

Abstract
Diabetes mellitus (DM) displays a high morbidity. The diabetic heart is susceptible to myocardial ischemia/reperfusion (MI/R) injury. Impaired activation of prosurvival pathways, endoplasmic reticulum (ER) stress, increased basal oxidative state, and decreased antioxidant defense and autophagy may render diabetic hearts more vulnerable to MI/R injury. Oxidative stress and mTOR signaling crucially regulate cardiometabolism, affecting MI/R injury under diabetes. Producing reactive oxygen species (ROS) and reactive nitrogen species (RNS), uncoupling nitric oxide synthase (NOS), and disturbing the mitochondrial quality control may be three major mechanisms of oxidative stress. mTOR signaling presents both cardioprotective and cardiotoxic effects on the diabetic heart, which interplays with oxidative stress directly or indirectly. Antihyperglycemic agent metformin and newly found free radicals scavengers, Sirt1 and CTRP9, may serve as promising pharmacological therapeutic targets. In this review, we will focus on the role of oxidative stress and mTOR signaling in the pathophysiology of MI/R injury in diabetes and discuss potential mechanisms and their interactions in an effort to provide some evidence for cardiometabolic targeted therapies for ischemic heart disease (IHD).
1. Introduction
Diabetes mellitus (DM) is a major risk factor for ischemic heart disease (IHD) [1]. The alteration of glucose metabolism leads to cardiac structural and functional perturbations, including left ventricular (LV) dysfunction, cardiac hypertrophy, and myocardial interstitial fibrosis. A number of diabetic subjects suffer from the impairments of diastolic dysfunction in an early stage without overt cardiovascular symptoms [2–4]. Cardiac hypertrophy is originally a compensatory response to pathological overload stress. However, the persistent DM-induced hypertrophy ultimately becomes maladaptive since it evolves into cardiac dysfunction, and finally develops into heart failure [5–7]. Hyperglycemia directly increases cardiac fibroblast and vascular smooth muscle cell proliferation and is associated with endothelial dysfunction, resulting in microvascular injury and hemodynamic alteration, which contribute to the vulnerability of tissue ischemia injury [8, 9]. Myocardial salvage after reperfusion may be limited by deleterious changes in the microcirculation of ischemic tissue [10]. All these pathophysiologic changes in the diabetic heart lead to a susceptibility to ischemia/reperfusion (I/R) injury [9, 11]. Consequences of increased cellular apoptosis and inflammation are present in the diabetic heart subjected to I/R injury [12–14]. It is truly different from the normotensive mechanisms since metabolic abnormalities and alteration of oxidative stress and autophagy. Among all these factors, oxidative stress and the mammalian target of rapamycin (mTOR) signaling are two critical ones [15, 16].
Oxidative stress is defined as an imbalance between free radicals production and destruction, which leads to multiple negative effects on cellular metabolism. mTOR kinase is also necessary for normal regulation of cardiac structure and cardiometabolic homeostasis. It promotes mitochondrial function in response to insulin resistance and affects cardiac energy deprivation and ischemia [17, 18]. Both of them participate in the pathogenesis and progression of myocardial ischemia/reperfusion (MI/R) injury under diabetes, acting as key regulators of cardiometabolism and cardiac function. However, the relationship between oxidative stress and mTOR signaling is complicated, since mTOR not only modulates oxidative stress but is also affected by reactive oxygen species (ROS) activation. In this review, we will focus on the role of oxidative stress and mTOR signaling in the pathophysiology of I/R injury in the diabetic heart and highlight their current interactions in an effort to provide some evidence for the potential cardiometabolic targeted therapies for IHD.
2. The Vulnerability of Diabetic Heart Subjected to MI/R Injury
DM severely damages cardiac energy homeostasis, leading to the cardiac dysfunction. It is well recognized that populations associated with DM were more likely to develop IHD and their long-term outcome is worsened [19]. Importantly, physical or pharmacologic ischemic preconditioning (IPC) and ischemic postconditioning (I-post) actions are ineffective under diabetic conditions [20–22], suggesting that the diabetic heart may be resistant to common cardioprotections.
2.1. Impaired Activation of Prosurvival Pathways
In the diabetic heart, the alteration of reperfusion injury salvage kinase (RISK) signaling significantly suppressed the cardioprotective effects of IPC [23, 24]. Studies demonstrated that glycogen synthase kinase-3β (GSK-3β) was activated by insulin resistance, thus inhibiting the prosurvival pathway of the phosphoinositide-3 kinase- (PI3k-) Akt signaling and the Janus-activated kinase- (JAK-) transcription 3 (STAT3) signaling, finally blunting the cardioprotective effects of I-post [25, 26]. Moreover, our previous study proved that adiponectin (APN) resistance existed in the diabetic cardiomyocytes and impaired APN's cardioprotection against MI/R injury [27]. APN resistance led to the dysfunctional APN-AMP-activated protein kinase (AMPK) axis and blocked the AMPK-independent antiperoxide/antinitration pathway, increasing the vulnerability of diabetic cardiomyocytes to I/R injury [27, 28].
2.2. Endoplasmic Reticulum (ER) Stress
Disturbed cardiometabolic homeostasis facilitates ER stress. The unfolded protein response (UPR) was proved to be involved in the pathogenesis of DM [29, 30]. Miki et al. demonstrated that DM-induced ER stress augmentation enhanced the mitochondrial permeability transition pore (mPTP) opening and increased mitochondrial calcium overload via inhibition of extracellular regulated MAP kinase (ERK) 1/2- GSK-3β pathway [31]. In contrast, suppression of ER stress could reduce myocardial infarction (MI) size in high fat diet- (HFD-) induced type 2 diabetes mellitus (T2DM) [32]. Our recent study found that preconditioning of C1q/TNF-related protein (CTRP) 9, a newly identified homologous of APN, protected the diabetic heart against I/R injury by reducing ER stress and inflammatory response [33].
2.3. Increased Basal Oxidative State and Impaired Antioxidant Signaling
Hyperglycemia enhances oxidative stress, promotes profibrogenic genes expression, and aggravates MI/R injury [34, 35]. ROS accumulation not only results from overproduction of free radicals, but also may be a consequence of decreased free radicals scavenger systems, including superoxide dismutase (Cu/Zn-SOD and Mn-SOD), catalase (CAT), and glutathione peroxidase (GPx) [36]. Cardiac expression of GPx levels is reduced in the diabetic apolipoprotein E-deficient mice [37]. Meanwhile, attempts to attenuate I/R injury using enzymatic and nonenzymatic antioxidants have not been universally successful in DM [38, 39].
2.4. Autophagy and mTOR Signaling
Autophagy is a cellular degradation pathway that crucially mediates cardiometabolism. It has been demonstrated that autophagy was required for IPC via mTOR signaling and Parkin-dependent pathway [40, 41]. However, mitochondrial biogenesis is impaired in the diabetic heart, following the alteration of autophagic activity. Hyperglycemia largely inhibited cardiac autophagosome and autolysosome formation by modulating mTOR-ULK1 signaling [42]. It deteriorated the cardioprotection of remote IPC (rIPC) because of the increase in nitrative stress and inhibition of autophagy via activation of mTOR signaling [43, 44].
3. The Role of Oxidative Stress in MI/R Injury under Diabetes
Oxidative stress is regarded as an imbalance between the generation and elimination of free radicals due to increased ROS and/or inadequate antioxidant defenses [45]. It develops directly or indirectly from hyperglycemia, hyperlipidemia, and insulin resistance under DM [15, 46] and in turn, disturbs metabolic hemostasis and impairs cardiac function. When available in appropriate amounts, free radicals act as signal transduction molecules while in large excess, they lead to DNA degeneration, lipid oxidation and membrane protein degeneration. However, in the diabetic heart, insulin resistance increases cardiomyocytes fatty acid oxidation together with a reduction of prostacyclin synthesis and endothelial nitric oxide (eNOS) synthase activity [47]. These changes lead to generation of ROS and reactive nitrogen species (RNS), endothelium dysfunction, formation of advanced glaciation end products, and alteration of the mitochondrial quality control, all of which contribute to the deleterious MI/R injury under diabetes [48]. Thus, DM-induced oxidative stress can be a primary component that initiates the onset and progression of cardiac dysfunction in MI/R injury (Figure 1).
Figure 1.
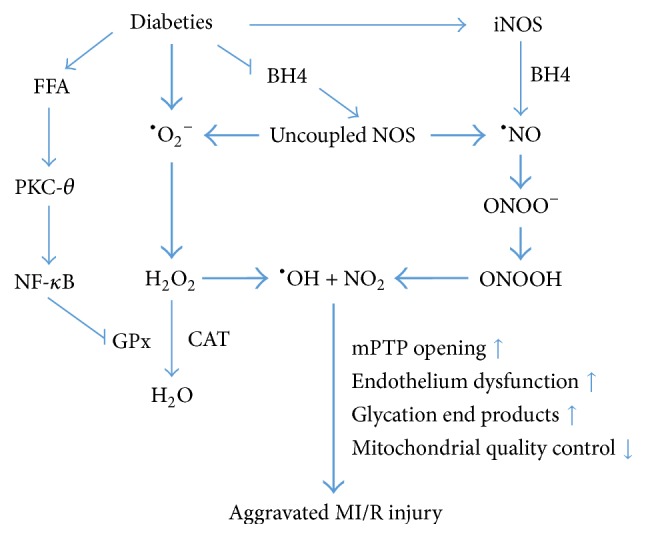
DM-induced higher basal oxidative state plays a master role in the progression of cardiometabolic disorders and negatively affects the MI/R injury. In this state, ROS and RNS accumulate dramatically. They initiate the reaction of ∙OH in parallel with the ONOO−/ONOOH generation, which becomes strong cytotoxic oxidant and causes oxidative damage and nitration. These then lead to endothelium dysfunction, formation of advanced glycation end products, and alteration of the mitochondrial quality control, all contributing to the deleterious MI/R injury in diabetic hearts. Free fatty acid (FFA); protein kinase C-θ (PKC-θ); nuclear factor of kappa light polypeptide gene (NF-κB); superoxide (∙O2−); hydrogen peroxide (H2O2); glutathione peroxidase (GPx); catalase (CAT); hydroxyl (∙OH); tetrahydrobiopterin (BH4); nitric oxide synthase (NOS); inducible NOS (iNOS); nitric oxide (∙NO); peroxynitrite (ONOO−); peroxynitrous acid (ONOOH); nitrogen dioxide (NO2); mitochondrial permeability transition pore (mPTP).
3.1. ROS and RNS Production
ROS are a group of short-lived, low-molecular-weight compounds derived from variety of reactions oxygen undergoes, including superoxide (∙O2−), hydroxyl (∙OH), hydrogen peroxide (H2O2), and hypochlorous acid (HOCl). The generation of ROS in the heart is few under physiologic conditions. ∙O2− leakages from mitochondrial electron transport chains (ETC) and soon be catalyzed into less cytotoxic H2O2 by SOD catalyzes, then finally be converted into water and molecular oxygen by either CAT or GPx system [45]. However, the homeostasis of cardiac oxidative state would be broken under DM since the generation of ∙O2− increased markedly. The accumulated ∙O2− is highly diffusible and damages cardiomyocytes [49]. H2O2 is more likely converted to ∙OH other than scavenged by CAT or GPx [50]. Moreover, hyperglycemia increases cardiac free fatty acid (FFA) levels, which extensively leads to a great rise of ROS formation and a reduction of GPx by activating nuclear factor of kappa light polypeptide gene (NF-κB) [51, 52] and its upstream mediator protein kinase C-θ (PKC-θ) [53]. ROS enhances mPTP opening, contributing to myocardial contractile dysfunction and tissue damage in ischemia-reperfused rat hearts [54].
Diabetic myocardial RNS production is also greatly increased, including radicals nitric oxide (∙NO) and nitrogen dioxide (∙NO2−). The rapid reaction of superoxide with nitric oxide (NO) forms a highly reactive intermediate, peroxynitrite (ONOO−), under MI/R injury. With increased intracellular acidification, ONOO− becomes more protonated to form peroxynitrous acid (ONOOH), which then rapidly turns into nitrogen dioxide (NO2) and ∙OH. The ONOO−/ONOOH becomes strong cytotoxic oxidant and causes oxidative damage and nitration, which contributes in parallel with the reaction of ∙OH generation during MI/R [45].
3.2. Uncoupled NOS
Diabetic mice exhibited increased risk of aggravated MI/R injury primarily because of impaired NO bioavailability. ONOO− may uncouple eNOS via oxidation of tetrahydrobiopterin (BH4), thus leads to further superoxide generation and an enhanced NO depletion [55]. However, reduced availability of BH4 was identified in diabetic rat vessels and endothelial cells. DM-induce NADPH increase further predisposes the heart to NOS uncoupling and ONOO− generation [56]. Maalouf et al. demonstrated that S-glutathionylation uncoupled eNOS and subsequently impaired endothelium-dependent vasodilation under oxidative stress [57]. Moreover, inducible NOS (iNOS) is activated in DM by inflammatory mediators, which makes iNOS uncoupling a predominant contributor for oxidative/nitrosative stress in diabetic myocardium [58].
3.3. Disturbing the Mitochondrial Quality Control
Mitochondria are the major sites of ROS production (0.2% to 2% of total oxygen taken by cells). These ROS can be scavenged by mitochondrial quality control to keep the mitochondria functional [59]. However, in the diabetic heart, mitochondrial quality control is damaged together with impaired mitochondrial respiratory capacity, leading to a dramatic accumulation of ROS [15]. Importantly, increased mitochondrial H2O2 emission then damages DNA, proteins, and lipid in membrane components and finally results in mitochondrial dysfunction [60]. The myocardium of db/db mice exhibited increased mitochondrial H2O2 generation, and overproduction of mitochondrial ROS occurring in conjunction with augmented electron delivery from increased fatty acid oxidation [51]. Taken together, these studies suggest that mitochondrial quality control regulates cellular oxidative stress, while, if damaged, oxidative stress in turn might affect mitochondrial dysfunction under DM.
4. The Dual Role of mTOR Signaling in MI/R Injury under Diabetes
mTOR is a 289 kDa serine/threonine kinase that crucially mediates energy metabolism [61]. It has two distinct multiprotein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [62–64]. mTORC1 regulates cellular homeostasis, stress responses, energy metabolism and autophagy by relying on the regulatory associated protein of mTOR (Raptor). In contrast, mTORC2 treats rapamycin-insensitive companion of mTOR (Rictor) as the component rather than Raptor and controls cell growth, survival, migration, and cell cycle progression [65]. mTOR kinase is necessary for normal regulation of cardiac structure and cardiometabolism. It also takes part in the maintenance of normal microvascular barrier function and endothelial permeability. However, the role of mTOR signaling in MI/R injury is still controversial. Researchers have found both cardioprotective and cardiotoxic effects of mTOR signaling when using its inhibitor-rapamycin or transgenic animals [66]. Besides, there is a complicated interplay between mTOR signaling and oxidative stress (Table 1).
Table 1.
Cardioprotective and cardiotoxic effects of mTOR signaling in MI/R injury under diabetes.
| Effect of mTOR | Study | Animal model | Interventions | Outcomes |
|---|---|---|---|---|
| Cardioprotective | Glazer et al. [67] | Transgenic mice | Overexpression of cardiac mTOR | Overexpression of cardiac mTOR reduced mortality in the acute phase and preserved cardiac function in the chronic phase after transient ischemia in vivo |
|
| ||||
| Cardioprotective | Land and Tee [68] | Transgenic mice | Overexpression of cardiac mTOR | mTOR-Tg mice performed better cardiac function recovery and had less of the necrotic markers CK and LDH subjected to I/R injury in high fat diet-induced obesity |
|
| ||||
| Cardioprotective | Park et al. [69] | Diabetic mice induced by STZ | Rapamycin (5 mg/kg i.v.) 10 min before I/R | Lin28a overexpression increased p-mTOR and p-p70s6k expression in myocardium exposed to I/R injury in diabetic mice while inhibition of mTOR reduced Lin28a's cardioprotective effects |
|
| ||||
| Cardioprotective | Schenkel et al. [70] | C57BL/6 mice | Torin1 (i.p.) immediately after MI with a short 2-day follow-up treatment to inhibit both mTORC1 and mTORC2 Rictor and Raptor siRNA (i.v.) to selectively inhibit mTORC1 or mTORC2 PRAS40 siRNA (i.v.) to inhibit mTOR1 |
Inhibition of both mTORC1 and mTORC2 with Torin1 led to increased cardiomyocyte apoptosis and tissue damage after MI. Predominant mTORC1 signaling by suppression of mTORC2 similarly increased cardiomyocyte apoptosis and tissue damage after myocardial infarction. In comparison, preferentially shifting toward mTORC2 signaling by inhibition of mTORC1 with PRAS40 led to decreased cardiomyocyte damage after MI |
|
| ||||
| Cardioprotective | Tanguy et al. [50] | Neonatal rat ventricular cardiomyocytes | Rapamycin: 50 nM Adenovirus overexpressing mTOR |
Inhibition of mTOR by rapamycin antagonized high glucose-induced inhibition of autophagy and enhanced cardiomyocyte death, while adenovirus-mediated overexpression of mTOR was sufficient to block autophagic flux regardless of glucose concentrations |
|
| ||||
| Cardioprotective | Rajapakse et al. [71] | Human umbilical vein endothelial cells (HUVECs) | Rapamycin: 25 nM | mTOR activation enhances the activity of HIF1α by inhibiting proteolytic degradation, resulting in elevated VEGF expression |
|
| ||||
| Cardioprotective | Chong et al. [72] | Human endothelial cells | Rapamycin: 5–10 ng/mL | Loss of mTOR blocks endothelial proliferation and angiogenesis as well as the proliferation of endothelial progenitor cells ex vivo |
|
| ||||
| Cardiotoxic | Yao et al. [73] | APN knockout mice | Rapamycin (2 mg/kg, i.p.) | Rapamycin reversed APN deficiency-induced drop of fat oxidation in high fat diet feeding |
|
| ||||
| Cardiotoxic | Si et al. [74] | Transgenic mice | Conditional mTOR knockout mice | Inhibition of mTORC1 reduced endoplasmic reticulum stress, thereby reducing cardiomyocytes death |
|
| ||||
| Cardiotoxic | Lemaître et al. [75] | CD-1 mice | Rapamycin (0.25 mg/kg, i.p.) | Inhibition of mTOR by rapamycin before ischemia reduced I/R-induced myocardial infarction via activating the JAK2 signal transducer and activator of transcription 3 (STAT3) signaling pathway |
|
| ||||
| Cardiotoxic | Fourcade et al. [76] | Transgenic mice | Cardiac-specific knockout Raptor to inhibit mTORC1 in vivo | In cardiac mTORC1 disrupted mice, fatty acid oxidation is significantly decreased, whereas glucose oxidation is increased subjected to transverse aortic constriction (TAC) |
|
| ||||
| Cardiotoxic | Maiese et al. [77] | Male WKY rats and HUVECs | Recombinant adenoviral (rAd) expressing short hairpin RNA (shRNA), S6K1 to inhibit mTORC1 | Inhibition of mTORC1/S6K1 signaling protected endothelial dysfunction related to eNOS uncoupling in vivo and in vitro |
|
| ||||
| Cardiotoxic | Wang et al. [78] | AMPKα2 knockout mice | Metformin (100 mg/kg/day, gavage) for 3 weeks | Administration of metformin was effective in attenuating TAC-induced LV remodeling in both wild-type and AMPKα2 knockout mice and reduced p-mTOR at Ser2448 and its downstream target p-p70S6K at Thr389 |
4.1. Cardiotoxic Effects of mTOR Signaling
Chronic increase of mTORC1 activity in T2DM causes insulin resistance, which contributes to hyperinsulinemia and hyperglycemia [79–81]. Evidences showed that mTORC1 was activated in the hearts of obese and diabetic animals during reperfusion, increasing the vulnerability of MI/R injury. In HFD-induced obesity mice, cardiac autophagosome formation was decreased, accompanied by cardiac dysfunction, which could be reversed by rapamycin (2 mg/kg, intraperitoneal injection, i.p.) and worsened by genetic APN disruption [82]. mTOR phosphorylates the mammalian homologue of autophagy related gene 13 (Atg13) and the mammalian Atg1 homologues UNC-51-like kinase 1 (ULK1) and ULK2 to prevent the progression of autophagy. Sciarretta et al. confirmed that rapamycin administration (1 mg/kg, i.p.) or partial mTOR deletion significantly reduced infarct size after ischemia through the restoration of autophagy [83]. Our previous studies also found that hypertension-induced mTOR activation altered cardiac morphology, function, and autophagy, which could be rescued by cardiac-specific overexpression of metallothionein [84, 85].
Importantly, activation of mTORC1 other than mTOR2 signaling affects cardiac metabolism and the susceptibility to ischemia injury [67]. A patient-level meta-analysis of randomized trials showed that selective activation of mTORC2 with concurrent inhibition of mTORC1 decreased cardiomyocytes apoptosis and tissue damage after MI [86]. It seems that different complex of mTOR performs different cardiac functions. Another cardiotoxic mTOR effect is altering STAT3 signaling pathway in the diabetic heart. Das et al. found that inhibition of mTOR by rapamycin (0.25 mg/kg, i.p.) before ischemia reduced I/R-induced MI in CD-1 mice via activating the JAK2-STAT3 signaling [87]. This was further proved in cardiac-specific STAT3-deficient mice [88].
4.2. Cardioprotections of mTOR Signaling
There are four major mTOR-related cardioprotective pathways: (1) insulin-mediated PI3K/Akt/mTOR signaling pathway; (2) GSK-3β inhibition signaling pathway; (3) mTOR-dependent angiogenesis signaling pathway; (4) mTORC2 activation signaling pathway. Cardiac PI3K/Akt causes insulin-stimulated glucose uptake and induces acute mTOR activation, thus improving cardiomyocytes survival and function [73]. Studies found that the PI3K/Akt/mTOR signaling pathway provided efficient cardioprotection against I/R injury induced by insulin [74]. Aoyagi et al. further observed that cardiac-specific transgenic mice overexpressing mTOR suppressed I/R-induced inflammation and necrosis, inhibited cardiac fibrosis in adverse LV remodeling in diet-induced obesity mice [89]. They demonstrated that Akt phosphorylation was higher in mTOR overexpressed mice than WT mice under HFD conditions and it was unlikely that mTOR's cardioprotective effects were mediated through autophagic activity. Zhang et al. also demonstrated that Lin28a overexpression protected against MI/R injury in diabetic mice through the insulin-PI3K-mTOR pathway [90].
mTOR's cardioprotection required the inhibition of GSK-3β to reduce the reperfusion injury through mTORC1 hyperactivation [85]. During periods of I-post, mTOR prevents cardiomyocytes apoptosis via mTOR-dependent GSK-3β inhibition mechanisms. mTORC1 regulates mPTP opening and promotes mitochondrial biogenesis, which may favor cardiac recovery after MI/R and promote the upregulation of antioxidant genes via the activation of proliferator-activated receptor γ coactivator-1α (PGC-1α) [85, 91, 92].
The altered lipid metabolism induced by insulin resistance results in a propensity for microvascular barrier dysfunction, accelerated atherosclerosis, increased vessel wall reactivity, and plaque complications. Angiogenesis is an important component of cardioprotection against I/R injury, which has been proved to be mechanically via mTOR-dependent pathway. Inhibition of mTOR signaling by rapamycin (2 μM) for 1 h leads to subsequent impaired angiogenesis in aortic endothelial cells [75]. Loss of mTOR activity by rapamycin (5–10 ng/mL) also blocks endothelial proliferation and angiogenesis [93] as well as the proliferation of endothelial progenitor cells ex vivo [94]. Hypoxia activates the mTOR pathway to promote angiogenesis and cell proliferation [93, 95]. mTOR activation enhances the activity of HIF1α by inhibiting proteolytic degradation, resulting in elevated VEGF expression. This effect is reversible by rapamycin (25 nM for human umbilical vein endothelial cells and 50 nM for HEK293 cells) [68, 69].
Study found that the cardioprotective effects mediated by mTOR overexpression were partly dependent on mTORC2 activation, which was beneficial to cardiomyocytes survival against I/R injury as well as chronic ischemic remodeling [96]. Considering that mTORC2 is rapamycin-insensitive, it is reasonable that using rapamycin to inhibit mTORC1 activity also presents cardioprotective actions against MI/R injury [97]. However, there was still little understanding of the complexity of mTORC2's regulation and its roles in cardiac functions.
4.3. Interactions between Oxidative Stress and mTOR Signaling
Cardiac mTOR is considered as an important regulator of oxidative stress by promoting mitochondrial biogenesis and oxidative metabolism through Ying-Yang 1- (YY1-) PGC-1α pathway [92]. Meanwhile, mTOR modulates autophagy, increases mitochondrial clearance and protects cardiomyocytes from oxidative stress-induced toxicity [72, 98] through the activation of protein kinase B (PKB) [99]. In contrast, other studies found that in cardiac mTOR disrupted mice, fatty acid oxidation is significantly decreased, whereas glucose oxidation is increased [100]. mTOR regulates oxidative stress-induced endothelium dysfunction. Inhibition of mTORC1 either with rapamycin or by S6K1 silencing recouples eNOS function, improves NO production, and inhibits O2− generation in the rat aortas [71]. mTOR also modulates cardiac fibrosis in the models of post-MI remodeling and cardiac hypertrophy [70, 101, 102] while treatment with rapamycin reduced ROS production in the myofibroblasts.
On the other hand, oxidative stress regulates mTORC1 ordinarily. ROS production contributes to the inhibition of GSK-3β and mTOR signaling [85]. Alternative origins of ROS, such as NADPH oxidase, may as well provoke mTOR activation and subsequent impair autophagy [76]. An intriguing link between peroxisomes, oxidative stress and autophagy has been recently described. Peroxisomal ROS has been shown to suppress mTORC1 activity, in models of the tuberous sclerosis complex signaling node TSC1 and TSC2 proteins [77, 103]. In contrast, Vigneron et al. found that, in the isolated-perfused mouse heart, IPC protected against I/R injury via inhibition of GSK-3β and a constant opening of mitoKATP with ROS generation to activate the mTOR pathway and induce cardioprotection [104].
5. Potential Cardiometabolic Target against Diabetic MI/R Injury
Although animal studies have found potential regulator aiming at oxidative stress or mTOR signaling under experimental diabetic conditions, clinical studies are still disappointing. Thus, new therapeutic targets as well as efficient cardioprotections against DM-induced MI/R injury are urgently needed.
5.1. Metformin
It is well recognized that metformin could reduce cardiovascular end points of T2DM independently from its glucose-lowering effects. Administration of metformin significantly attenuates I/R injury via relieving ER stress [105] and activating of AMPK-eNOS prosurvival pathway in both nondiabetic and diabetic mice [105, 106]. However, further research demonstrated that metformin effectively attenuated LV hypertrophy and dysfunction by activating mTOR, p70S6K (Thr389), and S6 phosphorylation in both wild-type and AMPKα2 KO mice, suggesting that metformin attenuated myocardial mTOR signaling independently of AMPKα2 activation [107]. Metformin reduces ROS generation and ameliorates oxidative stress-induced apoptosis and inflammation in cardiomyocytes [108] and endothelial cells [109]. It also protects against I/R-induced myocardial fibrosis by inhibiting fibrotic factors, including TGF-β1, TNF-α and basic fibroblast growth factor (bFGF) in the circulation and the myocardium [110, 111]. As a routine oral agent for T2DM, metformin might be a potential pharmacological therapeutic target to protect against MI/R injury under diabetes on the regulation of cardiac oxidative stress and mTOR signaling.
5.2. Sirtuin 1 (Sirt1)
Sirt1 is a member of Sirtuins family [77]. It controls cellular processes and maintains metabolic homeostasis by reducing apoptosis, attenuating inflammation, and modulating oxidative stress [112, 113]. It is a critical regulator in DM-induced MI/R injury. Sirt1-mediated PGC-1α activation could directly respond to H2O2-induced oxidative stress on the regulation of glutathione GPx1, CAT, and Mn-SOD [114]. Overexpression of Sirt1 inhibited oxidative stress and reduced MI/R injury via modulating eNOS activity under diabetic condition [113]. There is a crosstalk between AMPK, Sirt1 and mTOR signaling in the regulation of oxidative stress and cardiomyocytes autophagy [115–117]. Sirt1 deacetylates FoxO3a while mTORC1 can inhibit FoxO-mediated transcription of antioxidant gene targets, including the antioxidants Mn-SOD and catalase [116]. Meanwhile, Sirt1 positively regulates transcription of Rictor, activating the mTORC2 signaling by triggering a cascade of Akt and FoxO phosphorylation. Sirt1 deficiency mice performed increased ROS production and impaired mTORC2 signaling, leading to insulin resistance that could be largely reversed with antioxidant treatment [78]. Considering its specific functions in modulating oxidative stress, mTOR signaling, and mitochondrial dysfunction perturbed in the diabetic heart, Sirt1 may be a promising novel therapeutic target for MI/R injury under DM.
5.3. CTRP9
CTRP9 is a newly found APN paralog. It protects against obesity and T2DM through anti-inflammation and antiapoptotic actions. Increasing the circulating CTRP9 level is a beneficial action against HFD-induced obesity and glucose intolerance [118], whereas CTRP9-deficiency mice performed exacerbated insulin resistance [119]. Importantly, CTRP9 performs cardioprotective effects via inhibition of oxidative stress. Kambara et al. demonstrated that administration of exogenous CTRP9 inhibited oxidative stress, attenuated cardiomyocytes apoptosis, and suppressed inflammatory reactions in the ischemic heart [120, 121]. Su et al. observed the same results in the HFD-induced T2DM mice, implicating that differing from APN, there is no CTRP9 resistance in DM [122]. Our recent finding proved that CTRP9 protected the diabetic heart against I/R injury by reducing ER stress and inflammatory response [33]. Interestingly, compared to general pharmacologic antioxidants, the amount of cardiac endogenous CTRP9 is abundant, much higher than its expression in adipocytes and circulation, suggesting that CTRP9 may be a novel cardiokine. These findings indicate that CTRP9 may also be a potential therapeutic target for diabetic cardiac complications.
6. Conclusions
It is well established that DM aggravates MI/R injury and diabetic IHD patients experience worse clinical outcomes. Oxidative stress and mTOR signaling are master mediators of cardiometabolism and MI/R injury. ROS and RNS accumulation induces cardiomyocytes damage by direct oxidation of proteins, reactive lipid peroxidation products, and interaction with DNA. Uncoupling NOS tigers oxidation/nitration reaction and disturbing the mitochondrial quality control causes mitochondrial dysfunction. These may be mechanisms of oxidative stress impairing the diabetic heart.
When turning to mTOR signaling, it is still controversial to clearly understand the role of mTOR signaling in MI/R injury under DM since both cardioprotective and cardiotoxic effects were observed in vivo and in vitro. The conflicted outcomes could be explained by the following. (1) There is different duration of rapamycin treatment [123]. Study demonstrated that inhibition of mTORC1 before ischemia reduced the size of MI while rapamycin was not cardioprotective if administered before the reperfusion phase [87]. Moreover, different duration of rapamycin treatment contributes to the alteration of metabolic homeostasis. Houde et al. found that administration of rapamycin for two weeks could enhance the insulin level, leading to a glucose intolerance and insulin resistance in mice. However, more than six weeks treatment could improve insulin sensitivity [124]. (2) There is different phosphorylation site of mTORC1. mTORC1 predominately phosphorylated the specific site encompassing 4E-BP1 (T37) and (T46) that are rapamycin resistant. However, mTORC1 could phosphorylated S6K1 (T389), which is rapamycin sensitive under conditions. (3) There are degrees of mTOR activation in the regulation of autophagy. Yu et al. demonstrated that mTOR signaling was inhibited during autophagy initially, but reactivated with prolonged autophagy. The progress was autophagy-dependent and required the degradation of autolysosomal products. The enhanced mTOR activity in verse attenuated autophagy [125]. (4) There are different cardiac functions of mTORC1 and mTORC2. mTORC1 presents both beneficial and detrimental effects on MI/R injury while mTORC2 show mostly cardioprotective actions as its cellular survival functions [96, 97]. (5) There are inescapable defects of loss-of-function animal models. Conventional ablation of mTOR in mice results in embryonic death [126–128] while cardiac-specific mTOR knockout mouse also shows fatal, dilated cardiomyopathy [64]. Other deletions of mTOR downstream molecules including Raptor and S6K1 may partially inhibit mTOR signaling and also be detrimental since not only the maladaptive but also the physiological functions of the kinase are ablated.
The interplay between oxidative stress and mTOR signaling is complicated, since mTOR not only modulates oxidative stress but also is affected by oxidative stress activation [129]. However, it is unlikely that these fully explain what occurs in the diabetic heart, considering its complicated pathophysiological conditions. Further studies using appropriate in vivo models of DM are needed (Figure 2).
Figure 2.

The diabetic heart is susceptible to MI/R injury. Impaired activation of prosurvival pathways, endoplasmic reticulum (ER) stress, increased basal oxidative state, and decreased antioxidant defense and autophagy may render diabetic hearts to be more vulnerable to MI/R injury and be resistant to ischemic preconditioning (IPC) or ischemic postconditioning (I-post). Oxidative stress and mTOR signaling crucially regulate cardiometabolism, affecting MI/R injury under diabetes. Reperfusion injury salvage kinase (RISK); phosphoinositide-3 kinase (PI3k); glycogen synthase kinase-3β (GSK-3β); signal transducer and activator of transcription (STAT); autophagy related gene 13 (Atg13); mammalian Atg1 homologues UNC-51-like kinase (ULK); Janus kinase 2 (JAK2); extracellular regulated MAP kinase (ERK).
No therapeutic strategy has yet been demonstrated clinically effective against cardiac injury in diabetic population. Antihyperglycemic agent metformin and newly found free radicals scavengers, Sirt1 and CTRP9, may serve as promising pharmacological cardiometabolic targeted therapeutic genes.
Competing Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Mozaffarian D., Benjamin E. J., Go A. S., et al. Executive summary: heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133(4):447–454. doi: 10.1161/cir.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.Zoroufian A., Razmi T., Taghavi-Shavazi M., Lotfi-Tokaldany M., Jalali A. Evaluation of subclinical left ventricular dysfunction in diabetic patients: longitudinal strain velocities and left ventricular dyssynchrony by two-dimensional speckle tracking echocardiography study. Echocardiography. 2014;31(4):456–463. doi: 10.1111/echo.12389. [DOI] [PubMed] [Google Scholar]
- 3.Hamdani N., Hervent A.-S., Vandekerckhove L., et al. Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovascular Research. 2014;104(3):423–431. doi: 10.1093/cvr/cvu223. [DOI] [PubMed] [Google Scholar]
- 4.Shah A. M., Shin S. H., Takeuchi M., et al. Left ventricular systolic and diastolic function, remodelling, and clinical outcomes among patients with diabetes following myocardial infarction and the influence of direct renin inhibition with aliskiren. European Journal of Heart Failure. 2012;14(2):185–192. doi: 10.1093/eurjhf/hfr125. [DOI] [PubMed] [Google Scholar]
- 5.Frey N., Olson E. N. Cardiac hypertrophy: the good, the bad, and the ugly. Annual Review of Physiology. 2003;65:45–79. doi: 10.1146/annurev.physiol.65.092101.142243. [DOI] [PubMed] [Google Scholar]
- 6.Molkentin J. D., Dorn G. W., II Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annual Review of Physiology. 2001;63(1):391–426. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- 7.Frangogiannis N. G. Matricellular proteins in cardiac adaptation and disease. Physiological Reviews. 2012;92(2):635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert R. E. Endothelial loss and repair in the vascular complications of diabetes—mechanisms and therapeutic implications. Circulation Journal. 2013;77(4):849–856. doi: 10.1253/circj.cj-13-0236. [DOI] [PubMed] [Google Scholar]
- 9.Everett B. M., Brooks M. M., Vlachos H. E. A., Chaitman B. R., Frye R. L., Bhatt D. L. Troponin and cardiac events in stable ischemic heart disease and diabetes. The New England Journal of Medicine. 2015;373(7):610–620. doi: 10.1056/nejmoa1415921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emanueli C., Caporali A., Krankel N., Cristofaro B., Van Linthout S., Madeddu P. Type-2 diabetic Lepr(db/db) mice show a defective microvascular phenotype under basal conditions and an impaired response to angiogenesis gene therapy in the setting of limb ischemia. Frontiers in Bioscience. 2007;12:2003–2012. doi: 10.2741/2205. [DOI] [PubMed] [Google Scholar]
- 11.Pfeffer M. A., Claggett B., Diaz R., et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. The New England Journal of Medicine. 2015;373(23):2247–2257. doi: 10.1056/nejmoa1509225. [DOI] [PubMed] [Google Scholar]
- 12.Suzuki H., Kayama Y., Sakamoto M., et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes. 2015;64(2):618–630. doi: 10.2337/db13-1896. [DOI] [PubMed] [Google Scholar]
- 13.Connelly K. A., Kelly D. J., Zhang Y., et al. Functional, structural and molecular aspects of diastolic heart failure in the diabetic (mRen-2)27 rat. Cardiovascular Research. 2007;76(2):280–291. doi: 10.1016/j.cardiores.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 14.Rajesh M., Mukhopadhyay P., Btkai S., et al. Cannabidiol attenuates cardiac dysfunction, oxidative stress, fibrosis, and inflammatory and cell death signaling pathways in diabetic cardiomyopathy. Journal of the American College of Cardiology. 2010;56(25):2115–2125. doi: 10.1016/j.jacc.2010.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson E. J., Kypson A. P., Rodriguez E., Anderson C. A., Lehr E. J., Neufer P. D. substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. Journal of the American College of Cardiology. 2009;54(20):1891–1898. doi: 10.1016/j.jacc.2009.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Braunwald E. Biomarkers in heart failure. The New England Journal of Medicine. 2008;358(20):2148–2159. doi: 10.1056/nejmra0800239. [DOI] [PubMed] [Google Scholar]
- 17.Parra V., Verdejo H. E., Iglewski M., et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt-mTOR-NFκB-Opa-1 signaling pathway. Diabetes. 2014;63(1):75–88. doi: 10.2337/db13-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsui Y., Takagi H., Qu X., et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and beclin 1 in mediating autophagy. Circulation Research. 2007;100(6):914–922. doi: 10.1161/01.res.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 19.Nguyen Q. M., Sinivasan S. R., Xu J.-H., Chen W., Berenson G. S. Changes in risk variables of metabolic syndrome since childhood in pre-diabetic and type 2 diabetic subjects. The Bogalusa Heart Study. Diabetes Care. 2008;31(10):2044–2049. doi: 10.2337/dc08-0898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miki T., Itoh T., Sunaga D., Miura T. Effects of diabetes on myocardial infarct size and cardioprotection by preconditioning and postconditioning. Cardiovascular Diabetology. 2012;11, article 67 doi: 10.1186/1475-2840-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drenger B., Ostrovsky I. A., Barak M., Nechemia-Arbely Y., Ziv E., Axelrod J. H. Diabetes blockade of sevoflurane postconditioning is not restored by insulin in the rat heart: phosphorylated signal transducer and activator of transcription 3- and phosphatidylinositol 3-kinase-mediated inhibition. Anesthesiology—The Journal of the American Society of Anesthesiologists. 2011;114(6):1364–1372. doi: 10.1097/aln.0b013e31820efafd. [DOI] [PubMed] [Google Scholar]
- 22.Ferdinandy P., Hausenloy D. J., Heusch G., Baxter G. F., Schulz R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacological reviews. 2014;66(4):1142–1174. doi: 10.1124/pr.113.008300. [DOI] [PubMed] [Google Scholar]
- 23.Hausenloy D. J., Lecour S., Yellon D. M. Reperfusion injury salvage kinase and survivor activating factor enhancement prosurvival signaling pathways in ischemic postconditioning: two sides of the same coin. Antioxidants & Redox Signaling. 2011;14(5):893–907. doi: 10.1089/ars.2010.3360. [DOI] [PubMed] [Google Scholar]
- 24.Ghaboura N., Tamareille S., Ducluzeau P.-H., et al. Diabetes mellitus abrogates erythropoietin-induced cardioprotection against ischemic-reperfusion injury by alteration of the RISK/GSK-3β signaling. Basic Research in Cardiology. 2011;106(1):147–162. doi: 10.1007/s00395-010-0130-3. [DOI] [PubMed] [Google Scholar]
- 25.Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? Journal of Molecular and Cellular Cardiology. 2009;47(1):32–40. doi: 10.1016/j.yjmcc.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 26.Zhuo C., Wang Y., Wang X., Wang Y., Chen Y. Cardioprotection by ischemic postconditioning is abolished in depressed rats: role of Akt and signal transducer and activator of transcription-3. Molecular and Cellular Biochemistry. 2011;346(1-2):39–47. doi: 10.1007/s11010-010-0589-0. [DOI] [PubMed] [Google Scholar]
- 27.Yi W., Sun Y., Gao E., et al. Reduced cardioprotective action of adiponectin in high-fat diet–induced type II diabetic mice and its underlying mechanisms. Antioxidants & Redox Signaling. 2011;15(7):1779–1788. doi: 10.1089/ars.2010.3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y., Gao E., Tao L., et al. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation. 2009;119(6):835–844. doi: 10.1161/circulationaha.108.815043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eizirik D. L., Cardozo A. K., Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 30.Yang L., Zhao D., Ren J., Yang J. Endoplasmic reticulum stress and protein quality control in diabetic cardiomyopathy. Biochimica et Biophysica Acta—Molecular Basis of Disease. 2015;1852(2):209–218. doi: 10.1016/j.bbadis.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Miki T., Miura T., Hotta H., et al. Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho–glycogen synthase kinase-3β–mediated suppression of mitochondrial permeability transition. Diabetes. 2009;58(12):2863–2872. doi: 10.2337/db09-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barr L. A., Shimizu Y., Lambert J. P., Nicholson C. K., Calvert J. W. Hydrogen sulfide attenuates high fat diet-induced cardiac dysfunction via the suppression of endoplasmic reticulum stress. Nitric Oxide. 2015;46:145–156. doi: 10.1016/j.niox.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai S., Cheng L., Yang Y., et al. C1q/TNF-related protein 9 protects diabetic rat heart against ischemia reperfusion injury: role of endoplasmic reticulum stress. Oxidative Medicine and Cellular Longevity. 2016;2016:14. doi: 10.1155/2016/1902025.1902025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aragno M., Mastrocola R., Alloatti G., et al. Oxidative stress triggers cardiac fibrosis in the heart of diabetic rats. Endocrinology. 2008;149(1):380–388. doi: 10.1210/en.2007-0877. [DOI] [PubMed] [Google Scholar]
- 35.Su H., Ji L., Xing W., et al. Acute hyperglycaemia enhances oxidative stress and aggravates myocardial ischaemia/reperfusion injury: role of thioredoxin-interacting protein. Journal of Cellular and Molecular Medicine. 2013;17(1):181–191. doi: 10.1111/j.1582-4934.2012.01661.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghattas M. H., Abo-Elmatty D. M. Association of polymorphic markers of the catalase and superoxide dismutase genes with type 2 diabetes mellitus. DNA and Cell Biology. 2012;31(11):1598–1603. doi: 10.1089/dna.2012.1739. [DOI] [PubMed] [Google Scholar]
- 37.Lewis P., Stefanovic N., Pete J., et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation. 2007;115(16):2178–2187. doi: 10.1161/CIRCULATIONAHA.106.664250. [DOI] [PubMed] [Google Scholar]
- 38.Luan R., Liu S., Yin T., et al. High glucose sensitizes adult cardiomyocytes to ischaemia/reperfusion injury through nitrative thioredoxin inactivation. Cardiovascular Research. 2009;83(2):294–302. doi: 10.1093/cvr/cvp085. [DOI] [PubMed] [Google Scholar]
- 39.Kain V., Kumar S., Sitasawad S. L. Azelnidipine prevents cardiac dysfunction in streptozotocin-diabetic rats by reducing intracellular calcium accumulation, oxidative stress and apoptosis. Cardiovascular Diabetology. 2011;10, article no. 97 doi: 10.1186/1475-2840-10-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang C., Andres A. M., Ratliff E. P., Hernandez G., Lee P., Gottlieb R. A. Preconditioning involves selective mitophagy mediated by parkin and p62/SQSTM1. PLoS ONE. 2011;6(6) doi: 10.1371/journal.pone.0020975.e20975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andres A. M., Hernandez G., Lee P., et al. Mitophagy is required for acute cardioprotection by simvastatin. Antioxidants & Redox Signaling. 2014;21(14):1960–1973. doi: 10.1089/ars.2013.5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi S., Xu X., Chen K., Liang Q. Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy. 2012;8(4):577–592. doi: 10.4161/auto.18980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J., Rohailla S., Gelber N., et al. MicroRNA-144 is a circulating effector of remote ischemic preconditioning. Basic Research in Cardiology. 2014;109(5) doi: 10.1007/s00395-014-0423-z.423 [DOI] [PubMed] [Google Scholar]
- 44.Baranyai T., Nagy C. T., Koncsos G., et al. Acute hyperglycemia abolishes cardioprotection by remote ischemic perconditioning. Cardiovascular Diabetology. 2015;14(1, article 151) doi: 10.1186/s12933-015-0313-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raedschelders K., Ansley D. M., Chen D. D. Y. The cellular and molecular origin of reactive oxygen species generation during myocardial ischemia and reperfusion. Pharmacology and Therapeutics. 2012;133(2):230–255. doi: 10.1016/j.pharmthera.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Carson J. L., Scholz P. M., Chen A. Y., Peterson E. D., Gold J., Schneider S. H. Diabetes mellitus increases short-term mortality and morbidity in patients undergoing coronary artery bypass graft surgery. Journal of the American College of Cardiology. 2002;40(3):418–423. doi: 10.1016/S0735-1097(02)01969-1. [DOI] [PubMed] [Google Scholar]
- 47.Du X., Edelstein D., Obici S., Higham N., Zou M.-H., Brownlee M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. The Journal of Clinical Investigation. 2006;116(4):1071–1080. doi: 10.1172/jci23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Orasanu G., Plutzky J. The pathologic continuum of diabetic vascular disease. Journal of the American College of Cardiology. 2009;53(5):S35–S42. doi: 10.1016/j.jacc.2008.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yokota T., Kinugawa S., Hirabayashi K., et al. Oxidative stress in skeletal muscle impairs mitochondrial respiration and limits exercise capacity in type 2 diabetic mice. American Journal of Physiology—Heart and Circulatory Physiology. 2009;297(3):H1069–H1077. doi: 10.1152/ajpheart.00267.2009. [DOI] [PubMed] [Google Scholar]
- 50.Tanguy S., De Leiris J., Besse S., Boucher F. Ageing exacerbates the cardiotoxicity of hydrogen peroxide through the Fenton reaction in rats. Mechanisms of Ageing and Development. 2003;124(2):229–235. doi: 10.1016/S0047-6374(02)00185-9. [DOI] [PubMed] [Google Scholar]
- 51.Yamagishi S.-I., Edelstein D., Du X.-L., Kaneda Y., Guzmán M., Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. The Journal of Biological Chemistry. 2001;276(27):25096–25100. doi: 10.1074/jbc.m007383200. [DOI] [PubMed] [Google Scholar]
- 52.Lee J. Y., Sohn K. H., Rhee S. H., Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through toll-like receptor 4. Journal of Biological Chemistry. 2001;276(20):16683–16689. doi: 10.1074/jbc.m011695200. [DOI] [PubMed] [Google Scholar]
- 53.Coudronniere N., Villalba M., Englund N., Altman A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(7):3394–3399. doi: 10.1073/pnas.060028097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ananthakrishnan R., Kaneko M., Hwang Y. C., et al. Aldose reductase mediates myocardial ischemia-reperfusion injury in part by opening mitochondrial permeability transition pore. American Journal of Physiology—Heart and Circulatory Physiology. 2009;296(2):H333–H341. doi: 10.1152/ajpheart.01012.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang W. H., Kravtsov G. M., Sauert M., et al. Polyol pathway impairs the function of SERCA and RyR in ischemic-reperfused rat hearts by increasing oxidative modifications of these proteins. Journal of Molecular and Cellular Cardiology. 2010;49(1):58–69. doi: 10.1016/j.yjmcc.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen C.-A., Wang T.-Y., Varadharaj S., et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468(7327):1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maalouf R. M., Eid A. A., Gorin Y. C., et al. Nox4-derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. American Journal of Physiology—Cell Physiology. 2012;302(3):C597–C604. doi: 10.1152/ajpcell.00331.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Okazaki T., Otani H., Shimazu T., et al. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. Journal of Molecular and Cellular Cardiology. 2011;50(3):534–544. doi: 10.1016/j.yjmcc.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 59.Ashrafi G., Schwarz T. L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death & Differentiation. 2013;20(1):31–42. doi: 10.1038/cdd.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy M. P. Induction of mitochondrial ROS production by electrophilic lipids: a new pathway of redox signaling? American Journal of Physiology—Heart and Circulatory Physiology. 2006;290(5):H1754–H1755. doi: 10.1152/ajpheart.00040.2006. [DOI] [PubMed] [Google Scholar]
- 61.Wullschleger S., Loewith R., Hall M. N. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 62.Johnson S. C., Rabinovitch P. S., Kaeberlein M. MTOR is a key modulator of ageing and age-related disease. Nature. 2013;493(7432):338–345. doi: 10.1038/nature11861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Laplante M., Sabatini D. M. Regulation of mTORC1 and its impact on gene expression at a glance. Journal of Cell Science. 2013;126(8):1713–1719. doi: 10.1242/jcs.125773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang D., Contu R., Latronico M. V. G., et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. Journal of Clinical Investigation. 2010;120(10):p. 3735. doi: 10.1172/jci43008c1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Laplante M., Sabatini D. M. MTOR signaling in growth control and disease. Cell. 2012;149(2):274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aoyagi T., Kusakari Y., Xiao C.-Y., et al. Cardiac mTOR protects the heart against ischemia-reperfusion injury. American Journal of Physiology—Heart and Circulatory Physiology. 2012;303(1):H75–H85. doi: 10.1152/ajpheart.00241.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Glazer H. P., Osipov R. M., Clements R. T., Sellke F. W., Bianchi C. Hypercholesterolemia is associated with hyperactive cardiac mTORC1 and mTORC2 signaling. Cell Cycle. 2009;8(11):1738–1746. doi: 10.4161/cc.8.11.8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Land S. C., Tee A. R. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. Journal of Biological Chemistry. 2007;282(28):20534–20543. doi: 10.1074/jbc.M611782200. [DOI] [PubMed] [Google Scholar]
- 69.Park J.-W., Kim W.-H., Shin S.-H., et al. Visfatin exerts angiogenic effects on human umbilical vein endothelial cells through the mTOR signaling pathway. Biochimica et Biophysica Acta—Molecular Cell Research. 2011;1813(5):763–771. doi: 10.1016/j.bbamcr.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 70.Schenkel P. C., Tavares A. M. V., Fernandes R. O., et al. Time course of hydrogen peroxide-thioredoxin balance and its influence on the intracellular signalling in myocardial infarction. Experimental Physiology. 2012;97(6):741–749. doi: 10.1113/expphysiol.2012.064832. [DOI] [PubMed] [Google Scholar]
- 71.Rajapakse A. G., Yepuri G., Carvas J. M., et al. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS ONE. 2011;6(4) doi: 10.1371/journal.pone.0019237.e19237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chong Z. Z., Shang Y. C., Maiese K. Cardiovascular disease and mTOR signaling. Trends in Cardiovascular Medicine. 2011;21(5):151–155. doi: 10.1016/j.tcm.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yao H., Han X., Han X. The cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling pathway. American Journal of Cardiovascular Drugs. 2014;14(6):433–442. doi: 10.1007/s40256-014-0089-9. [DOI] [PubMed] [Google Scholar]
- 74.Si R., Tao L., Zhang H. F., et al. Survivin: a novel player in insulin cardioprotection against myocardial ischemia/reperfusion injury. Journal of Molecular and Cellular Cardiology. 2011;50(1):16–24. doi: 10.1016/j.yjmcc.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 75.Lemaître V., Dabo A. J., D'Armiento J. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicological Sciences. 2011;123(2):542–549. doi: 10.1093/toxsci/kfr181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fourcade S., Ferrer I., Pujol A. Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: a paradigm for axonal degeneration. Free Radical Biology and Medicine. 2015;88:18–29. doi: 10.1016/j.freeradbiomed.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 77.Maiese K., Chong Z. Z., Shang Y. C., Wang S. Translating cell survival and cell longevity into treatment strategies with SIRT1. Romanian Journal of Morphology and Embryology. 2011;52(4):1173–1185. [PMC free article] [PubMed] [Google Scholar]
- 78.Wang R.-H., Kim H.-S., Xiao C., Xu X., Gavrilova O., Deng C.-X. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. The Journal of Clinical Investigation. 2011;121(11):4477–4490. doi: 10.1172/jci46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ranieri S. C., Fusco S., Panieri E., et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(30):13420–13425. doi: 10.1073/pnas.1008647107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Drake J. C., Alway S. E., Hollander J. M., Williamson D. L. AICAR treatment for 14 days normalizes obesity-induced dysregulation of TORC1 signaling and translational capacity in fasted skeletal muscle. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2010;299(6):R1546–R1554. doi: 10.1152/ajpregu.00337.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Turdi S., Kandadi M. R., Zhao J., Huff A. F., Du M., Ren J. Deficiency in AMP-activated protein kinase exaggerates high fat diet-induced cardiac hypertrophy and contractile dysfunction. Journal of Molecular and Cellular Cardiology. 2011;50(4):712–722. doi: 10.1016/j.yjmcc.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo R., Zhang Y., Turdi S., Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2013;1832(8):1136–1148. doi: 10.1016/j.bbadis.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sciarretta S., Zhai P., Shao D., et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125(9):1134–1146. doi: 10.1161/circulationaha.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang L., Gao J.-Y., Ma J., et al. Cardiac-specific overexpression of metallothionein attenuates myocardial remodeling and contractile dysfunction in l-NAME-induced experimental hypertension: role of autophagy regulation. Toxicology Letters. 2015;237(2):121–132. doi: 10.1016/j.toxlet.2015.06.005. [DOI] [PubMed] [Google Scholar]
- 85.Jung C. H., Jun C. B., Ro S.-H., et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular Biology of the Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.De Waha A., Dibra A., Kufner S., et al. Long-term outcome after sirolimus-eluting stents versus bare metal stents in patients with Diabetes mellitus: a patient-level meta-analysis of randomized trials. Clinical Research in Cardiology. 2011;100(7):561–570. doi: 10.1007/s00392-010-0278-8. [DOI] [PubMed] [Google Scholar]
- 87.Das A., Salloum F. N., Durrant D., Ockaili R., Kukreja R. C. Rapamycin protects against myocardial ischemia–reperfusion injury through JAK2–STAT3 signaling pathway. Journal of Molecular and Cellular Cardiology. 2012;53(6):858–869. doi: 10.1016/j.yjmcc.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Das A., Salloum F. N., Filippone S. M., et al. Inhibition of mammalian target of rapamycin protects against reperfusion injury in diabetic heart through STAT3 signaling. Basic Research in Cardiology. 2015;110(3) doi: 10.1007/s00395-015-0486-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aoyagi T., Higa J. K., Aoyagi H., Yorichika N., Shimada B. K., Matsui T. Cardiac mTOR rescues the detrimental effects of diet-induced obesity in the heart after ischemia-reperfusion. American Journal of Physiology—Heart and Circulatory Physiology. 2015;308(12):H1530–H1539. doi: 10.1152/ajpheart.00008.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang M., Sun D., Li S., et al. Lin28a protects against cardiac ischaemia/reperfusion injury in diabetic mice through the insulin-PI3K-mTOR pathway. Journal of Cellular and Molecular Medicine. 2015;19(6):1174–1182. doi: 10.1111/jcmm.12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu Z., Xu X., Hu X., et al. PGC-1α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxidants & Redox Signaling. 2010;13(7):1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cunningham J. T., Rodgers J. T., Arlow D. H., Vazquez F., Mootha V. K., Puigserver P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature. 2007;450(7170):736–740. doi: 10.1038/nature06322. [DOI] [PubMed] [Google Scholar]
- 93.Humar R., Kiefer F. N., Berns H., Resink T. J., Battegay E. J. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB Journal. 2002;16(8):771–780. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 94.Miriuka S. G., Rao V., Peterson M., et al. mTOR inhibition induces endothelial progenitor cell death. American Journal of Transplantation. 2006;6(9):2069–2079. doi: 10.1111/j.1600-6143.2006.01433.x. [DOI] [PubMed] [Google Scholar]
- 95.Bernardi R., Guernah I., Jin D., et al. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature. 2006;442(7104):779–785. doi: 10.1038/nature05029. [DOI] [PubMed] [Google Scholar]
- 96.Völkers M., Konstandin M. H., Doroudgar S., et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128(19):2132–2144. doi: 10.1161/CIRCULATIONAHA.113.003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Thoreen C. C., Sabatini D. M. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009;5(5):725–726. doi: 10.4161/auto.5.5.8504. [DOI] [PubMed] [Google Scholar]
- 98.Wagner C., Tillack D., Simonis G., Strasser R. H., Weinbrenner C. Ischemic post-conditioning reduces infarct size of the in vivo rat heart: role of PI3-K, mTOR, GSK-3β, and apoptosis. Molecular and Cellular Biochemistry. 2010;339(1-2):135–147. doi: 10.1007/s11010-009-0377-x. [DOI] [PubMed] [Google Scholar]
- 99.Pal R., Palmieri M., Loehr J. A., et al. Src-dependent impairment of autophagy by oxidative stress in a mouse model of Duchenne muscular dystrophy. Nature Communications. 2014;5, article 4425 doi: 10.1038/ncomms5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shende P., Plaisance I., Morandi C., et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123(10):1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 101.Schenkel P. C., Tavares A. M. V., Fernandes R. O., et al. Redox-sensitive prosurvival and proapoptotic protein expression in the myocardial remodeling post-infarction in rats. Molecular and Cellular Biochemistry. 2010;341(1-2):1–8. doi: 10.1007/s11010-010-0431-8. [DOI] [PubMed] [Google Scholar]
- 102.Martínez-Martínez E., Jurado-López R., Valero-Muñoz M., et al. Leptin induces cardiac fibrosis through galectin-3, mTOR and oxidative stress: potential role in obesity. Journal of Hypertension. 2014;32(5):1104–1114. doi: 10.1097/hjh.0000000000000149. [DOI] [PubMed] [Google Scholar]
- 103.Zhang J., Kim J., Alexander A., et al. A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS. Nature Cell Biology. 2013;15(10):1186–1196. doi: 10.1038/ncb2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vigneron F., Dos Santos P., Lemoine S., et al. GSK-3β at the crossroads in the signalling of heart preconditioning: implication of mTOR and Wnt pathways. Cardiovascular Research. 2011;90(1):49–56. doi: 10.1093/cvr/cvr002. [DOI] [PubMed] [Google Scholar]
- 105.Sen S., Kundu B. K., Wu H. C., et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. Journal of the American Heart Association. 2013;2(3) doi: 10.1161/jaha.113.004796.e004796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Calvert J. W., Gundewar S., Jha S., et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS- mediated signaling. Diabetes. 2008;57(3):696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 107.Xu X., Lu Z., Fassett J., et al. Metformin protects against systolic overload-induced heart failure independent of AMP-activated protein kinase α2. Hypertension. 2014;63(4):723–728. doi: 10.1161/HYPERTENSIONAHA.113.02619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hu M., Ye P., Liao H., Chen M., Yang F. Metformin protects H9C2 cardiomyocytes from high-glucose and hypoxia/reoxygenation injury via inhibition of reactive oxygen species generation and inflammatory responses: role of AMPK and JNK. Journal of Diabetes Research. 2016;2016:9. doi: 10.1155/2016/2961954.2961954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kukidome D., Nishikawa T., Sonoda K., et al. Activation of AMP-activated protein kinase reduces hyperglycemia-induced mitochondrial reactive oxygen species production and promotes mitochondrial biogenesis in human umbilical vein endothelial cells. Diabetes. 2006;55(1):120–127. doi: 10.2337/diabetes.55.1.120. [DOI] [PubMed] [Google Scholar]
- 110.Wang X.-F., Zhang J.-Y., Li L., Zhao X.-Y., Tao H.-L., Zhang L. Metformin improves cardiac function in rats via activation of AMP-activated protein kinase. Clinical and Experimental Pharmacology and Physiology. 2011;38(2):94–101. doi: 10.1111/j.1440-1681.2010.05470.x. [DOI] [PubMed] [Google Scholar]
- 111.Daskalopoulos E. P., Dufeys C., Bertrand L., Beauloye C., Horman S. AMPK in cardiac fibrosis and repair: actions beyond metabolic regulation. Journal of Molecular and Cellular Cardiology. 2016;91:188–200. doi: 10.1016/j.yjmcc.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 112.Imai S.-I., Guarente L. NAD+ and sirtuins in aging and disease. Trends in Cell Biology. 2014;24(8):464–471. doi: 10.1016/j.tcb.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Donmez G., Guarente L. Aging and disease: connections to sirtuins. Aging Cell. 2010;9(2):285–290. doi: 10.1111/j.1474-9726.2010.00548.x. [DOI] [PubMed] [Google Scholar]
- 114.Zhong L., Mostoslavsky R. Fine tuning our cellular factories: sirtuins in mitochondrial biology. Cell Metabolism. 2011;13(6):621–626. doi: 10.1016/j.cmet.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu S.-B., Wu Y.-T., Wu T.-P., Wei Y.-H. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochimica et Biophysica Acta—General Subjects. 2014;1840(4):1331–1344. doi: 10.1016/j.bbagen.2013.10.034. [DOI] [PubMed] [Google Scholar]
- 116.Hong F., Larrea M. D., Doughty C., Kwiatkowski D. J., Squillace R., Slingerland J. M. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Molecular Cell. 2008;30(6):701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- 117.Maiese K. Targeting molecules to medicine with mTOR, autophagy and neurodegenerative disorders. British Journal of Clinical Pharmacology. 2016;82(5):1245–1266. doi: 10.1111/bcp.12804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Peterson J. M., Wei Z., Seldin M. M., Byerly M. S., Aja S., Wong G. W. CTRP9 transgenic mice are protected from diet-induced obesity and metabolic dysfunction. American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2013;305(5):R522–R533. doi: 10.1152/ajpregu.00110.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wei Z., Lei X., Petersen P. S., Aja S., Wong G. W. Targeted deletion of C1q/TNF-related protein 9 increases food intake, decreases insulin sensitivity, and promotes hepatic steatosis in mice. American Journal of Physiology—Endocrinology and Metabolism. 2014;306(7):E779–E790. doi: 10.1152/ajpendo.00593.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Su H., Yuan Y., Wang X.-M., et al. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFα-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Research in Cardiology. 2013;108(1, article no. 315) doi: 10.1007/s00395-012-0315-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kambara T., Shibata R., Ohashi K., et al. C1q/tumor necrosis factor-related protein 9 protects against acute myocardial injury through an adiponectin receptor I-AMPK-dependent mechanism. Molecular and Cellular Biology. 2015;35(12):2173–2185. doi: 10.1128/MCB.01518-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kambara T., Ohashi K., Shibata R., et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. Journal of Biological Chemistry. 2012;287(23):18965–18973. doi: 10.1074/jbc.M112.357939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fang Y., Westbrook R., Hill C., et al. Duration of rapamycin treatment has differential effects on metabolism in mice. Cell Metabolism. 2013;17(3):456–462. doi: 10.1016/j.cmet.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Houde V. P., Brûlé S., Festuccia W. T., et al. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59(6):1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yu L., McPhee C. K., Zheng L., et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gangloff Y.-G., Mueller M., Dann S. G., et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Molecular and Cellular Biology. 2004;24(21):9508–9516. doi: 10.1128/MCB.24.21.9508-9516.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Murakami M., Ichisaka T., Maeda M., et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Molecular and Cellular Biology. 2004;24(15):6710–6718. doi: 10.1128/MCB.24.15.6710-6718.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guertin D. A., Stevens D. M., Thoreen C. C., et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCα, but not S6K1. Developmental Cell. 2006;11(6):859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 129.Rong Y., McPhee C. K., Deng S., et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(19):7826–7831. doi: 10.1073/pnas.1013800108. [DOI] [PMC free article] [PubMed] [Google Scholar]