putative hydrolase YdeN (UniProtKB = P96671.1) from Bacillus subtilis. Bioinformatic analysis of ML-005 revealed a putative conserved alpha/beta hydrolase domain in this protein (Accession No. COG3545 at NCBI [19]) (Fig. 2a).
Fig. 2.
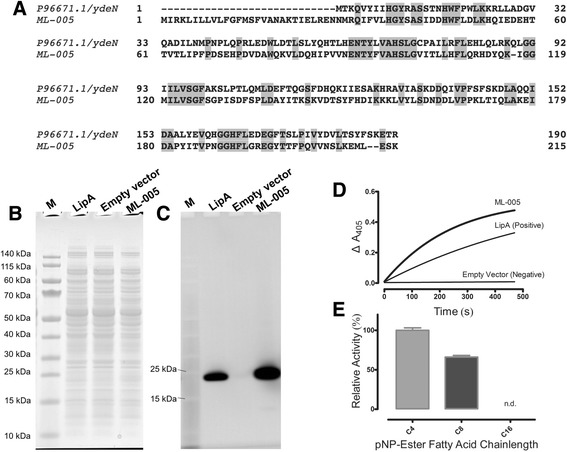
ML-005 is a novel esterase. a ML-005 is a distant relative (35% identity) of the uncharacterized putative hydrolase YdeN of B. subtilis. b Heterologous protein expression of ML-005 and lipase A LipA from B. subtilis (positive control) in E. coli from a plasmid was induced with 1 mM IPTG. E. coli carrying the empty vector served as negative control. Cells were disrupted by sonication and crude extracts were subjected to SDS PAGE, the protein content visualized by coomassie staining. c Lipid hydrolyzing activity was detected through in-gel zymography in the same crude cell lysates. The in-gel activity assay shows substrate conversion for positive control LipA from B. subtilis (23 kDa) and ML-005 (24.5 kDa) while a negative control of an extract of E. coli carrying the empty vector shows no activity. d Crude extract of E. coli expressing ML-005 hydrolyzes para-nitrophenyl-butyrate. Crude extract of E. coli expressing LipA from B. subtilis served as positive control, crude extract of E. coli containing the empty vector as negative control. Representative results are shown, results of all biological replicates can be found in Additional file 1: Figure S4. d Substrate specificity of purified ML-005 indicates a preference towards short-chain (C4) and medium-chain length (C8) para-nitrophenyl esters typical for esterases, no activity towards long-chain (C16) esters could be detected (n.d.). Specific activity of ML-005 towards para-nitrophenyl butyrate was 14.1 U mg−1
To verify the biocatalytic activity of the uncharacterized hydrolase ML-005, its DNA sequence was synthetized based on the metagenome data and cloned into Escherichia coli in an IPTG-inducible pBR322 -based expression vector with a tac promotor. The protein was then heterologously expressed in E. coli and its lipolytic activity confirmed through in-gel zymography (Fig. 2b, c). Furthermore, crude extract of E. coli expressing ML-005 showed high activity in a standard lipase/esterase enzyme assay, using p-nitrophenyl butyrate as a substrate (Fig. 2d). Lipid hydrolyzing enzymes can be categorized as lipase or esterases, with esterases typically preferring short-chain and lipases preferring long-chain fatty acid esters as substrates. We thus cloned the gene encoding ML-005 into a pET-based expression vector containing a T7-promotor, fusing a C-terminal His6-tag to the protein. We then expressed ML-005 in E. coli BL21 and purified it to homogeneity to test its reactivity towards para-nitrophenyl esters with fatty acids of differing chain-lengths. While ML-005 was effective in hydrolyzing short-chain (C4) and medium-chain length (C8) esters, we were not able detect any activity towards the long-chain p-nitrophenyl palmitate (C16), indicating that ML-005 is an esterase (Fig. 2e).
In conclusion, functional metaproteomics is an efficient tool to directly discover biocatalytic activity in the proteome of an environmental sample. The limitations of our approach pertain to the difficulties inherent in the isolation of proteins and DNA from environmental samples [20–24]. The complete phylogenetic diversity of a sample could only be harnessed if all DNA and all proteins expressed in the sample would be isolated. The method furthermore depends on an effective in-gel refolding of the biocatalyst and the availability of zymographic assays [25] that can be adapted to screen environmental samples for a certain biocatalytic activity.
Our results show that a simple workflow that combines 2D gel-based proteomics, functional screening, and metagenome-based protein identification makes it possible to identify novel lipolytic enzymes, an important class of biocatalysts, on the protein level, harnessing the phylogenetic diversity found in an environmental sample from a used cooking oil disposal site. We validated our approach by the heterologous expression and purification of the newly discovered and previously unknown esterase ML-005.
Acknowledgements
We thank Ulrich Kück for his support at the Department of General and Molecular Botany.
Funding
Principal funding was provided by the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007–2013)/ERC Grant agreement no 281384–FuMe to L.I.L. J.E.B. acknowledges funding from the State of North Rhine-Westphalia (Synapt G2-S mass spectrometer). M.N. acknowledges funding from the DFG (NO407/5-1).
Availability of data and materials
Sequencing data of the sample was submitted to the European Nucleotide Archive (www.ebi.ac.uk/ena) under project number PRJEB16064 and sample accession number ERP017906. The mass spectrometry proteomics data and the customized database have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005148 [26].
Authors’ contributions
LIL, JEB, and PS designed the experiments. PS and SS performed the experiments. PS and SS evaluated the mass spec data. MN, PS, and AK annotated the metagenomics data and assembled the database. PS and LIL wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- 2D
Two dimensional
- pMUB
para-methylumbelliferyl butyrate
Additional file
Supplementary methods [27–29], figures [30] and table [26]. (PDF 2013 kb)
Contributor Information
Premankur Sukul, Email: premankur.sukul@ruhr-uni-bochum.de.
Sina Schäkermann, Email: sina.schaekermann@ruhr-uni-bochum.de.
Julia E. Bandow, Email: julia.bandow@ruhr-uni-bochum.de
Anna Kusnezowa, Email: anna.kusnezowa@ruhr-uni-bochum.de.
Minou Nowrousian, Email: minou.nowrousian@ruhr-uni-bochum.de.
Lars I. Leichert, Phone: +49 234 3224585, Email: lars.leichert@ruhr-uni-bochum.de
References
- 1.Ogawa J. Microbial enzymes: new industrial applications from traditional screening methods. Trends Biotechnol. 1999;17:13–20. doi: 10.1016/S0167-7799(98)01227-X. [DOI] [PubMed] [Google Scholar]
- 2.Rappé MS, Giovannoni SJ. The uncultured microbial majority. Annu Rev Microbiol. 2003;57:369–94. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 3.Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol. 1998;5:R245–9. doi: 10.1016/S1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]
- 4.Nacke H, Will C, Herzog S, Nowka B, Engelhaupt M, Daniel R. Identification of novel lipolytic genes and gene families by screening of metagenomic libraries derived from soil samples of the German Biodiversity Exploratories. FEMS Microbiol Ecol. 2011;78:188–201. doi: 10.1111/j.1574-6941.2011.01088.x. [DOI] [PubMed] [Google Scholar]
- 5.Masuch T, Kusnezowa A, Nilewski S, Bautista JT, Kourist R, Leichert LI. A combined bioinformatics and functional metagenomics approach to discovering lipolytic biocatalysts. Front Microbiol. 2015;6:143. doi: 10.3389/fmicb.2015.01110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rashid M, Stingl U. Contemporary molecular tools in microbial ecology and their application to advancing biotechnology. Biotechnol Adv. 2015;33:1755–73. doi: 10.1016/j.biotechadv.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 7.Wilmes P, Heintz Buschart A, Bond PL. A decade of metaproteomics: where we stand and what the future holds. Proteomics. 2015;15:3409–17. doi: 10.1002/pmic.201500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keiblinger KM, Fuchs S, Zechmeister-Boltenstern S, Riedel K. Soil and leaf litter metaproteomics-a brief guideline from sampling to understanding. Muyzer G, editor. FEMS Microbiology Ecology. 2016;92:1-18. [DOI] [PMC free article] [PubMed]
- 9.Wilmes P, Bond PL. The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms. Environ Microbiol. 2004;6:911–20. doi: 10.1111/j.1462-2920.2004.00687.x. [DOI] [PubMed] [Google Scholar]
- 10.Benndorf D, Balcke GU, Harms H, von Bergen M. Functional metaproteome analysis of protein extracts from contaminated soil and groundwater. ISME J. 2007;1:224–34. doi: 10.1038/ismej.2007.39. [DOI] [PubMed] [Google Scholar]
- 11.Roberts IM. Hydrolysis of 4-methylumbelliferyl butyrate: A convenient and sensitive fluorescent assay for lipase activity. Lipids. 1985;20:243–7.
- 12.Ewing B, Green P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 1998;8:186–94. doi: 10.1101/gr.8.3.186. [DOI] [PubMed] [Google Scholar]
- 13.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 15.Hyatt D, Chen GL, LoCascio PF. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mølgaard A, Kauppinen S, Larsen S. Rhamnogalacturonan acetylesterase elucidates the structure and function of a new family of hydrolases. Structure. 2000;8:373–83. doi: 10.1016/S0969-2126(00)00118-0. [DOI] [PubMed] [Google Scholar]
- 17.Leščić Ašler I, Ivić N, Kovačić F, Schell S, Knorr J, Krauss U, et al. Probing enzyme promiscuity of SGNH hydrolases. Chem Eur J of Chem Bio. 2010;11:2158–67. doi: 10.1002/cbic.201000398. [DOI] [PubMed] [Google Scholar]
- 18.Kovačić F, Granzin J, Wilhelm S, Kojić-Prodić B, Batra-Safferling R, Jaeger K-E. Structural and functional characterisation of TesA—a novel lysophospholipase a from Pseudomonas aeruginosa. PLoS ONE. 2013;8:e69125. doi: 10.1371/journal.pone.0069125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI’s conserved domain database. Nucl Acids Res. 2015;43:D222–6. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks JP, Edwards DJ, Harwich MD, Rivera MC, Fettweis JM, Serrano MG, et al. The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 2015;15:66. doi: 10.1186/s12866-015-0351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keisam S, Romi W, Ahmed G, Jeyaram K. Quantifying the biases in metagenome mining for realistic assessment of microbial ecology of naturally fermented foods. Sci Rep. 2016;6:34155. doi: 10.1038/srep34155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nesme J, Achouak W, Agathos S, Bailey M, Baldrian P, Brunel D, et al. Back to the future of soil metagenomics. Front Microbiol. 2016;7:73. doi: 10.3389/fmicb.2016.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leary DH, Hervey WJ, IV, Deschamps JR, Kusterbeck AW, Vora GJ. Which metaproteome? The impact of protein extraction bias on metaproteomic analyses. Mol Cell Probes. 2013;27:193–9. doi: 10.1016/j.mcp.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Keiblinger KM, Wilhartitz IC, Schneider T, Roschitzki B, Schmid E, Eberl L, et al. Soil metaproteomics—comparative evaluation of protein extraction protocols. Soil Biol Biochem. 2012;54:14–24. doi: 10.1016/j.soilbio.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manchenko GP. Handbook of Detection of Enzymes on Electrophoretic Gels. Boca Raton: CRC Press; 2002.
- 26.Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, et al. ProteomeXchange providesglobally coordinated proteomics data submission and dissemination. Nat Biotechnol. 2014;32:223–6. [DOI] [PMC free article] [PubMed]
- 27.Stülke J, Hanschke R, Hecker M. Temporal activation of β-glucanase synthesis in Bacillus subtilis ismediated by the GTP pool. Microbiology. 1993;139:2041–5. [DOI] [PubMed]
- 28.Helling S, Vogt S, Rhiel A, Ramzan R, Wen L, Marcus K, et al. Phosphorylation and kinetics ofmammalian cytochrome c oxidase. Mol Cell Proteomics. 2008;7:1714–24. [DOI] [PMC free article] [PubMed]
- 29.Wenzel M, Patra M, Albrecht D, Chen DYK, Nicolaou KC, Metzler-Nolte N, et al. Proteomic signature offatty acid biosynthesis inhibition available for in vivo mechanism-of-action studies. Antimicrob Agents Chemother. 2011;55:2590–6. [DOI] [PMC free article] [PubMed]
- 30.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequencealignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Sequencing data of the sample was submitted to the European Nucleotide Archive (www.ebi.ac.uk/ena) under project number PRJEB16064 and sample accession number ERP017906. The mass spectrometry proteomics data and the customized database have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005148 [26].