

Abstract
γ-Aminobutyric acid type A receptors (GABAARs) are the principal mediators of fast synaptic inhibition in the brain as well as the low persistent extrasynaptic inhibition, both of which are fundamental to proper brain function. Thus unsurprisingly, deficits in GABAARs are implicated in a number of neurological disorders and diseases. The complexity of GABAAR regulation is determined not only by the heterogeneity of these receptors but also by its posttranslational modifications, the foremost, and best characterized of which is phosphorylation. This review will explore the details of this dynamic process, our understanding of which has barely scratched the surface. GABAARs are regulated by a number of kinases and phosphatases, and its phosphorylation plays an important role in governing its trafficking, expression, and interaction partners. Here, we summarize the progress in understanding the role phosphorylation plays in the regulation of GABAARs. This includes how phosphorylation can affect the allosteric modulation of GABAARs, as well as signaling pathways that affect GABAAR phosphorylation. Finally, we discuss the dysregulation of GABAAR phosphorylation and its implication in disease processes.
1. INTRODUCTION
With kinases alone constituting about 2% of the human genome, it is perhaps unsurprising that phosphorylation plays a key role in all aspects of cellular activity and is one of the best characterized of posttranslational modifications (Manning, Whyte, Martinez, Hunter, & Sudarsanam, 2002; Ubersax & Ferrell, 2007). Phosphorylation is dynamically executed by the opposing functions of kinases and phosphatases, which lead to changes in protein conformation and function. Protein kinases contain a common catalytic domain that catalyses the transfer of γ-ATP to a serine, threonine, or tyrosine residue of the intended protein. In contrast, phosphatases remove phosphate groups from their substrates. Kinases are primarily divided into two main groups: (1) the serine/threonine kinases which include cyclic-AMP dependent protein kinase (PKA), phospholipid-dependent protein kinase C (PKC), and Ca2+/calmodulin-dependent protein kinase II (CamKII) and (2) the tyrosine kinases which include Src family tyrosine kinases. The protein kinase family is considerably diverse, as exemplified by PKC, which consist of multiple isoforms with distinct responses to specific activation (Song & Messing, 2005; Tanaka & Nishizuka, 1994; Taylor, Buechler, & Yonemoto, 1990; Taylor, Knighton, Zheng, Ten Eyck, & Sowadski, 1992; Ubersax & Ferrell, 2007). PKC are composed of “classical” or “conventional” cPKC subgroup (isoforms α, β, and γ) which are activated by calcium, phosphatidylserine (PS), and diacylglycerol (DAG); the “novel” nPKC (δ, ε, η, and θ) which are activated by DAG and PS; and the “atypical” aPKC (ζ and λ/ι), activated by other lipid messengers. Phorbol esters, often used to activate PKC, predominantly stimulate cPKC and nPKC (Song & Messing, 2005; Tanaka & Nishizuka, 1994; Taylor et al., 1990, 1992; Ubersax & Ferrell, 2007).
Different kinases recognize specific consensus sequences in the target polypeptide. However, as exemplified below with GABAARs, the presence of consensus sequences for a specific kinase does not ensure that the protein is a substrate for the kinase in vivo. Likewise, bona fide phosphorylation sites may not correspond to the consensus sequence (Ubersax & Ferrell, 2007). GABAARs in particular are accepted phosphoproteins and their phosphorylation governs numerous processes, including directly varying channel function, regulating receptor trafficking, affecting receptor-interacting proteins, and their sensitivity to pharmacological agents (Brandon, Jovanovic, & Moss, 2002; Houston, He, & Smart, 2009; Jacob, Moss, & Jurd, 2008; Luscher, Fuchs, & Kilpatrick, 2011). Thus, the interplay between kinases and phosphatases can dynamically regulate neuronal excitability and ultimately shape brain function.
2. THE γ-AMINOBUTYRIC ACID TYPE A RECEPTORS
GABAARs are GABA-gated Cl−-channels responsible for the majority of inhibition in the mammalian brain and the major target for many clinically relevant drugs. Deficits in GABAAR function are increasingly implicated in numerous pathologies including anxiety (Lydiard, 2003; Rudolph & Mohler, 2004), cognitive deficits (D’Hulst & Kooy, 2007; DeLorey & Olsen, 1999; Thompson-Vest, Waldvogel, Rees, & Faull, 2003), depression (Luscher, Shen, & Sahir, 2011b), epilepsy (Benarroch, 2007; Fritschy, 2008), schizophrenia (Benes & Berretta, 2001; Charych, Liu, Moss, & Brandon, 2009), and substance abuse (Krystal et al., 2006). GABAARs are responsible for two forms of inhibition known as phasic (synaptic) and tonic (extrasynaptic) inhibition. Synaptic inhibition occurs via transient or “phasic” activation of GABAARs after the release of GABA from synaptic vesicles, whereas extrasynaptic inhibition necessitates low ambient levels of GABA for continual or “tonic” activation (Farrant & Nusser, 2005).
Structurally, GABAARs are heteropentameric channels (Fig. 1) composed from a pool of 19 possible subunits: α1–6, β1–3, γ1–3, δ, ε, θ, π, and ρ1–3 (Olsen & Sieghart, 2008). However, general consensus holds that the vast majority of receptors are assembled from two α, two β and one γ (or one δ) (Fig. 1). GABAARs composed of α1–3 and γ2 are largely synaptically located, whereas α4–6 and δ are predominantly expressed extrasynaptically (Farrant & Nusser, 2005; Luscher, Fuchs, & Kilpatrick, 2011; Nusser & Mody, 2002). In addition to the diversity of receptor subtypes, alternative splicing offers further heterogeneity. For instance, the γ2 subunits occur as short (γ2S) and long (γ2L) splice variants due to an additional eight amino acids in γ2L (Whiting, McKernan, & Iversen, 1990). Notably, differences in subunit compositions can have an impact on the physiological and pharmacological properties of these receptors (Jacob et al., 2008; Rudolph & Knoflach, 2011; Verdoorn, Draguhn, Ymer, Seeburg, & Sakmann, 1990). Moreover, specific subunits have distinct regional expression profiles which are developmentally regulated (Hortnagl et al., 2013; Laurie, Wisden, & Seeburg, 1992). Indeed, not only is GABAAR subunit expression brain region-specific, but the expression of particular subunits is also found to be specific at a subcellular level. Finally, on top of this staggering level of complexity, there are also the added intricacies of subunit-specific phosphorylation and dephosphorylation by multiple kinases and phosphatases.
Figure 1.

Schematic representation of the pentameric GABAAR. GABAARs are composed of a large extracellular N-terminal domain, four-transmembrane domains (TM1–4), and a major intracellular loop between TM3 and TM4 where phosphorylation (Ⓟ) primarily occurs. Binding of regions for benzodiazepines (BDZ) and GABA are depicted.
3. PHOSPHORYLATION SITES ON GABAAR
The major intracellular loop between TM3 and TM4 of GABAAR contains numerous consensus sites for phosphorylation by both serine/threonine and tyrosine protein kinases (Moss & Smart, 1996). Earlier work relied largely on glutathione S-transferase (GST) fusion proteins that encode for the large intracellular loop of specific GABAAR subunits. Several kinases were found to phosphorylate sites in this domain, the vast majority of which lie on β and γ2 subunits (Brandon, Delmas, Hill, Smart, & Moss, 2001; McDonald & Moss, 1994; McDonald & Moss, 1997; Moss, Doherty, & Huganir, 1992; Moss, Gorrie, Amato, & Smart, 1995; Table 1b and c). For example, a conserved serine residue found in β1–3 subunits can be phosphorylated by PKA, PKC, protein kinase G (PKG), and CamKII (McDonald & Moss, 1994, 1997; Moss, Doherty, et al., 1992). Both γ2S and γ2L can be phosphorylated on serine residue 327 (S327) by PKC, whereas γ2L has an additional residue at S343 that can be phosphorylated by PKC and CamKII (McDonald & Moss, 1994, 1997; Moss, Doherty, et al., 1992).
Table 1.
Known phosphorylation sites of GABAARs subunits
| (a) α and δ subunits | ||||||
|---|---|---|---|---|---|---|
| Subunit | Site | Kinase or phosphatase | Effect of phosphorylation, phosphomimetic | Selected publications | ||
| GST | Cell line | Neuron | ||||
| α1 | – | ☒ Akt ☒ PKA ☒ PKC ☑ Src |
HEK: α1β1γ2 ☒ PKA | FB synaptosome: ☑ CamKII | ☑– | HEK (Moss, Smart, et al., 1992) FB (Churn et al., 2002) GST (Moss, Doherty, et al., 1992; Vetiska et al., 2007; Wang, Liu, Pei, et al., 2003) |
| Putative T375 | – | – | – | Hip: Phosphomimetic ↓ gephyrin binding, ↓ synaptic clustering, and ↓ mIPSC amplitude. | (Mukherjee et al., 2011) | |
| α4 | S443 | – | COS7: α4β3 ☑ PKC | Hip: ☑ PKC | COS7 (α4β3): ↑ surface, ↑ insertion, ↑ surface stability, block IGABA rundown. Hip: ↑ surface, ↑ insertion, ↑ tonic current. |
(Abramian et al., 2014, 2010) |
| δ | – | – | COS7: ☒ PKC | – | – | (Abramian et al., 2010) |
| (b) β subunits | ||||||
|---|---|---|---|---|---|---|
| Subunit | Site | Kinase or phosphatase | Effect of phosphorylation | Selected publications | ||
| GST | Cell line | Neuron | ||||
| β1 | T227, Y230 | – | – | Hip | – | Mass spectrometry (Kang et al., 2011) |
| S384 | ☑ CamKII | – | – | – | GST (McDonald & Moss, 1994) | |
| S409 | ☑ CamKII ☑ PKA ☑ PKC ☑ PKG |
HEK:α1β1γ2 ☑ PKA; α1β1γ2S/L ☑ PKC | SCG |
HEK: ☑ PKA ↓ IGABA amplitude. HEK: ☑ PKC ↓ IGABA amplitude. |
GST (McDonald & Moss, 1994; Moss, Doherty, et al., 1992) HEK (Krishek et al., 1994; Moss, Smart, et al., 1992) SCG (Brandon, Jovanovic, Smart, & Moss, 2002) |
|
| β2 | – | – | – | Hip, Ctx: ☒ PKC (via BDNF) PFC: ☒ PKC |
Hip, Ctx (via BDNF): ☒ PKC no Ⓟ PFC (via 5HT2): ☒ PKC no change in Ⓟ |
BDNF (Jovanovic et al., 2004) PFC (Feng et al., 2001) |
| Y215, T439 | – | – | Hip | – | Mass spectrometry (Kang et al., 2011) | |
| Y372/Y379 | ☑ Src | HEK: α1β1γ2 | – | HEK (via insulin): ↑ surface. | (Vetiska et al., 2007) | |
| S410 | ☑ Akt ☑ CamKII ☑ PKA ☑ PKC ☑ PKG |
HEK: α1β2γ2 ☑ Akt, ☒ PKA Oocyte: α1β2γ2S ☑ PKC HEK: α4β2δ ☑ PKC |
Hip: ☑ Akt CGC: β2 KO ☒ CamKII |
GST: Phosphomimetic binds gephyrin. HEK, Hip: ☑ Akt ↑ surface, ↑ mIPSC amplitude & frequency. Oocyte: ☑ PKC ↓ IGABA amplitude (with γ2 S327 for full affect). CGC β 2 KO: No affect of CamKII activation compared to WT. HEK α4β2δ: ☑ PKC ↓ tonic inhibition, ↓ surface |
GST (Kowalczyk et al., 2013; McDonald & Moss, 1997) Akt (Wang, Liu, Pei, et al., 2003) Not PKA (McDonald et al., 1998) Oocyte (Kellenberger et al., 1992) β2 KO (Houston & Smart, 2006) Tonic (Bright & Smart, 2013) |
|
| β3 | T282, S406 | – | – | Hip | – | Mass spectrometry (Kang et al., 2011) |
| S383 | ☑ CamKII | HEK: α1β3 ☒ CamKII NG108-15: α1β3(γ2) ☑ CamKII |
Hip: ☑ CamKII |
NG108-15: ☑ CamKII ↑ IGABA amplitude. Hip (via VGCCs): ☑ CamKII ↑ surface, ↑ insertion, ↑ tonic current. Hip: ☑ CamKII LTPGABA ↑ synaptic clusters of gephyrin and GABAAR. |
GST (McDonald & Moss, 1997) NG108-15 (Houston et al., 2007) VGCC (Saliba et al., 2012) LTPGABA (Petrini et al., 2014) |
|
| S408 | ☑ PKC ☒ CamKII |
(see S408/S409) | (see S408/S409) | GST (McDonald & Moss, 1997) | ||
| S409 | ☑ CamKII ☑ PKA ☑ PKC ☑ PKG |
(see S408/S409) | (see S408/S409) | GST: Ⓟ-S409 ↓ binding to PKC. | GST (Brandon, Jovanovic, Smart, et al., 2002; McDonald & Moss, 1997) | |
| S408/S409 | (As above) | HEK: α1β3γ2 ☑ PKA NG108-15: α1β3 ☒ CamKII |
Ctx, Hip: RACK1-☑ PKC ☑ PP2A Ctx, Hip: PRIP-☑ PP2A Hip: ☑ PKA PRIP1-☑ PP1α |
HEK: ☑ PKA ↑ IGABA. CamKII NG108-15: Mutant does not change IGABA. Ⓟ inhibits binding to AP2. Ctx, Hip (via BDNF): ☑ PKC ☑ PP2A, transient ↑ then ↓ mIPSC amplitude and Ⓟ, ↑ surface but also reported to ↑ then ↓ surface in WT and ↑ surface in PRIP dKO Hip : ☑ PKA ↑ I GABA in WT but not in PRIP1 KO. PKA activity dissociates PRIP1-PP1 α which de Ⓟ S408/S409. |
HEK (McDonald et al., 1998) NG108-15 (Houston et al., 2007) AP2 (Kittler et al., 2005a) BDNF (Jovanovic et al., 2004; Kanematsu et al., 2006) PP1α (Terunuma et al., 2004) |
|
| (c) γ subunits | ||||||
|---|---|---|---|---|---|---|
| Subunit | Site | Kinase or phosphatase | Effect of phosphorylation | Selected publications | ||
| GST | Cell line | Neuron | ||||
| γ2S/L | – | ☒ Akt ☒ PKG ☒ PKA ☑ PKC γ2L ☒ PKC γ2S |
HEK: α1β1γ2 ☒ PKA | Hip, Ctx: ☒ PKC (via BDNF) PFC: RACK1-☑ PKC Hip Fyn: ☑ Fyn (KO ↓ phospho) |
Hip, Ctx (via BDNF): ☒ PKC no Ⓟ PFC (via 5HT2): ☑ PKC ↓ IGABA, γ↑ 2 Ⓟ |
GST (McDonald & Moss, 1994; Moss, Doherty, et al., 1992; Wang, Liu, Pei, et al., 2003; Whiting et al., 1990) HEK (Moss, Smart, et al., 1992), BDNF (Jovanovic et al., 2004). PFC (Feng et al., 2001), and Fyn (Jurd et al., 2010) |
| S327 | ☑ PKC ☑ PKCε ☑ CaN |
HEK: α1β1γ2S/L ☑ PKC ☑ PKCε Oocyte: α1β2γ2S ☑ PKC |
Hip: ☑ CaN Hip, Ctx: ☑ PKCε (PKCε KO ↓ phospho) |
HEK: ☑ PKC ↓ IGABA amplitude. HEK: ☑ PKCε modulates actions of benzodiazepine and ethanol Oocyte : ☑ PKC ↓ IGABA amplitude (with β2 S410 for full affect). Hip (via LTDGABA): ☑ CaN ↓ S327 Ⓟ required for LTDGABA, ↑ lateral diffusion |
GST (Moss, Doherty, et al., 1992) HEK (Krishek et al., 1994), PKCe (Qi et al., 2007), oocyte (Kellenberger et al., 1992), and LTDGABA (Muir et al., 2010; Wang, Liu, Haditsch, et al., 2003) |
|
| S348, T350 | ☑ CamKII | NG108-15: α1β3 ☒ CamKII |
NG108-15: ☒ CamKII Ⓟ-mutant does not change IGABA. |
GST (McDonald & Moss, 1994) NG108-15 (Houston et al., 2007) |
||
| Y365/Y367 | ☑ Fyn ☑ Src |
HEK: α1β1γ2L ☑ Src | Ctx, WB: basally Ⓟ |
HEK: ☑ Src ↑ IGABA amplitude. Ⓟ Y365/Y367 binds Src. Hip: Ⓟ Y367 binds Fyn. Ⓟ Y365/Y367 ↓ AP2 binding. Y365/7F+/+ : embryonic lethality. Y365/7F+/−: ↑ surface, ↑ size inhibitory synapse, ↑ mIPSC amplitude and frequency, ↓ AP2 binding, ↓ spatial object recognition |
HEK (Moss et al., 1995) GST, Ctx, and WB (Brandon et al., 2001) Fyn (Jurd et al., 2010) Y365/7F mouse (Tretter et al., 2009) AP2 (Kittler et al., 2008) |
|
| γ2L | S343 | ☑ CamKII ☑ PKC |
HEK: α1β1γ2S/L ☑ PKC | HEK: ☑ PKC ↓ IGABA amplitude | GST (McDonald & Moss, 1994; Moss, Doherty, et al., 1992); HEK (Krishek et al., 1994) | |
Whole-cell GABA-activated currents (IGABA); miniature inhibitory post synaptic current (mIPSC); Cerebellar granule cells (CGC); Cortex (Ctx); Forebrain (FB); Hippocampus (Hip); Prefrontal cortex (PFC); Superior cervical ganglion (SCG); Whole brain (WB); (↑) increase; (↓) decrease; (Ⓟ) phosphorylation; (deⓅ) dephosphor-ylation; (☑) phosphorylated/dephosphorylated by specific kinase/phosphatase; (☒) not phosphorylated/not dephosphorylated by specific kinase/phosphatase.
3.1. Phosphorylation in expression systems
The methods outlined above identified a number of sites that were later recapitulated in heterologous expression systems. Although studies of this manner may not account for the presence of proteins intimately associated with GABAARs, they allow for the direct examination of the effects of phosphorylation on specific residues with precise receptor combinations. These approaches have been carried out on the numerous phosphorylation sites first identified utilizing GST conjugated to the large intracellular loop of GABAARs (McDonald & Moss, 1994, 1997; Moss, Doherty, et al., 1992; Whiting et al., 1990). Human embryonic kidney (HEK293) cells lines have been extensively used as a vehicle for the expression of recombinant receptors for such studies, and generally these results have lent support to the in vitro studies, albeit with a few discrepancies (Table 1b and c).
3.1.1 PKA
As a specific example of such discrepancies, in vitro experiments have suggested that β1–3 subunits are phosphorylated by PKA (McDonald & Moss, 1997; Moss, Doherty, et al., 1992). In contrast, studies in HEK293 cells showed that β2-containing receptors were not phosphorylated and failed to be modulated by PKA activation (McDonald et al., 1998). However, PKA-induced phosphorylation did, in fact, differentially regulate β1 and β3 subunits expressed as α1β1/3γ2S in HEK293 cells. PKA-dependent phosphorylation of β1-containing receptors at S409 led to an inhibition of GABA-activated responses (McDonald et al., 1998; Moss, Smart, et al., 1992). In contrast, similar PKA activation in β3-containing receptors resulted in the potentiation of GABA-induced currents attributed to two adjacent serine residues at 408 and 409, both of which are phosphorylated by PKA. Moreover, mutation of S408 to an alanine (S408A) in the β3 subunit transformed the enhancement of activity to an inhibition analogous to β1 subunits that are exclusively phosphorylated on S409. The lack of β2 subunit regulation by PKA is most likely due to an inability of β2 to bind A-kinase anchoring proteins (AKAP) (see Section 4.3). These anchoring proteins are found endogenously in HEK293 cells (Gardner, Tavalin, Goehring, Scott, & Bahouth, 2006) and are crucial to subcellular targeting of PKA (Sanderson & Dell’Acqua, 2011).
Since PKA appears to exclusively phosphorylate β1 (S409) and β3 (S408/S409) subunits, it was generally expected that the regulation of GABAAR activity by PKA should depend on whether a population of receptors is largely β1- or β3-containing. However, contrary to experiments conducted with synaptically expressed α1 receptors, extrasynaptically expressed α4β3γ2 and α4β3δ receptors tell a different story. In HEK293 cells, PKA activation caused a larger increase of spontaneous activity in cells expressing α4β3δ receptors compared to α4β3γ2 receptors; however in the same cells, PKA activation did not affect currents that were activated by 1 μM GABA (Tang, Hernandez, & Macdonald, 2010). These results suggest that PKA could regulate GABAAR-mediated currents by an unknown process that depends on the GABA concentration.
3.1.2 CamKII and Src
Discrepancies also exist on residues that were thought to be phosphorylated by CamKII (McDonald & Moss, 1994, 1997). In these cases, it has been the choice of cell line that has appeared to underlie a particular kinase’s ability to modulate receptor function (Houston et al., 2009; Houston & Smart, 2006). CamKII did not regulate HEK293 cells transfected with α1β2 and α1β3 receptors. Contrary to this, in the neuroblastoma-glioma hybrid (NG108-15) cell line, CamKII activity enhanced GABA-evoked current amplitudes of α1β3 and α1β3γ2 receptor expressing cells, but not in cells expressing α1β2 and α1β2γ2 receptors (Houston &Smart, 2006). Moreover, similar enhancements were observed upon CamKII activation in cerebellar granule neurons from both wild-type mice and those in which the β2 subunit was specifically knocked out. This result suggests that in cerebellar granule neurons, CamKII effects are mediated primarily by the β3 subunit.
CamKII-dependent enhancement of GABA-evoked currents in α1β3-transfected NG108-15 cells (without γ2) was mediated solely by S383 on the β3 subunit (Houston, Lee, Hosie, Moss, & Smart, 2007). Other potential CamKII phosphorylation sites initially identified by GST pull-down experiments (McDonald & Moss, 1994, 1997) were not involved (Houston et al., 2007; Table 1b). Interestingly, this site not only deviates from the typical consensus site for CamKII, but, thus far, it is also a site exclusively phosphorylated by CamKII. Further, in α1β3γ2 transfected NG108-15 cells, CamKII-mediated enhancement of GABA-evoked currents were not exclusively mediated by S383 on the β3 subunit, but were also mediated by tyrosine phosphorylation sites on residues Y365/Y367 of the γ2 subunit. This suggests that activation of CamKII not only leads to the direct phosphorylation of S383 on β3 but also activates endogenous tyrosine kinases to phosphorylate sites on γ2 (Houston et al., 2007). The tyrosine kinase Src can specifically interact with β and γ2 subunits (Brandon et al., 2001). Moreover, Y365/Y367 in the γ2 subunit can be phosphorylated by Src kinase in α1β1γ2L-expressing A293 cells, which in agreement, also resulted in an enhancement of GABA-activated currents (Moss et al., 1995). These observations were attributed to the phosphorylation of γ2 residue at Y365/Y367 since mutations of these residues to phenylalanines (Y365/Y367F) ablated tyrosine phosphorylation and receptor modulation. Finally, while it is currently unknown why HEK293 cells expressing GABAARs lack modulation by CamKII, it has been postulated that the nonneuronal lineage of the cells might be the root. Alternatively, the lack of relevant anchoring proteins that would allow kinases to associate with GABAARs may also be a cause.
3.1.3 PKC
One of the first pieces of evidence that PKC modulates GABAARs came from experiments performed on Xenopus oocytes expressing chick brain mRNA. In this study, activation of PKC by phorbol esters decreased the amplitude of GABAAR-mediated currents (Sigel & Baur, 1988). The reduction in current amplitude was also observed in heterologous cells expressing α1β1γ2S/L and α1β2γ2S subunits. This effect was later discovered to be mediated by phosphorylation of S409/S410 in β1/β2 subunits, S327 in γ2S/γ2L subunits, and S343 in γ2L subunits (Kellenberger, Malherbe, & Sigel, 1992; Krishek et al., 1994). Moreover, the level of modulation by a particular phosphorylation site was site-specific. In particular, S343 in γ2 displayed the greatest effect on the GABA-induced response (Kellenberger et al., 1992; Krishek et al., 1994). In contrast, application of constitutively active PKC in L929 fibroblasts expressing α1β1γ2L enhanced GABAARs-mediated currents. These enhancements were prevented by mutations of either β1 S409 to alanine (S409A) or, similarly, γ2L S327A and γ2L S343A (Lin, Angelotti, Dudek, Browning, & Macdonald, 1996; Lin, Browning, Dudek, & Macdonald, 1994). Although the reason for the discrepancy is unclear, there may be numerous explanations: for example, the use of different experimental systems (Houston & Smart, 2006; Mercik, Pytel, & Mozrzymas, 2003), or differential activation of specific PKC isoforms due to the use of phorbol ester versus PKC isolated from bovine brain. Notably, the same phosphorylation sites abolished the observed decreases or increases in GABAAR currents, which perhaps indicates yet another PKC site may dictate the direction of the response.
Although the majority of studies generally concentrate on synaptic GABAARs, more recent evidence indicates that PKC activation may also affect extrasynaptic GABAARs. In COS7 cells, the activation of PKC by application of phorbol esters increases the phosphorylation of S443 on the α4 subunit and enhances surface levels of α4β3 receptors (Abramian et al., 2010). Phosphomimetic mutations revealed that the rise in surface levels were due to increased stability, concurrent with enhanced levels of insertion at the cell surface. On the contrary, activation of PKC has also been found to result in decreases in surface levels of GABAARs in α4β2δ-expressing HEK293 cells. In parallel, a decrease in tonic GABAAR inhibition was observed which was dependent on the phosphorylation of S410 on the β2 subunit. Further, PKC activation did not differentially affect S443 mutants of the α4 subunit when compared to wild type (Bright & Smart, 2013). The reasons for the apparent discrepancy are unclear, although they may be explained by the differential phosphorylation and/or recruitment of kinases and phosphatases to the β2 and β3 subunits as will be discussed below. This would suggest that PKC activity could bidirectionally regulate tonic inhibition depending on the β subunit subtype found in a given GABAAR.
3.1.4 Lessons from expression systems
The phosphorylation sites listed in Table 1a–c are by no means exhaustive; indeed, more recent work using mass spectrometric analysis has identified potential sites of phosphorylation on the β subunits of GABAARs (Kang, Heo, & Lubec, 2011). Moreover, the identification of the first phosphorylation site on an α subunit was more recently found on residue S443 of the α4 subunit (Abramian et al., 2010). Collectively, studies in vitro and in heterologous systems have highlighted important considerations when investigating phosphorylation. For example, the actions of kinases are not only receptor subunit specific, but also splice variants allow for additional sites of phosphorylation (McDonald & Moss, 1994; Moss, Doherty, et al., 1992; Whiting et al., 1990). Further, the determination of the existence of a phosphorylation site may depend on the experimental system utilized (Houston & Smart, 2006; Mercik et al., 2003) as well as the experimental conditions (Bright & Smart, 2013). Interestingly, there is the potential for multiple kinases to act on one site, or, alternatively, for a single kinase to be assigned to one particular site. Kinases can also phosphorylate neighboring sites to differentially regulate GABAARs (McDonald et al., 1998) and the phosphorylation on one site may affect phosphorylation of another site within the same subunit or on another subunit (Houston et al., 2007). Critically, these studies have made evident that subtype-specific phosphorylation of key residues can govern GABAAR activity and trafficking (Brandon, Jovanovic, et al., 2002; Kittler & Moss, 2003).
3.2. Divergent effects of kinases and phosphatases on neuronal GABAARs
Due to their diverse nature, the study of GABAARs in their neuronal environment has been, and still is, a challenge. Nevertheless, a large body of evidence suggests that phosphorylation can affect neuronal activity through the regulation of GABAARs. Similar to heterologous systems, β and γ2 subunits are the primary substrates for kinases in neurons. However, the effect of kinases on GABAARs in neuronal preparations is complex and often yields conflicting results. Disparate effects of phosphorylation on neurons should probably be expected due to the mixed populations of GABAARs composed of various subunits (Pirker, Schwarzer, Wieselthaler, Sieghart, & Sperk, 2000; Poisbeau, Cheney, Browning, & Mody, 1999). Indeed, and as exemplified with PKA, a single kinase can have divergent effects that are dependent on the β-subunit subtype. Moreover, kinases have a considerable number of effects on other substrates that may influence GABAAR activity, including the activation of signaling pathways and phosphorylation of associated proteins, which can make data interpretation challenging.
For example, PKA activation has been studied in a number of brain regions, and found to both increase and decrease GABA-evoked currents. Decreases in GABAAR-mediated currents were observed in cultured cerebellar granule cells, hippocampal pyramidal cells, neostriatal neurons, spinal cord neurons, and superior cervical ganglia (Flores-Hernandez et al., 2000; Moss, Smart, et al., 1992; Poisbeau et al., 1999; Porter, Twyman, Uhler, & Macdonald, 1990; Robello, Amico, & Cupello, 1993). However, increases in GABAAR activity were seen in cerebellar interneurons, hippocampal dentate granule cells and olfactory bulb granule cells (Kano & Konnerth, 1992; Nusser, Sieghart, & Mody, 1999; Poisbeau et al., 1999). Similar inconsistencies are apparent upon activation of PKC, with reports of decreased GABAAR activity in cortical neurons, hippocampal pyramidal neurons, retinal rod bipolar cells, and thalamic neurons (Brandon et al., 2000; Bright & Smart, 2013; Chou et al., 2010; Gillette & Dacheux, 1996), increases in hippocampal dentate granule cells and hippocampal neurons (Abramian et al., 2010; Poisbeau et al., 1999), and no effect in hippocampal pyramidal cells (Poisbeau et al., 1999).
There are a plethora of reasons why such differences may be observed, from the known heterogeneity of GABAARs in both receptor subunit combinations and overall regional distribution to the presence of numerous isoforms of PKC that may mediate differential effects (which may or may not be directed at GABAARs). Indeed, studies have reported that PKA and PKC elicit differential effects even within one region—that of the hippocampus. In CA1 pyramidal cells, PKA activation enhanced miniature inhibitory postsynaptic current (mIPSC) amplitudes, whereas PKC activation showed no observable effects. On the other hand, in dentate gyrus granule cells it was PKA that was unresponsive, whereas PKC activation led to increases in peak mIPSC amplitudes (Poisbeau et al., 1999). Although it is tempting to speculate that these observed effects correspond to the prominence of specific GABAAR subunits, the situation is unclear (Pirker et al., 2000; Wisden, Laurie, Monyer, & Seeburg, 1992), and such conclusions are perhaps even premature considering novel phosphorylation sites are still being discovered (Abramian et al., 2010; Kang et al., 2011).
As well as the aforementioned experiments performed utilizing GST fusion proteins and heterologous expression systems, PKA and PKC have both been demonstrated to phosphorylate purified neuronal GABAARs (Brandon et al., 2000; Moss & Smart, 1996). In neurons, β3 and γ2 are basally phosphorylated at S408/S409 and Y365/Y367, respectively (Brandon et al., 2001, 2000; Jovanovic, Thomas, Kittler, Smart, & Moss, 2004). These results support earlier studies utilizing GST fusion proteins as well as those in HEK293 cells (McDonald et al., 1998; McDonald & Moss, 1997). At β3 subunits, activation of PKC leads to increased phosphorylation at S408/S409, while inhibition of PKC led to concomitant decreases in receptor phosphorylation. Of particular note is that the enhancement of β3 phosphorylation by PKA activation in cortical neurons was only observed when PKC activity was inhibited (Brandon et al., 2000).
GABAARs constitutively cycle between the neuronal surface and intracellular compartments. The endocytosis of receptors from the neuronal surface is a clathrin-dependent process (Kittler et al., 2000, 2001). There is a wealth of evidence in expression systems that PKC activation also leads to a parallel loss of α1β2γ2 receptors from the surface (Chapell, Bueno, Alvarez-Hernandez, Robinson, & Leidenheimer, 1998; Connolly et al., 1999; Filippova, Sedelnikova, Zong, Fortinberry, & Weiss, 2000; Kittler et al., 2000). However, in heterologous cells, this is unlikely to be due to a direct phosphorylation of GABAARs since mutagenesis of all known PKC phosphorylation sites in recombinant receptors (α1, β2 S410A, γ2L S327A/S343A) did not inhibit PKC-mediated decreases in surface expression. Instead, PKC activation prevented receptors that are internalized constitutively from recycling back to the surface (Connolly et al., 1999; Filippova et al., 2000). Whether similar mechanisms are employed in PKC-mediated modifications of GABAARs in neurons is unclear. PKC activation resulting in decreases in GABAAR activity has reported to show variable effects in the level of surface receptors, with observed decreases in cerebellar granule cells (Balduzzi, Cupello, & Robello, 2002; Chou et al., 2010), increases in cortical and hippocampal neurons (Jovanovic et al., 2004), or no change in cortical neurons (Brandon et al., 2000). Additionally, the constitutive cycling of neuronal receptors was observed to occur at a significantly reduced rate when compared to HEK293 cells, which suggests additional receptor regulation and anchoring at the synapse (Connolly et al., 1999). Thus, differential effects observed in neurons may be dependent on regional differences as well as the activation of specific isozymes, which then leads to the regulation of various receptor associated proteins. Indeed, PKCε was observed to form a complex with N-ethylmaleimide sensitive factor (NSF) and γ2-containing GABAARs. The activation of PKCε was found to result in decreases in the surface levels of GABAAR, which was dependent upon PKCε phosphorylation of NSF and their recruitment to inhibitory synapses (Chou et al., 2010). Notably, NSF has also been shown to bind directly to β subunits of GABAARs at residues 395–415, which contains the major phosphorylation site conserved within β subunits (Goto et al., 2005). Whether phosphorylation of this site affects GABAAR–NSF interactions remains to be seen. Although NSF stabilizes excitatory AMPA receptors at the surface by disrupting endocytosis (Hanley, Khatri, Hanson, & Ziff, 2002), similar characterization of NSF and GABAARs interactions have not been performed.
4. GABAAR-INTERACTING PROTEINS AND PHOSPHORYLATION
GABAAR distribution and expression is under tight subtype-specific management, governed to some degree by its interacting partners. As with the phosphorylation of these receptors, GABAAR interactions primarily take place at the large intracellular loop, with the majority of interactions occurring through the β and γ2 subunits. These interactions affect trafficking and surface stability as well as the phosphorylation state of specific subunits (Charych et al., 2009; Chen & Olsen, 2007; Jacob et al., 2008; Kneussel & Loebrich, 2007).
4.1. Adaptor protein 2
The adaptor protein (AP2) is a heterotetrameric complex composed of α, β2, μ2, and σ2 subunits. AP2 binds membrane, cargo, and clathrin and is fundamental to clathrin-mediated endocytosis (McMahon & Boucrot, 2011). GABAARs β1–3, γ2, and δ subunits are directly associated with the μ2 subunit of AP2. As will be discussed below, thus far three mechanisms for the binding of AP2 to GABAARs have been identified (Kittler et al., 2005, 2000; Vithlani & Moss, 2009).
Binding sites important for AP2, clathrin and dynamin-mediated internalization of GABAARs were first identified on the β2 subunit. A dileucine motif on the β2 was reported to be required for PKC-mediated endocytosis in heterologous cells (Herring, Huang, Singh, Dillon, & Leidenheimer, 2005; Herring et al., 2003).
A second atypical AP2 binding motif on GABAARs was later determined as a region of highly basic amino acids, which contains a major phosphorylation site for PKA and PKC in β1 and β3 subunits, and PKC in β2 subunits (Brandon et al., 2003; Kittler et al., 2005). Phosphorylation of S408/S409 on the β3 subunit resulted in decreased association with the AP2 complex. Thus, when this site is dephosphorylated, AP2 binds GABAARs, thereby prompting receptor endocytosis (Fig. 2). Consequently, introducing dephosphorylated β3-derived peptides that compete for the AP2 interaction leads to an increase in mIPSC amplitude and whole-cell current (Kittler et al., 2005). Additionally, mutations of S408/S409 that decrease AP2 binding to the β3 subunit were found to result in a concomitant decrease in endocytosis and an increase in surface-expressed receptors. Interestingly, these mutations resulted in an enhancement in size and number of inhibitory synapses was observed with parallel decreases in the number of excitatory synapses (Jacob et al., 2009).
Figure 2.
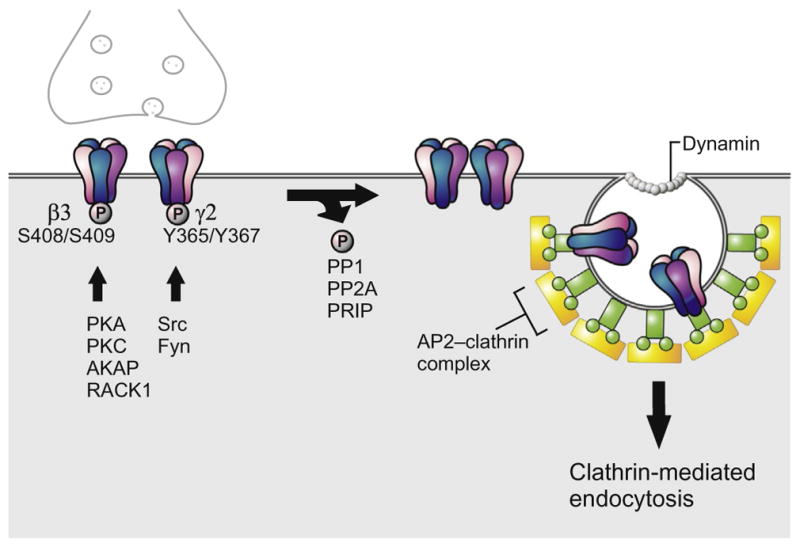
Phosphoregulation of GABAAR endocytosis. The endocytosis of GABAARs is regulated by the interaction of the AP2 complex with β and γ2 subunits. Phosphorylation of β3 (S408/S409) and γ2 (Y365/Y367) by PKA/PKC and Src/Fyn, respectively, prevents binding to AP2 and thus stabilizes these receptors at the cell surface. Phosphorylation of β1 and β3 subunits is facilitated by AKAP and RACK1. Dephosphorylation of β3 (S408/409) by PP1 and PP2A, and γ2 by an unknown phosphatase enables binding to AP2, triggering dynamin-dependent clathrin-mediated endocytosis. PRIP regulates dephosphorylation through binding PP1/PP2A and β subunits. The phosphorylation state of PRIP1 determines the release of active PP1α.
Finally, the third AP2 interaction motif on GABAARs comprises dual sites in the major intracellular domain of the γ2 subunit. A tyrosine motif Y-x-x-Φ (where x is any amino acid and Φ is hydrophobic) allows binding of AP2 to γ2-containing receptors on Y365GY367ECL. Phosphorylation of Y365/Y367 inhibits binding to AP2, leading to an enhancement in levels of surface receptors (Fig. 2). These motifs can work separately or together to decrease receptor numbers (Kittler et al., 2008). Indeed, phosphorylation within this motif (Y365/Y367) is mediated by Src family members (Brandon et al., 2001; Moss et al., 1995) including Fyn. Moreover, the phosphorylation of Y367 within the γ2 subunit facilitates the direct binding of Fyn kinase (Jurd, Tretter, Walker, Brandon, & Moss, 2010). To this end, Fyn knockout (KO) mice exhibit decreased phosphorylation at this residue in addition to altered GABAAR function (Boehm, Peden, Harris, & Blednov, 2004; Jurd et al., 2010). Infusion of peptides that block AP2–γ2 interaction in neurons resulted in an increase in mIPSC amplitude accompanied with an increase in receptors at the surface (Kittler et al., 2008). The importance of this site in vivo is highlighted by the embryonic lethality of knock-in mice where γ2 Y365/Y367 residues were mutated to phenylalanines (Y365F/Y367F). This observed phenotype is possibly due to increased levels of GABAergic excitation during early development (Ben-Ari, Khalilov, Kahle, & Cherubini, 2012). Experiments performed with viable heterozygous mice revealed increased surface levels of γ2, enhanced levels of GABAARs at specific subdomains of the hippocampus as well as deficits in spatial object recognition (Tretter et al., 2009).
Notably, δ subunits bind AP2 through sites containing both the classical Y-x-x-Φ motif as found in γ2 subunits and the atypical basic motif, which is found in β subunits. Since phosphorylation sites have not currently been identified within the δ subunits, whether this association is also mediated by phosphorylation within these motifs remains to be seen (Gonzalez, Moss, & Olsen, 2012).
4.2. Gephyrin
The important role of gephyrin as a postsynaptic scaffold protein underlying GABAAR synaptic clustering has been an area of intense study (Luscher, Fuchs, & Kilpatrick, 2011; Tretter et al., 2012; Tyagarajan & Fritschy, 2014). Gephyrin has been shown to bind GABAAR subunits α1–α3 (Mukherjee et al., 2011; Saiepour et al., 2010; Tretter et al., 2011) and more recently to β2 and β3 subunits (Kowalczyk et al., 2013). Importantly, this interaction may be regulated by the phosphorylation of specific subunits. For example, the mutation of a putative phosphorylation site, T375, in the α1subunit to a phosphomimetic, decreases its affinity togephyrin. The decrease in gephyrin affinity subsequently leads to decreases in synaptic α1-containing clusters and reductions in the amplitude of mIPSCs (Mukherjee et al., 2011). In addition, phospho-deficient mutations of S410 in the β2 subunit lead to a decrease in affinity for gephyrin (Kowalczyk et al., 2013). Whether putative sites such as T375 are altered by kinases and phosphatases are currently unknown, but it raises the intriguing possibility that phosphorylation may dynamically regulate the clustering of GABAARs at inhibitory synapses.
4.3. A-kinase anchoring protein
The proper targeting of kinases and phosphatases such as PKA, PKC, and calcineurin (CaN, also known as PP2B) to their appropriate substrates at both excitatory and inhibitory synapses require a family of AKAP scaffolding proteins (Brandon et al., 2003; Klauck et al., 1996; Sanderson & Dell’Acqua, 2011).
The AKAP Yotiao has been shown to bind PKA and the type 1 protein phosphatases (PP1) (Westphal et al., 1999). Furthermore, application of dopamine D4 receptor agonists resulted in decreases in GABAAR-mediated currents which required an intact Yotiao–PKA–PP1 complex (Wang, Zhong, & Yan, 2002). However, whether these actions are mediated by the direct phosphorylation or dephosphorylation of GABAARs is unknown.
AKAP79/150 has been demonstrated to directly bind to GABAAR β1 and β3, but not β2-subunits. PKA-mediated phosphorylation of the β3 subunit was found to be AKAP dependent (Brandon et al., 2003). However, there has been disagreement as to whether this occurs at the Golgi apparatus instead of at inhibitory synapses in the hippocampus (Lilly, Alvarez, & Tietz, 2005). Nevertheless, more recently, it has been suggested that the PKA–AKAP–CaN signaling complex residing at GABAergic synapses is required for the induction of GABAergic long-term depression in dopamine (DA) neurons of the ventral tegmental area (VTA) (Dacher, Gouty, Dash, Cox, & Nugent, 2013). Unfortunately, specific phosphorylation sites on GABAARs have not been characterized in this study, but it is tempting to speculate that direct phosphorylation of these receptors may play a role in DA signaling required for reward-related learning.
4.4. Phospholipase C-related inactive protein
The phospholipase C-related inactive protein (PRIP) family of proteins includes the ubiquitously expressed PRIP2, and PRIP1 that is principally expressed in the CNS. PRIPs were first discovered as proteins that bound to inositol 1,4,5-triphosphate (IP3) (Kanematsu, Mizokami, Watanabe, & Hirata, 2007; Kanematsu et al., 2000). PRIPs have multiple functions which include: (1) trafficking of γ2-containing GABAARs to the cell surface through the ternary binding of PRIPs to GABAAR and the GABAAR-associated protein (GABARAP), (2) regulating constitutive AP2- and clathrin-mediated endocytosis of GABAAR, and (3) modulating GABAAR phosphorylation (Kanematsu, Fujii, et al., 2007; Kanematsu, Mizokami, et al., 2007).
The ability of PRIP to regulate dephosphorylation occurs through its binding to protein phosphatases PP1α, PP2A, and the β subunits of GABAARs (Kanematsu, Mizokami, et al., 2007; Kanematsu et al., 2006; Terunuma et al., 2004; Yoshimura et al., 2001). The PRIP1–PP1α association renders the phosphatase catalytically inactive. However, upon PKA activation, PRIP1 phosphorylation facilitates the dissociation of the PRIP1–PP1α complex. The subsequent release of active PP1α dephosphorylates the β3 subunit specifically at the AP2 binding site, thereby resulting in GABAAR internalization (Kanematsu et al., 2006; Terunuma et al., 2004; Fig. 2). Further, PRIP1 KO mice displayed increased PP1α activity and functional deficits in PKA-mediated modulation of GABAARs (Terunuma et al., 2004). In contrast to PP1α, PRIP1–PP2A association does not alter PP2A activity. Ultimately, the level of β subunit phosphorylation will depend on the balance between the rate of direct phosphorylation of β subunits and the rate of phosphorylation of PRIP1 by PKA.
4.5. Receptor for activated C-kinase
The highly conserved, multifaceted adaptor protein receptor for activated C-kinase (RACK1) binds specifically to activated PKC and enables its trafficking to membrane locales, thereby allowing for PKC phosphorylation at precise receptor sites (Adams, Ron, & Kiely, 2011; Mochly-Rosen, Khaner, & Lopez, 1991; Ron et al., 1994). In addition to binding PKC, RACK1 also interacts with the major intracellular domain of GABAAR β1 and β3 subunits adjacent to a PKC binding site (Brandon, Jovanovic, Smart, et al., 2002; Brandon et al., 1999). The PKC isoform PKCβII binds directly to β1 and β3 subunits and phosphorylates residues S410 and S408/S409, respectively. Binding assays with GST-β1 fusion proteins revealed that RACK1 bound to residues 395–404, immediately upstream of the PKC binding site (residues 405–415) (Brandon, Jovanovic, Smart, et al., 2002; Brandon et al., 1999). Although association of RACK1 is not essential for PKC binding to β subunits, it potentiates the phosphorylation of β1 subunits at S409.
5. PHOSPHORYLATION AND ALLOSTERIC MODULATION
Pharmacological agents that target GABAAR for therapeutics are largely positive allosteric modulators used for their anesthetic, anticonvulsant, anxiolytic, or sedative-hypnotic actions. Positive allosteric modulators bind receptors at a site separate from the agonist binding site, enhancing the response of GABAARs to GABA. Importantly, allosteric modulation of GABAARs by barbiturates, benzodiazepines, and neurosteroids can be regulated by phosphorylation in a kinase- and subunit-specific manner. Further, allosteric modulators may also regulate the phosphorylation state of GABAARs.
5.1. Barbiturates and benzodiazepines
Benzodiazepines increase GABAAR current by increasing channel opening frequency, whereas barbiturates have dose-dependent differences in their effects. At low concentrations, barbiturates act to allosterically enhance the GABA response through increasing channel opening duration, while at higher concentrations they directly activate GABAARs (Korpi, Grunder, & Luddens, 2002; Macdonald & Olsen, 1994; MacDonald, Rogers, & Twyman, 1989).
Benzodiazepines bind the interface between α (1, 2, 3, or 5) and γ subunits and regulate GABAARs by increasing channel opening frequency upon GABA binding (Goldschen-Ohm, Wagner, Petrou, & Jones, 2010; Jacob et al., 2008; Macdonald & Olsen, 1994). Evidence from synaptosomal preparations suggests CamKII activation results in an increase in benzodiazepine binding to the receptor (Churn et al., 2002). Furthermore, a number of studies have reported that the effects of these drugs can be regulated via PKC activation. Pretreatment of α1β2γ2L-expressing oocytes with PKC activators resulted in increases in the ability of diazepam and pentobarbital to allosterically modulate GABA-induced currents (Leidenheimer, McQuilkin, Hahner, Whiting, & Harris, 1992). However, similar experiments performed on oocytes expressing α1β2γ2 by another group could not reproduce the effect of diazepam or pentobarbital (Ghansah & Weiss, 2001). Nonetheless, further support of PKC regulation of GABAAR allosteric modulation, albeit in the other direction, stems from PKCε KO mice. These studies showed that mice that lacked PKCε were more sensitive to benzodiazepines and barbiturates (Harris et al., 1995; Hodge et al., 1999). PKCε has been shown to phosphorylate the GABAAR γ2 subunit at S327. Indeed, in α1β2γ2-expressing HEK293 cells, phospho-null mutation of γ2 S327 enhances the actions of benzodiazepine (Qi et al., 2007). By comparison, PKCγ KO mice did not have altered sensitivity to barbiturates or benzodiazepines (Harris et al., 1995; Hodge et al., 1999). These studies outlined above highlight an added layer of complexity and sophistication, whereby different kinase isozymes specifically and differentially mediate the allosteric modulation of GABAARs, presenting a fertile topic for future studies.
5.2. Neurosteroids
Neurosteroids are steroids synthesized de novo in the brain by glia and neurons (Compagnone & Mellon, 2000; Herd, Belelli, & Lambert, 2007). Neurosteroids are potent and selective allosteric modulators of GABAARs that increase the channel open duration and frequency at lower concentrations and directly activate GABAARs at higher concentrations (Lambert, Cooper, Simmons, Weir, & Belelli, 2009). Besides their allosteric modulation of GABAARs, neurosteroids have also been shown to exert their effects through the potentiation of PKC phosphorylation on S443 within α4 subunits of GABAARs. Phosphorylation at this residue leads to a sustained up-regulation of α4-containing receptors through insertion of receptors into surface membranes, resulting in a selective enhancement of tonic current in hippocampal neurons (Abramian et al., 2014; Comenencia-Ortiz, Moss, & Davies, 2014). Although others have reported decreases in PKC-mediated tonic inhibition (Bright & Smart, 2013), this work nevertheless highlights a novel mechanism by which neurosteroids can alter neuronal inhibition.
The connection between PKC phosphorylation and neurosteroid modulation of GABAARs is complex. A number of studies have reported that neurosteroid modulation of GABAARs may be enhanced by PKC phosphorylation (Fáncsik, Linn, & Tasker, 2000; Harney, Frenguelli, & Lambert, 2003; Leidenheimer & Chapell, 1997; Vicini, Losi, & Homanics, 2002), while other studies report that the opposite is true (Hodge et al., 1999, 2002; Kia et al., 2011; Koksma et al., 2003). For example, GABAAR δ-subunit KO mice display reduced sensitivity to neurosteroids, which could be restored by activation of PKC with phorbol esters (Vicini et al., 2002). Further, inhibition of either PKA or PKC decreased the sensitivity of GABAARs to neurosteroids in hippocampal CA1 pyramidal neurons, whereas, in contrast, activation of PKC had no effect (Harney et al., 2003). Interestingly, and in the same preparation, PKC activation did enhance the sensitivity of GABAARs to neurosteroid in dentate gyrus granule cells. This suggests that even within a specific brain region, there are likely to be neuron-specific effects that depend upon both the circulating levels of neurosteroids as well as the phosphorylation status of GABAARs and/or their associated proteins. As with the enhanced response to benzodiazepine and barbiturate mentioned above, PKCε-deficient mice also exhibit increased sensitivity to neurosteroids (Hodge et al., 1999, 2002). Moreover, these mice were less anxious and had reduced response to stress hormones, consistent with the anxiolytic effects of neurosteroids (Hodge et al., 2002).
Further molecular mechanisms that impact GABAAR sensitivity to neurosteroids have emerged from studies of the hypothalamic oxytocin neurons of the supraoptic nuclei. These studies showed that substantial changes in activity around the time of parturition can be attributed to changes in circulating levels of progesterone and metabolites (Brussaard & Herbison, 2000; Concas et al., 1998). Recordings from these cells show that pregnant rats during late gestation are neurosteroid-sensitive, but that post-parturition, GABAARs become neurosteroid-insensitive (Brussaard, Wossink, Lodder, & Kits, 2000; Koksma et al., 2003). Moreover, these observations were ascribed to modifications in kinase and phosphatase activity. During late pregnancy, activation of PKC with phorbol esters or inhibition of phosphatases PP1 and PP2A reduced neurosteroid modulation of GABAAR. Conversely, after giving birth, when GABAARs are less sensitive to neurosteroid modulation, inhibitors of PP1 and PP2A or activation of PKC rescued neurosteroid sensitivity of GABAAR (Koksma et al., 2003).
Clearly, the impact of phosphorylation on allosteric modulation of GABAARs is complex, and information on the molecular mechanisms that regulate these processes is currently unavailable. Furthermore, it is still unclear from these studies whether kinases target receptors directly or one of its intimately associated proteins, and whether phosphorylation increases binding of allosteric modulators to receptors or whether it influences modulator-induced changes to channel gating. Nevertheless, it is evident that phosphorylation imparts further diversity to the interplay between GABAARs and allosteric modulators.
6. SIGNALING PATHWAYS THAT MODULATE GABAAR PHOSPHORYLATION
Excitation and inhibition in the brain is tightly and dynamically balanced and is crucial for proper brain function. Since GABAARs are the principal mediators of inhibition in the brain, it is unsurprising that multiple signaling pathways can regulate the phosphorylation status of these receptors.
6.1. Receptor tyrosine kinases
Receptor tyrosine kinases (RTKs) are an extensive class of cell-surface receptors that are essential to numerous cellular processes (Schlessinger, 2000). In particular, two ligand-receptor pairs of the RTK class include insulin and the insulin receptor, and brain-derived neurotrophic factor (BDNF) and its associated tyrosine kinase receptor (TrkB). Insulin and BDNF have both been implicated in the regulation of GABAARs, and will be discussed in more detail below.
6.1.1 Brain-derived neurotrophic factor
The neurotrophin tyrosine kinase receptor 2 (TrkB) exerts its effects through activation by BDNF (Klein et al., 1991). BDNF is abundant and ubiquitously expressed in the brain, playing key roles in neurogenesis, differentiation, survival, and synaptic plasticity (Boulle et al., 2012; Geral, Angelova, & Lesieur, 2013). More specifically, BDNF has been reported to be critical for the development of GABAergic synapses and GABAergic inhibition (Seil, 2003; Vicario-Abejón, Collin, McKay, & Segal, 1998). Studies have implicated BDNF in the increase in cell-surface expression of δ subunits of GABAARs. Although this effect was reported to be mediated by TrkB receptors, phospholipase C (PLCγ), and PKC, the substrate for the kinase was unidentified (Joshi & Kapur, 2009). Further, the actions of BDNF on GABAARs seem to be developmentally regulated and require the phosphorylation of residues on the β3 and γ2 subunits, as will be discussed below.
Exogenous application of BDNF led to an early, transient potentiation followed by a lasting decrease in GABAAR-mediated currents in cortical, hippocampal and superior colliculus neurons (Brünig, Penschuck, Berninger, Benson, & Fritschy, 2001; Henneberger, Jüttner, Rothe, & Grantyn, 2002; Jovanovic et al., 2004; Kanematsu et al., 2006). The biphasic nature of the response was attributed to the differential recruitment of PKC and RACK1 that was required for the initial rapid phosphorylation of GABAAR β3 subunit at S408/S409 (Jovanovic et al., 2004), followed by PRIP1/2 and PP2A mediated dephosphorylation of this site (Kanematsu et al., 2006). Intriguingly, the phosphorylation of S408/S409 has been shown to reduce association of PKC, whereas it increased binding of PP2A, which may explain the transient nature of phosphorylation that has been observed at this site (Brandon, Jovanovic, Smart, et al., 2002; Jovanovic et al., 2004; Kanematsu et al., 2006). In addition, application of BDNF to neurons from PRIP1/2 double knockout (dKO) mice showed a slow consistent increase of β3 phosphorylation complemented with an increase in GABA-induced current. This demonstrated that the initial PKC-dependent phosphorylation persisted, whereas the following PRIP-mediated PP2A dephosphorylation step was abrogated (Kanematsu et al., 2006). As mentioned previously, one would expect that the dephosphorylation of β3 at Ser408/S409 by PP2A would result in AP2 binding and subsequent endocytosis of receptors (Kittler et al., 2005), which would explain the subsequent decrease in mIPSC amplitude. However, the effects of BDNF on surface levels of GABAAR are in conflict, where both increases (Jovanovic et al., 2004; Kuczewski et al., 2011) and decreases (Kanematsu et al., 2006) have been observed. The reason for this discrepancy is unknown but may be mediated by a number of factors that include methodology, neuronal cell-type, age of neurons and the species that the neurons originated from. Nevertheless, these studies emphasize the importance of BDNF-mediated phosphorylation of GABAARs and bring further support to the prominence of RACK1 and PRIPs as facilitators of the phosphorylation and dephosphorylation of GABAARs.
In contrast to the regulation of GABAAR by BDNF in relatively young animals/culture described above, application of BDNF to slices from the prefrontal cortex (PFC) has been shown to cause enhancements in GABAAR-dependent inhibition to older (2–4 months) animals. BDNF led to increased phosphorylation of Y365/Y367 of γ2 subunits, a subsequent decrease in binding of AP2, and finally, an associated increase in levels of surface receptors. Further, BDNF was no longer able to modulate mIPSCs in mice where these residues had been mutated to Y365F/Y367F. Interestingly, these animals had increased antidepressant phenotype and increased neurogenesis. Although BDNF produced antidepressant-like actions and neurogenesis in wild-type animals, BDNF was unable to further modulate these effects in Y365F/Y367F mice (Vithlani et al., 2013).
Whether these mechanisms are truly different due to development, and not due to, for example, differences in experimental manipulations, deserves future attention. Indeed, BDNF application also increased surface levels of GABAARs (Porcher et al., 2011) in young developing cortical cultures at a stage when GABAARs are still excitatory (Ben-Ari et al., 2012; Owens, Boyce, Davis, & Kriegstein, 1996). Moreover, an additional complication that may be relevant to these studies is that BDNF also modulates K+–Cl− cotransporter activity, which could affect the efficacy of inhibitory transmission (Rivera et al., 2002; Shulga et al., 2008; Wardle & Poo, 2003).
6.1.2 Insulin
Although pancreatic insulin is well recognized for its regulation of blood glucose levels, insulin is also synthesized in the brain (Havrankova, Brownstein, & Roth, 1981) where it regulates synaptic plasticity, spine and dendritic morphogenesis, and neuronal survival (Bassil, Fernagut, Bezard, & Meissner, 2014; Chiu & Cline, 2010). Insulin treatment results in a rapid increase in surface levels of GABAARs and an enhancement of mIPSC amplitude (Fujii et al., 2010; Vetiska et al., 2007; Wan et al., 1997; Wang, Liu, Pei et al., 2003). Work on the mechanisms regulating these effects has so far concentrated on the β2 subunit of GABAARs. More specifically, the presence of insulin results in the phosphorylation at residues Y372 and Y379 of the β2 subunit of GABAARs by an unidentified kinase and these residues were critical for the increases in surface levels of GABAARs upon stimulation with insulin. Furthermore, enhancement of GABAAR-mediated currents was found to be phosphoinositide 3-kinase (PI3K) dependent and correlated with an increase in binding between PI3K p85 subunit SH2 domain and GABAARs specifically at these phosphorylated residues on the β2 subunit (Vetiska et al., 2007).
Elsewhere, other groups have shown that the observed increases in surface receptors required the well-documented activation by insulin of the PI3K-Akt pathway (Hemmings & Restuccia, 2012; Wan et al., 1997; Wang, Liu, Pei, et al., 2003). Activated Akt solely phosphorylated the S410 residue on the β2 subunit in α1β2γ2-containing HEK293 cells. Insulin stimulation of Akt in α1β2γ2-expressing HEK293 cells led to an enhancement in surface receptors and an increase in the amplitude of GABAAR-mediated mIPSCs (Wan et al., 1997). Indeed, the phosphorylation of this residue by Akt was also crucial for the observed translocation of GABAAR in cultured neurons (Wang, Liu, Pei, et al., 2003). Notably, upon insulin application, activated Akt translocated specifically to more distal dendritic locales where it colocalized with GABAARs. In addition, PRIP1 was required to traffic active Akt to GABAARs upon stimulation by insulin, since disruption of this complex in PRIP dKO mice or PRIP interference peptide in wild-type mice resulted in a dearth in phosphorylated β subunit. This was also paralleled with a lack of insulin-mediated potentiation of GABA-induced currents (Fujii et al., 2010). The S410 phosphorylation site in the β2 subunit is conserved (in β1 S409 and β3 S408/S409), and again one would presume that its phosphorylation may result in less binding to AP2, thereby increasing surface levels of receptor. Instead, inhibition of ER to Golgi trafficking by brefeldin A resulted in a decrease in GABA-evoked currents beyond that of controls. This suggests that increases in surface GABAAR are mediated by insertion of newly synthesized receptors concurrent and to a lesser degree with a decrease in endocytosis (Fujii et al., 2010). Whether these mechanisms also apply to other β subunits has not been explored.
Thus, the insulin-mediated effects hinge on the phosphorylation of GABAARs by two seemingly different mechanisms. How these processes may converge will be a fascinating topic for future investigations. Moreover, these studies advocate phosphorylation of a specific site that has differential consequences and is contingent on the kinase concerned.
6.2. Glutamate receptors
The vast majority of synapses use glutamate as their excitatory neurotransmitter. Glutamate release from presynaptic terminals triggers rapid activation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) and N-methyl-D-aspartate receptors (NMDARs). This activation leads to AMPAR and NMDAR opening to allow an influx of cations, and stimulation of the postsynaptic cell. The strength of synaptic transmission is extremely plastic and can be increased (long-term potentiation, LTP) or decreased (long-term depression, LTD) (Malenka & Bear, 2004). The polarity of this plasticity is largely defined by the kinetics and amount of Ca2+ influx through NMDARs. Strong, brief, high frequency stimulation (HFS) of the input neuron induces rapid and robust Ca2+ entry into cells, resulting in LTP. Additionally, weak, prolonged, low frequency stimulation, allowing for lower concentrations of Ca2+ entry, results in LTD (Bliss & Lømo, 1973; Dudek & Bear, 1992; Yang, Tang, & Zucker, 1999). The difference in the sensitivities of kinases and phosphatases to Ca2+ provides one of the mechanisms that underlie this bidirectional expression of plasticity (Lee, 2006). Together, LTP and LTD are thought to represent the cellular correlate for learning and memory in the CNS (Bear & Abraham, 1996; Bliss & Collingridge, 1993).
Likewise, similar long-term changes in synaptic strength occur at GABAergic synapses (Gaiarsa, Caillard, & Ben-Ari, 2002). NMDAR-dependent HFS that induces LTP at excitatory synapses in the hippocampus also results in the concomitant LTD (LTDGABA) at inhibitory synapses (Lu, Mansuy, Kandel, & Roder, 2000). The decrease in efficacy of GABAergic transmission was accompanied with the dephosphorylation at S327 in the γ2 subunit of GABAARs and was dependent upon the binding of the CaN catalytic domain to the intracellular loop of γ2 subunits (Wang, Liu, Haditsch, et al., 2003). Moreover, increases in excitatory activity resulting in NMDA-dependent Ca2+ influx lead to the fast and reversible diffusion of GABAARs from synaptic clusters and was found to be independent of receptor endocytosis (Bannai et al., 2009; Muir et al., 2010). The lateral diffusion of receptors was contingent upon CaN-dependent dephosphorylation of S327 on the γ2 subunit (Fig. 3) (Muir et al., 2010).
Figure 3.

Phosphoregulation of GABAAR trafficking. GABAAR are postsynaptically localized through its binding to gephyrin. (A) Strong NMDAR activation results in the influx of Ca2+. CamKII activation at excitatory synapses results in an increase of AMPARs at the cell surface. Calcineurin dependent dephosphorylation of GABAAR subunit γ2 (S327) enables diffusion of GABAARs away from the synapse. (B) Weaker stimuli induced by an NMDAR-dependent chemLTD protocol results in a moderate influx of Ca2+ and CamKII is targeted to inhibitory synapses. CamKII phosphorylation of β3 (S383) is critical for recruitment of gephyrin to synaptic sites, immobilization of GABAARs at synapses and an enhancement of cell surface levels of GABAARs.
Conversely, NMDAR-dependent chemically induced LTD (chemLTD) of excitatory transmission results in an increase of GABAARs at the surface and an enhancement of inhibitory transmission (LTPGABA). As has been found in excitatory synapses, the bidirectional regulation of GABAARs is dependent upon the strength of NMDAR activation and Ca2+ influx. Strong NMDAR activation leads to CamKIIα translocation to excitatory synapses, enhancing surface levels of AMPARs, whereas targeting of CamKIIα is inhibited at inhibitory synapses by CaN. Weaker stimuli resulted in CamKIIα localization at inhibitory synapses and an enhancement of surface levels of GABAARs (Marsden, Beattie, Friedenthal, & Carroll, 2007; Marsden, Shemesh, Bayer, & Carroll, 2010). More recent evidence lends further support to the role of GABAAR phosphorylation in these mechanisms. Similar induction of chemLTD resulted in a CamKII-dependent LTPGABA, marked by increases in surface levels of GABAARs and the potentiation in the mIPSC and spontaneous inhibitory synaptic current amplitude. During LTPGABA, GABAARs were immobilized at the synapse and the scaffold protein gephyrin was recruited from nearby extrasynaptic sites. Moreover, the phosphorylation of β3 at S383 increased during LTPGABA. Phosphorylation of β3 S383 was required for GABAA Rimmobilization at synapses and the recruitment of gephyrin to synaptic sites. Thus, the phosphorylation of β3 S383 is considered critical for the expression of LTPGABA. Finally, similar accumulation of GABAARs and gephyrin is observed in vivo following an experience-dependent plasticity protocol (Petrini et al., 2014; Fig. 3).
6.3. Voltage-gated Ca2+ channels
In cerebellar Purkinje neurons, activation of excitatory synapses induce long-lasting enhancement of GABAAR-mediated inhibitory current. This “rebound potentiation” (RP) occurs through activation of voltage-gated Ca2+ channels (VGCCs) resulting in a transient Ca2+ influx and is dependent upon CamKII (Kano, Kano, Fukunaga, & Konnerth, 1996) and γ2 subunit association with GABARAP (Kawaguchi & Hirano, 2007). Further, RP can be suppressed by GABA type B receptor (GABABR) activation through decreasing levels of PKA. This reduction of PKA ultimately leads to an increase in PP1 activity through the PKA/DARPP-32/PP1 signaling pathway resulting in the inhibition of CamKII (Hirano & Kawaguchi, 2014; Kawaguchi &Hirano, 2002). Since PKA and CamKII are both kinases of the β1 and β3 subunit, and because the PKA/DARPP-32/PP1 signaling pathways have previously been reported to increase the phosphorylation of β1 and β3 subunits (Flores-Hernandez et al., 2000), it is possible that direct phosphorylation of these subunits may underlie RP in these neurons.
Activation of L-type VGCCs in hippocampal preparations results in CamKII-dependent increases in cell surface α5β3-containing receptors and an enhancement of tonic current. Moreover, these observations were contingent on the phosphorylation of S383 on the β3 subunit, which subsequently led to the insertion of new receptors to the surface without affecting endocytosis. Increases or decreases in neuronal activity also resulted in respective increases and decreases in phosphorylated β3 and surface expression of GABAARs. Thus, neuronal activity can bidirectionally regulate surface-expressed GABAARs and tonic inhibition through the phosphorylation of a single residue on the β3 subunit (Saliba, Kretschmannova, & Moss, 2012).
6.4. Dopamine
Both fast synaptic glutamatergic (excitatory) and GABAergic (inhibitory) transmission can be modulated through G-protein-coupled mechanisms or by direct interactions with the comparatively slower neurotransmission of dopaminergic receptors. DA receptors are broadly classified into two distinct groups, the D1-like (D1 and D5) and D2-like (D2–4) classes of receptors. This distinction was based on the initial observations that DA could modulate adenylyl cyclase activity resulting in increased (D1-like) or decreased (D2-like) cyclic AMP (cAMP) production required for the subsequent activation of PKA (Beaulieu & Gainetdinov, 2011; Greengard, 2001). Since dopamine receptors affect PKA activity, evidence for a role of DA-mediated phosphorylation of GABAARs is largely derived from studies on the β subunits (Chen, Kittler, Moss, & Yan, 2006; Flores-Hernandez et al., 2000; Goffin et al., 2010; Terunuma et al., 2004).
DA can modulate the excitability of striatal medium spiny neurons (MSNs) expressing a tonic GABAAR current via a PKA and β3 subunit-dependent mechanism (Janssen, Ade, Fu, & Vicini, 2009; Janssen, Yasuda, & Vicini, 2011). In neostriatal neurons, DA activation of D1 and D5 receptors both decreased and enhanced GABA-evoked currents in MSNs (Flores-Hernandez et al., 2000) and cholinergic interneuron (Yan & Surmeier, 1997), respectively.
In particular, application of a D1 agonist in adult MSNs resulted in a PKA-dependent reduction in GABA-induced current. Membrane permeable analogues of cAMP mimicked the attenuation of the GABA-evoked response, which was further reduced by inhibition of PP1/PP2A. Additionally, DA was observed to increase the phosphorylation of β1 and β3 subunits (although β1 subunits were more prevalent), which was dependent upon DA- and cAMP-regulated phosphoprotein, 32 kDa (DARPP-32). Thus, the observed decrease in GABA-induced current requires activation of the PKA/DARPP-32/PP1 signaling pathway, leading to the modulation of GABAARs through the phosphorylation of β1 subunits (Flores-Hernandez et al., 2000).
In hippocampal cells, the activation of D1 receptors led to a PKA- and PRIP-dependent enhancement of GABA-evoked currents and a concomitant increase in phosphorylation of β3 residues S408/S409 (Terunuma et al., 2004). Thus, these studies lend support to aforementioned work in heterologous systems whereby PKA was observed to reduce GABA-induced currents by phosphorylation of S409 in the β1 subunit and increase currents by phosphorylation of two adjacent residues S408/S409 on the β3 subunit (McDonald et al., 1998).
In addition to D1-like receptors, D3 receptor activation in the MSNs of the nucleus accumbens was shown to reduce GABAAR-mediated currents concurrent with increased internalization and a decrease in surface receptors. These effects were dependent on cAMP, PKA, the β subunit of GABAAR as well as clathrin-dependent endocytosis (Chen et al., 2006). In this case, the activation of D3 receptors leads to reduced PKA activity resulting in the dephosphorylation of β subunits and the subsequent endocytosis of these receptors, which reduces GABAAR function.
Activation of D2-like receptors in the VTA induces LTD of GABAergic synapses and was dependent on the clathrin-mediated endocytosis of GABAARs. Moreover, these observations required an IP3-dependent rise in Ca2+, activation of CaN, and parallel inhibition of PKA. Finally, disrupting the AKAP–PKA complex mimicked LTDGABA (Dacher et al., 2013).
6.5. Others
In addition to its ability to suppress RP, GABABR activation increases α4βδ- and α6βδ-mediated tonic current in thalamocortical and dentate gyrus granule cells, respectively (Connelly, Errington, & Crunelli, 2013a). This effect was further dependent upon δ-containing receptors and PKA inhibitors mimicked GABABR-induced enhancements (Connelly, Errington, Di Giovanni, & Crunelli, 2013; Naylor, Liu, Niquet, & Wasterlain, 2013).
Studies of serotonergic neurotransmission in the PFC show that activation of 5-HT2 receptors decreases GABA-evoked currents, which is dependent upon PKC–RACK1 association and results in the PKC-dependent phosphorylation of GABAAR γ2 subunit (Feng, Cai, Zhao, & Yan, 2001). In contrast, activation of 5-HT4 receptors resulted in the reversible, bidirectional modulation of GABA-induced currents determined by basal PKA activity (Cai, Flores-Hernandez, Feng, & Yan, 2002).
Collectively, it is clear that multiple signaling pathways can, or have the potential to, orchestrate the phosphorylation state of specific residues on GABAARs. Further, these signaling molecules allows for specific modification determined by intricate cues and presents an additional layer of complexity in the regulation of GABAAR activity.
7. DYSREGULATION OF GABAAR PHOSPHORYLATION IN DISEASE
Although compromised trafficking of GABAARs are thought to play key roles in a number of pathological conditions (Benarroch, 2007; DeLorey & Olsen, 1999; Krystal et al., 2006; Rudolph & Mohler, 2004; Thompson-Vest et al., 2003), evidence for the importance of phosphorylation in these studies is relatively scarce.
7.1. Ischemia
Excessive release of glutamate in cerebral ischemia results in excitotoxicity and cell death (Lipton, 1999; Lo, Dalkara, & Moskowitz, 2003). Although the majority of research has focused on reducing the effects of the glutamatergic system, GABAAR trafficking is also considerably modified, which may exacerbate these effects (Schwartz-Bloom & Sah, 2001). Indeed, decreases in surface levels of GABAARs and an enhancement of ubiquitin-dependent lysosomal degradation have been reported in the in vitro oxygen-glucose deprivation (OGD) model of ischemia (Arancibia-Carcamo & Kittler, 2009; Liu et al., 2010; Mielke & Wang, 2005). Interestingly, the association of the α1 subunit to gephyrin decreased and could be rescued with a CaN inhibitor. Inhibitors of PP1α/PP2A rescued the loss of α1 subunit by OGD. Further, ischemic insult resulted in decreased phosphorylation of S408/S409 of the β3 subunit and phosphomimetic mutants of β3 or mutants that blocked AP2 binding protected cells from neuronal death (Mele, Ribeiro, Inacio, Wieloch, & Duarte, 2014; Smith et al., 2012).
7.2. Epilepsy
Epilepsy is a common and often devastating neurological disorder based on a striking imbalance between excitatory and inhibitory activity. Prolonged, continuous seizures (status epilepticus, SE) in animal models and humans can induce the development of temporal lobe epilepsy (TLE). Modifications in GABAAR trafficking and expression have been reported in patients and animal models of SE and TLE (Brooks-Kayal, Shumate, Jin, Rikhter, & Coulter, 1998; Goodkin, Joshi, Mtchedlishvili, Brar, & Kapur, 2008; Loup, Wieser, Yonekawa, Aguzzi, & Fritschy, 2000; Naylor, Liu, & Wasterlain, 2005; Sperk, Drexel, & Pirker, 2009; Terunuma et al., 2008). Increases and decreases in GABAAR expression have been observed, both of which are dependent upon specific subunits and the time period of epileptogenesis studied. Loss of GABAARs is thought to be one mechanism that underlies pharmacoresistance to benzodiazepines (Deeb, Maguire, & Moss, 2012). Furthermore, dephosphorylation of GABAARs has been implicated in the loss of these receptors during SE. In particular, induction of SE by pilocarpine resulted in the decrease of PKC-mediated phosphorylation of β3 S408/S409 residues. This dephosphorylation enhanced binding to AP2, resulting in receptor endocytosis during SE (Terunuma et al., 2008). Moreover, other reports have suggested kainate and pilocarpine-induced SE results in an NMDAR-dependent increase in CaN activity and expression resulting in the dephosphorylation of β2/3 subunits of GABAARs. Whether this phosphorylation status also resulted in changes to surface expression was not explored (Kurz et al., 2001; Wang, Chi, Wang, Wang, & Sun, 2009).
7.3. Drug abuse
Although ligands such as benzodiazepines may have clinically important uses, there is substantial evidence for a role of GABAARs in the regulation of the addictive properties of drugs of abuse (Kalivas, 2007; Tan, Rudolph, & Lüscher, 2011; Trudell, Messing, Mayfield, & Harris, 2014).
Increased BDNF levels after cocaine withdrawal results in decreased GABAergic inhibition and reduced GABAARs surface expression, concurrent with the facilitation of activity-induced LTP. These effects were mediated by the BDNF–TrkB–PP2A signaling pathway, which also resulted in decreases in phosphorylated β3 subunits. These effects have been suggested to underlie behavioral modifications after cocaine withdrawal (Lu, Cheng, Lim, Khoshnevisrad, & Poo, 2010). Application of BDNF has reported to lead to lasting decreases in GABAAR-mediated currents through the PRIP1/2 and PP2A mediated dephosphorylation of β3 S408/S409 (Kanematsu et al., 2006), but whether similar mechanisms apply here remains to be seen.
GABAAR subunits composition could be altered reversibly within minutes after a single intoxicating dose of alcohol, resulting in decreased surface levels of extrasynaptic α4 and δ-containing GABAARs (Gonzalez et al., 2012; Liang et al., 2007; Shen et al., 2011). This may contribute to the rapid tolerance observed after acute ethanol exposure (LeBlanc, Kalant, & Gibbins, 1975; Liang et al., 2007; Ludvig, George, Tang, Gonzales, & Bungay, 2001; Wallace et al., 2007). Further changes in subunit expression were observed at later time points with increases in α4 and γ2 subunits and decreases in α1 and δ subunits (Liang et al., 2007). Moreover, these changes persisted after chronic intermittent administration of alcohol followed by withdrawal (Liang et al., 2007, 2006). The ethanol-induced decreases in δ-containing receptors were attributed to increased AP2 binding and subsequent endocytosis (Gonzalez et al., 2012). Chronic ethanol exposure was also found to lead to changes in PKCγ association with GABAAR subunits, with decreases in α1 subunit association and enhancements in α4 subunit association. The reduction in PKCγ-α1 binding correlated with increased α1 subunit in clathrin-coated vesicles (Kumar, Kralic, O’Buckley, Grobin, & Morrow, 2003; Kumar, Sieghart, & Morrow, 2002). Further, inhibition of PKCγ (but not PKCβ) prevented ethanol-mediated modulations in α1 and α4 subunit expression (Kumar et al., 2010; Werner et al., 2011). Intriguingly, PKC phosphorylation of α4 at S443 has been shown to increase receptor numbers at the surface (Abramian et al., 2010). Whether this site can also be modulated through ethanol administration remains to be seen.
Phosphorylation of GABAAR may well underlie some of these changes, but evidence showing direct modifications of GABAAR phosphorylation states by ethanol comes from transgenic mice lacking specific isoforms of PKC. As well as altering the previously discussed positive allosteric effects of benzodiazepines, PKCε also modifies the allosteric effects of alcohol. As with benzodiazepines, PKCε KO mice display increased sensitivity to the effects of ethanol as well as a reduction in phosphorylated γ2 S327. In α1β2γ2 expressing HEK293 cells, phospho-null mutation of this site enhanced the effect of ethanol (Hodge et al., 1999; Qi et al., 2007). It is interesting to note that PKCε KO animals self-administered considerable less ethanol compared to their wild-type counterparts (Hodge et al., 1999; Olive, Mehmert, Messing, & Hodge, 2000). Strikingly, the modulation of ethanol sensitivity and self-administration in PKCε KO mice could be rescued to wild-type levels by suppression of the transgene (Choi, Wang, Dadgar, Chang, & Messing, 2002).
PKCδ KO mice are more resilient to the intoxicating effects of ethanol, an effect that is believed to be mediated by extrasynaptic, δ subunit-containing GABAARs. PKCδ KO mice showed no response to doses of ethanol which were shown to increase GABA-mediated tonic currents in thalamic and hippocampal neurons of wild-type animals (Choi et al., 2008). Although direct phosphorylation of δ subunits of GABAARs by PKCδ has been suggested, as yet, there is no direct evidence of δ subunit phosphorylation.
8. CONCLUSION
There has been a great deal of progress in understanding the molecular mechanisms that regulate GABAARs. The significance of the dynamic regulation of GABAARs by phosphorylation is unquestionable. Notwithstanding, there are still considerable gaps and inconsistencies in our knowledge as exemplified in the large number of conflicting results that have arisen from activation of kinases in neuronal preparations.
GABAAR may be phosphorylated on multiple sites and on multiple subunits, but our knowledge of how the phosphorylation of a specific site may impact on other sites within the same subunit, or on different subunits, is tenuous. Additionally, it is not certain how phosphorylation on multiple sites impacts the fate of receptors. Ultimately, addressing such gaps in our knowledge will provide much-needed clarity to the many differences that are observed in the roles of phosphorylation.
GABAAR-interacting proteins allow a further refinement of its regulation. Given the plethora of different GABAAR subunits, it is perhaps surprising that there is such a paucity of known interacting proteins. Thus, possible interactions that may be regulated by phosphorylation remain virtually unknown. In particular, α subunits are the most diverse of the GABAARs subtypes, and differentiating the particular interaction partners of specific α subunits may tell us, for example, how different subunits are targeted to particular subcellular regions. Of the α subtypes, only α4 has been recently shown to be phosphorylated at a particular residue. However, the established phosphorylation sites are unlikely to be comprehensive, and with this, it is likely that more phospho-dependent interactions will emerge. Indeed, more recently discovered sites (Abramian et al., 2010; Kang et al., 2011) suggest that seemingly disparate results in the activation of kinases could simply arise from gaps in our current knowledge. Therefore, further examination into this area is necessary in order to enhance our current knowledge on this topic.
Evidence suggests that phosphorylation can increase insertion of receptors to the membrane (Abramian et al., 2014, 2010) and although the mechanisms of GABAARs endocytosis are becoming more apparent, our understanding of receptor recycling and insertion is still in its infancy. It will be important to determine the consequence of these internalized receptors, and the phosphorylation of GABAARs seems ideally poised to mediate these trafficking decisions.
Studies on the impact of ethanol clearly demonstrate the need to selectively examine specific PKC isoforms to understand their role in the phosphorylation of GABAARs (Song & Messing, 2005). Moreover, the different effects of kinases and phosphatases could also be due to differences in development and/or regional expression of specific GABAAR subunits, kinases, phosphatases and/or associated proteins. Ultimately, entire pathways will have to be collectively examined to refine the full picture. Further challenges arise from the multiple signaling pathways that converge upon GABAARs, yielding another level of complexity. Indeed, the effects of BDNF on GABAARs are far from clear, probably due to developmental and region-specific expression of both proteins (Ip, Cheung, & Ip, 2001; Mizoguchi, Ishibashi, & Nabekura, 2003; Sathanoori et al., 2004; Webster, Herman, Kleinman, & Shannon, 2006).
The overwhelming complexity of GABAARs is multifaceted, owing to their diversity in subunit composition, differential localization and expression as well as posttranslational modifications. However, it is exactly this mechanistic complexity that makes GABAARs such opportune drug targets, since it favors the intricate tuning by pharmacological interventions. Accordingly, further research into this diversity through mechanisms of GABAAR phosphorylation will surely clarify matters of contention and provide new and exciting insights to understanding neuronal function in health and disease.
References
- Abramian AM, Comenencia-Ortiz E, Modgil A, Vien TN, Nakamura Y, Moore YE, et al. Neurosteroids promote phosphorylation and membrane insertion of extrasynaptic GABAA receptors. Proceedings of the National Academy of Sciences. 2014;111:7132–7137. doi: 10.1073/pnas.1403285111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramian AM, Comenencia-Ortiz E, Vithlani M, Tretter EV, Sieghart W, Davies PA, et al. Protein kinase C phosphorylation regulates membrane insertion of GABAA receptor subtypes that mediate tonic inhibition. Journal of Biological Chemistry. 2010;285:41795–41805. doi: 10.1074/jbc.M110.149229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams DR, Ron D, Kiely PA. RACK1, A multifaceted scaffolding protein: Structure and function. Cell Communication and Signaling: CCS. 2011;9:22. doi: 10.1186/1478-811X-9-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancibia-Carcamo IL, Kittler JT. Regulation of GABAA receptor membrane trafficking and synaptic localization. Pharmacology & Therapeutics. 2009;123:17–31. doi: 10.1016/j.pharmthera.2009.03.012. [DOI] [PubMed] [Google Scholar]
- Balduzzi R, Cupello A, Robello M. Modulation of the expression of GABAA receptors in rat cerebellar granule cells by protein tyrosine kinases and protein kinase C. Biochimica et Biophysica Acta (BBA): Biomembranes. 2002;1564:263–270. doi: 10.1016/s0005-2736(02)00460-1. [DOI] [PubMed] [Google Scholar]
- Bannai H, Lévi S, Schweizer C, Inoue T, Launey T, Racine V, et al. Activity-dependent tuning of inhibitory neurotransmission based on GABAAR diffusion dynamics. Neuron. 2009;62:670–682. doi: 10.1016/j.neuron.2009.04.023. [DOI] [PubMed] [Google Scholar]
- Bassil F, Fernagut PO, Bezard E, Meissner WG. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: Targets for disease modification? Progress in Neurobiology. 2014;118C:1–18. doi: 10.1016/j.pneurobio.2014.02.005. [DOI] [PubMed] [Google Scholar]
- Bear MF, Abraham WC. Long-term depression in hippocampus. Annual Review of Neuroscience. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacological Reviews. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. The Neuroscientist. 2012;18:467–486. doi: 10.1177/1073858412438697. [DOI] [PubMed] [Google Scholar]
- Benarroch EE. GABAA receptor heterogeneity, function, and implications for epilepsy. Neurology. 2007;68:612–614. doi: 10.1212/01.wnl.0000255669.83468.dd. [DOI] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Lømo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. The Journal of Physiology. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm SL, Peden L, Harris RA, Blednov YA. Deletion of the fyn-kinase gene alters sensitivity to GABAergic drugs: Dependence on β2/β3 GABAA receptor subunits. Journal of Pharmacology and Experimental Therapeutics. 2004;309:1154–1159. doi: 10.1124/jpet.103.064444. [DOI] [PubMed] [Google Scholar]
- Boulle F, Kenis G, Cazorla M, Hamon M, Steinbusch HWM, Lanfumey L, et al. TrkB inhibition as a therapeutic target for CNS-related disorders. Progress in Neurobiology. 2012;98:197–206. doi: 10.1016/j.pneurobio.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Hill J, Smart TG, Moss SJ. Constitutive tyrosine phosphorylation of the GABAA receptor γ2 subunit in rat brain. Neuropharmacology. 2001;41:745–752. doi: 10.1016/s0028-3908(01)00121-6. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Kittler JT, McDonald BJ, Sieghart W, Brown DA, et al. GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. Journal of Biological Chemistry. 2000;275:38856–38862. doi: 10.1074/jbc.M004910200. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Colledge M, Kittler JT, Brandon JM, Scott JD, et al. A-kinase anchoring protein 79/150 facilitates the phosphorylation of GABAA receptors by cAMP-dependent protein kinase via selective interaction with receptor β subunits. Molecular and Cellular Neuroscience. 2003;22:87–97. doi: 10.1016/s1044-7431(02)00017-9. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Moss SJ. Multiple roles of protein kinases in the modulation of γ-aminobutyric acid A receptor function and cell surface expression. Pharmacology & Therapeutics. 2002;94:113–122. doi: 10.1016/s0163-7258(02)00175-4. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Smart TG, Moss SJ. Receptor for activated C kinase-1 facilitates protein kinase C-dependent phosphorylation and functional modulation of GABAA receptors with the activation of G-protein-coupled receptors. Journal of Neuroscience. 2002;22:6353–6361. doi: 10.1523/JNEUROSCI.22-15-06353.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon NJ, Uren JM, Kittler JT, Wang H, Olsen R, Parker PJ, et al. Subunit-specific association of protein kinase C and the receptor for activated C kinase with GABA type A receptors. The Journal of Neuroscience. 1999;19:9228–9234. doi: 10.1523/JNEUROSCI.19-21-09228.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright DP, Smart TG. Protein kinase C regulates tonic GABAA receptor-mediated inhibition in the hippocampus and thalamus. European Journal of Neuroscience. 2013;38:3408–3423. doi: 10.1111/ejn.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA. Selective changes in single cell GABAA receptor subunit expression and function in temporal lobe epilepsy. Nature Medicine. 1998;4:1166–1172. doi: 10.1038/2661. [DOI] [PubMed] [Google Scholar]
- Brünig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABAA receptor surface expression. European Journal of Neuroscience. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Brussaard AB, Herbison AE. Long-term plasticity of postsynaptic GABAA-receptor function in the adult brain: Insights from the oxytocin neurone. Trends in Neurosciences. 2000;23:190–195. doi: 10.1016/s0166-2236(99)01540-4. [DOI] [PubMed] [Google Scholar]
- Brussaard AB, Wossink J, Lodder JC, Kits KS. Progesterone-metabolite prevents protein kinase C-dependent modulation of γ-aminobutyric acid type A receptors in oxytocin neurons. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:3625–3630. doi: 10.1073/pnas.050424697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Flores-Hernandez J, Feng J, Yan Z. Activity-dependent bidirectional regulation of GABAA receptor channels by the 5-HT4 receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. The Journal of Physiology. 2002;540:743–759. doi: 10.1113/jphysiol.2001.013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapell R, Bueno OF, Alvarez-Hernandez X, Robinson LC, Leidenheimer NJ. Activation of protein kinase C induces γ-aminobutyric acid type A receptor internalization in Xenopus oocytes. Journal of Biological Chemistry. 1998;273:32595–32601. doi: 10.1074/jbc.273.49.32595. [DOI] [PubMed] [Google Scholar]
- Charych EI, Liu F, Moss SJ, Brandon NJ. GABAA receptors and their associated proteins: Implications in the etiology and treatment of schizophrenia and related disorders. Neuropharmacology. 2009;57:481–495. doi: 10.1016/j.neuropharm.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GJ, Kittler JT, Moss SJ, Yan Z. Dopamine D-3 receptors regulate GABAA receptor function through a phospho-dependent endocytosis mechanism in nucleus accumbens. Journal of Neuroscience. 2006;26:2513–2521. doi: 10.1523/JNEUROSCI.4712-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZW, Olsen RW. GABAA receptor associated proteins: A key factor regulating GABAA receptor function. Journal of Neurochemistry. 2007;100:279–294. doi: 10.1111/j.1471-4159.2006.04206.x. [DOI] [PubMed] [Google Scholar]
- Chiu SL, Cline HT. Insulin receptor signaling in the development of neuronal structure and function. Neural Development. 2010;5:7. doi: 10.1186/1749-8104-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DS, Wang D, Dadgar J, Chang WS, Messing RO. Conditional rescue of protein kinase C epsilon regulates ethanol preference and hypnotic sensitivity in adult mice. Journal of Neuroscience. 2002;22:9905–9911. doi: 10.1523/JNEUROSCI.22-22-09905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DS, Wei W, Deitchman JK, Kharazia VN, Lesscher HMB, McMahon T, et al. Protein kinase C delta regulates ethanol intoxication and enhancement of GABA-stimulated tonic current. Journal of Neuroscience. 2008;28:11890–11899. doi: 10.1523/JNEUROSCI.3156-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou WH, Wang D, McMahon T, Qi ZH, Song M, Zhang C, et al. GABAA receptor trafficking is regulated by PKCε and the N-ethylmaleimide-sensitive factor. The Journal of Neuroscience. 2010;30:13955–13965. doi: 10.1523/JNEUROSCI.0270-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churn SB, Rana A, Lee K, Parsons JT, De Blas A, Delorenzo RJ. Calcium/calmodulin-dependent kinase II phosphorylation of the GABAA receptor α1 subunit modulates benzodiazepine binding. Journal of Neurochemistry. 2002;82:1065–1076. doi: 10.1046/j.1471-4159.2002.01032.x. [DOI] [PubMed] [Google Scholar]
- Comenencia-Ortiz E, Moss SJ, Davies PA. Phosphorylation of GABAA receptors influences receptor trafficking and neurosteroid actions. Psychopharmacology. 2014;231:3453–3465. doi: 10.1007/s00213-014-3617-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compagnone NA, Mellon SH. Neurosteroids: Biosynthesis and function of these novel neuromodulators. Frontiers in Neuroendocrinology. 2000;21:1–56. doi: 10.1006/frne.1999.0188. [DOI] [PubMed] [Google Scholar]
- Concas A, Mostallino MC, Porcu P, Follesa P, Barbaccia ML, Trabucchi M, et al. Role of brain allopregnanolone in the plasticity of γ-aminobutyric acid type A receptor in rat brain during pregnancy and after delivery. Proceedings of the National Academy of Sciences. 1998;95:13284–13289. doi: 10.1073/pnas.95.22.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly WM, Errington AC, Crunelli V. gamma-Hydroxybutyric acid (GHB) is not an agonist of extrasynaptic GABAA receptors. PLoS One. 2013;8:e79062. doi: 10.1371/journal.pone.0079062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly WM, Errington AC, Di Giovanni G, Crunelli V. Metabotropic regulation of extrasynaptic GABAA receptors. Frontiers in Neural Circuits. 2013;7 doi: 10.3389/fncir.2013.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly CN, Kittler JT, Thomas P, Uren JM, Brandon NJ, Smart TG, et al. Cell surface stability of γ-aminobutyric acid type A receptors. Journal of Biological Chemistry. 1999;274:36565–36572. doi: 10.1074/jbc.274.51.36565. [DOI] [PubMed] [Google Scholar]
- D’Hulst C, Kooy RF. The GABAA receptor: A novel target for treatment of fragile X? Trends in Neurosciences. 2007;30:425–431. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Dacher M, Gouty S, Dash S, Cox BM, Nugent FS. A-kinase anchoring protein–calcineurin signaling in long-term depression of GABAergic synapses. The Journal of Neuroscience. 2013;33:2650–2660. doi: 10.1523/JNEUROSCI.2037-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeb TZ, Maguire J, Moss SJ. Possible alterations in GABAA receptor signaling that underlie benzodiazepine-resistant seizures. Epilepsia. 2012;53:79–88. doi: 10.1111/epi.12037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorey TM, Olsen RW. GABA and epileptogenesis: Comparing gabrb3 gene-deficient mice with Angelman syndrome in man. Epilepsy Research. 1999;36:123–132. doi: 10.1016/s0920-1211(99)00046-7. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fáncsik A, Linn DM, Tasker JG. Neurosteroid modulation of GABA IPSCs is phosphorylation dependent. The Journal of Neuroscience. 2000;20:3067–3075. doi: 10.1523/JNEUROSCI.20-09-03067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: Phasic and tonic activation of GABAA receptors. Nature Reviews Neuroscience. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Feng J, Cai X, Zhao J, Yan Z. Serotonin receptors modulate GABAA receptor channels through activation of anchored protein kinase C in prefrontal cortical neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2001;21:6502–6511. doi: 10.1523/JNEUROSCI.21-17-06502.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippova N, Sedelnikova A, Zong Y, Fortinberry H, Weiss DS. Regulation of recombinant γ-aminobutyric acid (GABA)A and GABAC receptors by protein kinase C. Molecular Pharmacology. 2000;57:847–856. [PubMed] [Google Scholar]
- Flores-Hernandez J, Hernandez S, Snyder GL, Yan Z, Fienberg AA, Moss SJ, et al. D1 dopamine receptor activation reduces GABAA receptor currents in neostriatal neurons through a PKA/DARPP-32/PP1 signaling cascade. Journal of Neurophysiology. 2000;83:2996–3004. doi: 10.1152/jn.2000.83.5.2996. [DOI] [PubMed] [Google Scholar]
- Fritschy JM. Epilepsy, E/I balance and GABAA receptor plasticity. Frontiers in Molecular Neuroscience. 2008;1:5. doi: 10.3389/neuro.02.005.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii M, Kanematsu T, Ishibashi H, Fukami K, Takenawa T, Nakayama KI, et al. Phospholipase C-related but catalytically inactive protein is required for insulin-induced cell surface expression of gamma-aminobutyric acid type A receptors. Journal of Biological Chemistry. 2010;285:4837–4846. doi: 10.1074/jbc.M109.070045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiarsa JL, Caillard O, Ben-Ari Y. Long-term plasticity at GABAergic and glycinergic synapses: Mechanisms and functional significance. Trends in Neurosciences. 2002;25:564–570. doi: 10.1016/s0166-2236(02)02269-5. [DOI] [PubMed] [Google Scholar]
- Gardner LA, Tavalin SJ, Goehring AS, Scott JD, Bahouth SW. AKAP79-mediated targeting of the Cyclic AMP-dependent protein kinase to the β1-adrenergic receptor promotes recycling and functional resensitization of the receptor. Journal of Biological Chemistry. 2006;281:33537–33553. doi: 10.1074/jbc.M601809200. [DOI] [PubMed] [Google Scholar]
- Geral C, Angelova A, Lesieur S. From molecular to nanotechnology strategies for delivery of neurotrophins: Emphasis on brain-derived neurotrophic factor (BDNF) Pharmaceutics. 2013;5:127–167. doi: 10.3390/pharmaceutics5010127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghansah E, Weiss DS. Modulation of GABAA receptors by benzodiazepines and barbiturates is autonomous of PKC activation. Neuropharmacology. 2001;40:327–333. doi: 10.1016/s0028-3908(00)00166-0. [DOI] [PubMed] [Google Scholar]
- Gillette MA, Dacheux RF. Protein kinase modulation of GABAA currents in rabbit retinal rod bipolar cells. Journal of Neurophysiology. 1996;76:3070–3086. doi: 10.1152/jn.1996.76.5.3070. [DOI] [PubMed] [Google Scholar]
- Goffin D, Ali AB, Rampersaud N, Harkavyi A, Fuchs C, Whitton PS, et al. Dopamine-dependent Tuning of striatal inhibitory synaptogenesis. The Journal of Neuroscience. 2010;30:2935–2950. doi: 10.1523/JNEUROSCI.4411-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschen-Ohm MP, Wagner DA, Petrou S, Jones MV. An epilepsy-related region in the GABAA receptor mediates long-distance effects on GABA and benzodiazepine binding sites. Molecular pharmacology. 2010;77:35–45. doi: 10.1124/mol.109.058289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Moss SJ, Olsen RW. Ethanol promotes clathrin adaptor-Mediated endocytosis via the intracellular domain of delta-containing GABAA receptors. Journal of Neuroscience. 2012;32:17874–17881. doi: 10.1523/JNEUROSCI.2535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodkin HP, Joshi S, Mtchedlishvili Z, Brar J, Kapur J. Subunit-specific trafficking of GABAA receptors during status epilepticus. The Journal of Neuroscience. 2008;28:2527–2538. doi: 10.1523/JNEUROSCI.3426-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Terunuma M, Kanematsu T, Misumi Y, Moss SJ, Hirata M. Direct interaction of N-ethylmaleimide-sensitive factor with GABAA receptor β subunits. Molecular and Cellular Neuroscience. 2005;30:197–206. doi: 10.1016/j.mcn.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of slow synaptic transmission. Science. 2001;294:1024–1030. doi: 10.1126/science.294.5544.1024. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Khatri L, Hanson PI, Ziff EB. NSF ATPase and α-/β-SNAPs disassemble the AMPA receptor-PICK1 complex. Neuron. 2002;34:53–67. doi: 10.1016/s0896-6273(02)00638-4. [DOI] [PubMed] [Google Scholar]
- Harney SC, Frenguelli BG, Lambert JJ. Phosphorylation influences neurosteroid modulation of synaptic GABAA receptors in rat CA1 and dentate gyrus neurones. Neuropharmacology. 2003;45:873–883. doi: 10.1016/s0028-3908(03)00251-x. [DOI] [PubMed] [Google Scholar]
- Harris RA, McQuilkin SJ, Paylor R, Abeliovich A, Tonegawa S, Wehner JM. Mutant mice lacking the gamma isoform of protein kinase C show decreased behavioral actions of ethanol and altered function of gamma-aminobutyrate type A receptors. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3658–3662. doi: 10.1073/pnas.92.9.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havrankova J, Brownstein M, Roth J. Insulin and insulin receptors in rodent brain. Diabetologia. 1981;20(Suppl):268–273. [PubMed] [Google Scholar]
- Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harbor Perspectives in Biology. 2012;4:a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneberger C, Jüttner R, Rothe T, Grantyn R. Postsynaptic action of BDNF on GABAergic synaptic transmission in the superficial layers of the mouse superior colliculus. Journal of Neurophysiology. 2002;88:595–603. doi: 10.1152/jn.2002.88.2.595. [DOI] [PubMed] [Google Scholar]
- Herd MB, Belelli D, Lambert JJ. Neurosteroid modulation of synaptic and extrasynaptic GABAA receptors. Pharmacology & Therapeutics. 2007;116:20–34. doi: 10.1016/j.pharmthera.2007.03.007. [DOI] [PubMed] [Google Scholar]
- Herring D, Huang RQ, Singh M, Dillon GH, Leidenheimer NJ. PKC modulation of GABAA receptor endocytosis and function is inhibited by mutation of a dileucine motif within the receptor beta 2 subunit. Neuropharmacology. 2005;48:181–194. doi: 10.1016/j.neuropharm.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Herring D, Huang R, Singh M, Robinson LC, Dillon GH, Leidenheimer NJ. Constitutive GABAA receptor endocytosis is dynamin-mediated and dependent on a dileucine AP2 adaptin-binding motif within the β2 subunit of the receptor. Journal of Biological Chemistry. 2003;278:24046–24052. doi: 10.1074/jbc.M301420200. [DOI] [PubMed] [Google Scholar]
- Hirano T, Kawaguchi SY. Regulation and functional roles of rebound potentiation at cerebellar stellate cell-Purkinje cell synapses. Frontiers in Cellular Neuroscience. 2014;8:42. doi: 10.3389/fncel.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, et al. Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCε. Nature Neuroscience. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- Hodge CW, Raber J, McMahon T, Walter H, Sanchez-Perez AM, Olive MF, et al. Decreased anxiety-like behavior, reduced stress hormones, and neurosteroid supersensitivity in mice lacking protein kinase C epsilon. Journal of Clinical Investigation. 2002;110:1003–1010. doi: 10.1172/JCI15903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hortnagl H, Tasan RO, Wieselthaler A, Kirchmair E, Sieghart W, Sperk G. Patterns of mRNA and protein expression for 12 GABAA receptor subunits in the mouse brain. Neuroscience. 2013;236:345–372. doi: 10.1016/j.neuroscience.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston CM, He Q, Smart TG. CaMKII phosphorylation of the GABAA receptor: Receptor subtype- and synapse-specific modulation. The Journal of Physiology. 2009;587:2115–2125. doi: 10.1113/jphysiol.2009.171603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston CM, Lee HHC, Hosie AM, Moss SJ, Smart TG. Identification of the sites for CaMK-II-dependent phosphorylation of GABAA receptors. Journal of Biological Chemistry. 2007;282:17855–17865. doi: 10.1074/jbc.M611533200. [DOI] [PubMed] [Google Scholar]
- Houston CM, Smart TG. CaMK-II modulation of GABAA receptors expressed in HEK293, NG108-15 and rat cerebellar granule neurons. European Journal of Neuroscience. 2006;24:2504–2514. doi: 10.1111/j.1460-9568.2006.05145.x. [DOI] [PubMed] [Google Scholar]
- Ip FC, Cheung J, Ip NY. The expression profiles of neurotrophins and their receptors in rat and chicken tissues during development. Neuroscience Letters. 2001;301:107–110. doi: 10.1016/s0304-3940(01)01603-2. [DOI] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABAA receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nature reviews. Neuroscience. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Wan Q, Vithlani M, Saliba RS, Succol F, Pangalos MN, et al. GABAA receptor membrane trafficking regulates spine maturity. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:12500–12505. doi: 10.1073/pnas.0903943106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen MJ, Ade KK, Fu Z, Vicini S. Dopamine modulation of GABA tonic conductance in striatal output neurons. Journal of Neuroscience. 2009;29:5116–5126. doi: 10.1523/JNEUROSCI.4737-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen M, Yasuda RP, Vicini S. GABAA receptor β3 subunit expression regulates tonic current in developing striatopallidal medium spiny neurons. Frontiers in Cellular Neuroscience. 2011;5:15. doi: 10.3389/fncel.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi S, Kapur J. Slow intracellular accumulation of GABAA receptor δ subunit is modulated by BDNF. Neuroscience. 2009;164:507–519. doi: 10.1016/j.neuroscience.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABAA receptor phosphorylation, activity, and cell-surface stability. Journal of Neuroscience. 2004;24:522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Tretter V, Walker J, Brandon NJ, Moss SJ. Fyn kinase contributes to tyrosine phosphorylation of the GABAA receptor γ2 subunit. Molecular and cellular neurosciences. 2010;44:129–134. doi: 10.1016/j.mcn.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Neurobiology of cocaine addiction: Implications for new pharmacotherapy. American Journal on Addictions. 2007;16:71–78. doi: 10.1080/10550490601184142. [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Fujii M, Mizokami A, Kittler JT, Nabekura J, Moss SJ, et al. Phospholipase C-related inactive protein is implicated in the constitutive internalization of GABAA receptors mediated by clathrin and AP2 adaptor complex. Journal of Neurochemistry. 2007;101:898–905. doi: 10.1111/j.1471-4159.2006.04399.x. [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Mizokami A, Watanabe K, Hirata M. Regulation of GABAA-receptor surface expression with special reference to the involvement of GABARAP (GABAA receptor-associated protein) and PRIP (phospholipase C-related, but catalytically inactive protein) Journal of Pharmacological Sciences. 2007;104:285–292. doi: 10.1254/jphs.cp0070063. [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Yasunaga A, Mizoguchi Y, Kuratani A, Kittler JT, Jovanovic JN, et al. Modulation of GABAA receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complex underlying brain-derived neurotrophic factor-dependent regulation of GABAergic inhibition. Journal of Biological Chemistry. 2006;281:22180–22189. doi: 10.1074/jbc.M603118200. [DOI] [PubMed] [Google Scholar]
- Kanematsu T, Yoshimura K, Hidaka K, Takeuchi H, Katan M, Hirata M. Domain organization of p130, PLC-related catalytically inactive protein, and structural basis for the lack of enzyme activity. European Journal of Biochemistry. 2000;267:2731–2737. doi: 10.1046/j.1432-1327.2000.01291.x. [DOI] [PubMed] [Google Scholar]
- Kang SU, Heo S, Lubec G. Mass spectrometric analysis of GABAA receptor subtypes and phosphorylations from mouse hippocampus. Proteomics. 2011;11:2171–2181. doi: 10.1002/pmic.201000374. [DOI] [PubMed] [Google Scholar]
- Kano M, Kano M, Fukunaga K, Konnerth A. Ca2+-induced rebound potentiation of γ-aminobutyric acid-mediated currents requires activation of Ca2+/calmodulin-dependent kinase II. Proceedings of the National Academy of Sciences. 1996;93:13351–13356. doi: 10.1073/pnas.93.23.13351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Konnerth A. Potentiation of GABA-mediated currents by cAMP-dependent protein kinase. Neuroreport. 1992;3:563–566. doi: 10.1097/00001756-199207000-00004. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S, Hirano T. Signaling cascade regulating long-term potentiation of GABAA receptor responsiveness in cerebellar Purkinje neurons. Journal of Neuroscience. 2002;22:3969–3976. doi: 10.1523/JNEUROSCI.22-10-03969.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi S-y, Hirano T. Sustained structural change of GABAA receptor-associated protein underlies long-term potentiation at inhibitory synapses on a cerebellar purkinje neuron. The Journal of Neuroscience. 2007;27:6788–6799. doi: 10.1523/JNEUROSCI.1981-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, Malherbe P, Sigel E. Function of the alpha 1 beta 2 gamma 2S gamma-aminobutyric acid type A receptor is modulated by protein kinase C via multiple phosphorylation sites. Journal of Biological Chemistry. 1992;267:25660–25663. [PubMed] [Google Scholar]
- Kia A, Ribeiro F, Nelson R, Gavrilovici C, Ferguson SSG, Poulter MO. Kindling alters neurosteroid-induced modulation of phasic and tonic GABAA receptor-mediated currents: Role of phosphorylation. Journal of Neurochemistry. 2011;116:1043–1056. doi: 10.1111/j.1471-4159.2010.07156.x. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Chen G, Honing S, Bogdanov Y, McAinsh K, Arancibia-Carcamo IL, et al. Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:14871–14876. doi: 10.1073/pnas.0506653102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Chen G, Kukhtina V, Vahedi-Faridi A, Gu Z, Tretter V, et al. Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor gamma 2 subunit. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:3616–3621. doi: 10.1073/pnas.0707920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Delmas P, Jovanovic JN, Brown DA, Smart TG, Moss SJ. Constitutive endocytosis of GABAA receptors by an association with the Adaptin AP2 complex modulates inhibitory synaptic currents in hippocampal neurons. Journal of Neuroscience. 2000;20:7972–7977. doi: 10.1523/JNEUROSCI.20-21-07972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler JT, Moss SJ. Modulation of GABAA receptor activity by phosphorylation and receptor trafficking: Implications for the efficacy of synaptic inhibition. Current Opinion in Neurobiology. 2003;13:341–347. doi: 10.1016/s0959-4388(03)00064-3. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Rostaing P, Schiavo G, Fritschy JM, Olsen R, Triller A, et al. The subcellular distribution of GABARAP and its ability to interact with NSF suggest a role for this protein in the intracellular transport of GABAA receptors. Molecular and Cellular Neuroscience. 2001;18:13–25. doi: 10.1006/mcne.2001.1005. [DOI] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science (New York, NY) 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing S, Lamballe F, Tapley P, Bryant S, et al. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneussel M, Loebrich S. Trafficking and synaptic anchoring of ionotropic inhibitory neurotransmitter receptors. Biology of the Cell/Under the Auspices of the European Cell Biology Organization. 2007;99:297–309. doi: 10.1042/BC20060120. [DOI] [PubMed] [Google Scholar]
- Koksma JJ, van Kesteren RE, Rosahl TW, Zwart R, Smit AB, Luddens H, et al. Oxytocin regulates neurosteroid modulation of GABAA receptors in supraoptic nucleus around parturition. Journal of Neuroscience. 2003;23:788–797. doi: 10.1523/JNEUROSCI.23-03-00788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpi ER, Grunder G, Luddens H. Drug interactions at GABAA receptors. Progress in Neurobiology. 2002;67:113–159. doi: 10.1016/s0301-0082(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Kowalczyk S, Winkelmann A, Smolinsky B, Foerstera B, Neundorf I, Schwarz G, et al. Direct binding of GABAA receptor beta 2 and beta 3 subunits to gephyrin. European Journal of Neuroscience. 2013;37:544–554. doi: 10.1111/ejn.12078. [DOI] [PubMed] [Google Scholar]
- Krishek BJ, Xie X, Blackstone C, Huganir RL, Moss SJ, Smart TG. Regulation of GABAA receptor function by protein kinase C phosphorylation. Neuron. 1994;12:1081–1095. doi: 10.1016/0896-6273(94)90316-6. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Staley J, Mason G, Petrakis IL, Kaufman J, Harris RA, et al. gamma-Aminobutyric acid type A receptors and alcoholism - Intoxication, dependence, vulnerability, and treatment. Archives of General Psychiatry. 2006;63:957–968. doi: 10.1001/archpsyc.63.9.957. [DOI] [PubMed] [Google Scholar]
- Kuczewski N, Fuchs C, Ferrand N, Jovanovic JN, Gaiarsa JL, Porcher C. Mechanism of GABAB receptor-induced BDNF secretion and promotion of GABAA receptor membrane expression. Journal of Neurochemistry. 2011;118:533–545. doi: 10.1111/j.1471-4159.2011.07192.x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Kralic JE, O’Buckley TK, Grobin AC, Morrow AL. Chronic ethanol consumption enhances internalization of alpha 1 subunit-containing GABAA receptors in cerebral cortex. Journal of Neurochemistry. 2003;86:700–708. doi: 10.1046/j.1471-4159.2003.01894.x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Sieghart W, Morrow AL. Association of protein kinase C with GABAA receptors containing alpha 1 and alpha 4 subunits in the cerebral cortex: Selective effects of chronic ethanol consumption. Journal of Neurochemistry. 2002;82:110–117. doi: 10.1046/j.1471-4159.2002.00943.x. [DOI] [PubMed] [Google Scholar]
- Kumar S, Suryanarayanan A, Boyd KN, Comerford CE, Lai MA, Ren Q, et al. Ethanol reduces GABAA alpha 1 subunit receptor surface expression by a protein kinase C gamma-dependent mechanism in cultured cerebral cortical neurons. Molecular Pharmacology. 2010;77:793–803. doi: 10.1124/mol.109.063016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz JE, Sheets D, Parsons JT, Rana A, Delorenzo RJ, Churn SB. A significant increase in both basal and maximal calcineurin activity in the rat pilocarpine model of status epilepticus. Journal of Neurochemistry. 2001;78:304–315. doi: 10.1046/j.1471-4159.2001.00426.x. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Cooper MA, Simmons RDJ, Weir CJ, Belelli D. Neurosteroids: Endogenous allosteric modulators of GABAA receptors. Psychoneuroendocrinology. 2009;34(1 Supplement):S48–S58. doi: 10.1016/j.psyneuen.2009.08.009. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Wisden W, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. III. Embryonic and postnatal development. Journal of Neuroscience. 1992;12:4151–4172. doi: 10.1523/JNEUROSCI.12-11-04151.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc AE, Kalant H, Gibbins RJ. Acute tolerance to ethanol in the rat. Psychopharmacologia. 1975;41:43–46. doi: 10.1007/BF00421304. [DOI] [PubMed] [Google Scholar]
- Lee HK. Synaptic plasticity and phosphorylation. Pharmacology & Therapeutics. 2006;112:810–832. doi: 10.1016/j.pharmthera.2006.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidenheimer NJ, Chapell R. Effects of PKC activation and receptor desensitization on neurosteroid modulation of GABAA receptors. Molecular Brain Research. 1997;52:173–181. doi: 10.1016/s0169-328x(97)00255-6. [DOI] [PubMed] [Google Scholar]
- Leidenheimer NJ, McQuilkin SJ, Hahner LD, Whiting P, Harris RA. Activation of protein kinase C selectively inhibits the gamma-aminobutyric acidA receptor: Role of desensitization. Molecular Pharmacology. 1992;41:1116–1123. [PubMed] [Google Scholar]
- Liang J, Suryanarayanan A, Abriam A, Snyder B, Olsen RW, Spigelman I. Mechanisms of reversible GABAA receptor plasticity after ethanol intoxication. The Journal of Neuroscience. 2007;27:12367–12377. doi: 10.1523/JNEUROSCI.2786-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Zhang N, Cagetti E, Houser CR, Olsen RW, Spigelman I. Chronic intermittent ethanol-induced switch of ethanol actions from extrasynaptic to synaptic hippocampal GABAA receptors. The Journal of Neuroscience. 2006;26:1749–1758. doi: 10.1523/JNEUROSCI.4702-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly SM, Alvarez FJ, Tietz EI. Synaptic and subcellular localization of A-kinase anchoring protein 150 in rat hippocampal CA1 pyramidal cells: Co-localization with excitatory synaptic markers. Neuroscience. 2005;134:155–163. doi: 10.1016/j.neuroscience.2005.03.039. [DOI] [PubMed] [Google Scholar]
- Lin YF, Angelotti TP, Dudek EM, Browning MD, Macdonald RL. Enhancement of recombinant alpha 1 beta 1 gamma 2 L gamma-aminobutyric acidA receptor whole-cell currents by protein kinase C is mediated through phosphorylation of both beta 1 and gamma 2 L subunits. Molecular Pharmacology. 1996;50:185–195. [PubMed] [Google Scholar]
- Lin YF, Browning MD, Dudek EM, Macdonald RL. Protein kinase C enhances recombinant bovine α1β1γ2L GABAA receptor whole-cell currents expressed in L929 fibroblasts. Neuron. 1994;13:1421–1431. doi: 10.1016/0896-6273(94)90427-8. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiological Reviews. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu B, Li L, Zhang Q, Chang N, Wang D, Shan Y, et al. Preservation of GABAA receptor function by PTEN inhibition protects against neuronal death in ischemic. Stroke. 2010;41:1018–1026. doi: 10.1161/STROKEAHA.110.579011. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nature Reviews Neuroscience. 2003;4:399–414. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Loup F, Wieser HG, Yonekawa Y, Aguzzi A, Fritschy JM. Selective alterations in GABAA receptor subtypes in human temporal lobe epilepsy. The Journal of Neuroscience. 2000;20:5401–5419. doi: 10.1523/JNEUROSCI.20-14-05401.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng P-l, Lim BK, Khoshnevisrad N, Poo M-m. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Mansuy IM, Kandel ER, Roder J. Calcineurin-mediated LTD of GABAergic inhibition underlies the increased excitability of CA1 neurons associated with LTP. Neuron. 2000;26:197–205. doi: 10.1016/s0896-6273(00)81150-2. [DOI] [PubMed] [Google Scholar]
- Ludvig N, George MA, Tang HM, Gonzales RA, Bungay PM. Evidence for the ability of hippocampal neurons to develop acute tolerance to ethanol in behaving rats. Brain Research. 2001;900:252–260. doi: 10.1016/s0006-8993(01)02319-8. [DOI] [PubMed] [Google Scholar]
- Luscher B, Fuchs T, Kilpatrick CL. GABAAR trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011;70:385–409. doi: 10.1016/j.neuron.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Molecular Psychiatry. 2011;16:383–406. doi: 10.1038/mp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lydiard RB. The role of GABA in anxiety disorders. The Journal of Clinical Psychiatry. 2003;64(Suppl 3):21–27. [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW. Gabaa receptor channels. Annual Review of Neuroscience. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- MacDonald RL, Rogers CJ, Twyman RE. Barbiturate regulation of kinetic properties of the GABAA receptor channel of mouse spinal neurones in culture. Journal of Physiology. 1989;417:483–500. doi: 10.1113/jphysiol.1989.sp017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: An embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Marsden KC, Beattie JB, Friedenthal J, Carroll RC. NMDA receptor activation potentiates inhibitory transmission through GABA receptor-associated protein-dependent exocytosis of GABAA receptors. Journal of Neuroscience. 2007;27:14326–14337. doi: 10.1523/JNEUROSCI.4433-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsden KC, Shemesh A, Bayer KU, Carroll RC. Selective translocation of Ca2+/calmodulin protein kinase II alpha (CaMKII alpha) to inhibitory synapses. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:20559–20564. doi: 10.1073/pnas.1010346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG. Adjacent phosphorylation sites on GABAA receptor β subunits determine regulation by cAMP-dependent protein kinase. Nature Neuroscience. 1998;1:23–28. doi: 10.1038/223. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Moss SJ. Differential phosphorylation of intracellular domains of gamma-aminobutyric acid type A receptor subunits by calcium/calmodulin type 2-dependent protein kinase and cGMP-dependent protein kinase. Journal of Biological Chemistry. 1994;269:18111–18117. [PubMed] [Google Scholar]
- McDonald BJ, Moss SJ. Conserved phosphorylation of the intracellular domains of GABAA receptorβ2 and β3 subunits by cAMP-dependent protein kinase, cGMP-dependent protein kinase, protein kinase C and Ca2+/calmodulin type II-dependent protein kinase. Neuropharmacology. 1997;36:1377–1385. doi: 10.1016/s0028-3908(97)00111-1. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nature Reviews Molecular Cell Biology. 2011;12:517–533. doi: 10.1038/nrm3151. [DOI] [PubMed] [Google Scholar]
- Mele M, Ribeiro L, Inacio AR, Wieloch T, Duarte CB. GABAA receptor dephosphorylation followed by internalization is coupled to neuronal death in in vitro ischemia. Neurobiology of Disease. 2014;65:220–232. doi: 10.1016/j.nbd.2014.01.019. [DOI] [PubMed] [Google Scholar]
- Mercik K, Pytel M, Mozrzymas JW. Recombinant α1β2γ2 GABAA receptors expressed in HEK293 and in QT6 cells show different kinetics. Neuroscience Letters. 2003;352:195–198. doi: 10.1016/j.neulet.2003.08.060. [DOI] [PubMed] [Google Scholar]
- Mielke JG, Wang YT. Insulin exerts neuroprotection by counteracting the decrease in cell-surface GABAA receptors following oxygen-glucose deprivation in cultured cortical neurons. Journal of Neurochemistry. 2005;92:103–113. doi: 10.1111/j.1471-4159.2004.02841.x. [DOI] [PubMed] [Google Scholar]
- Mizoguchi Y, Ishibashi H, Nabekura J. The action of BDNF on GABAA currents changes from potentiating to suppressing during maturation of rat hippocampal CA1 pyramidal neurons. Journal of Physiology-London. 2003;548:703–709. doi: 10.1113/jphysiol.2003.038935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochly-Rosen D, Khaner H, Lopez J. Identification of intracellular receptor proteins for activated protein kinase C. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:3997–4000. doi: 10.1073/pnas.88.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss SJ, Doherty CA, Huganir RL. Identification of the cAMP-dependent protein kinase and protein kinase C phosphorylation sites within the major intracellular domains of the beta 1, gamma 2S, and gamma 2 L subunits of the gamma-aminobutyric acid type A receptor. Journal of Biological Chemistry. 1992;267:14470–14476. [PubMed] [Google Scholar]
- Moss SJ, Gorrie GH, Amato A, Smart TG. Modulation of GABAA receptors by tyrosine phosphorylation. Nature. 1995;377:344–348. doi: 10.1038/377344a0. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Modulation of amino acid-gated ion channels by protein phosphorylation. International Review of Neurobiology. 1996;39:1–52. doi: 10.1016/s0074-7742(08)60662-5. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG, Blackstone CD, Huganir RL. Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science (New York, NY) 1992;257:661–665. doi: 10.1126/science.1323140. [DOI] [PubMed] [Google Scholar]
- Muir J, Arancibia-Carcamo IL, MacAskill AF, Smith KR, Griffin LD, Kittler JT. NMDA receptors regulate GABAA receptor lateral mobility and clustering at inhibitory synapses through serine 327 on the gamma 2 subunit. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16679–16684. doi: 10.1073/pnas.1000589107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee J, Kretschmannova K, Gouzer G, Maric HM, Ramsden S, Tretter V, et al. The residence time of GABAARs at inhibitory synapses is determined by direct binding of the receptor alpha 1 subunit to gephyrin. Journal of Neuroscience. 2011;31:14677–14687. doi: 10.1523/JNEUROSCI.2001-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Liu H, Niquet J, Wasterlain CG. Rapid surface accumulation of NMDA receptors increases glutamatergic excitation during status epilepticus. Neurobiology of Disease. 2013;54:225–238. doi: 10.1016/j.nbd.2012.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor DE, Liu HT, Wasterlain CG. Trafficking of GABAA receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. Journal of Neuroscience. 2005;25:7724–7733. doi: 10.1523/JNEUROSCI.4944-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Mody I. Selective modulation of tonic and phasic inhibitions in dentate gyrus granule cells. Journal of Neurophysiology. 2002;87:2624–2628. doi: 10.1152/jn.2002.87.5.2624. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Sieghart W, Mody I. Differential regulation of synaptic GABAA receptors by cAMP-dependent protein kinase in mouse cerebellar and olfactory bulb neurones. The Journal of Physiology. 1999;521:421–435. doi: 10.1111/j.1469-7793.1999.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF, Mehmert KK, Messing RO, Hodge CW. Reduced operant ethanol self-administration and in vivo mesolimbic dopamine responses to ethanol in PKCepsilon-deficient mice. The European journal of neuroscience. 2000;12:4131–4140. doi: 10.1046/j.1460-9568.2000.00297.x. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W. International union of pharmacology. LXX. Subtypes of γ-aminobutyric acid A receptors: Classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacological Reviews. 2008;60:243–260. doi: 10.1124/pr.108.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens DF, Boyce LH, Davis MBE, Kriegstein AR. Excitatory GABA responses in embryonic and neonatal cortical slices demonstrated by gramicidin perforated-patch recordings and calcium imaging. The Journal of Neuroscience. 1996;16:6414–6423. doi: 10.1523/JNEUROSCI.16-20-06414.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini EM, Ravasenga T, Hausrat TJ, Iurilli G, Olcese U, Racine V, et al. Synaptic recruitment of gephyrin regulates surface GABAA receptor dynamics for the expression of inhibitory LTP. Nature Communications. 2014;5 doi: 10.1038/ncomms4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABAA receptors: Immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Poisbeau P, Cheney MC, Browning MD, Mody I. Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. The Journal of Neuroscience. 1999;19:674–683. doi: 10.1523/JNEUROSCI.19-02-00674.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcher C, Hatchett C, Longbottom RE, McAinch K, Sihra TS, Moss SJ, et al. Positive feedback regulation between γ-aminobutyric acid type A (GABAA) receptor signaling and brain-derived neurotrophic factor (BDNF) release in developing neurons. Journal of Biological Chemistry. 2011;286:21667–21677. doi: 10.1074/jbc.M110.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter NM, Twyman RE, Uhler MD, Macdonald RL. Cyclic AMP-dependent protein kinase decreases GABAA receptor current in mouse spinal neurons. Neuron. 1990;5:789–796. doi: 10.1016/0896-6273(90)90338-g. [DOI] [PubMed] [Google Scholar]
- Qi ZH, Song M, Wallace MJ, Wang D, Newton PM, McMahon T, et al. Protein kinase C epsilon regulates gamma-aminobutyrate type A receptor sensitivity to ethanol and benzodiazepines through phosphorylation of gamma 2 subunits. Journal of Biological Chemistry. 2007;282:33052–33063. doi: 10.1074/jbc.M707233200. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, et al. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. The Journal of Cell Biology. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robello M, Amico C, Cupello A. Regulation of GABAA receptor in cerebellar granule cells in culture: Differential involvement of kinase activities. Neuroscience. 1993;53:131–138. doi: 10.1016/0306-4522(93)90291-m. [DOI] [PubMed] [Google Scholar]
- Ron D, Chen CH, Caldwell J, Jamieson L, Orr E, Mochly-Rosen D. Cloning of an intracellular receptor for protein kinase C: A homolog of the beta subunit of G proteins. Proceedings of the National Academy of Sciences. 1994;91:839–843. doi: 10.1073/pnas.91.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Knoflach F. Beyond classical benzodiazepines: Novel therapeutic potential of GABAA receptor subtypes. Nature Reviews Drug Discovery. 2011;10:685–697. doi: 10.1038/nrd3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Mohler H. Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annual Review of Pharmacology and Toxicology. 2004;44:475–498. doi: 10.1146/annurev.pharmtox.44.101802.121429. [DOI] [PubMed] [Google Scholar]
- Saiepour L, Fuchs C, Patrizi A, Sassoè-Pognetto M, Harvey RJ, Harvey K. Complex role of collybistin and gephyrin in GABAA receptor clustering. Journal of Biological Chemistry. 2010;285:29623–29631. doi: 10.1074/jbc.M110.121368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba RS, Kretschmannova K, Moss SJ. Activity-dependent phosphorylation of GABAA receptors regulates receptor insertion and tonic current. Embo Journal. 2012;31:2937–2951. doi: 10.1038/emboj.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Dell’Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. The Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathanoori M, Dias BG, Nair AR, Banerjee SB, Tole S, Vaidya VA. Differential regulation of multiple brain-derived neurotrophic factor transcripts in the postnatal and adult rat hippocampus during development, and in response to kainate administration. Molecular Brain Research. 2004;130:170–177. doi: 10.1016/j.molbrainres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Schwartz-Bloom RD, Sah R. γ-Aminobutyric acidA neurotransmission and cerebral ischemia. Journal of Neurochemistry. 2001;77:353–371. doi: 10.1046/j.1471-4159.2001.00274.x. [DOI] [PubMed] [Google Scholar]
- Seil FJ. TrkB receptor signaling and activity-dependent inhibitory synaptogenesis. Histology and Histopathology. 2003;18:635–646. doi: 10.14670/HH-18.635. [DOI] [PubMed] [Google Scholar]
- Shen Y, Lindemeyer AK, Spigelman I, Sieghart W, Olsen RW, Liang J. Plasticity of GABAA receptors after ethanol pre-exposure in cultured hippocampal neurons. Molecular Pharmacology. 2011;79:432–442. doi: 10.1124/mol.110.068650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga A, Thomas-Crusells J, Sigl T, Blaesse A, Mestres P, Meyer M, et al. Posttraumatic GABAA-mediated [Ca2+]i increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience. 2008;28:6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E, Baur R. Activation of protein kinase C differentially modulates neuronal Na+, Ca2+, and gamma-aminobutyrate type A channels. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:6192–6196. doi: 10.1073/pnas.85.16.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KR, Muir J, Rao Y, Browarski M, Gruenig MC, Sheehan DF, et al. Stabilization of GABAA receptors at endocytic zones is mediated by an AP2 binding motif within the GABAA receptor β3 subunit. The Journal of Neuroscience. 2012;32:2485–2498. doi: 10.1523/JNEUROSCI.1622-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Messing RO. Protein kinase C regulation of GABAA receptors. Cellular and Molecular Life Sciences: CMLS. 2005;62:119–127. doi: 10.1007/s00018-004-4339-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperk G, Drexel M, Pirker S. Neuronal plasticity in animal models and the epileptic human hippocampus. Epilepsia. 2009;50:29–31. doi: 10.1111/j.1528-1167.2009.02365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan KR, Rudolph U, Lüscher C. Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends in Neurosciences. 2011;34:188–197. doi: 10.1016/j.tins.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annual Review of Neuroscience. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- Tang X, Hernandez CC, Macdonald RL. Modulation of spontaneous and GABA-evoked tonic α4β3δ and α4β3γ2L GABAA receptor currents by protein kinase A. Journal of Neurophysiology. 2010;103:1007–1019. doi: 10.1152/jn.00801.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Buechler JA, Yonemoto W. Camp-dependent protein kinase: Framework for a diverse family of regulatory enzymes. Annual Review of Biochemistry. 1990;59:971–1005. doi: 10.1146/annurev.bi.59.070190.004543. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Knighton DR, Zheng J, Ten Eyck LF, Sowadski JM. Structural framework for the protein kinase family. Annual Review of Cell Biology. 1992;8:429–462. doi: 10.1146/annurev.cb.08.110192.002241. [DOI] [PubMed] [Google Scholar]
- Terunuma M, Jang IS, Ha SH, Kittler JT, Kanematsu T, Jovanovic JN, et al. GABAA receptor phospho-dependent modulation is regulated by phospholipase C-related inactive protein type 1, a novel protein phosphatase 1 anchoring protein. Journal of Neuroscience. 2004;24:7074–7084. doi: 10.1523/JNEUROSCI.1323-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma M, Xu J, Vithlani M, Sieghart W, Kittler J, Pangalos M, et al. Deficits in phosphorylation of GABAA receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. Journal of Neuroscience. 2008;28:376–384. doi: 10.1523/JNEUROSCI.4346-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson-Vest NM, Waldvogel HJ, Rees MI, Faull RLM. GABAA receptor subunit and gephyrin protein changes differ in the globus pallidus in Huntington’s diseased brain. Brain Research. 2003;994:265–270. doi: 10.1016/j.brainres.2003.09.051. [DOI] [PubMed] [Google Scholar]
- Tretter V, Kerschner B, Milenkovic I, Ramsden SL, Ramerstorfer J, Saiepour L, et al. Molecular basis of the γ-aminobutyric acid a receptor α3 subunit interaction with the clustering protein gephyrin. Journal of Biological Chemistry. 2011;286:37702–37711. doi: 10.1074/jbc.M111.291336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter VE, Mukherjee J, Maric HM, Schindelin H, Sieghart W, Moss SJ. Gephyrin, the enigmatic organizer at GABAergic synapses. Frontiers in Cellular Neuroscience. 2012;6:23. doi: 10.3389/fncel.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Revilla-Sanchez R, Houston C, Terunuma M, Havekes R, Florian C, et al. Deficits in spatial memory correlate with modified gamma-aminobutyric acid type A receptor tyrosine phosphorylation in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20039–20044. doi: 10.1073/pnas.0908840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudell JR, Messing RO, Mayfield J, Harris RA. Alcohol dependence: Molecular and behavioral evidence. Trends in Pharmacological Sciences. 2014;35:317–323. doi: 10.1016/j.tips.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Fritschy JM. Gephyrin: A master regulator of neuronal function? Nature Reviews Neuroscience. 2014;15:141–156. doi: 10.1038/nrn3670. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nature Reviews Molecular Cell Biology. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Draguhn A, Ymer S, Seeburg PH, Sakmann B. Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron. 1990;4:919–928. doi: 10.1016/0896-6273(90)90145-6. [DOI] [PubMed] [Google Scholar]
- Vetiska SM, Ahmadian G, Ju W, Liu L, Wymann MP, Wang YT. GABAA receptor-associated phosphoinositide 3-kinase is required for insulin-induced recruitment of postsynaptic GABAA receptors. Neuropharmacology. 2007;52:146–155. doi: 10.1016/j.neuropharm.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejón C, Collin C, McKay RDG, Segal M. Neurotrophins induce formation of functional excitatory and inhibitory synapses between cultured hippocampal neurons. The Journal of Neuroscience. 1998;18:7256–7271. doi: 10.1523/JNEUROSCI.18-18-07256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Losi G, Homanics GE. GABAA receptor delta subunit deletion prevents neurosteroid modulation of inhibitory synaptic currents in cerebellar neurons. Neuropharmacology. 2002;43:646–650. doi: 10.1016/s0028-3908(02)00126-0. [DOI] [PubMed] [Google Scholar]
- Vithlani M, Hines RM, Zhong P, Terunuma M, Hines DJ, Revilla-Sanchez R, et al. The ability of BDNF to modify neurogenesis and depressive-like BEHAVIORS Is dependent UPON phosphorylation of tyrosine residues 365/367 in the GABAA-receptor gamma 2 subunit. Journal of Neuroscience. 2013;33:15567–15577. doi: 10.1523/JNEUROSCI.1845-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithlani M, Moss Stephen J. The role of GABAAR phosphorylation in the construction of inhibitory synapses and the efficacy of neuronal inhibition. Biochemical Society Transactions. 2009;37:1355. doi: 10.1042/BST0371355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MJ, Newton PM, Oyasu M, McMahon T, Chou WH, Connolly J, et al. Acute functional tolerance to ethanol mediated by protein kinase C epsilon. Neuropsychopharmacology. 2007;32:127–136. doi: 10.1038/sj.npp.1301059. [DOI] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, et al. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]
- Wang A, Chi Z, Wang S, Wang S, Sun Q. Calcineurin-mediated GABAA receptor dephosphorylation in rats after kainic acid-induced status epilepticus. Seizure. 2009;18:519–523. doi: 10.1016/j.seizure.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Haditsch U, Tu W, Cochrane K, Ahmadian G, et al. Interaction of calcineurin and type-A GABA receptor γ2 subunits produces long-term depression at CA1 inhibitory synapses. The Journal of Neuroscience. 2003;23:826–836. doi: 10.1523/JNEUROSCI.23-03-00826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, et al. Control of synaptic strength, a novel function of Akt. Neuron. 2003;38:915–928. doi: 10.1016/s0896-6273(03)00356-8. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhong P, Yan Z. Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. The Journal of Neuroscience. 2002;22:9185–9193. doi: 10.1523/JNEUROSCI.22-21-09185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle RA, Poo MM. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. The Journal of Neuroscience. 2003;23:8722–8732. doi: 10.1523/JNEUROSCI.23-25-08722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MJ, Herman MM, Kleinman JE, Shannon Weickert C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expression Patterns. 2006;6:941–951. doi: 10.1016/j.modgep.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Werner DF, Kumar S, Criswell HE, Suryanarayanan A, Fetzer JA, Comerford CE, et al. PKC gamma is required for ethanol-induced increases in GABAA receptor alpha 4 subunit expression in cultured cerebral cortical neurons. Journal of Neurochemistry. 2011;116:554–563. doi: 10.1111/j.1471-4159.2010.07140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser IDC, Langeberg LK, et al. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- Whiting P, McKernan RM, Iversen LL. Another mechanism for creating diversity in gamma-aminobutyrate type A receptors: RNA splicing directs expression of two forms of gamma 2 phosphorylation site. Proceedings of the National Academy of Sciences. 1990;87:9966–9970. doi: 10.1073/pnas.87.24.9966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisden W, Laurie DJ, Monyer H, Seeburg PH. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. The Journal of Neuroscience. 1992;12:1040–1062. doi: 10.1523/JNEUROSCI.12-03-01040.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABAA currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/s0896-6273(00)80402-x. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. Journal of Neurophysiology. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]
- Yoshimura K, Takeuchi H, Sato O, Hidaka K, Doira N, Terunuma M, et al. Interaction of p130 with, and consequent inhibition of, the catalytic subunit of protein phosphatase 1α. Journal of Biological Chemistry. 2001;276:17908–17913. doi: 10.1074/jbc.M009677200. [DOI] [PubMed] [Google Scholar]