

Abstract
Functional impact of cardiac ryanodine receptor (type 2 RyR or RyR2) phosphorylation by protein kinase A (PKA) remains highly controversial. In this study, we characterized a functional link between PKA-mediated RyR2 phosphorylation level and sarcoplasmic reticulum (SR) Ca2+ release and leak in permeabilized rabbit ventricular myocytes. Changes in cytosolic [Ca2+] and intra-SR [Ca2+]SR were measured with Fluo-4 and Fluo-5N, respectively. Changes in RyR2 phosphorylation at two PKA sites, serine-2031 and -2809, were measured with phospho-specific antibodies. cAMP (10 μM) increased Ca2+ spark frequency approximately two-fold. This effect was associated with an increase in SR Ca2+ load from 0.84 to 1.24 mM. PKA inhibitory peptide (PKI; 10 μM) abolished the cAMP-dependent increase of SR Ca2+ load and spark frequency. When SERCA was completely blocked by thapsigargin, cAMP did not affect RyR2-mediated Ca2+ leak. The lack of a cAMP effect on RyR2 function can be explained by almost maximal phosphorylation of RyR2 at serine-2809 after sarcolemma permeabilization. This high RyR2 phosphorylation level is likely the consequence of a balance shift between protein kinase and phosphatase activity after permeabilization. When RyR2 phosphorylation at serine-2809 was reduced to its “basal” level (i.e. RyR2 phosphorylation level in intact myocytes) using kinase inhibitor staurosporine, SR Ca2+ leak was significantly reduced. Surprisingly, further dephosphorylation of RyR2 with protein phosphatase 1 (PP1) markedly increased SR Ca2+ leak. At the same time, phosphorylation of RyR2 at serine 2031 did not significantly change under identical experimental conditions. These results suggest that RyR2 phosphorylation by PKA has a complex effect on SR Ca2+ leak in ventricular myocytes. At an intermediate level of RyR2 phosphorylation SR Ca2+ leak is minimal. However, complete dephosphorylation and maximal phosphorylation of RyR2 increases SR Ca2+ leak.
Keywords: Ca2+ spark, cardiomyocyte, protein kinase A, protein phosphatases, ryanodine receptor, sarcoplasmic reticulum Ca2+ leak
INTRODUCTION
Type-2 isoform of the ryanodine receptor (RyR2) is the primary Ca2+ release channel of the sarcoplasmic reticulum (SR) of cardiomyocytes. During systole, synchronized activation of RyR2s by the transarcolemmal inward Ca2+ current generates the global Ca2+ transient that activates contraction of the heart. During diastole, RyR2s are not completely quiescent, providing a pathway for SR Ca2+ leak. Although diastolic SR Ca2+ leak is relatively small in comparison with systolic SR Ca2+ release [1, 2], it might play a protective role against SR Ca2+ overload [3]. Thus, accurate RyR2 functioning during different phases of the cardiac cycle is essential for regular heart contraction [4]. RyR2 gating is controlled by several molecular mechanisms and defects in this control can cause SR Ca2+ mishandling and contractile dysfunction. For example, increased diastolic SR Ca2+ leak can contribute to cardiac arrhythmias in infarcted and failing hearts [5, 6].
RyR2 is a target of phosphorylation by different protein kinases, including protein kinase A (PKA). It has been suggested that RyR2 phosphorylation by PKA plays an important role in regulation of SR Ca2+ release during β-adrenergic receptor stimulation or in heart failure [5, 7–10]. While RyR2 can be phosphorylated at multiple sites [11], phosphorylation of two serines (Ser-2808 and Ser-2030 in small rodents or Ser-2809 and Ser-2031 in rabbit and human) by PKA was suggested to be of functional relevance [5, 12, 13]. Despite significant efforts, the role of PKA-dependent phosphorylation for the regulation of RyR2 function remains a highly debated topic [14, 15]. Originally, it has been suggested that RyR2 phosphorylation by PKA plays a critical role in SR Ca2+ mishandling in failing heart. It has been shown that chronic RyR2 phosphorylation at Ser-2808 during heart failure causes dissociation of the small regulatory protein FKBP12.6 from the channel. Such alteration in RyR2 structure leads to increased SR Ca2+ leak [16] as a result of prolonged subconductance openings of RyR2 [5]. Moreover, transgenic mice with Ser-2808 replaced with alanine (RyR2-S2808A mice) were more resistant to development of heart failure [17], whereas mice with a mutation that mimics RyR2 phosphorylation at Ser-2808 developed cardiomyopathy associated with SR Ca2+ mishandling [18]. However, other studies have shown little or no effect of PKA-mediated RyR2 phosphorylation on SR Ca2+ release and cardiac pump function. It has been shown that RyR2 phosphorylation had no effect on local SR Ca2+ release events (Ca2+ sparks) in transgenic animals lacking PKA effect on SERCA-mediated SR Ca2+ uptake [19]. Moreover, other studies in RyR2-S2808A mice did not find any significant difference in cardiac function during adrenergic receptor activation or heart failure progression [20, 21]. Several recent studies of SR Ca2+ release in cardiomyocytes and RyR2 gating in lipid bilayers suggest that the effect of PKA-mediated RyR2 phosphorylation on RyR2 function is more complex than previously assumed. It has been shown that RyR2 is significantly phosphorylated at Ser-2808/9 under “basal” condition (in the absence of adrenergic stimulation or PKA activation) [13, 20, 22, 23] and reduction in RyR2 phosphorylation increases SR Ca2+ release, causing SR Ca2+ depletion [24]. Therefore, it seems that RyR2 dephosphorylation, rather than phosphorylation, increases the channel activity. Studies of RyR2 activity in lipid bilayers revealed an even more complex pattern of RyR2 regulation by phosphorylation. It has been found that minimum and maximum RyR2 phosphorylation at Ser-2808 both increase RyR2 activity, suggesting a V-shaped dependence of RyR2 activity from its PKA-dependent phosphorylation level [22]. However, a study of a transgenic mouse model susceptible to cardiac arrhythmias suggested that 50% phosphorylation of RyR2 at Ser-2808 had a more detrimental effect on heart function and SR Ca2+ handling than complete dephosphorylation or maximum phosphorylation (i.e. suggesting a bell-shaped relationship between RyR2 activity and its phosphorylation level) [25]. Thus, it is unclear whether these opposite findings are due to significant differences between the acute effect of RyR2 phosphorylation observed in bilayer experiments or the chronic effect of RyR2 phosphorylation in transgenic animals.
We have recently developed an experimental approach for direct measurement of RyR2 function in the cellular environment under well-controlled cytosolic conditions [2, 3, 26]. Here we applied this approach to investigate a possible functional link between RyR2 phosphorylation by PKA at two sites (Ser-2809 and Ser-2031) and RyR2-mediated Ca2+ release in rabbit ventricular myocytes. The obtained results revealed that RyR2 phosphorylation by PKA has a complex effect on SR Ca2+ release. Between the two PKA sites on RyR2, Ser-2809 was more sensitive to PKA activation. We found that phosphorylation of Ser-2809 at ~75% results in the lowest SR Ca2+ leak rate. Maximum phosphorylation and maximum dephosphorylation of Ser-2809 both increase SR Ca2+ leak. Unless RyR2 is regulated by PKA via some other unidentified phosphorylation sites, these findings suggest that RyR2 has the lowest SR Ca2+ leak rates at a specific intermediate phosphorylation level at Ser-2809.
MATERIALS AND METHODS
Myocyte Isolation
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committees of Loyola University and Rush University Medical Center, and comply with USA regulations on animal experimentation. Ventricular myocytes were isolated from hearts of New Zealand White rabbits (31 animals were used in this studies) according to the procedure described previously [27]. Chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated. All experiments were performed at room temperature (20–24oC).
Intracellular Ca2+ Imaging in Permeabilized Myocytes
To record cytosolic [Ca2+] ([Ca2+]i) and intra-SR [Ca2+] ([Ca2+]SR) we used the high affinity Ca2+ indicator Fluo-4 and the low affinity Ca2+ indicator Fluo-5N, respectively (both indicators were obtained from Thermo Fisher Scientific). Changes in [Ca2+]i and [Ca2+]SR were measured with laser scanning confocal microscopy (Radiance 2000 MP, Bio-Rad, UK or LSM 410, Carl Zeiss, Germany) equipped with a ×40 oil-immersion objective lens (N.A.=1.3). Fluo-4 and Fluo-5N were excited with the 488 nm line of an argon ion laser and fluorescence was measured at >515 nm.
Measurements of Ca2+ sparks
Myocytes were permeabilized with 0.005% saponin [28, 29]. The experimental solution containing Fluo-4 pentapotassium salt (30 μM) was composed of (in mM): K-aspartate 100; KCl 15; KH2PO4 5; MgATP 5; EGTA 0.35; CaCl2 0.067; MgCl2 0.75; phosphocreatine 10; HEPES 10; creatine phosphokinase 5 U/ml; dextran (MW: 40,000) 4%, and pH 7.2 (KOH). Free [Ca2+] and [Mg2+] of this solution were 150 nM and 1 mM, respectively. Ca2+ sparks were recorded in line scan mode (3 ms per scan; pixel size 0.12 μm). Ca2+ sparks were detected and analyzed using SparkMaster [30]. The analysis included Ca2+ spark frequency (sparks × s−1 × (100 μm) −1), amplitude (ΔF/F0), full duration at half-maximal amplitude (FDHM; ms), and full width at half-maximal amplitude (FWHM; μm). F0 is the initial fluorescence recorded under steady-state conditions and ΔF=F − F0.
Measurements of SR Ca2+ leak
SR Ca2+ leak as a function of [Ca2+]SR was measured in permeabilized myocytes as described previously [2]. The SR was loaded with the low-affinity Ca2+ dye Fluo-5N after incubating myocytes with 5 μM of Fluo-5N/AM for 2.5 hours at 37°C followed by 1 hour of dye wash out [27]. Fluo-5N was excited with minimum laser energy of an argon ion laser (to minimize dye photobleaching). To improve signal-to-noise ratio of the low intensity Fluo-5N signal, fluorescence was collected with an open pinhole and averaged over the entire cellular width of an individual 2-D image (pixel size 0.2 μm). At the end of each experiment minimum Fluo-5N fluorescence (Fmin) was measured after depletion of the SR with 10 mM caffeine in the presence of 5 mM EGTA. Maximum Fluo-5N fluorescence (Fmax) was measured following an increase of [Ca2+] to 10 mM in the presence of caffeine [2]. Caffeine keeps RyRs open allowing [Ca2+] equilibration across the SR membrane [31]. The Fluo-5N signal was converted to [Ca2+] using the formula: [Ca2+]SR = Kd × (F − Fmin)/(Fmax − F), where Kd was 390 μM [2]. SR Ca2+ leak was measured as the changes of total [Ca2+]SR ([Ca2+]SRT) over time (d[Ca2+]SRT/dt) after complete SERCA inhibition with thapsigargin (TG; 10 μM). [Ca2+]SRT was calculated as: [Ca2+]SRT = Bmax/(1+Kd/[Ca2+]SR) + [Ca2+]SR; where Bmax and Kd were 2700 μM and 630 μM, respectively [32]. The rate of SR Ca2+ leak (d[Ca2+]SRT/dt) was plotted as a function of [Ca2+]SR for each time point during [Ca2+]SR decline.
Western Blot analysis
Equal amounts of freshly isolated myocyte suspension were treated with different compounds (e.g. cAMP, staurosporine) under the same experimental conditions as for [Ca2+] measurements. After treatment, cells were lysed in Laemmli buffer (Sigma-Aldrich). An equal amount of total lysate from each sample was subjected to 4–15% SDS-PAGE and transferred to nitrocellulose membranes for Western Blot analysis. Changes in RyR2 phosphorylation level at two PKA sites (Ser 2809 and 2031) and the CaMKII site (Ser 2815) were quantified using phospho-specific antibodies RyR-PS2809, RyR-PS2031 and RyR-PS2815 (Badrilla, Leeds, UK). The signal was normalized to total RyR2 level measured with the antibody C34 (DSHB, University of Iowa, USA). The secondary antibody was HRP-conjugated, therefore the RyR2 band was visualized using the Luminata Forte Western HRP Substrate (Millipore; USA) and the signal was quantified using the UVP EpiChemi3 imaging system and ImageJ software.
Statistics
Data are presented as mean ± SEM of n measurements. In case of single cell experiments (such as Ca measurements), n represents the number of cells isolated from at least 3 different animals. In case of R Western Blot analysis, n represents the number of animals used in these experiments. When only two groups were compared, statistical significance was determined by Student’s t-test. Significance between multiple groups was determined by one-way ANOVA followed by a Newman-Keuls post-hoc test. P<0.05 was considered statistically significant.
RESULTS
The effect of cAMP on Ca2+ sparks in permeabilized myocytes
To study the effect of PKA-mediated RyR2 phosphorylation on Ca2+ sparks, cAMP was introduced into the cytosol after sarcolemma permeabilization. After permeabilization in control conditions, Ca2+ sparks were detected at a stable frequency of 6.2±1.1 sparks × s−1 × (100 μm) −1. cAMP (10 μM) increased Ca2+ spark frequency to 13.9±1.6 sparks × s−1 × (100 μm) −1 (n=18 cells; Fig. 1). Fig 1A (top) shows Fluo-4 line scan images and corresponding F/F0 profiles of Ca2+ sparks recorded in control conditions, in the presence of cAMP and after cAMP wash out. The stimulatory effect of cAMP on spark frequency remained stable during cAMP application and was fully reversible after cAMP wash out (Fig 1A; bottom). cAMP also increased Ca2+ spark amplitude and width (Fig 1B), while spark duration was not affected by cAMP. Adding the PKA catalytic subunit (5 U/ml) together with cAMP (10 μM) did not result in a more pronounced effect on spark frequency than cAMP alone (data not shown), suggesting that endogenous PKA is sufficient for maximum stimulation of spark activity by cAMP. The selective inhibitor of PKA, PKA inhibitory peptide (PKI; 5 μM), abolished the activation of Ca2+ sparks by cAMP (Fig 2). Although, in several cases (3 out of 7 cells, including the one shown in Fig 2) an activation of sparks was detected only at the beginning of cAMP application. These results suggest that: 1) the augmentation of local SR Ca2+ release by cAMP is mainly mediated by a PKA-dependent mechanism, and 2) endogenous PKA is capable of producing a maximal effect on SR Ca2+ release.
Figure 1. Effect of cAMP on Ca2+ sparks in permeabilized rabbit ventricular myocytes.

A Top, representative confocal line scan images with corresponding F/F0 profiles of Ca2+ sparks in control conditions, during addition of cAMP (10 μM), and following washout of cAMP. F/F0 profiles were obtained by averaging the fluorescence over the 1 μm wide regions indicated by the black bars. Bottom, representative example of cAMP effect on Ca2+ spark frequency and on SR Ca load estimated with caffeine. The black arrows indicate applications of caffeine in control conditions and during cAMP application. B, Ca2+ spark properties (frequency, amplitude, spatial width) and SR Ca2+ load in control conditions, during cAMP application, and after wash out of cAMP. *P<0.05 vs Control.
Figure 2. Effect of cAMP on Ca2+ sparks during inhibition of PKA.

Top, representative confocal line scan images with corresponding F/F0 profiles of Ca2+ sparks during treatment with PKI, followed by application of cAMP in the presence of PKI. F/F0 profiles were obtained by averaging the fluorescence signal over the 1 μm wide regions indicated by the black bars. Bottom, changes of Ca2+ spark frequency in the presence of PKI with and without cAMP present.
We also found that the increased Ca2+ spark activity during cAMP application was associated with an increase in SR Ca2+ load (Fig 1A and B). In these experiments, SR Ca2+ load was measured as the amplitude of cytosolic Ca2+ transient during a fast caffeine (10 mM) application (Fig 1A; bottom). Because RyR2 activity highly depends on [Ca2+]SR, the observed effect of cAMP on Ca2+ sparks is consistent with an elevated SR Ca2+ load (presumably due to activation of SERCA-dependent Ca2+ uptake).
The effect of cAMP on SR Ca2+ leak in permeabilized myocytes
To separate the effect of the cAMP-PKA pathway on RyR2-mediated Ca2+ release from its effect on SERCA-dependent Ca2+ uptake, we studied the effect of cAMP on [Ca2+]SR decline during SERCA inhibition. Under these conditions the rate of [Ca2+]SR decline represents the SR Ca2+ leak rate [2]. In the following experiments we used the low affinity Ca2+ dye Fluo-5N entrapped within the SR to directly measure changes in [Ca2+]SR. Fig 3A shows a representative example of [Ca2+]SR recording from a permeabilized ventricular myocyte in control conditions, during SERCA inhibition by thapsigargin (TG; 10 μM) and following RyR2 activation by caffeine (10 mM). The application of cAMP (10 μM) significantly increased [Ca2+]SR measured before SERCA inhibition (Fig 3B; black circles). On average, cAMP increased initial [Ca2+]SR from 811±15 to 1209±39 μM (n=12 cells). This effect of cAMP on SR Ca2+ load was entirely dependent on PKA activity, because PKI completely abolished this effect (Fig 2B; gray circles). At the same time, cAMP did not affect the rate of SR Ca2+ leak. In these experiments, SR Ca2+ leak was quantified as the time constant (τ) of [Ca2+]SR decline during SERCA inhibition (Fig 3C). Together, these data suggest that the cAMP effect on Ca2+ spark activity (Fig 1) was mainly due to an augmentation of SERCA-dependent Ca2+ uptake and SR Ca2+ load, and not due to RyR-mediated Ca2+ release.
Figure 3. Effect of cAMP on SR Ca2+ load and leak.

A, changes of [Ca2+]SR after SERCA inhibition with thapsigargin (TG). Application of caffeine at the end of experiment indicates complete Ca2+ depletion of the SR. B, changes of [Ca2+]SR during cAMP application (black circles) and during cAMP applications in the presence of PKI (grey circles). In both cases, thapsigargin (TG) was applied to inhibit SERCA. The dashed curves show fit of [Ca2+]SR decay during SERCA inhibition. C, time constant (τ) of [Ca2+]SR decay during SERCA inhibition in control conditions, during cAMP application and during cAMP applications in the presence of PKI.
Changes in RyR2 phosphorylation at PKA sites after membrane permeabilization
Using phospho-specific antibodies, we analyzed changes in RyR2 phosphorylation during PKA activation by cAMP. First, we measured RyR2 phosphorylation at Ser-2809, because RyR2 phosphorylation at this site has been suggested to play a key role in regulation of SR Ca2+ release during PKA activation, adrenergic stimulation and in heart failure [5, 17, 18, 33]. The specificity of the antibody for Ser-2809 (PS2809) has been characterized in detail previously [13]. Similarly to previously published work [13, 23, 34, 35], we found a significant level of RyR2 phosphorylation at Ser-2809 in intact myocytes in control conditions (Fig 4A). By comparing this “basal” phosphorylation level to the maximum RyR2 phosphorylation during PKA activation, we estimated that ~65% of RyR2 subunits are phosphorylated at Ser-2809 in intact myocytes in control conditions. More important, we found that the phosphorylation level at Ser-2809 progressively increased after membrane permeabilization (Fig 4A). 15 min after membrane permeabilization, RyR2 phosphorylation at Ser-2809 reached ~95% of its maximal level (Fig 4B). Thus, the lack of cAMP effect on SR Ca2+ leak (Fig 3) could be explained by the possibility that membrane permeabilization causes almost maximal phosphorylation of RyR2 at Ser-2809 and, thus, application of cAMP is unable to generate any further effect on RyR2 function.
Figure 4. Changes in RyR2 phosphorylation at the PKA site.
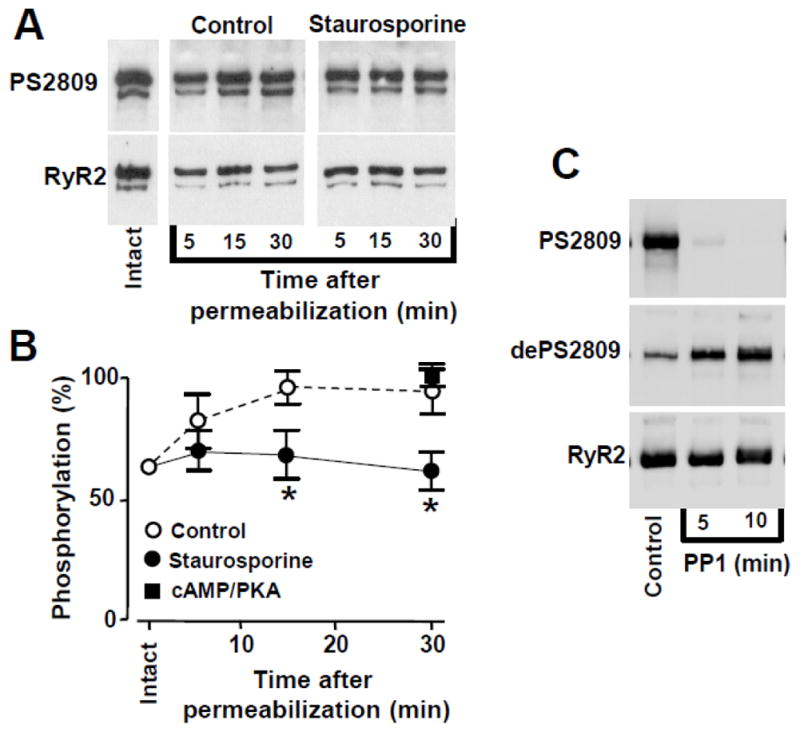
A, representative Western blot showing the effect of membrane permeabilization on RyR2 phosphorylation at the PKA site (Ser-2809) at three time points (5, 15, 30 min) in control conditions and in the presence of staurosporine. Changes in phosphorylation were compared to the level of phosphorylation in intact cardiomyocytes and were normalized to the total level of RyR2. B, averaged normalized data of phosphorylation level at Ser-2809 before and at different time points after permeabilization in control conditions (open circles), with staurosporine (black circles), and in the presence of PKA and cAMP (black squares). C, representative Western blot showing the effect of PP1 on RyR2 phosphorylation at the PKA site (Ser-2809) at two time points (5 and 10 min). The effect PP1 on RyR2 phosphorylation was determined with antibodies against phosphorylated (PS2809) and unphosphorylated (dePS2809) Sr-2809. *P<0.05 vs Control.
We hypothesized that the increase in RyR2 phosphorylation at Ser-2809 in permeabilized myocytes was the result of a shift in the balance between protein kinase and phosphatase activity towards enhanced RyR2 phosphorylation. Indeed, we found that incubation of myocytes with the broad-spectrum protein kinase inhibitor staurosporine (1 μM) before and after membrane permeabilization prevented the increase of RyR2 phosphorylation at Ser-2809 after membrane permeabilization (Fig 4B), i.e. staurosporine maintained Ser-2809 phosphorylation at ~65% of its maximum level. Incubation of myocytes with protein phosphatase 1 (PP1; 2 U/ml) completely dephosphorylated RyR2 at Ser-2809 (Fig 4C). The lack of phosphorylation of RyR2 was also confirmed by using antibodies against both phosphorylated (PS2809) and unphosphorylated (dePS2809) Ser-2809. Unlike Ser-2809, RyR2 phosphorylation at another PKA site Ser-2031 did not significantly change after membrane permeabilization. Moreover, cAMP, staurosporine and PP1 had negligible effects on RyR2 phosphorylation at this site under our experimental conditions (online supplement).
The pharmacological approach used here to manipulate RyR2 phosphorylation at PKA-specific sites can potentially also affect RyR2 phosphorylation at the CaMKII site. Thus, we analyzed changes in RyR2 phosphorylation at Ser-2815 under identical experimental conditions. We found that ~70% of RyR2 subunits were phosphorylated at CaMKII site (online supplement). However, the phosphorylation level at this site did not change significantly 15 min after membrane permeabilization in control conditions as well as in the presence of staurosporine. PP1 (2 U/ml) decreased phosphorylation of RyR2 at Ser-2815 by 21% (online supplement).
The effect of RyR2 phosphorylation on SR Ca2+ leak
Since staurosporine did not significantly affect RyR2 phosphorylation at Ser-2031and Ser-2815, we used this protein kinase inhibitor to prevent the increase in RyR2 phosphorylation at Ser-2809 after membrane permeabilization. When the RyR2 phosphorylation level at Ser-2809 was kept at its “basal” level (~65% of maximal phosphorylation obtained after incubation with cAMP/PKA; Fig 4B), SR Ca2+ leak rate was significantly slowed compared to control (Fig 5A). Concomitantly, staurosporine increased SR Ca2+ load (estimated from [Ca2+]SR measured before SERCA inhibition). However, complete dephosphorylation of RyR2 at Ser-2809 (Fig 4C) accelerated SR Ca2+ leak and depleted SR Ca2+ (Fig 5B). Partial dephosphorylation at the CaMKII site might also contribute to these effects. In Fig 5A and 5B, SR Ca2+ leak in control conditions is shown as black dashed line. Averaged results of staurosporine and PP1 effects on the rate of [Ca2+]SR decay (τ) during SERCA inhibition are shown in Fig 5C.
Figure 5. Effect of staurosporine and PP1 on SR Ca2+ load and leak.

Changes of [Ca2+]SR during staurosporine (A) and PP1 application (B). In both cases, thapsigargin (TG) was applied to inhibit SERCA. The dashed grey curve shows a fit of [Ca2+]SR decay during SERCA inhibition in the presence of staurosporine (A) or PP1 (B). The dashed black curves show fit of [Ca2+]SR decay during SERCA inhibition in control conditions (original data are shown in Fig 3A). C, time constant (τ) of [Ca2+]SR decay after SERCA inhibition in control conditions, during staurosporine and PP1 application. *P<0.05 vs Control.
For each experimental group we determined SR Ca2+ leak as a function of SR Ca2+ load. Free [Ca2+]SR after SERCA inhibition was converted to total [Ca2+]SR ([Ca2+]TSR) based on the known intra-SR Ca2+-buffer capacity (see Methods and [32]). SR Ca2+ leak rate, which was measured as changes of [Ca2+]TSR over time (d[Ca2+]TSR/dt), was plotted against the corresponding free [Ca2+]SR to obtain the Ca2+ leak-load relationship (Fig. 6A). We also analyzed SR Ca2+ leak in the presence of the RyR2 inhibitor ruthenium red (RuR; 20 μM). Thus, RyR2-mediated Ca2+ leak can be separated from other leak components. Fig 6B summarizes the effect of Sr-2809 phosphorylation on RyR2-mediated SR Ca2+ leak measured at [Ca2+]SR=750 μM, a level representative for diastolic SR Ca2+ load. Unless some other unidentified PKA phosphorylation sites affect RyR2 function, RyR2 phosphorylation at Ser-2809 has a complex and opposite effects on SR Ca2+ leak, dependent on phosphorylation level. At an intermediate level of RyR2 phosphorylation SR Ca2+ leak reaches a minimum. However, two extreme conditions associated with RyR2 phosphorylation (complete dephosphorylation or maximal phosphorylation of RyR2) drastically increase SR Ca2+ leak. Moreover, this characteristic effect of RyR2 phosphorylation was observed over a broad range of [Ca2+]SR (~50 to >1,000 μM), and thus appears to be independent of SR Ca2+ load.
Figure 6. Effect of RyR2 phosphorylation at the PKA site on SR Ca2+ leak.

A, RyR2-mediated SR Ca2+ leak rate plotted as a function of [Ca2+]SR measured under different experimental conditions: PP1, cAMP, control, staurosporine and Ruthenium Red (RuR). B, average SR Ca2+ leak rates measured at [Ca2+]SR = 750 μM under different experimental conditions. The RyR2-mediated Ca2+ leak was calculated by subtracting SR Ca2+ leak in the presence of RuR (marked by the black bars and the white dotted line) from the total SR Ca2+ leak. *P<0.05 vs Control.
DISCUSSION
In the heart, activation of the cAMP-PKA signaling pathway causes phosphorylation of several proteins that play a key role in the regulation of intracellular Ca2+ homeostasis and myocardial contraction. For example, phosphorylation of the dihydropyridine receptor (DHPR) and phospholamban (PLB) by PKA during adrenergic stimulation increases the transarcolemmal inward Ca2+ current and SR Ca2+ load [36]. Eventually, activation of these molecular mechanisms increases the action potential-induced Ca2+ transient, causing stronger myocardial contraction during systole (i.e. exerting a positive inotropic effect). RyR2 is also a target for PKA-mediated phosphorylation [4, 37, 38]. However, the contribution of RyR2 phosphorylation to the positive inotropic response is not well defined. It is not even clear whether this post-translational modification of RyR2 causes activation or inhibition of SR Ca2+ release. Such contradictory findings arose from several factors, including that RyR2-mediated Ca2+ release highly depends on SR Ca2+ load. Because PLB phosphorylation by PKA causes SERCA activation with subsequent increase of SR Ca2+ load, it remains challenging to separate the indirect effect of PKA on SR Ca2+ release via SR Ca2+ load from its direct effect on RyR2 activity. In this study, we used a novel approach that allows to simultaneously measure SR Ca2+ load and SR Ca2+ leak in ventricular myocytes [2]. Thus, we were able to define how RyR2 phosphorylation by PKA affects SR Ca2+ leak independently of PKA effects on SR Ca2+ load.
In agreement with previous work [19], we found that cAMP significantly increased Ca2+ spark amplitude and frequency in permeabilized ventricular myocytes (Fig 1). The catalytic subunit of PKA produced a similar effect on Ca2+ sparks as cAMP alone, suggesting that endogenous PKA is capable to activate SR Ca2+ release in the presence of cAMP. Inhibition of PKA with a potent and selective PKA inhibitor (PKI) prevented activation of Ca2+ spark by cAMP (Fig 2). These results suggest that PKA-independent cellular mechanisms (such as Epac) play a minor role in activation of SR Ca2+ release during cAMP accumulation. Moreover, the positive effect of cAMP on Ca2+ spark amplitude and frequency was associated with an increase in SR Ca2+ load. To determine whether PKA activation increased Ca2+ sparks directly by activating RyR2 or indirectly by activating SERCA, we measured the decline of [Ca2+]SR after SERCA inhibition (i.e. SR Ca2+ leak) in control conditions and during PKA activation. Results of these experiments revealed that SR Ca2+ leak was not affected by PKA activation (Fig 3). Because Ca2+ sparks are an important component of SR Ca2+ leak [2], the observed effect of cAMP/PKA on Ca2+ sparks was likely mediated by an increase in SR Ca2+ load.
In parallel experiments, we analyzed changes in RyR2 phosphorylation levels at two PKA sites (Ser-2809 and Ser-2031). Similarly to previously published work [13, 20, 22, 23], we found a significant level of RyR2 phosphorylation at Ser-2809 in intact myocytes (Fig 4). We estimated that ~65% of RyR2 subunits are phosphorylated at Ser-2809 in control conditions. It has been shown previously that kinases other than PKA are responsible for this “basal” RyR2 phosphorylation [13]. Importantly, however, the phosphorylation level at Ser-2809 progressively increased after membrane permeabilization (even without PKA activation), reaching almost maximal level within 15 min. At the same time, RyR2 at Ser-2031 was not significantly phosphorylated in control conditions (similar to [10, 13, 20]), after membrane permeabilization and during PKA activation (online supplement). Thus, the lack of cAMP effect on SR Ca2+ leak in permeabilized myocytes (Fig 3) can be explained by the fact that RyR2 at Ser-2809 was maximally phosphorylated after membrane permeabilization. These findings can explain some negative results reported previously regarding the lack of PKA effect on RyR2 in permeabilized cells [19].
Phosphorylation status of RyR2 is regulated by the balanced activities of protein kinases and phosphatases, which are bound to the channel [37]. It seems that membrane permeabilization causes a shift in this balance toward RyR2 phosphorylation at Ser-2809 (but not at Ser-2031). Inhibition of “basal” protein kinase activity with staurosporine reduced RyR2 phosphorylation at Ser-2809 to level observed in intact myocytes (~65%). At the same time, staurosporine significantly reduced SR Ca2+ leak (Fig 5). Interestingly, complete dephosphorylation of RyR2 at Ser-2809 accelerated SR Ca2+ leak and depleted SR Ca2+ load. These interventions, however, did not alter the Ser-2031 phosphorylation status. Unless RyR2 can be regulated by PKA via some other unidentified phosphorylation sites, these results suggest that RyR2 phosphorylation at Ser-2809 has a complex effect on SR Ca2+ leak in ventricular myocytes. At an intermediate level of RyR2 phosphorylation SR Ca2+ leak is minimal. However, complete dephosphorylation and maximal phosphorylation of RyR2 increases SR Ca2+ leak. These results are in agreement with a previous bilayer study showing a V-shaped dependence of RyR2 activity on its phosphorylation at Ser-2809 [22]. The pharmacological approach used here to manipulate RyR2 phosphorylation at PKA sites would potentially also affect CaMKII-dependent RyR2 phosphorylation. Although RyR2 is phosphorylated at the CaMKII site in control conditions, this level did not change after membrane permeabilization and during staurosporine application. The decrease in RyR2 phosphorylation at the CaMKII site during PP1 application might also contribute to an increase in SR Ca2+ leak.
The fact that 65% of RyR2 subunits are phosphorylated at Ser-2809 in intact myocytes in control conditions suggests that activation of cAMP/PKA signaling pathway will always activate SR Ca2+ leak by phosphorylating the remaining RyR2 subunits. Although the physiological importance of RyR2-mediated Ca2+ leak during adrenergic stimulation is not quite clear, the increased SR Ca2+ leak might work as a protective mechanism against SR Ca2+ overload during SERCA activation. However, in conditions of congestive heart failure when SR Ca2+ leak is already increased due to CaMKII activation, a further increase in SR Ca2+ leak can be detrimental for SR Ca2+ handling and electrical stability of ventricular myocytes.
Supplementary Material
Highlights.
The results of this study illustrate that phosphorylation of ryanodine receptor (RyR2) by protein kinase A (PKA) regulates sarcoplasmic reticulum (SR) Ca2+ leak in ventricular myocytes.
Complete dephosphorylation and maximal phosphorylation of RyR2 increase SR Ca2+ leak. However, an intermediate level of RyR2 phosphorylation reduces SR Ca2+ leak to its minimal level.
Among several phosphorylation sites on RyR2 tested, phosphorylation of serine 2809 has the most profound effect on RyR2 function.
Acknowledgments
This work was supported by the National Institutes of Health Grants HL130231 (to A.V.Z.) and HL62231, HL80101 and HL101235 (to L.A.B.) and the Leducq Foundation (to L.A.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 2.Zima AV, Bovo E, Bers DM, Blatter LA. Ca2+ spark-dependent and -independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol. 2010;588:4743–57. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bovo E, Mazurek SR, Blatter LA, Zima AV. Regulation of sarcoplasmic reticulum Ca2+ leak by cytosolic Ca2+ in rabbit ventricular myocytes. J Physiol. 2011;589:6039–50. doi: 10.1113/jphysiol.2011.214171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch. 2014;466:1129–37. doi: 10.1007/s00424-014-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 6.Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, et al. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res. 2009;84:387–95. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht: Kluwer Academic Publishers; 2001. [Google Scholar]
- 8.El-Armouche A, Eschenhagen T. Beta-adrenergic stimulation and myocardial function in the failing heart. Heart Fail Rev. 2009;14:225–41. doi: 10.1007/s10741-008-9132-8. [DOI] [PubMed] [Google Scholar]
- 9.Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–12. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- 10.Xiao B, Zhong G, Obayashi M, Yang D, Chen K, Walsh MP, et al. Ser-2030, but not Ser-2808, is the major phosphorylation site in cardiac ryanodine receptors responding to protein kinase A activation upon beta-adrenergic stimulation in normal and failing hearts. Biochem J. 2006;396:7–16. doi: 10.1042/BJ20060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takasago T, Imagawa T, Furukawa K, Ogurusu T, Shigekawa M. Regulation of the cardiac ryanodine receptor by protein kinase-dependent phosphorylation. J Biochem. 1991;109:163–70. doi: 10.1093/oxfordjournals.jbchem.a123339. [DOI] [PubMed] [Google Scholar]
- 12.Xiao B, Jiang MT, Zhao M, Yang D, Sutherland C, Lai FA, et al. Characterization of a novel PKA phosphorylation site, serine-2030, reveals no PKA hyperphosphorylation of the cardiac ryanodine receptor in canine heart failure. Circ Res. 2005;96:847–55. doi: 10.1161/01.RES.0000163276.26083.e8. [DOI] [PubMed] [Google Scholar]
- 13.Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376:80–5. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bers DM. Ryanodine receptor S2808 phosphorylation in heart failure: smoking gun or red herring. Circ Res. 2012;110:796–9. doi: 10.1161/CIRCRESAHA.112.265579. [DOI] [PubMed] [Google Scholar]
- 15.Valdivia HH. Ryanodine receptor phosphorylation and heart failure: phasing out S2808 and "criminalizing" S2814. Circ Res. 2012;110:1398–402. doi: 10.1161/CIRCRESAHA.112.270876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, et al. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–6. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 17.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, et al. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–98. doi: 10.1172/JCI32726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, et al. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J Clin Invest. 2010;120:4375–87. doi: 10.1172/JCI37649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Kranias EG, Mignery GA, Bers DM. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ Res. 2002;90:309–16. doi: 10.1161/hh0302.105660. [DOI] [PubMed] [Google Scholar]
- 20.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, et al. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–29. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Makarewich CA, Kubo H, Wang W, Duran JM, Li Y, et al. Hyperphosphorylation of the cardiac ryanodine receptor at serine 2808 is not involved in cardiac dysfunction after myocardial infarction. Circ Res. 2012;110:831–40. doi: 10.1161/CIRCRESAHA.111.255158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98:1506–13. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- 23.Bovo E, Lipsius SL, Zima AV. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during beta-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol. 2012;590:3291–304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Gyorke S. Protein phosphatases decrease sarcoplasmic reticulum calcium content by stimulating calcium release in cardiac myocytes. J Physiol. 2003;552:109–18. doi: 10.1113/jphysiol.2003.046367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu B, Ho HT, Velez-Cortes F, Lou Q, Valdivia CR, Knollmann BC, et al. Genetic ablation of ryanodine receptor 2 phosphorylation at Ser-2808 aggravates Ca(2+)-dependent cardiomyopathy by exacerbating diastolic Ca2+ release. J Physiol. 2014;592:1957–73. doi: 10.1113/jphysiol.2013.264689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zima AV, Picht E, Bers DM, Blatter LA. Termination of cardiac Ca2+ sparks: role of intra-SR [Ca2+], release flux, and intra-SR Ca2+ diffusion. Circ Res. 2008;103:e105–e115. doi: 10.1161/CIRCRESAHA.107.183236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domeier TL, Blatter LA, Zima AV. Alteration of sarcoplasmic reticulum Ca2+ release termination by ryanodine receptor sensitization and in heart failure. J Physiol. 2009;587:5197–209. doi: 10.1113/jphysiol.2009.177576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zima AV, Picht E, Bers DM, Blatter LA. Partial inhibition of sarcoplasmic reticulum ca release evokes long-lasting ca release events in ventricular myocytes: role of luminal ca in termination of ca release. Biophys J. 2008;94:1867–79. doi: 10.1529/biophysj.107.114694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zima AV, Copello JA, Blatter LA. Effects of cytosolic NADH/NAD(+) levels on sarcoplasmic reticulum Ca(2+) release in permeabilized rat ventricular myocytes. J Physiol. 2004;555:727–41. doi: 10.1113/jphysiol.2003.055848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster - Automated Calcium Spark Analysis with ImageJ. Am J Physiol Cell Physiol. 2007 doi: 10.1152/ajpcell.00586.2006. [DOI] [PubMed] [Google Scholar]
- 31.Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free [Ca2+] in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–5. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 32.Shannon TR, Ginsburg KS, Bers DM. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys J. 2000;78:322–33. doi: 10.1016/S0006-3495(00)76595-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511–8. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang MT, Lokuta AJ, Farrell EF, Wolff MR, Haworth RA, Valdivia HH. Abnormal Ca2+ release, but normal ryanodine receptors, in canine and human heart failure. Circ Res. 2002;91:1015–22. doi: 10.1161/01.res.0000043663.08689.05. [DOI] [PubMed] [Google Scholar]
- 35.Carter S, Colyer J, Sitsapesan R. Maximum phosphorylation of the cardiac ryanodine receptor at serine-2809 by protein kinase a produces unique modifications to channel gating and conductance not observed at lower levels of phosphorylation. Circ Res. 2006;98:1506–13. doi: 10.1161/01.RES.0000227506.43292.df. [DOI] [PubMed] [Google Scholar]
- 36.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 37.Marks AR. Cardiac intracellular calcium release channels: role in heart failure. Circ Res. 2000;87:8–11. doi: 10.1161/01.res.87.1.8. [DOI] [PubMed] [Google Scholar]
- 38.George CH. Sarcoplasmic reticulum Ca2+ leak in heart failure: mere observation or functional relevance? Cardiovasc Res. 2008;77:302–14. doi: 10.1093/cvr/cvm006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.