

Programmable crystallization of thin films produces patterns and bespoke microstructures for semiconductor applications.
Keywords: crystallization, polycrystalline thin films, amorphous thin films, small-molecule organic semiconductors, Organic Electronics, organic thin film transistors, homogeneous nucleation, patterning, classical nucleation theory, nucleation and growth
Abstract
The functional properties and technological utility of polycrystalline materials are largely determined by the structure, geometry, and spatial distribution of their multitude of crystals. However, crystallization is seeded through stochastic and incoherent nucleation events, limiting the ability to control or pattern the microstructure, texture, and functional properties of polycrystalline materials. We present a universal approach that can program the microstructure of materials through the coherent seeding of otherwise stochastic homogeneous nucleation events. The method relies on creating topographic variations to seed nucleation and growth at designated locations while delaying nucleation elsewhere. Each seed can thus produce a coherent growth front of crystallization with a geometry designated by the shape and arrangement of seeds. Periodic and aperiodic crystalline arrays of functional materials, such as semiconductors, can thus be created on demand and with unprecedented sophistication and ease by patterning the location and shape of the seeds. This approach is used to demonstrate printed arrays of organic thin-film transistors with remarkable performance and reproducibility owing to their demonstrated spatial control over the microstructure of organic and inorganic polycrystalline semiconductors.
INTRODUCTION
The long-range crystalline order of atoms or molecules in a single crystal material dictates its physical, chemical, electronic, and/or biological properties. In polycrystalline materials, the phase, size, shape, and relative spatial arrangement of an ensemble of crystals and their boundaries dictate many of the functional properties and the technological utility of these materials. Processing materials with well-controlled microstructure has been the aim of research and development efforts spanning areas as broad and varied as metallurgy, food, pharmaceuticals, energy, and electronics.
The crystallization of materials begins with the stochastic nucleation and growth of the crystalline phase, irrespective of how it is formed, such as from vapor or liquid sources, from drying ink, or by the annealing of a glassy solid (1, 2). The random nature of nucleation implies that extrinsic properties of any polycrystalline material are subject to the ensemble behavior of all grains, as well as their boundaries, phases, polymorphs, and associated defects. In semiconducting materials, grain boundary defects produce detrimental trap states within the band gap. The coexistence of different phases, textures, and polymorphs can compound this problem, creating barriers to charge transport and causing charge recombination, with important detrimental effects on the optoelectronic properties of the semiconductor (3–6). Conventional semiconductor manufacturing has mitigated the formation of such defects (for example, in cases of Si and III-V semiconductors) by relying on epitaxial growth on monocrystalline substrates under ultrahigh vacuum conditions. However, these remedies are not applicable to conventional polycrystalline films deposited on glass or plastic substrates or in the context of solution-processable semiconductors, such as printed conjugated organic molecules, organohalide perovskites, and transition metal oxides, which can be fabricated in ambient air with the promise of delivering high-performance electronic, optoelectronic, and photovoltaic devices at low cost (7–10). Among these emerging semiconductors, polycrystalline organic small molecules have had the distinction of achieving field-effect hole mobilities over 1 cm2/V∙s (11, 12), with several recent reports of mobilities higher than 10 cm2/V∙s, easily surpassing the field-effect hole mobilities of hydrogenated amorphous silicon (a-Si:H) in p-channel field-effect transistors (13). However, the crystallization of organic semiconductors suffers from important intrinsic limitations—similar to other soft matter, such as pharmaceutical molecules and food plastics. These limitations include pervasive polymorphism, coexistence of ordered and disordered regions, and paracrystallinity, which are largely due to weak van der Waals interactions (14). The stochastic nature of nucleation and the incoherent growth of a large population of crystals leading up to the formation of polycrystalline materials compounds this problem, with the high anisotropy of properties in organic semiconductors making them particularly sensitive to texture and to the relative orientation of domain boundaries (12). A more coherent form of crystallization should mitigate many of the microstructural heterogeneities associated with stochastic nucleation and may enable a new degree of control and tailoring of the properties of polycrystalline materials.
Here, we show that homogeneous nucleation and subsequent growth of highly textured crystals can be entirely programmed in terms of location and time, enabling the production of bespoke and patterned microstructures. The demonstrated capability is derived from a classical nucleation theory (CNT) prediction, which stipulates that the effective rate of homogeneous nucleation is proportional to the volume of material. The crystallization behavior of amorphous films is thus shown to be entirely controllable by introducing thickness variations, which locally change the volume of material. Crystalline domains formed early in thick regions can seed the lateral epitaxial growth of directional and textured domains, even on disordered surfaces. Polycrystalline films are thus produced with an entirely programmed microstructure, including the location, size, shape, texture, and in-plane crystallographic orientation of domains, as well as the location and orientation of domain boundaries. We use this approach to demonstrate bespoke printed organic semiconductor arrays, which are devoid of common microstructural deficiencies, including mixed texture, polymorphism, and disorder, and we demonstrate remarkable device-to-device performance reproducibility compared to conventional crystallization methods. The ability to create bespoke structures with ease is extended to many other material systems, including metal oxide and hybrid perovskite semiconductors, and is expected to affect material processing in areas of electronics, optoelectronics, photonics, energy, pharmaceuticals, biosciences, and beyond (8, 15–19).
RESULTS
Thickness-dependent nucleation behavior
We made the intriguing observation that crystallization occurs sooner in thick parts of an amorphous film, as shown in Fig. 1A. A solidified droplet of an amorphous organic semiconductor, known as 5,11-bis(triethylsilylethynyl) anthradithiophene (TES ADT) (20), was cast and left to dry on a stationary substrate forming the well-known “coffee ring” feature (21, 22), a common observation in nature as well as in solution casting and inkjet printing of materials (23, 24). When the amorphous sample was annealed by exposing it to a small dose of solvent vapor (25, 26), it crystallized first in the ~4-μm-thick outer rim (Fig. 1A and fig. S1), whereas the thinner center (~100 nm thick) remained amorphous for an extended period. The center eventually crystallized through the radial and coherent crystallization from the outer rim, resulting in a highly textured core not subject to random nucleation. We generalize the observation of thickness-dependent crystallization by preparing more classical thin films by vacuum deposition. In one instance, a thin film of 5,6,11,12-tetraphenylnaphthacene (rubrene) was deposited while retracting a mask stepwise to create an intentional height step in the film from 10 to 50 nm (Fig. 1B). Upon thermal annealing for a short duration, we observed nucleation and growth of crystalline spherulites in the thicker part of the sample, whereas the thinner section remained amorphous (Fig. 1B) and did not exhibit any evidence of nucleation in the thin section even after the thick region has fully crystallized (Fig. 1C). Similar experiments were performed on vacuum-deposited inorganic metal oxide films of molybdenum oxide (MoOx) prepared with different thicknesses (27). The thinner films required more time to crystallize and exhibited a substantially smaller nucleation density than the thicker films (fig. S2). These experiments confirm qualitatively that the onset of nucleation of the crystalline phase in amorphous thin films is thickness-dependent.
Fig. 1. Crystallization in amorphous films tends to be initiated in thicker regions through homogeneous nucleation.
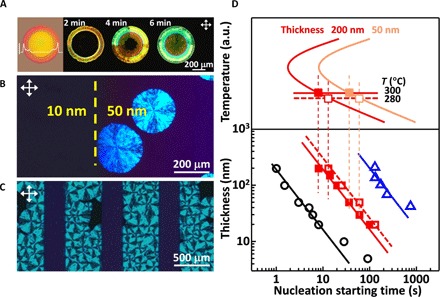
(A) Optical micrographs of TES ADT films during solvent vapor annealing. The leftmost figure shows the height profile (see fig. S1). (B and C) Polarized optical micrographs of a vacuum-deposited rubrene film with a deliberate thickness step (yellow dashed line) after thermal annealing for only ~10 s (B) and thicker ribbons fully crystallized (while thin sections remain amorphous) after ~60 s (C). (D) Plots of the estimated nucleation starting time at different temperatures (top) for MoOx films (□) of different thicknesses. The line fits at the bottom have a fixed slope of −1. Evaporated rubrene thin films (○) were annealed at T = 150°C. Evaporated MoOx thin films were annealed at T = 280°C (□) and T = 300°C (■). Spin-coated TES ADT thin films (Δ) were crystallized by solvent vapor annealing. The onset of nucleation was identified visually for all samples by inspection using polarized optical microscopy (see fig. S4).
Nucleation events occurring during vacuum deposition of thin films and solution deposition of thin films under slow drying conditions are typically heterogeneous in nature because they are initiated at the surface of the substrate and therefore do not tend to be related to film thickness (28, 29). To explain the thickness-dependent nucleation behavior observed in amorphous films, we turn instead to homogeneous nucleation as the most plausible explanation. Homogenous nucleation can produce highly textured crystals in thin films because of thickness confinement effects (see fig. S2 for further discussion) (30). CNT typically treats the rate of homogeneous nucleation (R) as a volumetric term defined as the number of nuclei (N) formed per unit volume (ΔV) in a given time (Δt) [R = N/(ΔV⋅Δt)] (see fig. S3 for further discussion) (31). If we apply bulk CNT theory in the context of a film of finite thickness (d) and ignore heterogeneous nucleation at the interfaces, we will see that the number of atoms or molecules available to form critical nuclei per unit area (ΔA) of film scales with d. The effective nucleation rate in thicker parts of the sample should therefore increase, even as the volumetric homogeneous nucleation rate remains thickness-invariant. This leads us to define an effective homogeneous nucleation rate per unit area and time, which we call the areal nucleation rate (RA)
| (1) |
Accordingly, the onset of nucleation, corresponding approximately to the time (t1) when the first nucleation event occurs (N = 1), is inversely proportional to the thickness of the film, such that
| (2) |
We have experimentally evaluated t1 versus d for different material systems, as summarized in Fig. 1D. The onset of nucleation, for instance, in vacuum-deposited MoOx thin films with respect to thickness is remarkably consistent with Eq. 2 (the line in the double logarithm has a fixed slope of −1), even when considering different annealing temperatures (fig. S4). Similarly, t1 ∝ d−1 is experimentally observed for thermal annealing of amorphous rubrene films prepared by vacuum deposition and for solvent vapor annealing of amorphous TES ADT films prepared by spin coating (Fig. 1D and fig. S5). The experimental validation of Eq. 2 for both organic and inorganic semiconductor materials and thermal and solvent vapor annealing processes provides material support to the notion that crystallization in these amorphous films is initiated through homogeneous nucleation, irrespective of preparation method. Thickness-dependent nucleation can have significant implications for thin-film processing because it provides a new handle on the crystallization behavior and microstructure of initially amorphous films and, by extension, a way to control the properties and functionality of polycrystalline materials both globally and locally.
Topographic seeding of crystallization and production of bespoke microstructures
As the first practical implementation of thickness-dependent onset of nucleation, we have sought to create and use thick regions as seeds from which the remainder of the thin film might be crystallized coherently while preventing stochastic nucleation outside the seeds. We have created height variations at specific locations of an amorphous film of rubrene by vacuum depositing rubrene through a shadow mask with rectangular ribbon patterns on top of a uniform layer of rubrene (Fig. 2, A and B). Upon annealing the sample for a short duration, spherulites nucleate randomly in the thick regions and propagate radially to the edges of the rectangular ribbons, where they appear to stop, effectively converting only the thick ribbon and leaving the remainder of the film in the amorphous phase (Fig. 1C). With continued annealing, crystallization propagates linearly and coherently from the edges of the ribbon and into the thin sections, where nucleation is less likely to occur by design (Fig. 2C). The edge of the ribbon thus becomes a linear seed for coherent and directional crystallization through a linear growth front into the surrounding amorphous sample. This example demonstrates how the location of crystalline seeds, their shape, and the orientation and direction of propagation of crystallization can be programmed locally to transform parts or—potentially— the entire amorphous film into a polycrystalline film with bespoke microstructure.
Fig. 2. Programming crystallization and producing bespoke microstructures by manipulating thin-film topography.
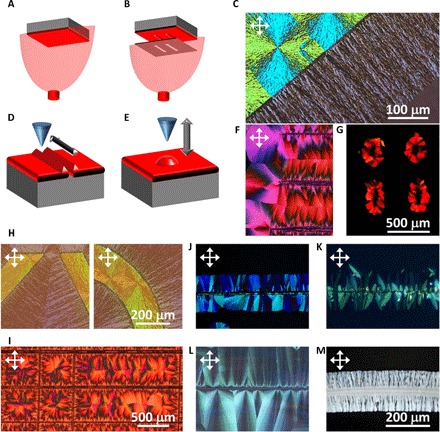
(A and B) Schematics representing a two-step vacuum deposition method in which a shadow mask creates a thicker region of arbitrary pattern (for example, rectangular strips) on a uniform film to subsequently pattern and seed the crystallization coherently. (C) Polarized optical micrograph of thermally annealed rubrene films using the two-step deposition method for ~60 s (200 nm for the top left brighter part and 40 nm for the dimmer part). (D and E) Schematics representing the mechanical patterning method, which uses a solid object to stamp or to scratch the film with the aim of creating thickness variations at designated locations. (F and G) Polarized optical micrograph of crystallized TES ADT samples with periodically imprinted lines (F) and dots (G). The imprint lines/dots seed the linear/spherulitic crystallization. (H) Polarized optical micrographs of a rubrene film with the same thickness contrast as (C) but with more sophisticated patterns. (I) Polarized optical micrograph of solvent vapor annealed of a TES ADT thin film with various grid-like patterns. (J to M) Polarized optical micrographs of amorphous thin films crystallized using imprinted lines, including vacuum-deposited rubrene (J), vacuum-deposited MoOx (K), solution-processed PEO (L), and solution-processed PbI2 hybrid perovskite DMF solvate film (M).
In a second approach, we have explored scratching and stamping methods to create bands of thick material through displacement. This can be carried out in ambient air without resorting to vacuum deposition, making it compatible with solution-based coating and printing methods. The topographic image of a solution-processed amorphous TES ADT film after scratching (fig. S6) confirms that this method can create rough ridges that are two to three times taller than the surrounding film. Annealing the sample crystallizes the ridges (produced by scratching or stamping) first, creating seeds for the coherent and directional crystallization of the surrounding amorphous material, as shown in Fig. 2F for linear crystallization and in Fig. 2G for radial crystallization of laterally pinned spherulite arrays.
We leveraged these approaches to program more sophisticated structures. In Fig. 2H, rubrene crystallization was tailored by the two-step vacuum deposition method to orient the growth front at two different angles, forming a predictably straight domain boundary with a fixed misalignment angle (Fig. 2H, left). A similar approach was also used to create a curved growth front (Fig. 2H, right). Using the stamping method in combination with a printed layer of TES ADT, we demonstrate a combination of square and rectangular arrays (Fig. 2I) wherein straight grid lines act as the seeding lines for two-dimensional coherent and directional crystallization, resulting in inverted “squarulites” and “rectangulites” of TES ADT at designated locations (see the supplementary movies). The working principle of programmable crystallization in rubrene (Fig. 2J), TES ADT (fig. S7A), and MoOx (Fig. 2K) has been extended to other material systems, as summarized in Fig. 2 (L and M) and fig. S7B, including poly(ethylene oxide) (PEO; an insulating polymer that was melted at 100°C and then scratched while cooling down; Fig. 2L), methylammonium lead iodine perovskite (CH3NH3PbI3; a metal-organic hybrid semiconductor that was solution-processed and then scratched while drying at room temperature to crystallize the precursor solvate phase before annealing at 100°C; Fig. 2M) (32), and phenyl-C61-butyric acid methyl ester (PCBM; an organic small molecule that was solution-processed and thermally annealed at 260°C; fig. S7B).
Semiconductor heterogeneities and OTFT device reproducibility
Encouraged by the ability to produce bespoke microstructures on demand, we turn our attention to investigating the potential benefits of this approach in the context of polycrystalline semiconductor materials and, more specifically, in printed organic electronics. The microstructural heterogeneities of polycrystalline organic semiconductors are notorious for causing a great deal of device-to-device performance variability in circuits (4, 33). As an illustrative example of this problem, we show in Fig. 3 close-up images of an array of bottom-gate top-contact organic thin-film transistors (OTFTs) (Fig. 3A) fabricated using TES ADT films prepared by conventional (stochastic) crystallization (Fig. 3B) and periodic linear (coherent) crystallization (Fig. 3C). Microstructural heterogeneities in these two types of TES ADT films have been evaluated by spatially mapping the (011) reflection of the α phase of TES ADT using microbeam-grazing incidence wide-angle x-ray scattering (μGIWAXS; Fig. 3D and fig. S8) (34). The structural map of the conventional sample reveals the shifting position of the (011) reflection as a result of changing polymorphism in two adjacent spherulites (35, 36). By contrast, linearly crystallized TES ADT reveals no polymorphic variations when mapped across the entire area between two linear seeds. We summarize in Fig. 4B the associated carrier mobilities and their distributions for devices fabricated using conventionally and periodic linearly crystallized TES ADT films (36). The conventional film has an average mobility of 0.5 cm2/(V·s) but exhibits a very large device-to-device spread ranging from 0.06 to 1 cm2/(V·s), resulting in a whopping ±100% relative error. The linear crystallization strategy (αP) with random placement of OTFT channels reduces the spread in performance, demonstrating an improvement in the uniformity of semiconductor properties and a global increase in transport properties across the entire substrate. The impact of reduced microstructural heterogeneities on the trap states in the organic semiconductor was studied by photothermal deflection spectroscopy (PDS; Fig. 3D), which helps evaluate the Urbach energy (Eu) of the semiconductor film (Fig. 3D, inset) (37). The αp phase displayed Eu that was ~1 meV lower than that of the conventionally crystallized α phase. Despite the small difference, this is expected to significantly affect transport properties because Eu ~ kT at room temperature, where k is the Boltzmann constant and T is temperature (38).
Fig. 3. Improved microstructural homogeneity by linear patterning of thin-film crystallization.
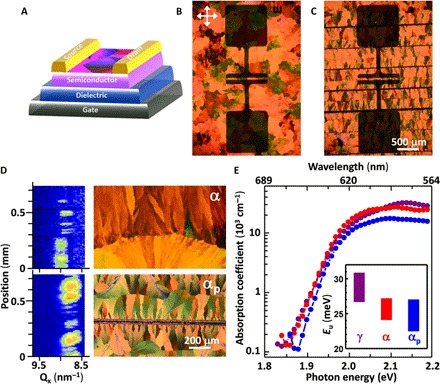
(A) Schematic illustration of a bottom-gate top-contact OTFT device. (B and C) Polarized optical micrographs of an OTFT using a conventionally crystallized TES ADT film (α) (B) and an OTFT using a periodic linear crystallization of TES ADT (αp) (C). (D) μGIWAXS maps focusing on the Q value of the (011) reflection at the x-axis of the detector (Qx) of TES ADT in the area associated with the polarized micrograph. (E) Absorption spectra of TES ADT films in γ, α, and αp phases and microstructures, as measured by PDS. Inset to (E) plots the Urbach energy extracted from the exponential tail of the absorption spectra.
Fig. 4. Improved OTFT device performance reproducibility by linear patterning of thin-film microstructure with respect to OTFT channel.
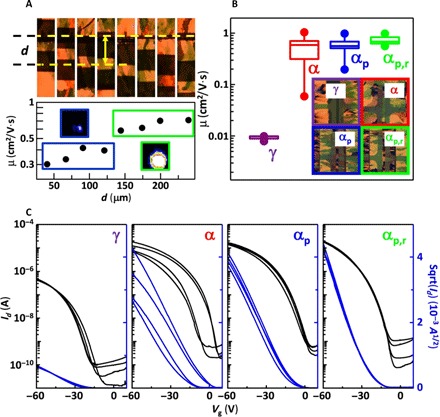
(A) Close-up view of the channel of OTFTs with respect to the distance (d) of the linear crystallization seed line (indicated by the top yellow line). Hole mobility (saturation) of OTFT devices with respect to d. The insets in the bottom panel of (A) shows a representative (011) reflection for d < 120 μm (□) and d > 120 μm (□). (B) Hole mobility and its distribution over >10 devices prepared using different scenarios. The filled circles represent the maximum and minimum values, whereas the boxes represent 20, 50, and 75% of the values. Insets to (B) show polarized optical micrographs of TES ADT thin films in various microstructural states in the channel area of the OTFT devices. (C) Four representative transfer characteristics in the saturation regime of OTFT devices prepared following each scenario.
The periodic seeding of TES ADT offers the opportunity to preprogram and design the microstructure of the film with respect to the location of transistor channels. In this vein, the structural map of the periodic linearly crystallized film (Fig. 4A) reveals periodic variations in the intensity of the (011) reflection, indicating that some regions exhibit a long-range crystalline order that is more pronounced than that of others. Normally, such structural variations are presumed to be randomly distributed in a conventionally crystallized sample. However, it appears that the periodic patterning approach produces such deficiencies periodically too, which means that the impact of these regions on transport can be studied and their effects on devices can be potentially avoided. To do so, we have carefully aligned the periodic linear patterns with the OTFT channel and systematically changed the distance between the channel of the OTFT and the linear seeding line. We find in Fig. 4A that devices placed >120 μm from the nucleation line coincide with the highly ordered region and exhibit consistently higher mobility by a factor of 2 to 3. This may be an additional source of a significant lack of reproducibility in conventional devices. We take advantage of this insight to devise a linear patterning strategy that consistently registers all OTFT device channels (αp,r) within the high-mobility bands in the TES ADT film, thus excluding low-mobility regions from playing a role in the circuit components. In doing so, we achieved an increase in the overall mobility toward 1 cm2/(V·s) and further reduced the spread of carrier mobility substantially (Fig. 4B). The improved reproducibility is highlighted by the reduced scatter of representative transfer characteristics of OTFT devices (Fig. 4C). Other important figures of merit, such as threshold voltage and subthreshold swing, also exhibited much improved device-to-device reproducibility (see fig. S9, B and C). The above example demonstrates how programmable crystallization can allow OTFT devices to be fabricated with a consistent and reproducible microstructure such that they always achieve the high-end performance potential of the material without any compromises.
DISCUSSION
We have demonstrated that crystallization of polycrystalline thin films can be entirely programmed with unprecedented ease and versatility. This capability stems from the fact that crystallization of amorphous films appears to be dominated by homogeneous nucleation of the crystalline phase, rather than by heterogeneous nucleation. This means that the effective nucleation behavior in a film of finite thickness is closely linked to the thickness of the film. We demonstrate that thickness variation–produced patterned thin films can serve as seeds to program crystallization behavior, including location, onset, orientation, and growth front geometry. We see tremendous opportunities and benefits emerging from this insight and from related methods for a wide range of fields reliant on thin-film manufacturing.
We have investigated the impact of these new opportunities in the context of printed electronics, where microstructural variations often translate into very significant spreads in device performance on the same substrate, owing to microstructural deficiencies, including disorder, texture, polymorphism, and misorientation. The practical demonstrations involving OTFTs can be extrapolated to other material systems and reveal primarily two material-agnostic benefits of the programmable crystallization approach: (i) the ability to reduce unwanted microstructural heterogeneities in large-area polycrystalline films through a more controlled crystallization method can produce higher-quality polycrystalline thin films; and (ii) the ability to spatially program and pattern the microstructure and to modulate the functional properties of a material can help design and manufacture much better printed integrated circuits. Both of these are universally desirable in polycrystalline semiconductors and should find favor with polycrystalline materials in general.
The demonstrated ability to overcome the inherent and stubborn limitations associated with polycrystalline organic semiconductors, which can crystallize during deposition or are crystallized subsequent to amorphous film deposition, should help this important class of materials meet the strict device uniformity and reproducibility standards for their implementation in large-area printed electronics and optoelectronics applications (39).
MATERIALS AND METHODS
Materials and chemicals
Rubrene, molybdenum trioxide (MoO3), PEO, PCBM, toluene, 1,2-dichloroethane (DCE), chlorobenzene, dimethylformamide (DMF), and PbI2 were purchased from Sigma-Aldrich and used as received. TES ADT was provided by J. Anthony from the University of Kentucky. Methylammonium iodide (MAI) was purchased from Dyenamo and baked overnight at 70°C in a vacuum oven.
Film fabrication
Scratching/stamping of thin films
Mechanical contact of stainless steel needles was conducted by hand for stamping/scratching on the thin films. Because the hardness of the substrate (glass or Si/SiO2 substrate used in this work) was significantly higher than that of the needles, we did not control the scratching/stamping force and speed in this work.
5,11-bis(Triethylsilylethynyl) anthradithiophene
Amorphous TES ADT thin films were deposited from a 2 weight % (wt %) toluene solution on top of thermally grown SiO2 by spin coating or drop casting at 40°C in air. The films were annealed at 50° and 100°C for 5 s each to prevent dewetting in the subsequent solvent vapor annealing step. After annealing, the films were patterned by scratching with needles or stamping with a die at room temperature and subsequently transferred to a small chamber with controlled gas flow and an open container filled with DCE to create a solvent vapor environment for solvent vapor annealing at room temperature, according to Dickey et al. (25). The chamber was purged with air to interrupt the growth propagation of the crystals, at the required time or after crystallization is complete.
Rubrene
Amorphous rubrene thin films were vacuum-deposited by thermal sublimation in a high-vacuum chamber with a base pressure of 10−7 mbar and connected to a nitrogen glove box. To fabricate patterned structures, we performed two depositions, with the first deposition forming a uniform film and with the second deposition forming thicker stripes of the material through a shadow mask. The films were thermally annealed inside the nitrogen glove box at 150°C for different times. The rubrene samples were not exposed to air because this was found to inhibit crystallization, possibly due to photooxidation.
Poly(ethylene oxide)
PEO thin films were fabricated by spin coating an aqueous PEO solution (50 mg/ml) in air. Spherulitic crystal structures were observed in the as-cast thin films. The thin films were subsequently melted by heating to 100°C, and patterns were made by scratching while the sample was being melted. The samples were subsequently removed from the hot plate, and crystallization appeared from the scratch line during cooling.
Molybdenum oxide
MoOx thin films were deposited by vacuum deposition. The thin films were thermally annealed in air to generate crystal structures. The nucleation starting time was recorded for annealing at 300°C by cross-polarized optical microscopy. Scratched films (50 nm thick) crystallized from the edge of the patterns by annealing at 230°C. This reduced competitive nucleation away from the edge of the patterns.
Phenyl-C61-butyric acid methyl ester
PCBM thin films were deposited from a 1 wt % chlorobenzene solution by spin coating in air. The thin films were heated at 130°C to remove any residual solvent and patterned by scratching at the same temperature when the material was soft enough to create differences in thickness at the location of the scratch. The thin films were subsequently annealed at 260°C to generate crystal structures.
Hybrid perovskite
MAI and PbI2 were mixed in a 1:1 M ratio and dissolved in anhydrous DMF to form a 1 M solution that was stirred overnight at 60°C. The solution was filtered with a 0.2-μm polytetrafluoroethylene filter and spin-coated for 10 s in air on top of a Si wafer with thermal oxide. Scratch patterns were created in the precursor state before the film was fully dried. The film was subsequently heated to 100°C to initiate precursor solvate formation and achieve full conversion into perovskite crystals (32).
Optical microscopy
Optical microscopy was performed with a Nikon FN-1 polarized optical microscope fitted with a Nikon D4S camera to record still images and videos that are available in the Supplementary Materials.
Profilometry
Height profiles were obtained with a KLA-TENCOR P6 Surface Profiler.
Quartz crystal microbalance with dissipation monitoring measurements
Quartz crystal microbalance with dissipation monitoring (QCM-D) sensors are made of AT-cut piezoelectric quartz crystals (active area, 12 mm2; resonance frequency, 5 MHz) with a 10-nm Au layer and a 100-nm SiO2 layer on top. Thin films of TES ADT were fabricated by spin coating (for thinner films) and drop casting (for thicker films) directly on the sensors. TES ADT–coated quartz sensors were loaded into the QCM-D open module (E4 model, Q-Sense, Biolin Scientific) mounted in a custom-built environmental chamber equipped with a solvent liquid reservoir and nitrogen flow. The QCM-D technique was used to detect the onset of crystallization of TES ADT during solvent vapor annealing. Because TES ADT films were exposed to solvent vapor, they took up solvent and subsequently crystallized. However, as soon as the sample crystallized, a substantial fraction of the solvent was expelled from the growing crystalline phase, providing an indirect and approximate indication of the onset of TES ADT crystallization.
Atomic force microscopy
Surface analysis of the samples was carried out using an Agilent 5400 scanning probe microscope in tapping mode.
Organic field-effect transistors
Transistors fabricated in this work were of bottom-gate top-contact architecture on a Si/SiO2 substrate, with the 300-nm SiO2 as insulator. The substrate was cleaned in an ultrasonic bath of acetone, isopropanol, ethanol, and deionized water for 15 min each. Then, it was washed in a solution containing deionized water (five parts), aqueous NH4OH (one part), and aqueous H2O2 (one part) at 80°C for 30 min. Before thin-film deposition, the substrate was cleaned in ultraviolet (UV)–ozone for 10 min. Top contacts were thermally evaporated using shadow masks with a typical channel length of 80 μm and a width of 1 mm. All statistics were derived with at least 10 devices.
Electrical measurements were performed in a nitrogen atmosphere using a semiconductor parameter analyzer (Kiethley 4200-SCS). Field-effect mobilities were calculated using the standard thin-film model in the saturation regime of the device using , where L, W, and C are the channel length, channel width, and geometric capacitance of the dielectric, respectively.
μGIWAXS mapping
μGIWAXS measurements were performed at the D-line of Cornell High Energy Synchrotron Source (CHESS) at Cornell University. The x-ray beam with a wavelength of 1.155 Å and a wide band-pass (1.47%) was focused into a 15 μm × 15 μm spot using a single-bounce capillary. The samples were placed on the focal point of the capillary (55 mm away from the capillary tip) with an incidence angle of 0.15°. An optical microscope was located vertically on top of the sample and was used to monitor the beam and sample locations. A Pilatus 300K detector located 213.5 mm from the sample was used for an exposure time of 1 s and at a lateral sample scan step of 5 μm according to a mapping procedure previously developed elsewhere (35).
Photothermal deflection spectroscopy
PDS measurements were performed using a home-built PDS setup. The light from a 250 W quartz tungsten-halogen lamp (Newport 66996-250Q-R1) was sent through a monochromator (LOT MSH-300) and used as pump, allowing excitation across the UV–to–near-infrared spectral region. The pump light was modulated by a chopper operating at a constant frequency of a few hertz and focused on the sample, which itself was immersed in a chemically inert liquid [perfluorohexane (C6F14); Sigma-Aldrich] during the measurement. A small fraction of the monochromatic pump light was split off as intensity reference and measured by lock-in detection (Stanford Research Systems SR830 lock-in amplifier) using a pyroelectric detector (Newport DET-L-PYC5-R-P). Thin-film samples for PDS were prepared on cleaned quartz substrates by spin coating from the solution. A stabilized continuous-wave laser (Thorlabs HR S015 HeNe, 633 nm) was used as a probe beam source focused closely on the sample surface. The deviation of the probe beam was detected by a Si quadrant detector (Thorlabs PDP90A) using lock-in detection (Stanford Research Systems SR830). The entire setup was controlled by a home-built LabVIEW-based data acquisition and device control code. The PDS spectra were set to absolute scale by matching the spectra with integrating sphere measurements on a Varian Cary 6000 spectrophotometer. The Urbach energy was extracted from α = α0exp(E/Eu) (40), where α is the absorption coefficient, α0 is a pre-exponential factor, and E is the photon energy.
Supplementary Material
Acknowledgments
We thank N. Treat, N. Stingelin, and E.H. Sargent for helpful discussions, J.E. Anthony for providing us with TES ADT and H. Hu for assistance with atomic force microscopy measurements. A.H.B. and F.L. thank K. Vandewal for his contribution to the PDS setup and M. Baier for help with the experiments. Funding: Part of this work was performed at CHESS, which was supported by the NSF and the NIH/National Institutes of General Medical Science via NSF award DMR-1332208. The research reported here was supported by the King Abdullah University of Science and Technology. A.A. is grateful to Saudi Arabian Basic Industries Corporation (SABIC) for the Career Development SABIC Chair. Author contributions: L.Y. and A.A. conceived and designed the project. G.O.N.N. assisted with the vacuum deposition and crystallization studies of rubrene and MoOx films. M.R.N. assisted with the fabrication and crystallization of TES ADT thin films and transistors. R.L. assisted with μGIWAXS mapping experiments. A.R.K. conducted the QCM-D measurements. R.M. assisted with the fabrication and crystallization of organohalide perovskite films. A.H.B. and F.L. conducted the PDS measurements. L.Y. performed all other experiments. L.Y. and A.A. wrote the manuscript. A.A. supervised the project. All authors read and commented on the final version of the manuscript. Competing interests: A.A. and L.Y. have a U.S. patent application related to this work (WO 2016/199093; 10 June 2016). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/3/e1602462/DC1
fig. S1. Height profile of the solidified droplet of TES ADT.
fig. S2. Polarized optical micrographs of MoOx thin films with different thicknesses.
fig. S3. Illustrative plots of the volumetric and areal nucleation rates with respect to temperature.
fig. S4. Polarized optical micrographs of MoOx thin films (100 nm) at different annealing times.
fig. S5. QCM-D measurements during solvent vapor annealing.
fig. S6. Atomic force microscopy topography image of a TES ADT film scratched with a needle.
fig. S7. Optical micrographs of TES ADT and PCBM films crystallized using linear seeding.
fig. S8. μGIWAXS images of the αp and α phases.
fig. S9. Typical OTFT output characteristics of TES ADT thin films using conventional and programmable crystallization.
movie S1. Conventional crystallization of a TES ADT film proceeding stochastically and incoherently.
movie S2. Linear programmed crystallization of TES ADT proceeding simultaneously and coherently from horizontal seeding lines.
movie S3. Periodic dot array crystallization of TES ADT proceeding simultaneously and coherently from an array of imprinted seed dots.
movie S4. Square programmed crystallization of TES ADT proceeding simultaneously and coherently from horizontal and vertical seeding lines.
movie S5. Square and rectangular programmed crystallization of TES ADT proceeding simultaneously and coherently from horizontal and vertical seeding lines.
REFERENCES AND NOTES
- 1.McCulloch I., Heeney M., Bailey C., Genevicius K., MacDonald I., Shkunov M., Sparrowe D., Tierney S., Wagner R., Zhang W., Chabinyc M. L., Kline R. J., McGehee M. D., Toney M. F., Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nat. Mater. 5, 328–333 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Nomura K., Ohta H., Ueda K., Kamiya T., Hirano M., Hosono H., Thin-film transistor fabricated in single-crystalline transparent oxide semiconductor. Science 300, 1269–1272 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Yang Y., Zheng Y., Cao W., Titov A., Hyvonen J., Manders J. R., Xue J., Holloway P. H., Qian L., High-efficiency light-emitting devices based on quantum dots with tailored nanostructures. Nat. Photonics 9, 259–266 (2015). [Google Scholar]
- 4.Lee S. S., Loo Y.-L., Structural complexities in the active layers of organic electronics. Annu. Rev. Chem. Biomol. Eng. 1, 59–78 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Jurchescu O., Mourey D. A., Subramanian S., Parkin S. R., Vogel B. M., Anthony J. E., Jackson T. N., Gundlach D. J., Effects of polymorphism on charge transport in organic semiconductors. Phys. Rev. B 80, 085201–085207 (2009). [Google Scholar]
- 6.Li X., Kadashchuk A., Fishchuk I. I., Smaal W. T. T., Gelinck G., Broer D. J., Genoe J., Heremans P., Bässler H., Electric field confinement effect on charge transport in organic field-effect transistors. Phys. Rev. Lett. 108, 066601–066605 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Hwang K., Jung Y.-S., Heo Y.-J., Scholes F. H., Watkins S. E., Subbiah J., Jones D. J., Kim D.-Y., Vak D., Toward large scale roll‐to‐roll production of fully printed perovskite solar cells. Adv. Mater. 27, 1241–1247 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Minemawari H., Yamada T., Matsui H., Tsutsumi J., Haas S., Chiba R., Kumai R., Hasegawa T., Inkjet printing of single-crystal films. Nature 475, 364–367 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Yu X., Marks T. J., Facchetti A., Metal oxides for optoelectronic applications. Nat. Mater. 15, 383–396 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Kim T.-H., Cho K.-S., Lee E. K., Lee S. J., Chae J., Kim J. W., Kim D. H., Kwon J.-Y., Amaratunga G., Lee S. Y., Choi B. L., Kuk Y., Kim J. M., Kim K., Full-colour quantum dot displays fabricated by transfer printing. Nat. Photonics 5, 176–182 (2011). [Google Scholar]
- 11.Niazi M. R., Li R., Li E. Q., Kirmani A. R., Abdelsamie M., Wang Q., Pan W., Payne M. M., Anthony J. E., Smilgies D.-M., Thoroddsen S. T., Giannelis E. P., Amassian A., Solution-printed organic semiconductor blends exhibiting transport properties on par with single crystals. Nat. Commun. 6, 8598 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giri G., Li R., Smilgies D.-M., Li E. Q., Diao Y., Lenn K. M., Chiu M., Lin D. W., Allen R., Reinspach J., Mannsfeld S. C. B., Thoroddsen S. T., Clancy P., Bao Z., Amassian A., One-dimensional self-confinement promotes polymorph selection in large-area organic semiconductor thin films. Nat. Commun. 5, 3573 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Mei J., Diao Y., Appleton A. L., Fang L., Bao Z., Integrated materials design of organic semiconductors for field-effect transistors. J. Am. Chem. Soc. 135, 6724–6746 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Fratini S., Ciuchi S., Bandlike motion and mobility saturation in organic molecular semiconductors. Phys. Rev. Lett. 103, 266601 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Villani F., Vacca P., Nenna G., Valentino O., Burrasca G., Fasolino T., Minarini C., della Sala D., Inkjet printed polymer layer on flexible substrate for OLED applications. J. Phys. Chem. C 113, 13398–13402 (2009). [Google Scholar]
- 16.Briseno A. L., Mannsfeld S. C. B., Ling M. M., Liu S., Tseng R. J., Reese C., Roberts M. E., Yang Y., Wudl F., Bao Z., Patterning organic single-crystal transistor arrays. Nature 444, 913–917 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Campbell M., Sharp D. N., Harrison M. T., Denning R. G., Turberfield A. J., Fabrication of photonic crystals for the visible spectrum by holographic lithography. Nature 404, 53–56 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Ko D.-H., Tumbleston J. R., Zhang L., Williams S., DeSimone J. M., Lopez R., Samulski E. T., Photonic crystal geometry for organic solar cells. Nano Lett. 9, 2742–2746 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Théry M., Racine V., Pépin A., Piel M., Chen Y., Sibarita J.-B., Bornens M., The extracellular matrix guides the orientation of the cell division axis. Nat. Cell Biol. 7, 947–953 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Payne M. M., Parkin S. R., Anthony J. E., Kuo C.-C., Jackson T. N., Organic field-effect transistors from solution-deposited functionalized acenes with mobilities as high as 1 cm2/V·s. J. Am. Chem. Soc. 127, 4986–4987 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Deegan R. D., Bakajin O., Dupont T. F., Huber G., Nagel S. R., Witten T. A., Capillary flow as the cause of ring stains from dried liquid drops. Nature 389, 827–829 (1997). [Google Scholar]
- 22.Kajiya T., Kaneko D., Doi M., Dynamical visualization of “coffee stain phenomenon” in droplets of polymer solution via fluorescent microscopy. Langmuir 24, 12369–12374 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Graddage N., Chu T.-Y., Ding H., Py C., Dadvand A., Tao Y., Inkjet printed thin and uniform dielectrics for capacitors and organic thin film transistors enabled by the coffee ring effect. Org. Electron. 29, 114–119 (2016). [Google Scholar]
- 24.Chen F.-C., Lu J.-P., Huang W.-K., Using ink-jet printing and coffee ring effect to fabricate refractive microlens arrays. IEEE Photonics Technol. Lett. 21, 648–650 (2009). [Google Scholar]
- 25.Dickey K. C., Anthony J. E., Loo Y.-L., Improving organic thin‐film transistor performance through solvent‐vapor annealing of solution‐processable triethylsilylethynyl anthradithiophene. Adv. Mater. 18, 1721–1726 (2006). [Google Scholar]
- 26.Sheldon R. P., Blakey P. R., Liquid-induced crystallization in polymers. Nature 195, 172–173 (1962). [Google Scholar]
- 27.Anwar M., Hogarth C. A., Structural investigations and colour centres in MoO3 films deposited by vacuum evaporation. Int. J. Electron. 67, 567–576 (1989). [Google Scholar]
- 28.Li R., Khan H. U., Payne M. M., Smilgies D.-M., Anthony J. E., Amassian A., Heterogeneous nucleation promotes carrier transport in solution‐processed organic field‐effect transistors. Adv. Funct. Mater. 23, 291–297 (2013). [Google Scholar]
- 29.Neff J. L., Milde P., León C. P., Kundrat M. D., Eng L. M., Jacob C. R., Hoffmann-Vogel R., Epitaxial growth of pentacene on alkali halide surfaces studied by Kelvin probe force microscopy. ACS Nano 8, 3294–3301 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Mencik Z., Plummer H. K., Van Oene H., Confined-growth crystallization of polyethylene. J. Polym. Sci. 10, 507–517 (1972). [Google Scholar]
- 31.Zanotto E. D., James P. F., Experimental tests of the classical nucleation theory for glasses. J. Non-Cryst. Solids 74, 373–394 (1985). [Google Scholar]
- 32.Munir R., Sheikh A. D., Abdelsamie M., Hu H., Yu L., Zhao K., Kim T., Tall O. E., Li R., Smilgies D. M., Amassian A., Hybrid Perovskite Thin-Film Photovoltaics: In situ diagnostics and importance of the precursor solvate phases. Adv. Mater. 29, 1604113 (2017). 10.1002/adma.201604113 [DOI] [PubMed] [Google Scholar]
- 33.Chen J., Shao M., Xiao K., Rondinone A. J., Loo Y.-L., Kent P. R. C., Sumpter B. G., Li D., Keum J. K., Diemer P. J., Anthony J. E., Jurchescu O. D., Huang J., Solvent-type-dependent polymorphism and charge transport in a long fused-ring organic semiconductor. Nanoscale 6, 449–456 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Li R., Ward J. W., Smilgies D.-M., Payne M. M., Anthony J. E., Jurchescu O. D., Amassian A., Direct structural mapping of organic field‐effect transistors reveals bottlenecks to carrier transport. Adv. Mater. 24, 5553–5558 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Yu L., Li X., Pavlica E., Koch F. P. V., Portale G., da Silva I., Loth M. A., Anthony J. E., Smith P., Bratina G., Kjellander B. K. C., Bastiaansen C. W. M., Broer D. J., Gelinck G. H., Stingelin N., Influence of solid-state microstructure on the electronic performance of 5,11-bis(triethylsilylethynyl) anthradithiophene. Chem. Mater. 25, 1823–1828 (2013). [Google Scholar]
- 36.Ueno S., Nishida T., Sato K., Synchrotron radiation microbeam X-ray analysis of microstructures and the polymorphic transformation of spherulite crystals of trilaurin. Cryst. Growth Des. 8, 751–754 (2008). [Google Scholar]
- 37.Venkateshvaran D., Nikolka M., Sadhanala A., Lemaur V., Zelazny M., Kepa M., Hurhangee M., Kronemeijer A. J., Pecunia V., Nasrallah I., Romanov I., Broch K., McCulloch I., Emin D., Olivier Y., Cornil J., Beljonne D., Sirringhaus H., Approaching disorder-free transport in high-mobility conjugated polymers. Nature 515, 384–388 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Podzorov V., Menard E., Borissov A., Kiryukhin V., Rogers J. A., Gershenson M. E., Intrinsic charge transport on the surface of organic semiconductors. Phys. Rev. Lett. 93, 086602 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Sheats J. R., Manufacturing and commercialization issues in organic electronics. J. Mater. Res. 19, 1974–1989 (2004). [Google Scholar]
- 40.Yakuphanoglu F., Sekerci M., Optical characterization of an amorphous organic thin film. Opt. Appl. 35, 209–214 (2005). [Google Scholar]
- 41.Siegmann A., Geil P. H., Crystallization of polycarbonate from the glassy state. Part II. Thin films melted and quenched. J. Macromol. Sci. Part B 4, 273–291 (1970). [Google Scholar]
- 42.W. Callister, D. Rethwisch, Materials Science and Engineering: An Introduction (John Wiley and Sons, New York, 2007). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/3/e1602462/DC1
fig. S1. Height profile of the solidified droplet of TES ADT.
fig. S2. Polarized optical micrographs of MoOx thin films with different thicknesses.
fig. S3. Illustrative plots of the volumetric and areal nucleation rates with respect to temperature.
fig. S4. Polarized optical micrographs of MoOx thin films (100 nm) at different annealing times.
fig. S5. QCM-D measurements during solvent vapor annealing.
fig. S6. Atomic force microscopy topography image of a TES ADT film scratched with a needle.
fig. S7. Optical micrographs of TES ADT and PCBM films crystallized using linear seeding.
fig. S8. μGIWAXS images of the αp and α phases.
fig. S9. Typical OTFT output characteristics of TES ADT thin films using conventional and programmable crystallization.
movie S1. Conventional crystallization of a TES ADT film proceeding stochastically and incoherently.
movie S2. Linear programmed crystallization of TES ADT proceeding simultaneously and coherently from horizontal seeding lines.
movie S3. Periodic dot array crystallization of TES ADT proceeding simultaneously and coherently from an array of imprinted seed dots.
movie S4. Square programmed crystallization of TES ADT proceeding simultaneously and coherently from horizontal and vertical seeding lines.
movie S5. Square and rectangular programmed crystallization of TES ADT proceeding simultaneously and coherently from horizontal and vertical seeding lines.