

Abstract
The ExoU type III secretion enzyme is a potent phospholipase A2 secreted by the Gram-negative opportunistic pathogen, Pseudomonas aeruginosa. Activation of phospholipase activity is induced by protein-protein interactions with ubiquitin in the cytosol of a targeted eukaryotic cell, leading to destruction of host cell membranes. Previous work in our laboratory suggested that conformational changes within a C-terminal domain of the toxin might be involved in the activation mechanism. In this study, we use site-directed spin-labeling electron paramagnetic resonance spectroscopy to investigate conformational changes in a C-terminal four-helical bundle region of ExoU as it interacts with lipid substrates and ubiquitin, and to examine the localization of this domain with respect to the lipid bilayer. In the absence of ubiquitin or substrate liposomes, the overall structure of the C-terminal domain is in good agreement with crystallographic models derived from ExoU in complex with its chaperone, SpcU. Significant conformational changes are observed throughout the domain in the presence of ubiquitin and liposomes combined that are not observed with either liposomes or ubiquitin alone. In the presence of ubiquitin, two interhelical loops of the C-terminal four-helix bundle appear to penetrate the membrane bilayer, stabilizing ExoU-membrane association. Thus, ubiquitin and the substrate lipid bilayer act synergistically to induce a conformational rearrangement in the C-terminal domain of ExoU.
Keywords: electron paramagnetic resonance (EPR), phospholipase, protein structure, Pseudomonas aeruginosa (P. aeruginosa), ubiquitin
Introduction
A wide variety of bacterial pathogens express effector proteins that influence or modify the host environment in a way that facilitates their survival and dissemination. A particularly effective mechanism used by several important Gram-negative pathogens is the type III secretion system (T3SS),4 a complex multi-protein apparatus that introduces bacterial effector proteins directly into the cytosol of targeted eukaryotic cells (1). The Gram-negative opportunistic pathogen Pseudomonas aeruginosa synthesizes four effector proteins (termed ExoS, ExoT, ExoU, and ExoY) that are injected into host cells via the T3SS(2–5), of which ExoU is the most strongly correlated with virulence and negative clinical outcome (6, 7).
ExoU is a 74-kDa (687-amino acid) soluble protein that exhibits phospholipase activity in the presence of ubiquitin and ubiquitinylated proteins (8). The catalytic domain of ExoU exhibits structural similarity to the patatin group of plant phospholipases, as well as to eukaryotic phospholipases (9). Unlike their eukaryotic counterparts, activation by ubiquitin appears to be a property of many bacterial patatin-like proteins (10). ExoU is the best-characterized member of the family and serves as a model system to understand the mechanisms of activation and substrate recognition. The enzyme is capable of hydrolyzing a wide variety of phospholipid and lysophospholipid substrates (11, 12), including those found in the bacterial inner membrane (e.g. phosphatidylethanolamine and phosphatidylglycerol). Indeed, co-expression of ExoU and ubiquitin in Escherichia coli results in rapid degradation of the cytoplasmic membrane and cell death (8, 10). Consequently, the requirement of a eukaryotic cofactor is essential in protecting P. aeruginosa and perhaps other bacterial genera from membrane degradation by their own toxins.
Two X-ray crystal structures of ExoU in complex with its cognate chaperone, SpcU, have been solved (13, 14). These structures are in excellent agreement and reveal ExoU to be a multi-domain protein containing a chaperone-binding domain, a catalytic domain, a bridging domain, and a C-terminal four-helix bundle domain. Although overall sequence homology with other phospholipases is weak, catalytic serine and aspartate residues and a glycine-rich motif thought to stabilize the transition state complex are well conserved (9, 15–18). In contrast, the C-terminal bridging and four-helix bundle domains do not appear to have close sequence or structural equivalents in either the patatins or the eukaryotic phospholipases.
At present the structure of ExoU in the absence of SpcU, the mechanism by which ubiquitin binding activates ExoU, and the manner in which ExoU associates with target membranes are unknown. Previous studies in our lab indicated that ExoU is a highly flexible protein that samples multiple conformational states in the absence of its protein cofactor (19). Consistent with this observation, the previous crystallographic studies found that nearly 25% of the amino acid residues in ExoU could not be localized from the electron density map (13, 14). Structural flexibility may be a necessary characteristic of type III secreted proteins, because they must unfold to pass through the relatively narrow (∼25–40 Å diameter) lumen of the needle-like T3SS apparatus (1).
In this study, we have used site-directed spin labeling in conjunction with EPR spectroscopy to examine the structure and membrane interactions of ExoU's C-terminal four-helix bundle domain (residues 587–687). Mutagenesis and C-terminal deletion studies have implicated this region of the protein in membrane localization (20–23), and it has been suggested that the four-helix bundle may be a common structural element in targeting a wide variety of diverse bacterial toxins to the membrane interface (24, 25). Forty-one single-cysteine mutants in this domain were constructed, purified, and spin-labeled with the sulfhydryl-specific spin label, MTSL (1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl methanethiosulfonate). Analysis of spin label side-chain dynamics and accessibilities to paramagnetic relaxation agents in the presence and absence of ubiquitin and substrate membranes indicates that the C-terminal four-helix bundle of ExoU undergoes a synergistic conformational change in the presence of both ubiquitin and substrate liposomes that is not observed with either ubiquitin or liposomes alone.
Results
Phospholipase Activity of Spin-labeled ExoU Mutants
Forty-one single-cysteine substitutions were individually constructed within ExoU, a natively cysteine-free protein, focusing on its C-terminal four-helix bundle (residues 587–687, Fig. 1). The cysteine mutants were purified and labeled with MTSL, and kinetic analysis of phospholipase activity was performed using the fluorescent phospholipid analog PED6 in the presence of recombinant linear diubiquitin (diUb). Diubiquitin was chosen for this study as it has been shown that ubiquitin polymers of several linkage types (including linear diubiquitin) have a greatly enhanced ability to bind and activate ExoU as compared with monomeric forms (8). A large majority of the spin-labeled derivatives retained substantial phospholipase activity, with only two sites (P616R1 and L651R1)5 having less than 10% catalytic activity as compared with WT ExoU (supplemental Fig. S1). Four derivatives with impaired phospholipase activity (<25% WT activity) were located on the first helix (N608R1, K611R1, F615R1, and P616R1), suggesting that this helix may play an important role in ubiquitin-mediated activation. Two additional sites with diminished activity are centrally located in loops one and three (Q623R1 and F666R1, respectively). Far-UV CD spectra indicated that each of these low activity derivatives retained native-like overall secondary structure content (supplemental Fig. S2). Overall, these results indicate that the structure of the C-terminal helical bundle is generally tolerant to site-specific cysteine substitution and spin labeling with MTSL.
FIGURE 1.
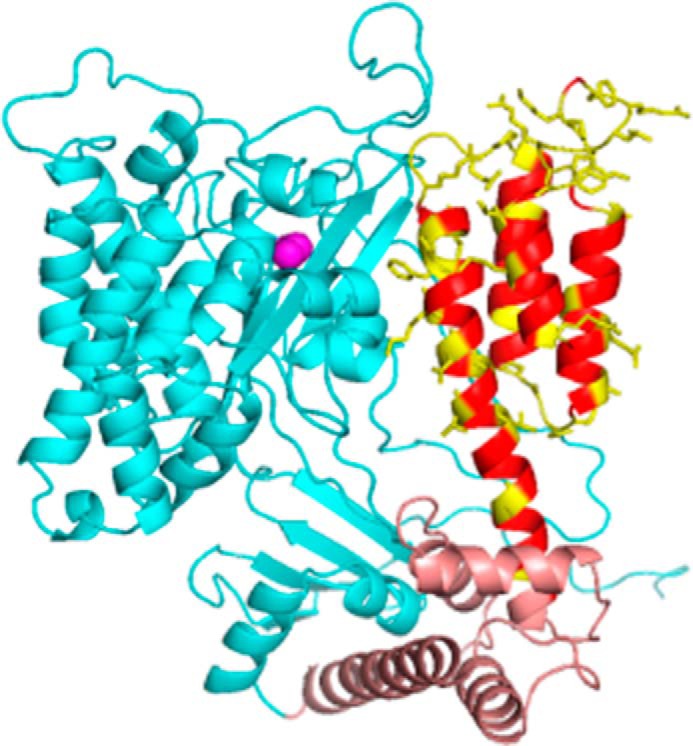
Structural model of ExoU. Shown is a model based on PDB 4AKX (13) displaying the catalytic domain (cyan), bridging domain (brown), and C-terminal four-helix bundle (red/yellow). Native side chains at sites of spin label attachment are shown in yellow. The catalytic serine (Ser-142) is indicated by magenta spheres. Loops were modeled using Rosetta (see “Experimental Procedures”).
Synergistic Conformational Changes in the Presence of Ubiquitin and Substrate Liposomes
Shown in Fig. 2 are superimposed EPR spectra for ExoU alone in solution (the inactive apoenzyme state, Apo), ExoU mixed with liposomes (Lipo), ExoU mixed with diUb only (Ub), and ExoU in the presence of both liposomes and diUb (the activated holoenzyme state, Holo) at sites representing different structural elements of the C-terminal helical bundle. The EPR spectral line shape is exquisitely sensitive to the rotational mobility, and hence local environment, of the nitroxide side chain. Increased spin label motion is indicated by a narrowing and increase in amplitude of the lines, whereas decreased motion leads to line broadening and an overall decrease in amplitude. Tertiary contacts with neighboring side chains can lead to highly anisotropic motion of the spin label and/or the superposition of spectra corresponding to multiple conformational states, producing complex line shapes that are most evident in the low-field region of the spectra (Fig. 2, insets). It is apparent that for many of these sites, the EPR spectra in the apo state and in the presence of either diUb or liposomes alone are essentially identical, with marked differences observed only in the presence of both membranes and ubiquitin. EPR spectra for all 41 spin-labeled variants of ExoU in the presence of liposomes alone or in the presence of liposomes and linear diUb are shown in supplemental Figs. S3 and S4, respectively. None of the sites examined in this study exhibited changes in R1 side-chain motion upon the addition of diUb alone (data not shown).
FIGURE 2.
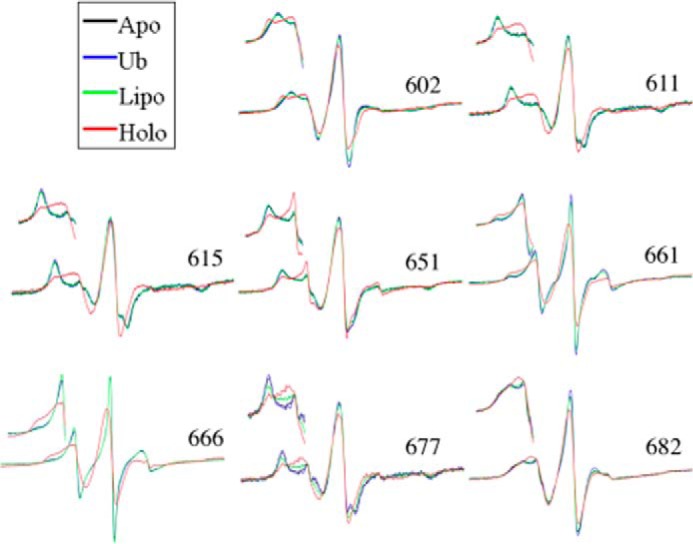
Continuous-wave EPR spectral overlays of several MTSL spin-labeled ExoU derivatives. Continuous-wave EPR spectra at representative sites along the four-helix bundle (site indicated by the residue number) are shown. Each overlay shows four conditions: ExoU alone (Apo); ExoU with a 12-fold excess diUb (Ub); ExoU in the presence of 100-fold molar excess liposomes (Lipo); and ExoU in the presence of both diUb and liposomes (Holo). Scan widths of the full spectra are 100 G. Insets show the low-field region for each spectrum.
ExoU has been shown to bind strongly to l-α-phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) in lipid-overlay experiments (13), and membrane destruction following intoxication of eukaryotic cells appears to involve interaction with and hydrolysis of PI(4,5)P2 on the cytosolic membrane surface (26). ExoU associates with PI(4,5)P2-containing eukaryotic model membranes with an apparent dissociation constant on the order of 0.2 × 10−3 m (10). Thus, under our experimental conditions (5–20 mm lipid, 0.05–0.2 mm ExoU), the protein is almost fully membrane-bound. Nonetheless, for the majority of sites, the addition of liposomes alone had little or no effect on the rotational mobility of the R1 side chain (supplemental Fig. S3). In contrast, the addition of liposomes and diUb in combination (the holo state) caused pronounced changes in spin label dynamics throughout the entire C-terminal domain (supplemental Fig. S4).
In comparing apo and holo states, we observed both increased and decreased mobility of the spin label side chain, depending on the labeling site (supplemental Figs. 2, S3, and S4). For example, sites 611, 615, 651, and 677, shown in Fig. 2, all exhibit increased spin label mobility in the holo state, whereas sites 661 and 666 show decreased mobility in the holo state (Fig. 2). EPR spectra for V602R1 in the apo and holo states are particularly interesting. For V602R1 in the holo state, the EPR spectrum indicates the presence of two conformational states in slow exchange that are modified or not resolved in the apo state. The V602R1 apo state spectrum could be fit with a single motional component having a rotational correlation time of 6.7 ns (supplemental Fig. S5). In contrast, simulation of the V602R1 holo state indicated the presence of two motional components having correlation times of 2.3 and 16.5 ns (supplemental Fig. S5). Whether these represent two conformational states of the protein or two rotameric states of the nitroxide is currently under investigation. For comparison, the EPR spectra for R661R1 in either the apo or the holo state were fit with two components having rotational correlation times of 1.2 and 3.2 ns in the apo state and 5.9 and 11.4 ns in the holo state (supplemental Fig. S6), consistent with a large decrease in mobility of the third interhelical loop upon formation of the holo state (discussed below).
Although spectral simulation can provide important insights into the rotational modes of the spin label side chain at a given site, for comparison over a given region of protein structure, the scaled mobility parameter, Ms (see “Experimental Procedures,” Equation 1), provides a simplified measure of spin label dynamics that more readily reflects structural trends. Ms is normalized to 1 for sites with the greatest rotational mobility (27), and has proven extremely useful for comparing relative mobility as a function of the labeling site (27–29). Shown in Fig. 3 is a plot of Ms for R1 at each of the spin-labeled sites for ExoU in the apo and holoenzyme states. In the apo state (Fig. 3A, open circles), rotational mobility is greatest in the interhelical loops and is relatively suppressed at sites located within the C-terminal helices (Fig. 3A, shaded regions), as reflected by mapping Ms onto the ExoU crystal structure (Fig. 3B). These results suggest that the overall structure of the C-terminal four-helix bundle for ExoU in the apo state is in good agreement with that observed in crystallographic models of the ExoU-SpcU complex. As noted above, adoption of the holo state induces structural changes that alter the scaled mobility of the spin label at sites throughout the entire C-terminal bundle (Fig. 3A, filled triangles).
FIGURE 3.
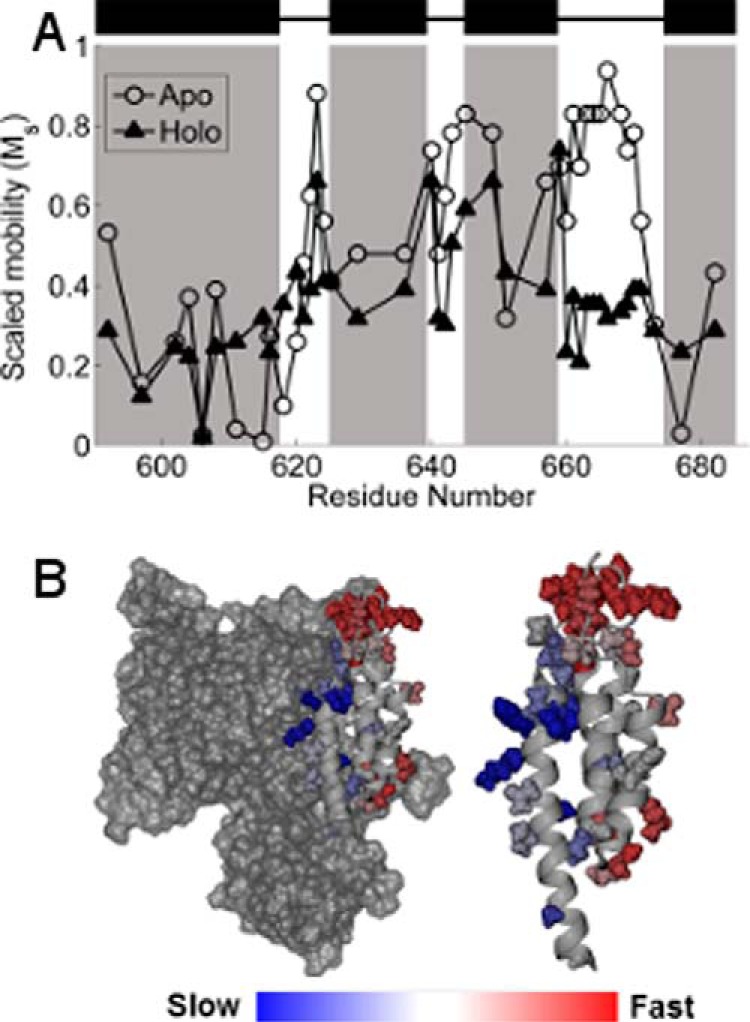
A, scaled mobility of the R1 side chain. Open circles indicate the apo state, and filled triangles indicate the holo state. Higher values of Ms indicate greater mobility of the R1 side chain. Black rectangles and shaded regions indicate helices in the C-terminal domain as defined by the crystal structures (13, 14). B, Ms in the apo state mapped onto the crystal structure of ExoU. Color changes from blue to red indicate increasing motion of the R1 side chain. Left, the catalytic domain is shown in surface rendering. Right, the isolated C-terminal four-helix bundle.
Comparison of the scaled mobility parameter in the apo and holo states indicates two regions of the C-terminal domain where changes in spin label motion are particularly pronounced: the C-terminal half of the first helix and the third interhelical loop (Figs. 3 and 4). Sites in the C-terminal half of the first helix (residues 611–618) become substantially more mobile in the holo state, indicating a loss of tertiary contacts in this region as ExoU adopts its active conformational state. The relatively low mobility of these sites in the apo state likely reflect close packing with the catalytic domain (Fig. 3B), and the increase in motion suggests that the two domains may separate upon catalytic activation. This interface, although distal from the catalytic site, appears to play an important role in the activation process, as four of the spin-labeled variants in this region show reduced catalytic activity. Conversely, the addition of diUb and liposomes resulted in a large decrease in the mobility of the MTSL side chain throughout the entire third interhelical loop (residues 660–670), a region of the C-terminal domain that is highly exposed in the apo state (Fig. 3B). Reduced spin label mobility in this loop could reflect the formation of new tertiary contacts, or an overall decrease in backbone dynamics due to interaction with the membrane.
FIGURE 4.
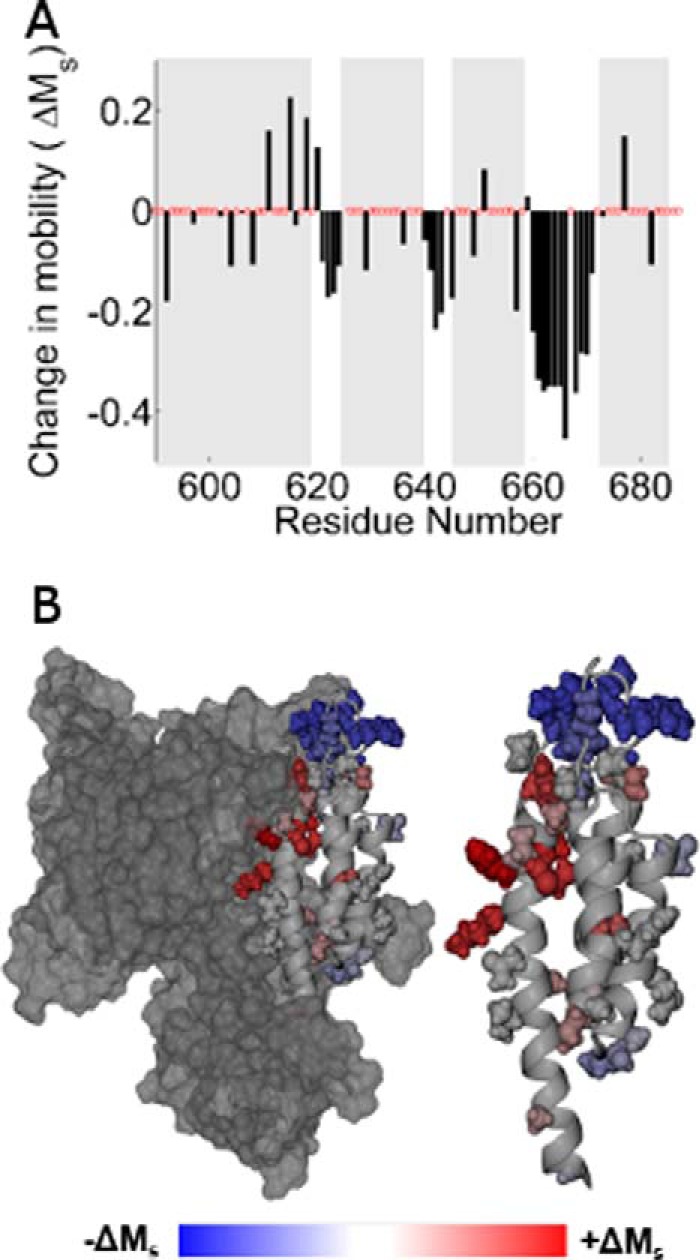
A, histogram showing the change in scaled mobility, Ms(apo) − Ms(holo), as a function of the labeling site. Clusters of negative values indicating decreased mobility in the holo state correspond to the three interhelical loops. B, the change in Ms mapped onto the ExoU crystal structure. Sites that increased motion upon adoption of the holo state are shown in red, and sites that exhibit decreased motion are shown in blue.
Localization of the C-terminal Domain Relative to the Membrane Bilayer
To further examine the interaction of the of the C-terminal domain with the membrane, we carried out power saturation studies in the presence of oxygen and nickel ethylenediaminediacetic acid (NiEDDA) as relaxation agents that probe primarily the membranous and aqueous phases, respectively. Shown in Fig. 5A is the EPR accessibility parameter (π) for NiEDDA as a function of labeling position in the apo state, in the presence of liposomes, and in the holo state. The accessibility parameter is directly proportional to bimolecular collision rate for a given paramagnetic probe (30), and in soluble proteins, it exhibits good correlation with the fraction of accessible surface area (31). In agreement with the scaled mobility results described above, π(NiEDDA) values for the apo state again parallel the crystallographic secondary structure with the highest bimolecular collision rates observed in the interhelical loops. In the holo state, interaction with NiEDDA is strongly suppressed throughout the C-terminal domain, with striking decreases relative to the apo state in each of the interhelical loops. Accessibilities to NiEDDA in the presence of liposomes alone were generally intermediate to the apo and holo states, although some regions (e.g. helix 1, the second interhelical loop) were more similar to the holo state. Oxygen accessibility parameters are shown in Fig. 5B. Interaction with O2 is significantly increased upon adoption of the holo state for several regions of the four-helix bundle, including sites within the first and third helices and in interhelical loops one and three. In the presence of liposomes alone, O2 accessibility more closely resembles the apo state for most sites, with intermediate values observed in the third interhelical loop. Together, these data indicate that ubiquitin binding is an important trigger facilitating membrane association of the C-terminal four-helix bundle domain.
FIGURE 5.

Accessibilities and membrane localization as a function of labeling position for ExoU variants alone in solution (Apo, open circles), in the presence of liposomes (Lipo, filled circles), and in the presence of liposomes and diUb (Holo, red triangles). Shaded areas indicate helices in the C-terminal domain as defined by the crystal structures (13, 14). A, accessibility to NiEDDA. B, accessibility to oxygen. C, the EPR depth parameter. D, localization of the spin label side chain relative to the membrane bilayer. Negative values indicate exposure to the aqueous phase.
Fig. 5C shows the EPR depth parameter (Φ) as a function of labeling position for the apo state, in the presence of liposomes alone, and in the presence of both liposomes and diUb. This parameter, derived from the relative bimolecular collision rates with oxygen and NiEDDA (see Equation 4 under “Experimental Procedures”), increases with increasing penetration into the membrane and is proportional to membrane depth for those sites exposed to the hydrophobic phase of the bilayer (32). In the apo state, Φ values were all negative, as expected in the absence of a membrane bilayer, with limiting values of approximately −2.8. In the presence of liposomes alone, Φ values increased modestly but remained negative for all but one site (N608R1). In contrast, in the holo state, Φ increased for almost every site examined and several sites had positive values indicative of insertion into the membrane. A large number of sites exhibited Φ values in the holo state from −1.5 to near 0, typical of labeling sites located in close proximity to the aqueous membrane interface. Even greater Φ values were observed in the holo state for sites within the first helix and in the first and third interhelical loops (Fig. 5C), reflecting deeper penetration into the lipid bilayer.
To localize the C-terminal four-helix bundle with regard to the membrane bilayer, we used Equation 5 (see “Experimental Procedures”) to convert Φ values to distances relative to the phospholipid phosphate moieties (Fig. 5D). The parameters for Equation 5 were determined based on interaction of the C2 domain of cPLA2 with bilayers having a very similar composition to those used here (33), and gave very good agreement with our own lipid standards (supplemental Fig. S7). The data in Fig. 5D indicate increased penetration into the membrane in the presence of diUb. Immersion into the bilayer is particularly notable for sites in the first helix, and for sites in the first and third interhelical loops. Residues A606R1, K608R1, F611R1, and F615R1 in helix 1, residues P621R1 and G622R1 in the first interhelical loop, and residues G662R1–F666R1 in the third interhelical loop all reach depths of ∼ 8 Å or greater below the lipid phosphates. In addition, a large number of sites in the holo state localized to a region 4–6 Å below the lipid phosphates. In contrast, the second interhelical loop (centered around residue Thr-642) appears to remain exposed to the aqueous phase.
Discussion
ExoU is a potent virulence factor exported through the type III secretion system of P. aeruginosa that, remarkably, requires non-covalent interactions with ubiquitin for catalytic activation. ExoU has broad substrate specificity, such that the regulation of activity by a required interaction with a eukaryotic cofactor is necessary to ensure that ExoU is inactive within the bacterium. Crystallographic models of ExoU, determined in complex with its cognate chaperone SpcU, indicate that it contains a catalytic domain with structural characteristics similar to plant and eukaryotic Type IVA phospholipases, along with novel C-terminal bridging and four-helix bundle domains (13, 14). The C-terminal four-helix bundle of ExoU has been implicated in both membrane binding and interaction with ubiquitin (20, 22, 34). The goal of this study was to investigate conformational changes and membrane interactions of the C-terminal four-helix bundle domain in the presence and absence of a ubiquitin cofactor.
Our data on the rotational mobility and solvent accessibility of the R1 side chain as a function of the labeling site indicate that the tertiary structure of the C-terminal domain of ExoU in solution is in good agreement with that expected based on the crystal structure of the ExoU-SpcU complex. In general, we found that rotational mobility at sites within the crystallographic helices was more restricted than at residues located in connecting loops (Fig. 3). Similarly, accessibility to the aqueous relaxation agent, NiEDDA, was greater in the interhelical loops (Fig. 5A). Mapping spin label mobility onto the crystal structure supports the concept that reduced mobility occurs at tertiary contact sites, especially in the first helix of the bundle where restricted motion of the R1 side chain appears to correlate with packing against the catalytic domain (Fig. 3B).
Relatively few sites exhibited changes in spin label motion in the presence of liposomes alone. Similarly, membrane association in the presence of liposomes alone resulted in relatively modest increases in oxygen accessibility and the EPR depth parameter. However, Φ increased substantially in the combined presence of liposomes and diUb throughout the C-terminal domain. These results suggest that in the absence of ubiquitin, ExoU associates with target membranes with little change in the tertiary structure of its C-terminal domain. Similarly, we observed that the addition of diUb alone also had little to no effect on either spin label motion or accessibility throughout the four-helix bundle.
In contrast to the modest effects of either diUb or liposomes alone, the addition of diUb and liposomes in combination induced marked changes in both spin label mobility and accessibility throughout the C-terminal domain. The observed effects included regions of increased mobility, as in the first helix of the bundle, as well as decreased mobility at sites located in the interhelical loops. In particular, striking changes in rotational mobility and accessibility between apo and holo states were observed for labeling sites in the third interhelical loop. These sites, encompassing residues 660–670, were among the most dynamic in the apo state and the most motionally restricted in the holo state. The amino acid sequence of loop 3, ARGFLRFGKPL, contains three cationic and eight nonpolar residues. This “basic-hydrophobic” motif is well suited to mediate interaction with the negatively charged surface of the phospholipid bilayer (25, 35–37). Previous studies have shown that a minimal deletion of the last eight residues of ExoU (679–687) inactivates the enzyme and abrogates membrane binding (22). Our data support the notion that this C-terminal truncation may disrupt packing of the four-helix bundle so that the preceding third interhelical loop is not properly displayed for membrane insertion.
Accessibility to NiEDDA indicated that the third interhelical loop is highly solvent-exposed in the apo state, but is inaccessible to this polar relaxation agent in the holo state. Membrane insertion by this loop reached depths of 8–10 Å, comparable with membrane insertion depths observed for loop residues in the C2 domains of human cytosolic phospholipase A2 (33, 38). The relatively short first interhelical loop (residues 620–624) and adjacent sites within the first helix of the bundle also appear to penetrate the bilayer to a similar depth, whereas labeling sites within the remaining C-terminal helices are localized near the membrane surface.
These studies provide new insights into the structural characteristics of ExoU-membrane interaction. Phospholipases and other proteins that function at the membrane interface utilize a variety of mechanisms to facilitate association with the lipid bilayer (39, 40). In many of these proteins, membrane association is mediated by a domain that is distal from the active site. We postulate that membrane integration of the C-terminal four-helix bundle of ExoU, which is well removed from the site of phospholipase activity, stabilizes association of the catalytic domain with the target lipid bilayer and may help position the active site in proximity to its lipid substrate. A rough schematic of the proposed interaction is shown in Fig. 6. This conceptual model depicts the observed interaction of the C-terminal four-helix bundle with the membrane and the resultant positioning of the catalytic site near the bilayer interface.
FIGURE 6.

Schematic of ExoU membrane association and conformational changes. The ExoU four-helix bundle (cylinders) is depicted as dissociating and being displaced from the catalytic domain (rectangles) upon adoption of the holo state (upper right), accompanied by conformational changes in the bridging domain (oval) and catalytic domain that expose the catalytic site (star) to the membrane surface (located at the top of the figure). The binding of ubiquitin (octagon) and membrane association in the absence of ubiquitin (upper left) likely result in conformational changes relative to the apo state (lower left) that remain to be elucidated, and the location of the ubiquitin-binding site has not been fully identified.
Our studies demonstrate that adoption of the catalytically active “holo” state of ExoU requires interaction with both a ubiquitin cofactor and a lipid substrate. This is in agreement with the recent suggestion that, for proteins that traffic between the membrane and aqueous phases, the membrane should be considered as an allosteric regulator of the protein's conformational state (41, 42). Additional studies in progress will be required to fully define the membrane interaction interface of ExoU, the likely conformational changes that accompany ubiquitin binding, identification of the ubiquitin-binding site, and the degree to which membrane association alters the structure of the catalytic domain.
Experimental Procedures
Protein Expression, Spin Labeling, and Purification
The ExoU gene (GenBankTM: U97065.1) was expressed from a pET15b vector as an N-terminal histidine-tagged protein from E. coli BL21 (DE3) pLysS in Terrific Broth. Recombinant expression of the full-length protein was initiated at A600 of 0.8 with 0.25 mm isopropyl-d-thiogalactopyranoside followed by incubation at 30 °C, 225 rpm for 3 h. Cultures were harvested by a 20-min centrifugation at 6,000 × g, 4 °C and stored at −80 °C prior to lysis by French pressure cell. Cellular debris was removed by centrifugation for 20 min at 30,000 × g, 4 °C before application of the supernatant to Talon cobalt affinity resin (Clontech). Protein was purified according to the manufacturer's recommendation before elution in 50 mm sodium phosphate buffer, 150 mm NaCl, pH 7.0, %0.02 sodium azide (Buffer A) with 50 mm EDTA. The eluate was desalted into Buffer A via FPLC with a HiPrep 26/10 desalting column, and the histidine tag was removed by thrombin cleavage. The sample was passed through a HiTrap benzamidine FF column and cobalt affinity resin before spin labeling overnight with a 5-fold molar excess of MTSL (Santa Cruz Biotechnology) in Buffer A and 20% glycerol, followed by concentration at 4 °C with centrifugal filters (Amicon Ultra-15, 30,000 molecular weight cut-off). The concentrate was purified by size exclusion chromatography (HiLoad 16/600 Superdex 200 prep grade column) in Buffer A supplemented with 20% glycerol, and peak fractions were concentrated, assessed for concentration by measuring absorbance at 280 nm, and aliquoted for flash-freezing and storage at −80 °C. Recombinant diubiquitin was expressed and purified in a similar fashion. All site-specific mutagenesis was conducted with Change-IT kits (Affymetrix).
Analysis of Enzymatic Activity
In vitro phospholipase activity assays were conducted at room temperature in a SpectraMax M5 plate reader as described previously (43). Assays contained 10 nm ExoU, 60 μm PED6 (Molecular Probes), and various amounts of recombinant diubiquitin (1–45 μm). Kinetic constants were determined by non-linear regression analyses with a one-site specific binding model using GraphPad Prism 5.0 software (GraphPad Software Inc.). The conversion of relative fluorescence units to nmol of PED6 was 59,300 relative fluorescence units = 1 nmol of PED6.
Liposome Preparation
Lipids and lipid analog spin labels were acquired as chloroform stock solutions from Avanti Polar Lipids. Model eukaryotic membrane liposomes were prepared similarly to those described previously (10). Briefly, a mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylserine (POPS), and l-α-PI(4,5)P2 was combined in a molar ratio of 40:40:20:1, dried under a stream of nitrogen gas, and stored overnight under vacuum. The dried lipid film was suspended in 50 mm MOPS, pH 7.0, 150 mm NaCl, and 20% glycerol, subjected to 10 freeze-thaw cycles (liquid N2 to 37 °C), and extruded 15 times through a 0.1 micron filter using a syringe mini-extrusion apparatus (Avanti Polar Lipids). Final lipid concentration was determined by Stewart assay (44).
Circular Dichroism
Recombinant ExoU was diluted to 0.1 mg/ml in 20 mm sodium phosphate, pH 7.0, and data were collected on a JASCO J-710 spectropolarimeter in a 1-mm quartz cell at room temperature. Readings were obtained at 1-nm intervals from 250 to 190 nm at a sweep rate of 50 nm/min, typically signal averaging for 8–16 scans.
EPR Spectroscopy
Continuous wave (CW) EPR spectra for analysis of spin label motion were collected on an Elexsys E500 spectrometer (Bruker Biospin, Billerica, MA) equipped with a high-Q cavity operating at X-band. Samples consisted of 50 μm spin-labeled ExoU either in buffer alone (20 mm MOPS, pH 7.0, 150 mm NaCl, and 20% glycerol) or with 5 mm liposomes and/or 600 μm recombinant diubiquitin. Spectrometer conditions consisted of a time constant of 10.28 ms; conversion time, 20.48 s; scan time, 20.97 s; a 100-kHz field modulation amplitude of 1.0 G; 10-milliwatt microwave power; and a sweep width of 100 G. Spectra were baseline corrected and normalized using MATLAB and EasySpin (45). Simulations of CW EPR spectra (supplemental Figs. S5 and S6) were carried out using the LabView Multicomponent program written by Christian Altenbach.
The scaled mobility parameter (Ms) was calculated according to (27)
| (Eq. 1) |
where δ is the peak-peak width of the center EPR line and δm and δi are the center line widths of the most mobile site (apo Q623R1) and most immobile sites (apo K611R1 and apo F615R1) examined, respectively.
CW power saturation EPR was carried out on a Varian Century Series X-Band spectrometer equipped with a two-loop, one-gap resonator (XP-0201, Molecular Specialties). Samples (0.1–0.15 mm spin-labeled ExoU with and without 20 mm liposomes and/or 1 mm diubiquitin in a final volume of 4 μl) were contained in a gas-permeable 0.6-mm inner diameter TPX capillary. Power saturation curves were determined by measuring the peak-peak amplitude of the center line and fitting data to the function (32)
| (Eq. 2) |
where A is the signal amplitude, P is the microwave power, I and ϵ are floating variables, and P½ is the half-saturation parameter. Data were obtained under N2 gas flow, in equilibrium with a flow of compressed air (20% O2), and under N2 in the presence of 5–20 mm NiEDDA, or 20–100 mm NiEDDA for phosphatidylcholine spin label standards (Avanti Polar Lipids). Accessibility parameters for oxygen and NiEDDA (π(O2) and π(NiEDDA), respectively) were calculated according to Ref. 46
| (Eq. 3) |
where x represents the respective relaxation agent, and ΔHpp is the peak-peak width of the center line. Parameters for DPPH (α,α′-diphenyl-β-picrylhydrazyl) were obtained with a powder sample for standardization of resonator performance. π(NiEDDA) values were normalized to 20 mm NiEDDA (33). The EPR depth parameter (Φ) was calculated according to Refs. 32, 33, and 46.
| (Eq. 4) |
EPR depth parameters were used to calculate position relative to membrane lipid phosphates using the relationship (33)
| (Eq. 5) |
where x is the position of the spin label (in Å), A and D are set bulk values of Φ in the water and hydrophobic phases, respectively, B determines the slope in the linear region of the curve, and C is the inflection point. Parameters A = 3.81, B = 0.11, C = 8.2, and D = 1.0 were used, as determined previously for a very similar lipid composition (33), adjusted to our limiting value in the bulk aqueous phase of −2.8. These parameters gave good agreement with experimentally measured standards (see supplemental Fig. S7).
Enzyme Loop Modeling and Molecular Dynamics Simulation
Kinematic loop modeling utilizing Rosetta 3.5 (release rosetta_2014.20.56838) was conducted on the ExoU input structure (Protein Data Bank (PDB): 3TU3) to construct loop structures absent from the crystallographic coordinates (47). Three hundred structures were produced through a remodeling stage (-loops:remodel perturn_kic) and a refinement stage (-loops:refine refine_kic), with extra χ rotamer flags (-ex1 and -ex2) included. Cluster analysis using Calibur (48) was then conducted to select the decoy with the most neighboring structures from the larger of the two clusters generated. This structure was then selected for a 10-ns molecular dynamics simulation in Gromacs 5.0 (49). A CHARMM27 force field and TIP3P water model were selected prior to simulation in a charge-neutral solvent box consisting of water and sodium ions (50). A velocity rescaling thermostat was used for temperature equilibration (100 ps at 300 K), followed by a 100-ps pressure equilibration. A Verlet cutoff scheme of neighbor searching was used along with particle mesh Ewald summation of long-range electrostatics (51). A 10-ns simulation was conducted using leapfrog integration, and the final structure pose is shown in Fig. 1.
Author Contributions
M. H. T., D. M. A., D. W. F., and J. B. F. conceived the study, designed and performed experiments, and analyzed the data. A. B. designed and performed experiments. M. H. T., D. M. A., D. W. F., and J. B. F. wrote the paper. All authors evaluated the results and approved the final version of the paper.
Supplementary Material
Acknowledgments
We thank Molly Riegert and Jennifer Brayshaw for technical assistance.
This work was supported by National Institutes of Health Grants GM114234 (to J. B. F.) and AI104922 (to D. W. F). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S7.
P616R1, L651R1, etc. indicate the native amino acid using the standard single letter code, the amino acid number, and R1 to indicate mutation to cysteine and addition of the spin-label side chain.
- T3SS
- type III secretion system
- MTSL
- 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl methanethiosulfonate
- diUb
- diubiquitin
- PI(4,5)P2
- l-α-phosphatidylinositol 4,5-bisphosphate
- NiEDDA
- nickel ethylenediaminediacetic acid
- CW
- continuous wave
- G
- Gauss
- DPPH
- α,α′-diphenyl-β-picrylhydrazyl.
References
- 1. Galán J. E., Lara-Tejero M., Marlovits T. C., and Wagner S. (2014) Bacterial type III secretion systems: specialized nanomachines for protein delivery into target cells. Annu. Rev. Microbiol. 68, 415–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yahr T. L., Goranson J., and Frank D. W. (1996) Exoenzyme S of Pseudomonas aeruginosa is secreted by a type III pathway. Mol. Microbiol. 22, 991–1003 [DOI] [PubMed] [Google Scholar]
- 3. Yahr T. L., Mende-Mueller L. M., Friese M. B., and Frank D. W. (1997) Identification of type III secreted products of the Pseudomonas aeruginosa exoenzyme S regulon. J. Bacteriol. 179, 7165–7168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yahr T. L., Vallis A. J., Hancock M. K., Barbieri J. T., and Frank D. W. (1998) ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. U.S.A. 95, 13899–13904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Finck-Barbançon V., Goranson J., Zhu L., Sawa T., Wiener-Kronish J. P., Fleiszig S. M., Wu C., Mende-Mueller L., and Frank D. W. (1997) ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol. Microbiol. 25, 547–557 [DOI] [PubMed] [Google Scholar]
- 6. Roy-Burman A., Savel R. H., Racine S., Swanson B. L., Revadigar N. S., Fujimoto J., Sawa T., Frank D. W., and Wiener-Kronish J. P. (2001) Type III protein secretion is associated with death in lower respiratory and systemic Pseudomonas aeruginosa infections. J. Infect. Dis. 183, 1767–1774 [DOI] [PubMed] [Google Scholar]
- 7. Hauser A. R., Cobb E., Bodi M., Mariscal D., Vallés J., Engel J. N., and Rello J. (2002) Type III protein secretion is associated with poor clinical outcomes in patients with ventilator-associated pneumonia caused by Pseudomonas aeruginosa. Crit. Care Med. 30, 521–528 [DOI] [PubMed] [Google Scholar]
- 8. Anderson D. M., Schmalzer K. M., Sato H., Casey M., Terhune S. S., Haas A. L., Feix J. B., and Frank D. W. (2011) Ubiquitin and ubiquitin-modified proteins activate the Pseudomonas aeruginosa T3SS cytotoxin, ExoU. Mol. Microbiol. 82, 1454–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sato H., Frank D. W., Hillard C. J., Feix J. B., Pankhaniya R. R., Moriyama K., Finck-Barbançon V., Buchaklian A., Lei M., Long R. M., Wiener-Kronish J., and Sawa T. (2003) The mechanism of action of the Pseudomonas aeruginosa-encoded type III cytotoxin, ExoU. EMBO J. 22, 2959–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Anderson D. M., Sato H., Dirck A. T., Feix J. B., and Frank D. W. (2015) Ubiquitin activates patatin-like phospholipases from multiple bacterial species. J. Bacteriol. 197, 529–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sato H., Feix J. B., Hillard C. J., and Frank D. W. (2005) Characterization of phospholipase activity of the Pseudomonas aeruginosa type III cytotoxin, ExoU. J. Bacteriol. 187, 1192–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tamura M., Ajayi T., Allmond L. R., Moriyama K., Wiener-Kronish J. P., and Sawa T. (2004) Lysophospholipase A activity of Pseudomonas aeruginosa type III secretory toxin ExoU. Biochem. Biophys. Res. Commun. 316, 323–331 [DOI] [PubMed] [Google Scholar]
- 13. Gendrin C., Contreras-Martel C., Bouillot S., Elsen S., Lemaire D., Skoufias D. A., Huber P., Attree I., and Dessen A. (2012) Structural basis of cytotoxicity mediated by the type III secretion toxin ExoU from Pseudomonas aeruginosa. PLoS Pathog. 8, e1002637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Halavaty A. S., Borek D., Tyson G. H., Veesenmeyer J. L., Shuvalova L., Minasov G., Otwinowski Z., Hauser A. R., and Anderson W. F. (2012) Structure of the type III secretion effector protein ExoU in complex with its chaperone SpcU. PLoS One 7, e49388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rydel T. J., Williams J. M., Krieger E., Moshiri F., Stallings W. C., Brown S. M., Pershing J. C., Purcell J. P., and Alibhai M. F. (2003) The crystal structure, mutagenesis, and activity studies reveal that patatin is a lipid acyl hydrolase with a Ser-Asp catalytic dyad. Biochemistry 42, 6696–6708 [DOI] [PubMed] [Google Scholar]
- 16. Hirschberg H. J., Simons J. W., Dekker N., and Egmond M. R. (2001) Cloning, expression, purification and characterization of patatin, a novel phospholipase A. Eur. J. Biochem. 268, 5037–5044 [DOI] [PubMed] [Google Scholar]
- 17. Dessen A., Tang J., Schmidt H., Stahl M., Clark J. D., Seehra J., and Somers W. S. (1999) Crystal structure of human cytosolic phospholipase A2 reveals a novel topology and catalytic mechanism. Cell 97, 349–360 [DOI] [PubMed] [Google Scholar]
- 18. Phillips R. M., Six D. A., Dennis E. A., and Ghosh P. (2003) In vivo phospholipase activity of the Pseudomonas aeruginosa cytotoxin ExoU and protection of mammalian cells with phospholipase A2 inhibitors. J. Biol. Chem. 278, 41326–41332 [DOI] [PubMed] [Google Scholar]
- 19. Benson M. A., Komas S. M., Schmalzer K. M., Casey M. S., Frank D. W., and Feix J. B. (2011) Induced conformational changes in the activation of the Pseudomonas aeruginosa type III toxin, ExoU. Biophys. J. 100, 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Veesenmeyer J. L., Howell H., Halavaty A. S., Ahrens S., Anderson W. F., and Hauser A. R. (2010) Role of the membrane localization domain of the Pseudomonas aeruginosa effector protein ExoU in cytotoxicity. Infect. Immun. 78, 3346–3357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tyson G. H., and Hauser A. R. (2013) Phosphatidylinositol 4,5-bisphosphate is a novel coactivator of the Pseudomonas aeruginosa cytotoxin ExoU. Infect. Immun. 81, 2873–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stirling F. R., Cuzick A., Kelly S. M., Oxley D., and Evans T. J. (2006) Eukaryotic localization, activation and ubiquitinylation of a bacterial type III secreted toxin. Cell. Microbiol. 8, 1294–1309 [DOI] [PubMed] [Google Scholar]
- 23. Rabin S. D., Veesenmeyer J. L., Bieging K. T., and Hauser A. R. (2006) A C-terminal domain targets the Pseudomonas aeruginosa cytotoxin ExoU to the plasma membrane of host cells. Infect. Immun. 74, 2552–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geissler B., Tungekar R., and Satchell K. J. (2010) Identification of a conserved membrane localization domain within numerous large bacterial protein toxins. Proc. Natl. Acad. Sci. U.S.A. 107, 5581–5586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Geissler B., Ahrens S., and Satchell K. J. (2012) Plasma membrane association of three classes of bacterial toxins is mediated by a basic-hydrophobic motif. Cell. Microbiol. 14, 286–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sato H., and Frank D. W. (2014) Intoxication of host cells by the T3SS phospholipase ExoU: PI(4,5)P2-associated, cytoskeletal collapse and late phase membrane blebbing. PLoS ONE 9, e103127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Columbus L., and Hubbell W. L. (2004) Mapping backbone dynamics in solution with site-directed spin labeling: GCN4–58 bZip free and bound to DNA. Biochemistry 43, 7273–7287 [DOI] [PubMed] [Google Scholar]
- 28. Columbus L., and Hubbell W. L. (2002) A new spin on protein dynamics. Trends Biochem. Sci. 27, 288–295 [DOI] [PubMed] [Google Scholar]
- 29. Klug C. S., and Feix J. B. (2008) Methods and applications of site-directed spin labeling EPR spectroscopy. Methods Cell Biol. 84, 617–658 [DOI] [PubMed] [Google Scholar]
- 30. Altenbach C., Froncisz W., Hemker R., McHaourab H., and Hubbell W. L. (2005) Accessibility of nitroxide side chains: absolute Heisenberg exchange rates from power saturation EPR. Biophys. J. 89, 2103–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Isas J. M., Langen R., Haigler H. T., and Hubbell W. L. (2002) Structure and dynamics of a helical hairpin and loop region in annexin 12: a site-directed spin labeling study. Biochemistry 41, 1464–1473 [DOI] [PubMed] [Google Scholar]
- 32. Altenbach C., Greenhalgh D. A., Khorana H. G., and Hubbell W. L. (1994) A collision gradient method to determine the immersion depth of nitroxides in lipid bilayers: application to spin-labeled mutants of bacteriorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 91, 1667–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Frazier A. A., Wisner M. A., Malmberg N. J., Victor K. G., Fanucci G. E., Nalefski E. A., Falke J. J., and Cafiso D. S. (2002) Membrane orientation and position of the C2 domain from cPLA2 by site-directed spin labeling. Biochemistry 41, 6282–6292 [DOI] [PubMed] [Google Scholar]
- 34. Anderson D. M., Feix J. B., Monroe A. L., Peterson F. C., Volkman B. F., Haas A. L., and Frank D. W. (2013) Identification of the major ubiquitin-binding domain of the Pseudomonas aeruginosa ExoU A2 phospholipase. J. Biol. Chem. 288, 26741–26752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang W., Crocker E., McLaughlin S., and Smith S. O. (2003) Binding of peptides with basic and aromatic residues to bilayer membranes: phenylalanine in the myristoylated alanine-rich C kinase substrate effector domain penetrates into the hydrophobic core of the bilayer. J. Biol. Chem. 278, 21459–21466 [DOI] [PubMed] [Google Scholar]
- 36. Victor K. G., and Cafiso D. S. (2001) Location and dynamics of basic peptides at the membrane interface: electron paramagnetic resonance spectroscopy of tetramethyl-piperidine-N-oxyl-4-amino-4-carboxylic acid-labeled peptides. Biophys. J. 81, 2241–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ellena J. F., Moulthrop J., Wu J., Rauch M., Jaysinghne S., Castle J. D., and Cafiso D. S. (2004) Membrane position of a basic aromatic peptide that sequesters phosphatidylinositol 4,5 bisphosphate determined by site-directed spin labeling and high-resolution NMR. Biophys J. 87, 3221–3233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Malmberg N. J., Van Buskirk D. R., and Falke J. J. (2003) Membrane-docking loops of the cPLA2 C2 domain: detailed structural analysis of the protein-membrane interface via site-directed spin-labeling. Biochemistry 42, 13227–13240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao J., Burke J. E., and Dennis E. A. (2013) Using hydrogen/deuterium exchange mass spectrometry to define the specific interactions of the phospholipase A2 superfamily with lipid substrates, inhibitors, and membranes. J. Biol. Chem. 288, 1806–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 41. Bucher D., Hsu Y. H., Mouchlis V. D., Dennis E. A., and McCammon J. A. (2013) Insertion of the Ca2+-independent phospholipase A2 into a phospholipid bilayer via coarse-grained and atomistic molecular dynamics simulations. PLoS Comput. Biol. 9, e1003156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mouchlis V. D., Bucher D., McCammon J. A., and Dennis E. A. (2015) Membranes serve as allosteric activators of phospholipase A2, enabling it to extract, bind, and hydrolyze phospholipid substrates. Proc. Natl. Acad. Sci. U.S.A. 112, E516–E525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Benson M. A., Schmalzer K. M., and Frank D. W. (2010) A sensitive fluorescence-based assay for the detection of ExoU-mediated PLA2 activity. Clin. Chim. Acta 411, 190–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stewart J. C. M. (1980) Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal. Biochem. 104, 10–14 [DOI] [PubMed] [Google Scholar]
- 45. Stoll S., and Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 [DOI] [PubMed] [Google Scholar]
- 46. Klug C. S., Su W., and Feix J. B. (1997) Mapping of the residues involved in a proposed β-strand located in the ferric enterobactin receptor FepA using site-directed spin-labeling. Biochemistry 36, 13027–13033 [DOI] [PubMed] [Google Scholar]
- 47. Mandell D. J., Coutsias E. A., and Kortemme T. (2009) Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Methods 6, 551–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li S. C., and Ng Y. K. (2010) Calibur: a tool for clustering large numbers of protein decoys. BMC Bioinformatics 11, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Foloppe N., and MacKerell J. A. D. (2000) All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 21, 86–104 [Google Scholar]
- 51. Páll S., and Hess B. (2013) A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 184, 2641–2650 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.