

Abstract
Analysis of human chromosomal translocation sequence locations has yielded valuable clues about the mechanism of the translocations and their timing. Biochemical work on the mechanisms of DNA breakage and rejoining permits formulation of a detailed model of the human chromosomal translocation process in lymphoid neoplasms. Most human lymphomas are derived from B cells in which the break is initiated by activation-induced deaminase (AID). The partner locus in many cases is located at one of the antigen receptor loci, and this break is generated by the RAG complex or by AID. After breakage, the joining process typically occurs by nonhomologous DNA end joining (NHEJ). Some of the insights into this mechanism also apply to nonlymphoid neoplasms.
Keywords: lymphoma, DNA repair, lymphoid malignancy, activation-induced deaminase (AID), RAG (recombination-activating gene), DNA recombination, V(D)J recombination, immunoglobulin class switch recombination (CSR), nonhomologous DNA end joining (NHEJ), switch region (S region)
Introduction to Neoplastic Translocations
Most chromosomal translocations consist of a breakage phase involving two chromosome breaks and then a joining phase 1(Fig. 1). Each of the two chromosome breaks generates two DNA ends, resulting in a total of four DNA ends, which all must be rejoined if the translocation is to be a reciprocal exchange of the two chromosomal arms. Reciprocal translocations are the most common type of translocation in lymphoid neoplasms (the translocation process is less well understood for carcinomas and sarcomas, which are often found to be nonreciprocal by the time that they become clinically apparent). The joining phase defines the sequence window in which the original break occurred, and we must work backward from this window to determine the sequence motifs that contributed to the original breakage mechanism. For this reason, we begin at the end – namely, with the joining phase, before devoting the remainder of our discussion on the more complex subject of how the chromosome breaks occur in the first place. Our emphasis is on human lymphoid translocations because, the breakage phase for these translocations is better understood than for other neoplasms. However, our summary of the joining phase applies to all human chromosomal translocations, regardless of the tissue of origin. For lymphoid translocations, the chromosome breaks are usually caused by either AID or the RAG complex 1–4. For the small subset of lymphoid translocations where AID or RAG are not involved, there is a short list of causes that also apply to nonlymphoid cells (see Fig. 1, upper right).
Figure 1. Causes and Repair of Double-Strand DNA Breaks.

Physiologic and pathologic causes of double-strand breaks in mammalian somatic cells are listed at the top. During S and G2 of the cell cycle, homology-directed repair is common because the two sister chromatids are in close proximity, providing a nearby homology donor. Homology-directed repair includes homologous recombination (HR) and single-strand annealing (SSA). At any time in the cell cycle, double-strand breaks can be repaired by nonhomologous DNA end joining (NHEJ). Proteins involved in the repair pathways are listed.
The Joining Phase of Human Lymphoid Chromosomal Translocations
After two chromosomal breaks arise, a new configuration generates two derivative chromosomes (each with its own centromere) in a stable reciprocal translocation 5, 6. The newly formed junctions generated in most human lymphoid translocations have the canonical features of nonhomologous DNA end joining (NHEJ)(Suppl. Fig. 1)7, 8. These typically include (a) nucleotide loss from the DNA ends (Suppl. Fig. 2); (b) alignment of terminal microhomology (1 or 2 bp) between the two DNA ends to guide the rejoining process; and (c) template-independent nucleotide insertion by either polymerase (pol) mu or pol lamda, or by TdT polymerase in lymphoid cells. TdT polymerase activity is strictly template-independent whereas pol mu and pol lambda activities are predominantly template-dependent, but have a limited degree of template-independent activity (usually < 3nt)5. Such nucleotide additions are called N-nucleotides or N-regions, which was the original designation given for TdT additions. TdT is only expressed in early T and B cells (pro-/pre-B and -T) in humans to increase junctional diversity during the normal V(D)J recombination process (INSET Box 1). The presence of extensive N-nts in many human lymphoid chromosomal translocation junctions is one indication that most patient lymphoid translocations occur during the pro-B and pre-B phases of B cell differentiation, and correspondingly in early T cells 1.
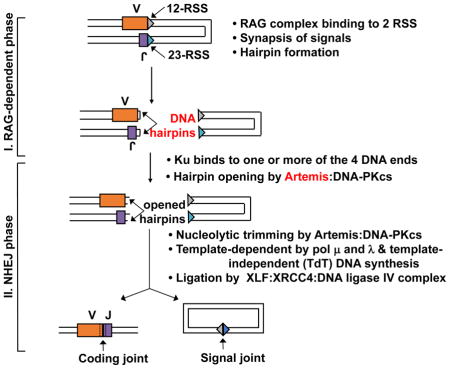
INSET BOX 1. Mechanism of V(D)J Recombination.
V(D)J recombination occurs at sequences called 12-RSS and 23-RSS (triangles in the figure), where RSS designates recombination signal sequence. An RSS contains conserved heptamer and nonamer sequence elements, separated by either 12 or 23 nonconserved base pairs, and hence the designation 12-RSS and 23-RSS. One recombination event requires one 12-RSS and one 23-RSS, and this is called the 12/23 rule. Along with the constitutively expressed HMGB1 protein, the early lymphoid-specific RAG1 and RAG2 proteins form a complex, designated the RAG complex, which nicks and then hairpins the DNA ends at the V and J segments in the figure. Ku can bind to any of the four DNA ends. The Artemis:DNA-PKcs complex then binds to the V and J hairpin ends and nicks the hairpins in a manner that usually results in a 3′ overhang. The Artemis:DNA-PKcs complex can then further endonucleolytically resect at any 3′ or 5′ overhang. Pol μ and λ can fill-in in a template-independent manner. The ligase complex includes XLF (Cernunnos), XRCC4, and DNA ligase IV. Some antigen receptor loci have not only V and J segments, but also D segments; hence, the name V(D)J recombination.
Terminal microhomology usage arises when nucleotides that are the same between two DNA termini align with one another, followed by resection of the excess DNA flaps, resulting in only one block of microhomology being preserved in the final junction. The apparent frequency of this is not as high in human lymphoid chromosomal translocations as it is in other instances of double-strand break (DSB) repair by NHEJ. This might be because TdT addition to DNA ends may generate new terminal microhomology that was not present in the original DNA ends, and thus, such alignment may not be recognizable as microhomology usage. In contrast, a process that is known as alternative DNA end joining (alt-EJ) is noted for having >3 nts of terminal microhomology alignment. In murine translocations, it is possible that there is a more robust a-EJ pathway that is more apparent in experimentally mutant mice that lack NHEJ components 6. For naturally-arising human chromosomal translocations, however, the joining phase is due to NHEJ [sometimes called c-NHEJ, where the c designates classical or canonical, to distinguish it from the less defined a-EJ (microhomology-mediated end joining, or MMEJ, is another name for a-EJ)]9.
One unusual feature of human lymphoid translocation NHEJ events is something called templated nucleotides (T-nts)10, 11. This is where nucleotides appear to be copied from either of the two DNA ends and then incorporated into the final junction in either a direct or inverted orientation to create a short duplication or a short inversion. We have described previously how T-nts are likely to arise due to slippage and template-dependent synthesis by either pol mu or pol lambda 5. T-nts are present in only a small subset of translocation junctions, whereas the other features mentioned above are present in most of the lymphoid translocation junctions. T-nts are rarely found in murine NHEJ or in human nonlymphoid translocations 10, 11.
Causes of Chromosome Breaks
We now turn to the more complex topic of how chromosomal breaks arise in the first place. The list of general causes of chromosomal breaks applies to all living cells and includes (a) ionizing radiation, (b) oxidative free radicals, (c) replication across a nick, (d) pathologic action by nuclear enzymes such as nucleases acting at particularly fragile sites, (e) failed topoisomerase II reactions, and (f) mechanical stress. These same general causes occur in lymphoid cells 5(Fig. 1). In addition to these general mechanisms, in lymphoid cells, the physiologic functions of the RAG complex or of activation-induced deaminase (AID) can go awry and contribute substantially to translocations 1–4. The normal V(D)J recombination process catalyzed by the RAG complex is shown in INSET BOX 1. Simple models for the role of AID in immunoglobulin heavy chain class switch recombination (IgH CSR) and somatic hypermutation (Ig SHM) are shown in INSET BOX 2 and 3, respectively. The developmental times of expression of RAG and AID are depicted in Suppl. Fig. 3.
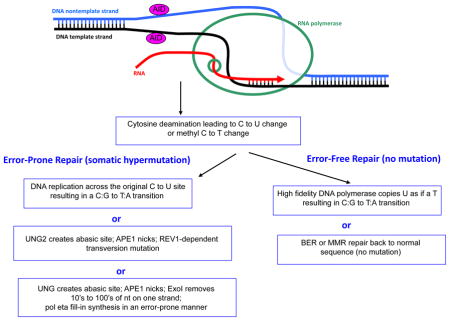
INSET BOX 3. Mechanistic Aspects of Ig Somatic Hypermutation.
Though not fully understood, some of the known elements of the somatic hypermutation (SHM) process are depicted. AID requires ssDNA 20, 37. Unlike for IgH CSR, where stable kilobase length R-loops provide ssDNA, for Ig SHM at the physiologic loci (primarily IgH and IgL V segments), the ssDNA may arise simply due to transcription, which is accompanied by transient underwinding of the duplex DNA as the RNA polymerase passes through a region. After AID deamination of C to U on either strand of the DNA (or methyl C to T), then either of two error-free mechanisms may repair the site, without mutation. But in Ig SHM, any of three error-prone pathways may operate. First, an error-free DNA polymerase may simply copy the U, which is read as a T, resulting in a C:G to T:A transition. Second, uracil glycosylase (UNG2) will remove the U, and apurinic/apyrimidinic endonuclease 1 (APE1) will nick 5′ of the abasic site. This will then allow a REV1-dependent transversion to result. Third, after UNG2 and APE1 action, Exo1 can resect downstream of the nick site, providing a long gap that is filled-in by the error-prone pol eta.
Before discussing the role of the RAG complex and AID in lymphoid malignancies, it is useful to note that all lymphoid chromosomal translocations require two DSBs. These two DSBs may have independent causes. In the following sections, we will consider events where the RAG complex causes the breaks on both chromosomes. We will refer to these as RAG-type events at both chromosomal locations (e.g., RAG-type/RAG-type translocation or deletion to indicate the role of RAGs at both DSBs)(Fig. 2). In other cases, AID can initiate damage causing a DSB on one chromosome (an AID-type break), and the RAG complex would cause the break at the other chromosome (e.g., RAG-type/AID-type translocation or deletion). Or both breaks can be AID-type, as discussed below. In yet other cases, a break may be of uncertain origin: not RAG or AID, but perhaps due to one of the general causes mentioned above such as oxidative damage or replication across a nick. We will refer to these as simply ‘uncertain-type’ to indicate DSBs of uncertain origin. To summarize, the two DSBs required for lymphoid translocations may be a combination of any of these: RAG, AID or DSBs of uncertain origin.
Figure 2. Common Chromosomal Translocations in T and B Cells.


A. Some lymphoid translocations involve a DSB that is a RAG-type event at each of the two participating chromosomes (top left). Such RAG-type/RAG-type translocations are common in T cell lymphomas and a subset of B cell lymphomas. More commonly among B cell lymphomas, the initial translocation involves a RAG-type event at one chromosome, often the normal IgH locus [or one of the other antigen receptor loci, such as IgL (κ or λ) or one of the TCR loci] and an AID-type event at the other chromosome (bottom right). After exchange of the chromosomal arms, the DNA ends are joined by NHEJ to generate the two derivative chromosomes. Such events are the predominant mechanism found in most human B cell lymphomas. Centromeres are shown as a filled black oval.
B. AID-Type events can cause translocations in early B cells or in germinal center B cells. In the upper left and right, an AID-type event and a RAG-type break give rise to translocations in pro-/pre-B cells. In the lower left and right, an AID-type event on one chromosome can give rise to a translocation involving a failed IgH class switch recombination (CSR) event, which is a physiologic AID-type event. Centromeres are shown as a filled black oval.
Features of Human B Cell Translocations
Pro-/Pre-B Cell Translocations of the BCL-1 Locus, BCL-2 Locus, the E2A Gene, the MALT1 Gene, and the CRLF2 Gene
Many human B cell malignancies involve translocations between the IgH locus and non-Ig loci on other chromosomes 1, 12–15. The breaks at the IgH locus are usually directly adjacent to V, D or J segments, if the breaks arise due to the RAG complex. The non-Ig loci most often include human translocations involving BCL-1, BCL-2, E2A, MALT1 and CRLF2, and these translocations occur when the incipient neoplastic cell is a pro-B or pre-B cell (Fig. 2).
It is important to note that the neoplasms arising from these pro-B or pre-B translocations often differentiate into mature B cells before they give rise to the tumor cells. This is the case for follicular lymphoma (FL), mantle cell lymphoma (MCL), mucosal-associated lymphoid tumor (MALT) lymphoma, and diffuse large B cell lymphoma (DLBCL), which have a cell surface phenotype of a mature B cell rather than that of an early B cell. Thus, the initial characteristic translocation occurs at the early B cell stage (pro-/pre-B), but the cells differentiate further along the B cell pathway before transforming into a mature B cell neoplasm 1.
The RAG-type event at the IgH locus in these translocations is most often created by a ‘failed’ DH to JH recombination event. The event ‘failed’ in the sense that normally the DH and JH join to one another but, in these pathologic cases, the DH and JH join instead to the DNA ends at a DSB in a non-Ig locus. The DSB at the non-Ig locus is usually not at a recombination signal sequences (RSS), and hence, is not a RAG complex initiated DSB. Based on inferences described in later sections, these breaks occur in a process initiated by AID (AID-type events). Thus, these translocations are RAG-type/AID-type events; that is, a RAG-type event occurs at the IgH locus and an AID-type event occurs at the non-Ig locus, resulting in two DSBs.
Mature B Cell Translocations of the c-MYC and BCL-6 Genes
Some translocations involving the IgH locus do not break at the V, D or J segments but rather break at the IgH class switch regions (IgH S) during a failed class switch recombination (CSR) event. IgH CSR is a process that is initiated by AID (Inset Box 2). Hence, DSBs in the IgH S regions are AID-type events. The IgH CSR process occurs not in pro-/pre-B cells but rather in mature B cells in the germinal centers of the lymph nodes, spleen, and Peyer’s patches of the GI tract, as reviewed elsewhere 16–18.
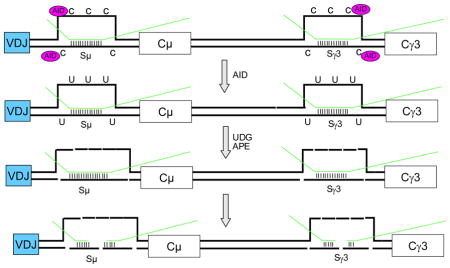
INSET BOX 2. Mechanism of Mammalian Ig Heavy Chain Class Switch Recombination.
Mammalian IgH switch regions form R-loops upon transcription, and these have now been demonstrated to be important for the efficiency of IgH class switch recombination (CSR)39, 40. AID requires single-stranded DNA in order to recognized cytosine (C) as a substrate, and R-loops provide a fully single-stranded nontemplate strand. AID has a preference for C that is surrounded by the sequence WGCW, where W = A or T. But AID can deaminate any C to a U (and any methyl C to a T, though at an efficiency that is somewhat lower)37. Once AID has converted some of the C’s in the switch region to U, then uracil glycosylase (UNG2) can remove the U to create an abasic site [depicted as short gaps in figure]. Next, AP endonuclease (APE1) can create a nick at the abasic site. This explains nicks on the nontemplate strand. RNase H can remove portions of the RNA [RNA depicted as thin green lines] that are annealed to the template DNA strand, thereby exposing single-stranded DNA regions– thus allowing AID action there as well [depicted as large gaps in bottom image]. AID can also act on the template strand at the edge of the R-loop.
For human IgH locus translocations that involve the IgH class switch regions, a failure of the DNA ends at Sμ to be joined to the ends of a DSB at a downstream switch region (e.g, Sγ) allows the two Sμ ends to be joined to the two ends of a break on another chromosome, such as at c-MYC or BCL-6 (Suppl. Fig. 4)15. Thus, the IgH locus breaks in these events are usually created during IgH CSR.
Studies in murine systems have shown that the c-MYC break is dependent on AID 19. For human translocations, our work has shown that the c-MYC and BCL-6 translocations occur at DNA sequence motifs for which the AID enzyme has a known predilection, as discussed below in detail 15. Thus, the breaks at BCL-6 and c-MYC are AID-type events, and the breaks at the IgH switch regions are AID-dependent. These human translocations between IgH S regions and BCL-6 and c-MYC are AID-type/AID-type events.
Focal DNA Sequence Motifs at Human B and T Cell Chromosomal Breaks
AID-Type Breaks at CpG and WGCW Motifs
One important way to determine the cause of spontaneous chromosome breaks is to determine if the breaks preferentially occur near any specific DNA sequence. In human patient translocations, for a variety of B cell malignancies, two DNA sequence motifs are located at or near breaks at an elevated frequency which is highly statistically significant (p < 10−3). The two motifs are CG (also called CpG) and WGCW, where W = A or T (Suppl. Fig. 5)1, 12–15.
The palindromic C’s within WGCW are well-known as the optimal site of cytosine deamination by AID among all possible sequences 20–22. Like many enzymes for which DNA is a substrate, even though C is the site of action, the adjacent nucleotides in the DNA have an influence on the site-selection the AID enzyme. The C’s within CG sites are also distinctive for being potential sites of DNA methylation (also called methyl CpG sites). AID can deaminate at CG as well as the methylated form of CG (designated mCpG most commonly). It is noteworthy that CG and GC (the core of WGCW) are the only two sequences that pack C’s as close as possible in a configuration where these are opposite one another on the two strands of the DNA duplex. This probably favors the formation of DSBs during repair of each deaminated C because repair is occurring at two sites across from one another in the DNA helix.
The identification of CG and WGCW as sequence motifs is the strongest evidence implicating AID as the cause of DSBs in these human lymphoid chromosomal translocations 1, 12–15. A role for AID explains why C is the site of initial breakage; namely because AID is a cytosine deaminase. A direct molecular test using replicating human minichromosomes documented the role of AID and the methylation of CG sites in this proposed series of biochemical steps for the translocation break process 23. There are three mechanisms by which a mismatch site created by AID could lead to a DSB: (a) DNA glycosylase creation of an abasic site, followed by action by APE1; (b) action by the Artemis:DNA-PKcs nuclease complex; or (c) endonuclease action by the RAG complex (Suppl. Fig. 6). We have proposed that the Artemis:DNA-PKcs complex may account for most of the DNA cutting events because this would account for how breaks in two different locations of in the genome (e.g., one initiated by AID and one by the RAG complex) could both occur within a constrained time window (Suppl. Fig. 6) 23.
Direct evidence in mice and humans for AID action in these translocations showed that (a) cytosine-deamination at CpGs is enriched in patient-derived ALL samples throughout the genome; (b) human fetal liver and bone marrow B cell precursors have both active AID and RAG1/2 proteins; and (c) concurrent enzyme expression of RAG1/2 and AID occurs in single murine pre-B cell clones and in human pre-B ALL clones; and (d) inflammatory cytokine stimuli increase the chromosomal damage caused by simultaneous expression of AID and RAG1/2 [the damage includes chromosomal deletions, insertions, inversions, in addition to chromosomal translocations 24].
The molecular and cellular data provides compelling support for the concept that AID-initated lesions explain a large number of human chromosomal breaks. Future work in this area could focus on the balance of the various causes: AID-type to RAG-type translocations versus RAG-type to RAG-type or AID-type to AID-type translocations. Additionally, greater insight into the types of viral or bacterial infection that raise AID levels in human pre-B cells would be valuable for understanding how these contribute to increases in lymphoid malignancies in children 24.
RAG-Type Breaks at CAC Motifs
RAG-type breaks virtually always occur at CAC sequences, because this is the essential portion of the heptamer of all RSS. In human T cell malignancies, the RAG complex can cause interstitial deletions quite commonly. Most notably, in pre-T acute lymphoblastic leukemia/lymphoma (ALL), the RAG complex causes breaks at both the SCL (also called TAL1) and SIL (SCL-Interrupting Locus) locations 25, 26. Deletions due to the RAG complex also occur at the p16 gene in human pre-T lymphomas 27. In the designation system used here, these are all RAG-type/RAG-type deletions. This propensity for interstitial deletions may simply reflect that the RAG complex can bind the RSS more readily when they are tethered together -- that is, on the same DNA molecule 28. This is because the synaptic assembly of the RAG complex with two RSS or pseudo-RSS sites will be faster for RSSs that are nearby on the same chromosome 29. In murine models that involve aberrant expression or mutant forms of the RAG complex, action at CAC motifs is unusually common 30–32.
Less common than the interstitial (intrachromosomal) deletions just mentioned, there are RAG-type/RAG-type translocations in human pre-T cell lymphomas 33. Examples include T-cell receptor locus translocations with the SCL (TAL1), LMO2, HOX11(TLX1), and TTG1 loci in pre-T lymphomas (Fig. 2A).
A small percentage of human B cell malignancies also appear to involve interstitial deletions caused by the RAGs acting at both DSBs (RAG-type/RAG-type events), such as the CRLF2-P2RY8 event on the X-chromosome in some human pre-B ALL 34.
In pre-B ALL cells that already have a ETV6-RUNX1 (TEL-AML1) translocation by a yet undefined mechanism, the resulting cells subsequently acquire secondary deletions, inversions, insertions and translocations at off-target CAC-motifs due to continuous production of the RAG complex in the pre-B cells 35.
A common inversion on human chromosome 7 occurs between the physiologic RSS sites located at the TCRβ locus (chr 7q35) and the TCRγ locus (chr 7p14)33. This inversion can be seen in the peripheral blood of normal individuals and is of no known clinical significance 36.
The Single-Stranded DNA Requirement for AID Action at Cytosine in Human B Cells
AID only acts at cytosines that are within regions of single-stranded DNA (ssDNA)20, 37. Large zones of ssDNA can be created when an R-loop is formed 38–40. We have shown that R-loops form at IgH switch regions and that this is physiologically important for the IgH CSR process to occur 39–41. The stability and kilobase-length of R-loops provides ample ssDNA for efficient AID action during IgH class switch recombination. In contrast, Ig somatic hypermutation (SHM, also called affinity maturation) is a slower process, perhaps in part because the generation of ssDNA during transcription is very short and transient, and does not involve stable R-loop formation (see Inset Box 3).
Like the physiologic sites for AID action, pathologic or off-target sites of AID action must also have at least transient ssDNA character. The most common off-target oncogene hit by AID is c-MYC, accounting for >90% of lymphomas in most B-cell lymphoma mouse models [~3% in humans, see below]. One study using a monoclonal antibody (called S9.6) against RNA:DNA duplexes, has raised the possibility of a genomic R-loop at the exon1/intron1 junction of c-MYC, where most translocations occur 42; however, the S9.6 antibody is cross-reactive with RNA:RNA duplexes, and this currently precludes a conclusive statement about R-loops at this location in the genome 43, 44. If further studies demonstrate an R-loop at the exon1/intron1boundary of c-MYC, it would explain the action of AID at this location.
Divergently directed transcription is known to generate transient ssDNA between the diverging RNA polymerases 45, 46. The vulnerability of transcriptionally active regions to AID activity has been demonstrated in murine models 47, 48. This is highly relevant to the human BCL-6 gene 47, which undergoes translocations to the IgH S region by an AID-type/AID-type mechanism (Suppl. Fig. 7)15. The 2 kb BCL-6 fragile zone is delimited by the BCL-6 promoter and a noncoding RNA (ncRNA) region so precisely that it suggests a causal role that limits the translocations to this 2 kb zone rather than anywhere else within the remaining 9 kb of intron 1 of the BCL-6 gene 49. These two promoters are oriented in a convergent or opposing manner. But importantly, once the two RNA polymerases have passed one another, the physical impact on the DNA topology is the same as for divergent promoters 49. Divergent versus convergent transcriptional effects on chromosome breakage have now been formally tested in S. cerevisiae, and the results supports the importance of divergent transcription and the importance of topological tension for convergent transcription 50. Thus, relatively large zones with a high frequency of translocation such as at BCL-6 may be caused by divergent or closely positioned convergent transcription promoters.
Though fragile zones of a few kilobases may be explained by transient ssDNA generated at R-loops or by zones of divergent transcription, the reason for AID action at smaller zones of 20 to 150 bp is much less clear 1, 12–15. The BCL-1 major translocation cluster (MTC) region is only 100–150 bp and lies within a transcriptionally inactive region. The BCL-2 major breakpoint region (MBR) is not a site of R-loop or of divergent transcription. The same applies to the BCL-2 intermediate and minor cluster regions (icr and mcr) and to the MALT1, the CRLF2 and the E2A fragile zones (Fig. 3). We currently do not understand how these zones acquire a ssDNA state, but we have identified direct repeats at some of these sites, which could give rise to strand slippage 1. Further studies are needed to elucidate the underlying cause(s) of the elevated frequency of DSBs in these regions.
Figure 3. A High Proportion of Breakpoints on BCL-2, BCL-1, and E2A Fall into Fragile Regions that are Less than 600 bp.

Schematics of the BCL-1 [near CCND1 gene], E2A (also called TCF3), and BCL-2 regions illustrate clustering of breakpoints within the various identified cluster regions [green or black starbursts]. The breakpoints that do not fall into cluster regions are plotted randomly for illustrative purposes [short vertical lines]. In the top line (green horizontal line), the MTC is located about 110 kb from the gene for the cyclin D1 oncoprotein. The 150 bp MTC contains about 30% of breakpoints, whereas the remaining 70% of events are distributed widely over the surrounding 340 kb as recently mapped and sequenced 14. The second line shows a diagram of intron 13 of the E2A gene, taken from 84, which showed that 75% of breakpoints occur in the 23 bp E2A cluster, while the surrounding 3 kb only account for 25%. The third line depicts relative proportions of breakpoints at the BCL-2 MBR, icr, and mcr cluster regions. The third exon of the BCL-2 gene, black box on left, contains the MBR [major breakpoint region (or zone)] within the 3′ UTR region, while the centromeric 29 kb contains the icr (intermediate cluster region/zone) and mcr (minor cluster region/zone). Short vertical lines in the gene diagram mark the approximate locations and relative abundance of patient breakpoints. The 175 bp MBR, 105 bp icr, and 561 bp mcr account for about 50%, 13%, and 5% of bcl-2 translocation breakpoints. Every CG sequence motif in each of the three fragile zones the ~29kb downstream of the BCL-2 gene (MBR, icr, mcr) is a hotspot for human translocation. The figure shows the location of nearly all published BCL-2 translocations with expanded detail of the MBR in the very bottom line of the figure. Fragile regions or zone for one of the oncogenes in this review, BCL-2, are shown, but the principles apply to the other fragile zones as well. Within each fragile zone, the actual translocations occur at DNA sequence motifs, consisting of either the sequence CG (CpG) or mCG (when methylated) or WGCW where W=A or T. Human lymphomas are clinically indistinguishable regardless of the position of the breakpoint within the 29 kb. We are trying to determine why these BCL-2 and related zones at other loci (such as BCL-1, MTC, E2A, MALT1, and CRLF2) are highly preferred for DNA breakage and translocation. In the expanded MBR DNA sequence diagram (bottom line of figure), each small black triangle marks the breakpoint of a single, patient translocation. The breakage frequency within this 175 bp region is 300-fold higher than what one would expect to occur at random. Patient breaks within the MBR are not uniformly distributed across the entire 175 bp, but rather are focused in 3 peaks (bell-shaped gray areas), and there are CG sequence motifs (red bases) located near the center of each peak 1. The p values for the proximity of the MBR breaks to CG sites are between 10−42 and 10−96 (highly significant). The importance of CG and WGCW as translocation sequence motifs applies not only to the BCL-2 gene, but also to most other human B cell neoplastic chromosomal translocations. For example, breaks at the major translocation cluster (MTC) of the BCL-1 gene are also located near CG sites (p values of 10−9 to 10−13), and breaks on the telomeric side of the MTC are significantly near WGCW sites. In the MALT1 gene translocations to IgH in human MALT lymphomas (Fig. 2B), the breaks are also located at CG sites (p = 0.002). The proximity of CG to breaks applies also to the E2A gene translocations (p = 10−4) and the CRLF2 gene translocations to IgH (p = 0.0004) in human pre-B ALL.
B Cell Development and the Timing of the Translocations
The patient translocation sequence motifs, the junctions, and the Ig partners provide information on several facets of the translocation process. First, for any translocation in which one of the partners is directly adjacent to a V, D, or J segment of an Ig locus, the translocation likely occurred during pro-B or pre-B cell development because this is the phase when V(D)J recombination occurs (Fig. 4 and Suppl. Fig. 3). As mentioned above, when we analyzed the junctions of these translocations, regardless of the partner with which the Ig locus was translocated, they frequently included evidence of TdT nuclease activity 1, 12–15. Because TdT expression is specific to pro-/pre-B cells (and pro-/pre-T cells), this confirms that these B cell translocation events occurred during early B cell development.
Figure 4. Timing of chromosomal translocations as a function of B-cell development.

The three long rectangles in the middle of the figure (labeled BCL6, AID, and RAG) depict expression levels. Breakpoint motif analysis suggests that most lymphoma translocations occur either in germinal center B-cells, when AID and BCL-6 are highly expressed, or in pro-B/pre-B cells, when the RAG complex is highly expressed and AID is expressed at low levels (see text). The Ig breaks in most Ig-MYC and Ig-BCL6 translocations are in SH regions, which contain hundreds of WGCW repeats, and the MYC and BCL6 breaks occur near WGCW motifs scattered throughout these partner loci (red text in figure). The Ig breaks in most pro-B/pre-B cell translocations are generated at JH and DH segments by the RAG complex as part of the V(D)J recombination process (blue text). Most CpG breaks occur at Ig partner loci in pro-B/pre-B cell translocations but also in a subset of non-Ig–BCL6 rearrangements that probably occur in germinal center B-cells (orange text)14, 15.
Second, the CG and WGCW motifs at the non-Ig partner loci indicate that AID is being expressed at the same time as RAG-mediated Ig gene rearrangement in the pro-/pre-B cells. There have been many reports demonstrating AID expression in both murine and human pre-B cells 24, 51–59. The level of expression is low compared to that in germinal center B cells, but it appears to be sufficient to catalyze the low but important incidence of human lymphoid translocations 1, 7, 12–15, 24. Translocations involving at least one chromosome with an AID-type break account for nearly half of all human hematopoietic malignances (Fig. 5).
Figure 5. Fraction of Human Hematopoietic Malignancies Explained by AID-Type Breaks.

Each human hematopoietic malignancy is shown to reflect the fraction of all hematopoietic malignances. Numbers of events reflect the incidence of all events per year in the USA, including all ages (adults and children)85. The translocations and their percentage in each malignancy are estimated from Swerdlow et al 36. The portion in RED are translocations that involve at least one AID-type event, often at the oncogene. Some are AID-type/AID-type events, such as translocations between BCL-6 or c-MYC and the IgH switch regions. But most are AID-type/RAG-type events, such as BCL-1, BCL-2, MALT1, or CRLF2 to the IgH locus during failed DH to JH joining. The small fraction colored blue are RAG-type/RAG-type events. We note that many other translocations are not listed (e.g., BCR-ABL1 translocations also occur in B cell lineage ALL; and MLL-AF9 is seen in some cases of AML). This figure is not intended to be comprehensive, and readers are referred to current ACS statistics and Swerdlow et al. for further details.
Comparison of Human Translocations in Patients and Murine Model Systems
Several important studies in murine and cellular model systems have provided interesting information about the translocation process 3, 4, 47, 48, 60–62. In mice that spontaneously develop B cell lymphoma or develop lymphoma after being injected intraperitoneally with the immunologic adjuvant oil called pristane, the translocations of c-MYC account for nearly all of the tumors (>90%) 63. This is in contrast to humans where c-MYC is translocated in only 3% of human B cell lymphomas (and almost never in other neoplasms).
Several studies have shown that murine c-MYC translocations require AID 19. Unlike in patient translocations, no AID preferred DNA sequence motifs were initially identified at the break sites within c-MYC in the murine studies. Our re-analysis of c-MYC breaks in spontaneous and pristane-induced murine lymphomas identified AID motifs, specifically WGCW. But this was possible only after we had identified the WGCW motif in human patient c-MYC translocations, thereby allowing us to search for the same motif at the murine junctions 15.
Many murine lymphoid translocation models use the I-SceI endonuclease (which cleaves dsDNA at long recognition sequences) to create a defined break at a specific location (for example in c-MYC), and then this DSB is used to do a genome-wide search to determine the location of preferred spontaneously-breaking partners 64. From these studies, it was determined that gene promoters are preferred sites of breakage, perhaps because these regions may have open chromatin and some degree of ssDNA character due to the RNA polymerase II melting of the promoter(s). Further studies have shown that murine promoters are often sites of noncoding RNA transcription that might cause changes in local DNA topology accounting for the observations in mice 47, 48. Of note, the most common translocation fragile zones in humans (BCL-2, BCL-1, E2A, MALT1 and CRLF2) are not at promoters (or enhancers), illustrating a key difference between murine models and human translocations.
Replication stress has been raised as one factor in murine models involving use of replication poisons or repair inhibitors 65. Breakage zones in such models cover megabase zones, just as they do for the common fragile sites in humans, when replication poisons are used for cultured human cells 65, 66. The breakage zone around the murine BCL-2 gene covers a zone of 100,000 to 1 million bp in one murine model 65. This is quite distinct from the naturally-arising human translocation breakage zones at the BCL-2 gene, which are a few hundred bp in size 1.
Biological Factors Relevant to Neoplastic Chromosomal Translocations
Some translocations create a neoplastic cellular proliferation advantage 67, 68. In this review, the focus has been on the factors that make some very localized (20 to 600 bp) DNA sequences particularly prone to translocation events across many patients (e.g., BCL-2, BCL-1, E2A, MALT1 and CRLF2). These very small or localized zones can be 10 to 1000-fold more likely to be the site of translocation than sites in immediately adjacent DNA or within larger zones (e.g., 29–31 kb, as in the case of the BCL-2 gene, or 400 kb, as in the BCL-1 translocation). The malignancies arising from DSBs and translocations outside of the fragile zones versus within the fragile zones are indistinguishable.
Some rearrangements result in the formation of chimeric fusion transcripts from two genes, such as the t(1;19) translocation involving E2A and PBX1 69–74. In this case, the DSB could occur across much of the length of a given intron within each gene so as to create the neoplastic fusion transcript. This is the case or the PBX1 gene, where breaks occur randomly, at multiple locations in one intron. These DSBs are likely random and due to mechanisms such as free radical damage (i.e., ROS), ionizing radiation (IR), or aborted topoisomerase II reactions. But at the E2A gene, an AID-type break is localized to a 23 bp zone that is <1% of the size of the 3.2 kb intron in which it occurred.
Similar principles apply to the t(9;22) translocation that occurs in chronic myeloid leukemia (CML) and the t(9;22) translocation in a subset of acute lymphoblastic lymphoma (ALL) in which the BCR and ABL1 genes fuse, producing a fused transcript encoding the oncogenic p210 75. DSB formation at both the BCR and ABL1 genes occur over broad translocation zones (5.8 kb in the BCR gene and ~200 kb in the ABL1 gene) 76–79. Among the various BCR-ABL1 translocations that give rise to malignancies, the resulting fusion gene contains at least the first exon of BCR, which encodes an oligomerization domain, and frequently contains exons 2 to 11 of ABL1, which encode the tyrosine kinase. The 200 kb breakage zone in ABL1 contains translocation break sites distributed randomly within alternative exons 1a and 1b. The 5.8 kb breakage zone in BCR is the major breakpoint cluster region (M-BCR), encompassing exons 13 and 14. A minor BCR (m-BCR), which is ~130 kb in length in intron 1, can give rise to a shorter fusion gene encoding p190. Leukemia arises from this event only when the resulting mRNA encodes a functional protein. Thus, only some splice combinations produce a functional BCR/ABL1 fusion protein. Therefore, it is not the higher breakage frequency in BCR that makes it a translocation zone; rather, it is the growth advantage provided by the resulting functional BCR-ABL1 fusion protein which determines the boundaries of the translocation zone.
Again, the DSBs in both BCR and ABL1 are likely due to random causes (ROS, IR, topoisomerase reaction failures). Therefore, it is useful to distinguish between localized hotspots (i.e., high concentrations of breaks in small zones within the total breakage zone, usually <600 bp), as in the BCL-2, BCL-1 or E2A examples, versus broad translocation zones that often span the length of an entire intron (usually a few kilobases or even 10–100 kilobases in length).
For translocations involving small localized hotspots (20–600 bp), there are two major factors that make the translocation biologically apparent -- the increased propensity for DSB at the fragile zone and the resulting cellular growth advantage provided by the gene fusion product. In contrast, translocations that are not the result of “hotspot propensity” give rise to a malignancy when the growth advantage alone brings the translocation to the level of clinical attention 1.
Future Questions
Molecular Basis for the Fragile Zones in Human B Cell Lymphoma
We do not know why the CG motif breaks are clustered very tightly in zones of 20 to 600 bp, and yet CG sites located only 20 to 100 bp away are not vulnerable. There are many possible explanations, such as the strand slippage between repeats mentioned above or unusual interactions between certain DNA sequences and the histone octamer. Further studies are needed to experimentally examine these possibilities.
WGCW versus CG Motifs in Human B Cell Lymphoma
We can not yet explain why some human B cell lymphomas (BCL-2; BCL-1 MTC, CRLF2, MALT1 and E2A) occur at CG motifs but not WGCW motifs, whereas the bcl-1 breaks that occur outside of the MTC are located at either CG or WGCW motifs.
Similarly, we do not know why the c-MYC and BCL-6 translocations occur at the 4 bp WGCW motifs, but not at the 2 bp CG motifs. Of special note, the c-MYC and BCL-6 translocations usually also have point mutations near the translocation junction, but the CG motifs at the BCL-2, BCL-1, CRLF2, MALT1 and E2A translocations only rarely have such mutations. The WGCW motifs are within transcribed zones and are much larger regions (~2kb), whereas the CG motifs are not in transcribed zones and are found in smaller clusters of 20 to 600 bp.
Other Aspects
Some studies have suggested that the nuclear proximity of the two chromosomes involved in the translocation may be important for the translocation event 61, 80–82. Clearly, after the breaks have occurred, the DNA ends must come into proximity in order to be joined. But does chromosomal proximity predispose to translocation? There is no evidence in human lymphoid translocations that proximity is an important factor. Rather, the DNA breakage process appears to be the rate-limiting step, and studies in murine systems support this interpretation 83.
Concluding Comments
We have established the biochemical mechanism for a major fraction of human lymphomas. For T cell lymphomas and for B ALL, RAG-type/RAG-type combinations are common. For most B cell lymphomas, RAG-type/AID-type combinations are most frequent, such as at the BCL-2 gene, the bcl-1 MTC, CRLF2, and MALT1. For AID-type breaks that use the CG motif, we do not yet know the basis for their occurrence within precise 20 to 600 bp zones.
Supplementary Material
At-a-glance Summary.
Neoplastic chromosomal translocations (and other pathologic chromosomal rearrangements) can be considered in terms of a breakage phase, in which broken duplex DNA ends are generated, and then a rejoining phase. The rejoining phase is done by the nonhomologous DNA end joining (NHEJ) pathway.
In B lymphoid neoplasms, the breakage phase is often initiated by either the activation-induced deaminase, AID, or the RAG complex (RAG1 and RAG2). In T lymphoid neoplasms, the RAG complex is often responsible.
For B lymphoid neoplasms, the chromosome break sites are often in hotspot zones, and the breaks are often near CG (CpG) or WGCW sites, which are sites where AID can initiate lesions that lead to double-strand breaks. AID is known to only deaminate cytosines that are within single-stranded DNA (ssDNA) regions.
The ssDNA in the hotspot zones may arise due to a variety of causes that are still under investigation, such as transcriptionally-induced topological tension, R-loop formation, or some type of strand slippage at short repeat sequences. Concurrent expression of a low level of AID in pre-B cells, along with the usual high level of RAG expression, permits human translocation events in which a RAG-generated break joins to an AID-initiated break.
Examples of this include the human t(14;18) translocation involving the IgH locus and the BCL-2 gene. The t(11;14) translocation involving the IgH locus and the BCL-1 locus (CCND1 gene) is another example.
The principles discerned from lymphoid translocations are useful for considering the mechanism of chromosomal rearrangements and translocations generally. In particular, the sequence motif analysis may be generally useful for nonlymphoid cells.
Acknowledgments
The author thanks Dr. Raymond Mosteller for comments on the manuscript. Work in the author’s lab is supported by NIH. Many collaborators as well as members of the Lieber lab were central to the work leading to this review. Dr. Zhengfei Lu compiled the information leading to Figure 5.
GLOSSARY
- Chromosomal rearrangement
Pathologic rearrangements can involve deletions, inversions, insertions, duplications, or translocations.
- Chromosomal translocation
Translocations can be reciprocal or nonreciprocal, and the results are called derivative chromosomes.
- Reciprocal translocations
These are only stable if each derivative chromosome has one centromere; otherwise, one chromosome is lost due to lack of a centromere, and the other chromosome suffers repeated breakage-fusion-bridge cycles.
- RAG complex
Antigen receptors of the vertebrate immune system rely on RAG1 and RAG2, which form the RAG complex and which creates double-strand breaks at specific sequences called recombination signal sequences (RSS).
- RSS
Recombination signal sequences are the targeting sequences next to V, D and J segments for RAG binding, and an RSS consists of a palindromic heptamer (CACAGTG) and an AT-rich nonamer (ACAAAAACA) separated by 12 or 23 bp.
- AID
Activation-induced deaminase is a cytidine deaminase that converts C to T (or methyl C to U) in B cells of all vertebrates when it is expressed (predominantly in germinal centers, but at low levels in pre-B cells).
- DSB
Double-strand DNA breaks are repaired predominantly by NHEJ (throughout the cell cycle) or by homologous recombination (in late S/G2).
- NHEJ
Nonhomologous DNA end joining is major pathway for repairing double-strand DNA breaks that do not have homology between them (shared microhomology of 1 to 3 bp between the two DNA ends is often utilized during the joining process).
- V(D)J recombination
The DNA recombination process in pro-/pre-B and pro-/pre-T cells by which the antigen receptors variable domain exons are assembled from sub-exonic segments called V, D and J to generate a complete T-cell receptor or immunoglobulin.
- IgH CSR
Immunoglobulin class switch recombination is the DNA recombination process by which the heavy chain isotype is changed from making IgM to making IgG, IgA, or IgE.
- Methyl CpG
The 5′-CG-3′ dinucleotide is methylated at some (not all) locations in the vertebrate genome.
Biography
Michael Lieber is a professor at the University of Southern California. His laboratory has focused on chromosomal translocations, V(D)J recombination, IgH class switch recombination and NHEJ since its inception in 1989 and has discovered the CpG motif of translocations, the R-loops in IgH CSR, and the ligase of NHEJ.
References
- 1.Tsai AG, et al. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135:1130–42. doi: 10.1016/j.cell.2008.10.035. The CpG sequence motif is decribed here as a site of translocation breakage. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahowald GK, Baron JM, Sleckman BP. Collateral damage from antigen receptor gene diversification. Cell. 2008;135:1009–12. doi: 10.1016/j.cell.2008.11.024. [DOI] [PubMed] [Google Scholar]
- 3.Gostissa M, Alt FW, Chiarle R. Mechanisms that promote and suppress chromosomal translocations in lymphocytes. Annu Rev Immunol. 2011;29:319–50. doi: 10.1146/annurev-immunol-031210-101329. [DOI] [PubMed] [Google Scholar]
- 4.Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141:27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lieber MR. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 7.Tsai AG, Lieber MR. Mechanisms of chromosomal rearrangement in the human genome. BMC Genomics. 2010;11(Suppl 1):S1. doi: 10.1186/1471-2164-11-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lieber MR, Gu J, Lu H, Shimazaki N, Tsai AG. Nonhomologous DNA End Joining (NHEJ) and Chromosomal Translocations in Humans. Subcell Biochem. 2010;50:279–96. doi: 10.1007/978-90-481-3471-7_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghezraoui H, et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol Cell. 2014;55:829–42. doi: 10.1016/j.molcel.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaeger U, et al. Follicular lymphomas BCL-2/IgH junctions contain templated nucleotide insertions: novel insights into the mechanism of t(14;18) translocation. Blood. 2000;95:3520–3529. This is the first description of templated nucleotides at chromosomal translocation junctions. [PubMed] [Google Scholar]
- 11.Welzel N, et al. Templated nucleotide addition and immunoglobulin JH-gene utilization in t(11;14) junctions: implications for the mechanism of translocation and the origin of mantle cell lymphoma. Cancer Res. 2001;61:1629–1636. [PubMed] [Google Scholar]
- 12.Tsai AG, Lu Z, Lieber MR. The t(14;18)(q32;q21)/IGH-MALT1 translocation in MALT lymphomas is a CpG-type translocation, but the t(11;18)(q21;q21)/API2-MALT1 translocation in MALT lymphomas is not. Blood. 2010;115:3640–1. doi: 10.1182/blood-2010-01-265157. author reply 3641–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsai AG, Yoda A, Weinstock DM, Lieber MR. t(X;14)(p22;q32)/t(Y;14)(p11;q32) CRLF2-IGH translocations from human B-lineage ALLs involve CpG-type breaks at CRLF2, but CRLF2/P2RY8 intrachromosomal deletions do not. Blood. 2010;116:1993–4. doi: 10.1182/blood-2010-05-286492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Greisman HA, et al. IgH Partner Breakpoint Sequences Provide Evidence that AID Initiates t(11;14) and t(8;14) Chromosomal Breaks in Mantle Cell and Burkitt Lymphomas. Blood. 2012;120:2864–2867. doi: 10.1182/blood-2012-02-412791. This study describes the occurrence of WGCW motifs at human translocations, and thus directly implicates AID as initiating the lesions that lead to the breaks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu Z, et al. BCL6 breaks occur at different AID sequence motifs in Ig-BCL6 and non-Ig-BCL6 rearrangements. Blood. 2013;121:4551–4. doi: 10.1182/blood-2012-10-464958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annu Rev Immunol. 2008;26:261–92. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaudhuri J, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- 18.Yu K, Lieber MR. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair. 2003;2:1163–1174. doi: 10.1016/j.dnarep.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 19.Robbiani DF, et al. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135:1028–38. doi: 10.1016/j.cell.2008.09.062. This is the first paper to implicate AID as the cause of c-myc translocations in mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalyzed cytosine deamination on single-stranded DNA stimulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. This paper describes the behavior of AID on single-stranded DNA templates. [DOI] [PubMed] [Google Scholar]
- 21.Yu K, Huang FT, Lieber MR. DNA substrate length and surrounding sequence affect the activation induced deaminase activity at cytidine. J Biol Chem. 2004;279:6496–6500. doi: 10.1074/jbc.M311616200. [DOI] [PubMed] [Google Scholar]
- 22.Han L, Masani S, Yu K. Overlapping activation-induced cytidine deaminase hotspot motifs in Ig class-switch recombination. Proc Natl Acad Sci U S A. 2011;108:11584–9. doi: 10.1073/pnas.1018726108. This is the first paper to demonstrate that Ig CSR breaks occur preferentially at WGCW sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui X, et al. Both CpG Methylation and AID are Required for the Fragility of the Human Bcl-2 Major Breakpoint Region: Implications for the Timing of the Breaks in the t(14;18) Mol Cell Biol. 2013;33:947–957. doi: 10.1128/MCB.01436-12. This paper provides molecular support for a model in which AID acts at methylated CpG sites within the BCL-2 MBR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swaminathan S, et al. Mechanisms of clonal evolution in childhood acute lymphoblastic leukemia. Nat Immunol. 2015;16:766–74. doi: 10.1038/ni.3160. This paper provides evidence in both human and mouse pre-B cells for concurrent expression and action of AID and the RAG complex to cause chromosomal rearrangements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aplan PD, et al. Disruption of the human SCL locus by illegitimate V-(D)-J recombinase activity. Science. 1990;250:1426–9. doi: 10.1126/science.2255914. [DOI] [PubMed] [Google Scholar]
- 26.Kirsch IR, editor. The Causes and Consequences of Chromosomal Translocations. CRC; Boca Raton, FL: 1993. [Google Scholar]
- 27.Kitagawa Y, et al. Prevalent involvement of illegitimate V(D)J recombination in chromosome 9p21 deletions in lymphoid leukemia. J Biol Chem. 2002;277:46289–46297. doi: 10.1074/jbc.M208353200. [DOI] [PubMed] [Google Scholar]
- 28.Mahowald GK, et al. Aberrantly resolved RAG-mediated DNA breaks in Atm-deficient lymphocytes target chromosomal breakpoints in cis. Proc Natl Acad Sci U S A. 2009;106:18339–44. doi: 10.1073/pnas.0902545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Askary A, Shimazaki N, Bayat N, Lieber MR. Modeling of the RAG reaction mechanism. Cell Rep. 2014;7:307–15. doi: 10.1016/j.celrep.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu J, et al. Chromosomal Loop Domains Direct the Recombination of Antigen Receptor Genes. Cell. 2015;163:947–59. doi: 10.1016/j.cell.2015.10.016. This murine study describes the chromatin conditions necessary for RAG-mediated chromosomal rearrangements. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helmink BA, Sleckman BP. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol. 2012;30:175–202. doi: 10.1146/annurev-immunol-030409-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deriano L, et al. The RAG2 C terminus suppresses genomic instability and lymphomagenesis. Nature. 2011;471:119–23. doi: 10.1038/nature09755. This paper describes how the C-terminus of RAG2 is important to minimize off-target action by the RAG complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghavan SC, Kirsch IR, Lieber MR. Analysis of the V(D)J recombination efficiency at lymphoid chromosomal translocation breakpoints. J Biol Chem. 2001;276:29126–29133. doi: 10.1074/jbc.M103797200. This paper analyzes human fragile sites where RAG complex cuts. [DOI] [PubMed] [Google Scholar]
- 34.Weigert O, Weinstock DM. The evolving contribution of hematopoietic progenitor cells to lymphomagenesis. Blood. 2012;120:2553–61. doi: 10.1182/blood-2012-05-414995. [DOI] [PubMed] [Google Scholar]
- 35.Papaemmanuil E, et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat Genet. 2014;46:116–25. doi: 10.1038/ng.2874. This paper shows that on-going RAG expression in human cells containing an initial genetic lesion (ETV6-RUNX1) can result in chromosomal rearrangements that cause progression to pre-B ALL malignancy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swerdlow SH, et al., editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. WHO Press; Lyon: 2008. This is the gold standard for human hematopathology. [Google Scholar]
- 37.Bransteitter R, Pham P, Scharff MD, Goodman MF. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc Natl Acad Sci. 2003;100:4102–4107. doi: 10.1073/pnas.0730835100. This is the first paper to desmonstrate that cytosine deamination by AID requires that the cytosine be within single-stranded DNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shinkura R, et al. The influence of transcriptional orientation on endogenous switch region function. Nature Immunol. 2003;4:435–441. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- 39.Zhang ZZ, et al. The Strength of an Ig Switch Region Is Determined by Its Ability to Drive R Loop Formation and Its Number of WGCW Sites. Cell Rep. 2014;8:557–569. doi: 10.1016/j.celrep.2014.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang ZZ, Pannunzio NR, Hsieh CL, Yu K, Lieber MR. The role of G-density in switch region repeats for immunoglobulin class switch recombination. Nucleic Acids Res. 2014;42:13186–93. doi: 10.1093/nar/gku1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu K, Chedin F, Hsieh C-L, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunol. 2003;4:442–451. doi: 10.1038/ni919. The first kilobase nuclear R-loops in eukaryotes are described. [DOI] [PubMed] [Google Scholar]
- 42.Yang Y, et al. Arginine Methylation Facilitates the Recruitment of TOP3B to Chromatin to Prevent R Loop Accumulation. Mol Cell. 2014;53:484–97. doi: 10.1016/j.molcel.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips DD, et al. The sub-nanomolar binding of DNA-RNA hybrids by the single-chain Fv fragment of antibody S9.6. J Mol Recognit. 2013;26:376–81. doi: 10.1002/jmr.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang ZZ, Pannunzio NR, Hsieh CL, Yu K, Lieber MR. Complexities Due to Single-Stranded RNA During Antibody Detection of Genomic RNA:DNA Hybrids. BMC Res Notes. 2015;8:127. doi: 10.1186/s13104-015-1092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu LF, Wang JC. Supercoiling of the DNA template during transcription. Proc Natl Acad Sci USA. 1987;84:7024–7027. doi: 10.1073/pnas.84.20.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kouzine F, Sanford S, Elisha-Feil Z, Levens D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat Struct Mol Biol. 2008;15:146–54. doi: 10.1038/nsmb.1372. Transcription-driven topological effects in the genome of mammalian cells are described, supporting the twin-domain model for transcription in supercoiling of DNA. [DOI] [PubMed] [Google Scholar]
- 47.Meng FL, et al. Convergent Transcription at Intragenic Super-Enhancers Targets AID-Initiated Genomic Instability. Cell. 2014;159:1538–48. doi: 10.1016/j.cell.2014.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pefanis E, et al. Noncoding RNA transcription targets AID to divergently transcribed loci in murine B cells. Nature. 2014;514:389–393. doi: 10.1038/nature13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu Z, et al. Convergent BCL6 and lncRNA promoters demarcate the major breakpoint region for BCL6 translocations. Blood. 2015;126:1730–1. doi: 10.1182/blood-2015-07-657999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pannunzio NR, Lieber MR. Dissecting the Roles of Divergent and Convergent Transcription in Chromosome Instability. Cell Rep. 2016;14:1025–31. doi: 10.1016/j.celrep.2015.12.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mao C, et al. T cell-independent somatic hypermutation in murine B cells with an immature phenotype. Immunity. 2004;20:133–44. doi: 10.1016/s1074-7613(04)00019-6. [DOI] [PubMed] [Google Scholar]
- 52.Han JH, et al. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity. 2007;27:64–75. doi: 10.1016/j.immuni.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ueda Y, Liao D, Yang K, Patel A, Kelsoe G. T-independent activation-induced cytidine deaminase expression, class-switch recombination, and antibody production by immature/transitional 1 B cells. J Immunol. 2007;178:3593–601. doi: 10.4049/jimmunol.178.6.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuraoka M, et al. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc Natl Acad Sci U S A. 2011;108:11560–5. doi: 10.1073/pnas.1102571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuraoka M, et al. Activation-induced cytidine deaminase expression and activity in the absence of germinal centers: insights into hyper-IgM syndrome. J Immunol. 2009;183:3237–48. doi: 10.4049/jimmunol.0901548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumar S, et al. Flexible ordering of antibody class switch and V(D)J joining during B-cell ontogeny. Genes Dev. 2013;27:2439–44. doi: 10.1101/gad.227165.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Umiker BR, et al. Production of IgG autoantibody requires expression of activation-induced deaminase in early-developing B cells in a mouse model of SLE. Eur J Immunol. 2014;44:3093–108. doi: 10.1002/eji.201344282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelsoe G. Curiouser and curiouser: the role(s) of AID expression in self-tolerance. Eur J Immunol. 2014;44:2876–9. doi: 10.1002/eji.201445102. [DOI] [PubMed] [Google Scholar]
- 59.Cantaert T, et al. Activation-Induced Cytidine Deaminase Expression in Human B Cell Precursors Is Essential for Central B Cell Tolerance. Immunity. 2015;43:884–895. doi: 10.1016/j.immuni.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152:417–29. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roukos V, Misteli T. The biogenesis of chromosome translocations. Nat Cell Biol. 2014;16:293–300. doi: 10.1038/ncb2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qian J, et al. B Cell Super-Enhancers and Regulatory Clusters Recruit AID Tumorigenic Activity. Cell. 2014;159:1524–37. doi: 10.1016/j.cell.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kovalchuk AL, et al. Mouse model of endemic Burkitt translocations reveals the long-range boundaries of Ig-mediated oncogene deregulation. Proc Natl Acad Sci U S A. 2012;109:10972–7. doi: 10.1073/pnas.1200106109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chiarle R, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–19. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barlow JH, et al. Identification of early replicating fragile sites that contribute to genome instability. Cell. 2013;152:620–32. doi: 10.1016/j.cell.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet. 2007;41:169–92. doi: 10.1146/annurev.genet.41.042007.165900. [DOI] [PubMed] [Google Scholar]
- 67.Look AT. Oncogenic Transcription Factors in Human Acute Leukemias. Science. 1997:278. doi: 10.1126/science.278.5340.1059. [DOI] [PubMed] [Google Scholar]
- 68.Korsmeyer SJ. Chromosomal translocations in lymphoid malignancies reveal novel proto-oncogenes. Annu Rev Immunol. 1992;10:785–807. doi: 10.1146/annurev.iy.10.040192.004033. [DOI] [PubMed] [Google Scholar]
- 69.Hunger SP, et al. The t(1;19)(q23;p13) results in consistent fusion of E2A and PBX1 coding sequences in acute lymphoblastic leukemias. Blood. 1991;77:687–93. [PubMed] [Google Scholar]
- 70.LeBrun DP, Cleary ML. Fusion with E2A alters the transcriptional properties of the homeodomain protein PBX1 in t(1;19) leukemias. Oncogene. 1994;9:1641–7. [PubMed] [Google Scholar]
- 71.Monica K, LeBrun DP, Dedera DA, Brown R, Cleary ML. Transformation properties of the E2a-Pbx1 chimeric oncoprotein: fusion with E2a is essential, but the Pbx1 homeodomain is dispensable. Mol Cell Biol. 1994;14:8304–14. doi: 10.1128/mcb.14.12.8304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nourse J, et al. Chromosomal translocation t(1;19) results in synthesis of a homeobox fusion mRNA that codes for a potential chimeric transcription factor. Cell. 1990;60:535–45. doi: 10.1016/0092-8674(90)90657-z. [DOI] [PubMed] [Google Scholar]
- 73.Kamps MP, Murre C, Sun XH, Baltimore D. A new homeobox gene contributes the DNA binding domain of the t(1;19) translocation protein in pre-B ALL. Cell. 1990;60:547–55. doi: 10.1016/0092-8674(90)90658-2. [DOI] [PubMed] [Google Scholar]
- 74.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56:777–83. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 75.Heisterkamp N, Groffen J. Philadelphia-positive leukemia: a personal perspective. Oncogene. 2002;21:8536–40. doi: 10.1038/sj.onc.1206080. [DOI] [PubMed] [Google Scholar]
- 76.Groffen J, Heisterkamp NC. Philadelphia chromosome translocation. Crit Rev Oncog. 1989;1:53–64. [PubMed] [Google Scholar]
- 77.Heisterkamp N, Stam K, Groffen J, de Klein A, Grosveld G. Structural organization of the bcr gene and its role in the Ph’ translocation. Nature. 1985;315:758–61. doi: 10.1038/315758a0. [DOI] [PubMed] [Google Scholar]
- 78.Heisterkamp N, et al. Localization of the c-ab1 oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature. 1983;306:239–42. doi: 10.1038/306239a0. [DOI] [PubMed] [Google Scholar]
- 79.van der Feltz MJ, et al. Nucleotide sequence of both reciprocal translocation junction regions in a patient with Ph positive acute lymphoblastic leukaemia, with a breakpoint within the first intron of the BCR gene. Nucleic Acids Res. 1989;17:1–10. doi: 10.1093/nar/17.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roukos V, et al. Spatial dynamics of chromosome translocations in living cells. Science. 2013;341:660–4. doi: 10.1126/science.1237150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rocha PP, et al. Close proximity to Igh is a contributing factor to AID-mediated translocations. Mol Cell. 2012;47:873–85. doi: 10.1016/j.molcel.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rocha PP, Micsinai M, Kluger Y, Skok JA. Response to Casellas et al. Mol Cell. 2013;51:277–8. doi: 10.1016/j.molcel.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Casellas R, Resch W, Hakim O, Nussenzweig MC. The origin of B cell recurrent chromosomal translocations: proximity versus DNA damage. Mol Cell. 2013;51:275–6. doi: 10.1016/j.molcel.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wiemels JL, et al. Site-specific translocation and evidence of postnatal origin of the t(1;19) E2A-PBX1 fusion in childhood acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2002;99:15101–6. doi: 10.1073/pnas.222481199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.AmericanCancerSociety. 2009 [Google Scholar]
- 86.Downs JA, Jackson SP. A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol. 2004;5:367–378. doi: 10.1038/nrm1367. [DOI] [PubMed] [Google Scholar]
- 87.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 88.Falzon M, Fewell J, Kuff EL. EBP-80, a Transcription Factor Closely Resembling the Human Autoantigen Ku, Recognizes Single- to Double-Strand Transitions in DNA. J Biol Chem. 1993;268:10546–52. [PubMed] [Google Scholar]
- 89.Yaneva M, Kowalewski T, Lieber MR. Interaction of DNA-dependent protein kinase with DNA and with Ku: biochemical and atomic-force microscopy. EMBO J. 1997;16:5098–5112. doi: 10.1093/emboj/16.16.5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Grundy GJ, et al. APLF promotes the assembly and activity of non-homologous end joining protein complexes. Embo J. 2013;32:112–25. doi: 10.1038/emboj.2012.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Niewolik D, et al. DNA-PKcs dependence of artemis endonucleolytic activity: differences between hairpins and 5′ or 3′ overhangs. J Biol Chem. 2006;281:33900–33909. doi: 10.1074/jbc.M606023200. [DOI] [PubMed] [Google Scholar]
- 92.Goodarzi AA, et al. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. Embo J. 2006;25:3880–9. doi: 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Povirk LF. Biochemical mechanisms of chromosomal translocations resulting from DNA double-strand breaks. DNA Repair (Amst) 2006;5:1199–212. doi: 10.1016/j.dnarep.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 94.Buck D, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 95.Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 96.Chappell C, Hanakahi LA, Karimi-Busheri F, Weinfeld M, West SC. Involvement of human polynucleotide kinase in double-strand break repair by non-homologous end joining. EMBO J. 2003;21:2837–2832. doi: 10.1093/emboj/21.11.2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pannunzio NR, Li S, Watanabe G, Lieber MR. Non-homologous end joining often uses microhomology: implications for alternative end joining. DNA Repair (Amst) 2014;17:74–80. doi: 10.1016/j.dnarep.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol Cell Biol. 2013;33:1468–73. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.