

Summary
Despite the impressive impact of CTLA4 and PD1-PDL1 targeted cancer immunotherapy, a large proportion of patients with many tumor types fail to respond. Consequently, the focus has shifted to targeting alternative inhibitory receptors (IRs) and suppressive mechanisms within the tumor microenvironment. LAG3 (CD223) is the third IR to be targeted in the clinic, consequently garnering considerable interest and scrutiny. LAG3 up-regulation is required to control overt activation and prevent the onset of autoimmunity. However, persistent antigen exposure in the tumor microenvironment results in sustained LAG3 expression, contributing to a state of exhaustion manifest in impaired proliferation and cytokine production. The exact signaling mechanisms downstream of LAG3 and interplay with other IRs remains largely unknown. However, the striking synergy between LAG3 and PD1 observed in multiple settings, coupled with the contrasting intracellular cytoplasmic domain of LAG3 as compared with other IRs, highlights the potential uniqueness of LAG3. There are now four LAG3-targeted therapies in the clinic with many more in preclinical development, emphasizing the broad interest in this IR. Given the translational relevance of LAG3 and the heightened interest in the impact of dual LAG3/PD1 targeting in the clinic, the outcome of these trials could serve as a nexus; significantly increasing or dampening enthusiasm for subsequent targets in the cancer immunotherapeutic pipeline.
Keywords: LAG3, CD223, cancer immunotherapy, immune regulation, regulatory T cells, inhibitory receptors, monoclonal antibodies
Introduction
Upregulated expression of inhibitory receptors (IRs) is essential to balance co-stimulatory receptor activity and limit T cell activation, thereby preventing autoimmunity, autoinflammation and tissue damage. However, tumors can hijack these so-called immune checkpoint mechanisms as protection against anti-tumor immune responses elicited by CD4+ and CD8+ T cells. First described in a chronic viral setting, tolerized antigen-specific T cells display elevated expression of IRs within the tumor microenvironment, corresponding to functional unresponsiveness as measured by reduced proliferation and cytokine release (1, 2). Along with recruitment of regulatory T cells (Tregs), the resulting immune tolerance creates multiple barriers for effective tumor elimination (3, 4). Thus, recent cancer immunotherapeutic approaches have aimed to reverse such exhaustion by targeting IRs “to release the brakes” allowing re-invigoration of cytotoxic T cells to attack tumors.
Early success targeting CTLA4 and PD1 resulted in impressive tumor remission in patients with several tumor subtypes, which led to cancer immunotherapy being highlighted as a significant breakthrough by the leading scientific journals in 2013 (5, 6). Although Ipilimumab (Bristol-Myers Squibb, humanized anti-CTLA4 IgG1 monoclonal antibody [mAb]), Pembrolizumab (Merck, humanized anti-PD1 IgG4 mAb) and Nivolumab (Bristol-Myers Squibb, humanized anti-PD1 IgG4 mAb) can yield objective responses in a significant proportion of patients with a broad spectrum of tumor types, the majority of patients do not respond (7, 8). For example, only 28% of patients with advanced melanoma and 21% of patients with renal cell carcinoma (RCC) receiving Nivolumab demonstrated objective responses (8). While some tumor subtypes previously thought to be difficult to treat, such as non-small cell lung cancer (NSCLC), are responsive to PD1 blockade, other less immunologically-inflamed tumors, such as pancreatic and prostate cancers, remain relatively resistant (9). Thus, identification of additional IR targets, and a clear mechanistic understanding of their function, is essential to increase the activity of combinatorial cancer immunotherapeutic approaches.
With the U.S. FDA-approval of multiple therapeutics that target the PD1/PDL1 pathways, attention has turned to other IRs such as LAG3, TIGIT and TIM3 (10–12). Moreover, clinical trials are ongoing to determine the optimal combination of immunotherapeutics; not only combining antagonistic mAbs against different IRs, but also with other front-line modalities including chemotherapy, radiation therapy and/or cancer vaccines. As a result, the number of possible immunotherapy regimens has grown exponentially, giving rise to the rationale that synergistic combinations should yield significant clinical improvement compared with current monotherapies (13).
Lymphocyte Activation Gene-3 (LAG3; CD223) is a potential cancer immunotherapeutic target due to its negative regulatory role on T cells and its capacity, in combination with PD1, to mediate a state of exhaustion (10). LAG3 was first targeted in the clinic in 2006 using a LAG3-Ig fusion protein (IMP321). Phase I clinical trials were initiated with anti-LAG3 (BMS-986016) in 2013, making it the third IR to be targeted in the clinic with an antagonistic mAb. There are now several LAG3 modulating immunotherapeutics at various stages of clinical and pre-clinical development. As a result of the increased interest in LAG3 as a therapeutic target, there is considerable interest in gaining a greater understanding of the biology and mechanism of action of LAG3. In this review, we will provide an in-depth description of the functional role of LAG3, and highlight its expression pattern, interacting ligands and regulation. The translational rationale for targeting LAG3, deriving from exciting pre-clinical data in a number of murine models, will be discussed followed by a complete analysis of the historical and current state of clinical trials utilizing LAG3 immunomodulators. While a thorough understanding of LAG3 biology is important for the optimal design and interpretation of clinical trials, there are still many questions that remain regarding the biology and function of LAG3, along with the mechanistic interplay with other IRs, which will be highlighted in this review.
LAG3 Discovery and Structure
LAG3 was discovered by Triebel and colleagues in 1990 as a novel 498-amino acid type I transmembrane protein identified on activated human NK and T cell lines (14). The LAG3 gene is located adjacent to CD4 on chromosome 12 in human (chromosome 6 in mouse). Further, LAG3 was predicted to be highly structurally homologous to CD4 with four extracellular immunoglobulin superfamily (IgSF)-like domains (D1–D4) (Figure 1). LAG3 and CD4 also exhibit rigidity between the D1 and D2 domains and between the D3 and D4 domains due to a long, continuous β-strand that extends between these domains, but with relative flexibility between the D1–D2 and D3–D4 substructures (15, 16).
Figure 1. Ligand interaction and structural similarities between LAG3 and CD4.
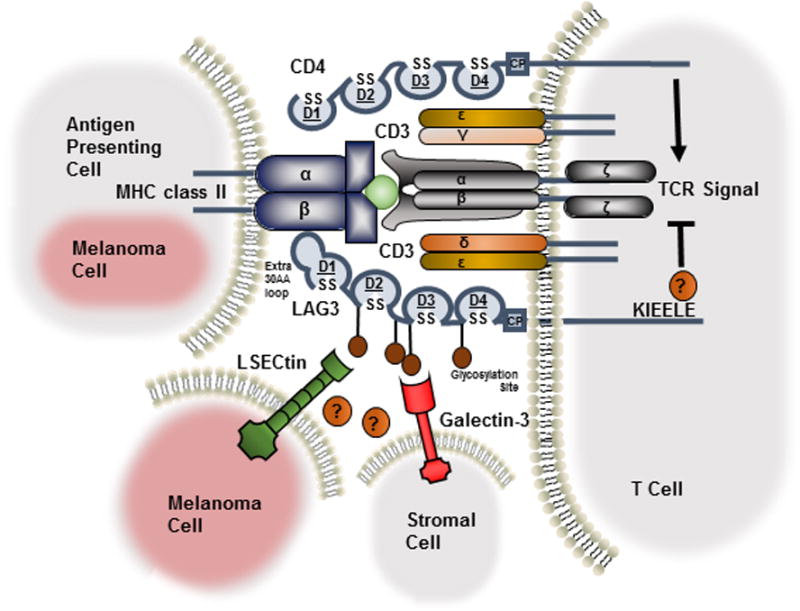
LAG3, like CD4, consists of four extracellular immunoglobulin superfamily-like domains (D1-D4). LAG3 utilizes an additional 30 amino acid loop in D1 to bind to MHC class II with greater affinity. Ligation of MHC class II, expressed by antigen presenting cells or aberrantly by melanoma cells, with LAG3 mediates an intrinsic negative inhibitory signal, in which the KIEELE motif in the cytoplasmic domain is indispensable. LAG3 is highly glycosylated with two additional ligands postulated, LSECtin, expressed on melanoma cells, and Galectin-3, expressed on stromal cells and CD8+ T cells in the tumor microenvironment.
Although these two genes are evolutionarily similar with adjacent chromosomal localization and the same intron/exon organization (both have eight exons), the two molecules share only ~20% amino acid sequence homology (14). Despite this, structural motifs are highly conserved between LAG3 and CD4, translating to the same extracellular folding patterns resulting in LAG3 being able to bind MHC class II, albeit at a distinct site and with a greater affinity than CD4 (15). There is little sequence similarity in the transmembrane and cytoplasmic regions, indicating the two molecules diverged early in evolution, resulting in LAG3 having a distinct yet undefined mechanism of downstream signaling and consequent inhibitory function.
Unlike CD4, which binds MHC class II over an extended contact surface area consisting of a relatively large number of amino acid residues (17), LAG3 utilizes an additional loop consisting of only 30 amino acids in the D1 domain between the C and C′ β strands that is not present in CD4 (15). The D2 domain has been shown to be essential for LAG3-MHC class II ligation, and is perhaps involved in positioning the D1 domain (15). While the D3 and D4 domains are dispensable for binding, they may be required to extend the D1 domain within reach of MHC class II.
Biochemical analyses suggested that LAG3 molecules expressed on the cell surface dimerize via the D1 domain (18). In contrast, a soluble monomeric form of LAG3 (sLAG3), generated by alternative splicing, was suggested to be released by IFNγ-producing CD4+ T cell clones (19). More recently, it was shown that sLAG3 (containing D1–D4) can be shed from the cell surface via proteolytic cleavage of the membrane-proximal connecting peptide (18). Although LAG3 cell surface oligomerization is relatively weak, it may be required for stable MHC class II interaction. Indeed, LAG3-Ig fusion proteins (which are divalent), bind to cell surface MHC class II with mAb-like efficiency, yet cell surface-shed sLAG3 is a monomer and does not bind MHC class II with any appreciable affinity.
LAG3 was suggested to be spatially associated with the TCR:CD3 complex clustered in lipid raft microdomains to allow for clustering of signaling molecules and the formation of the immunological synapse (20). However, a detailed understanding of the association of LAG3 with the TCR:CD3 complex and its mechanism of action is currently lacking.
LAG3 and CD4 possess some unique structural and molecular differences in their cytoplasmic domains that likely mediate their divergent functions. LAG3 lacks a binding site in the cytoplasmic tail for the tyrosine kinase p56Lck, which CD4 utilizes to facilitate signal transduction downstream of the T cell receptor (TCR) (15). Instead, the LAG3 cytoplasmic domain appears to have three definable motifs, which are largely conserved phylogenetically (Figure 2). First, LAG3 possesses a serine-based motif which could act as a PKC substrate (two serine residues in humans; one in mice [Ser454]). Interestingly, there are no tyrosine (or threonine) residues, limiting phosphorylation events that might initiate signal transduction via protein kinases or phosphatases. Consequently, the intracellular tail of LAG3 does not have immunoreceptor tyrosine-based inhibition motifs (ITIM) or immunoreceptor tyrosine-based switch motifs (ITSM). These motifs are often used by other IRs to engage Src homology 2 (SH2) domain-containing protein tyrosine phosphatases, such as (SHP)-1/2 that are used by PD1 to limit TCR signaling (21). Mutation of Ser454 does not abrogate LAG3 function in CD4+ T cells (22). LAG3 does, however, possess two distinct motifs in the cytoplasmic tail first noted when LAG3 was cloned (23), which will be further discussed with respect to LAG3 signaling later in this review. One is a repetitive “EP” motif consisting of a series of glutamic acid-proline dipeptide repeats. The second is a relatively unique “KIEELE” motif, highlighted by an essential lysine residue. Both regions are conserved between human and mouse with LAG3 overall sharing 70% protein sequence identity (15). The conserved features of the cytoplasmic domain are demonstrated by alignment of the human LAG3 sequence with many other species (Figure 2).
Figure 2. Alignment of the LAG3 cytoplasmic domain.

The cytoplasmic domain of the species indicated is shown. Boxes: Blue = potential serine phosphorylation site; Green = ‘KIEELE’ motif region; Red = ‘EP’ repetitive motif. Residues: Red = EP residues within the EP motif region; Blue = conserved residues (100%); Purple = semi-conserved residues (>85% – 25/29); Green = dominant residues (>60% – 18/29).
LAG3 Ligands
MHC Class II
Given the structural similarity between LAG3 and CD4, it is perhaps not surprising that MHC class II molecules are also ligands for LAG3 (Figure 1). COS-7 cells transfected with human LAG3 were shown to form rosettes with MHC class II-expressing human B lymphoblastic cell lines, these rosettes were disrupted with blocking antibodies against LAG3 or HLA-DR (15, 24). Mutagenesis studies confirmed that LAG3’s interaction with MHC class II involves a restricted number of amino acid residues found in the proline-rich D1 loop of LAG3, a structural feature that is not present in CD4 (15). Moreover, the binding affinity of LAG3 for MHC class II was found to be ~100-fold greater than CD4 (Kd: 60nM) (25, 26). This led to the hypothesis that LAG3 competes with CD4 for MHC class II binding, thus negatively impacting CD4 function. While LAG3 mutants that do not bind to MHC class II exhibit reduced function (15, 22), LAG3 cytoplasmic tailless mutants neither compete with CD4 nor mediate the inhibitory effects of LAG3, suggesting that transmission of a inhibitory signal via its cytoplasmic domain is a critical aspect of its function (22). Furthermore, rat anti-mouse LAG3 (C9B7W) does not block the LAG3:MHC class II interaction and yet is a potent inhibitor of LAG3 function in vitro and in vivo (27–29). Taken together, these data suggest that while MHC class II binding by LAG3 may be involved in its function, LAG3 probably does not function primarily by disrupting CD4:MHC class II interactions. Rather, it is likely that LAG3 transmits inhibitory signals via its cytoplasmic domain (22), although the precise nature of this signaling remains unknown.
Human melanoma often expresses MHC class II molecules and this expression is associated with poor prognosis (30). Thus, MHC class II ligation of LAG3, which is highly expressed on melanoma-infiltrating T cells, may facilitate their clonal exhaustion. As demonstrated in vitro, such an interaction may contribute to an escape mechanism by tumor cells as protection against apoptosis, as this was not evident in MHC class II-negative tumor cell lines (31). In fact, a recent study showed that MHC class II-expressing melanoma cells attracts an infiltration of tumor-specific CD4+ T cells, perhaps mediated by interaction with LAG3, which in turn negatively influences CD8+ T cell responses (32).
Alternative Ligands
Since LAG3 impacts CD8+ T cell function to a comparable extent as CD4+ T cells, it is puzzling how LAG3 binding to MHC Class II could influence the function of CD8+ T cells. While this interaction might impact CD8+ T cell interaction with APCs, LAG3 may not mediate the same effects when it interacts with target cells that don’t express Class II MHC. Thus, some have speculated that there may be additional LAG3 ligands (Figure 1).
In terms of alternative ligands, LAG3 is extensively glycosylated and it has been suggested that it can interact with Galectin-3, a 31-kDa lectin that has been shown to modulate T cell responses through a variety of mechanisms (24). LAG3 was also shown to be essential for Galectin-3-mediated suppression of CD8+ T cell-secreted IFNγ in vitro (33). Galectin-3 is expressed by many cells within the tumor microenvironment, albeit not the tumor itself, thus interaction with LAG3 on tumor-specific CD8+ T cells may regulate anti-tumor immune responses (34).
Another potential ligand that may bind to LAG3 in a similar manner is liver sinusoidal endothelial cell lectin (LSECtin), a cell surface lectin that is a member of the DC-SIGN family (35). LSECtin is expressed in the liver and has also been identified in human melanoma tissues where it promotes growth by inhibiting anti-tumor T-cell dependent responses (36). The interaction between LAG3 and LSECtin in melanoma cells was found to inhibit IFNγ production by antigen-specific effector T cells (36).
Interactions with these two potential alternative ligands may serve to broaden LAG3’s impact on T cell function, particularly with regard to an intrinsic role for LAG3 on CD8+ T cells in the tumor microenvironment. Lastly, one cannot rule out the possibility that there may be additional ligands for LAG3 that remain undiscovered. Interestingly, it was recently shown that LAG3 binds α-synuclein preformed fibrils in the central nervous system, thereby facilitating pathogenesis in a mouse model of Parkinson’s disease (37). These data highlight the potential for other immunologically relevant ligands and also suggests a role for LAG3 outside the immune system.
LAG3 Expression
LAG3 is expressed on activated human T and NK cells (14). Under physiological conditions, LAG3 is an activation marker for CD4+ and CD8+ T cells, and like other IRs including PD1, it is first detectable 24 hours after stimulation in vitro, with expression peaking at 48 hours before declining by day 8 in mice (27). Persistent T cell activation in a chronic inflammatory environment, as in a tumor or during chronic viral infection, results in sustained co-expression of LAG3 on T cells that frequently co-express additional IRs (PD1, TIGIT, TIM3, CD160, 2B4) resulting in a state of T cell dysfunction, which will be discussed later in this review (1).
Unlike effector T cells, thymic-derived tTregs constitutively express low levels of LAG3 in the resting state, which is enhanced upon activation. In a model in which hemagglutinin (HA) TCR transgenic-CD4+ specific T cells (clone 6.5) were transferred into C3-HA mice that express HA in multiple epithelial tissues, tolerized HA-CD4+ T cells became induced Tregs, which also showed high LAG3 expression with potent regulatory activity (38). In human cancer patients, LAG3 has been found to be preferentially expressed on tumor infiltrating Tregs in a number of tumor subtypes as compared to the periphery. The role of LAG3 (and other IRs) on Tregs in mediating exhaustion and/or functionality is somewhat controversial in this setting and will be discussed with respect to the translational relevance of these findings.
LAG3, when co-expressed with CD49b, was suggested to define a subset of peripherally induced IL10+CD4+ type 1 T regulatory (Tr1) cells in both mice and humans (39). Although Tr1 cells secrete high amounts of IL-10, surface markers that define this population have been ambiguous. Thus, the use of LAG3/CD49b as surrogate markers would allow phenotypic analysis of this suppressive cell type. However, a more recent study showed that expression of LAG3, amongst other IRs, is dynamic and not exclusive in defining these IL-10 producing cells (40). Nevertheless, LAG3 contributes to Tr1 function as blocking LAG3 abrogated their suppressive activity (39).
A small percentage (~18%) of γδ T cells also constitutively express LAG3, with significant levels of LAG3 mRNA present in TCRγδ intraepithelial lymphocytes (IEL) (27, 41). However, the clinical and functional significance of LAG3 expression on this cell type is unknown. Additionally, TCRαβ+CD8αα+ IEL express significantly increased levels of LAG3 as compared with TCRαβ+CD8αβ+ IEL or the spleen (42). LAG3 expression was also reported on activated B cells in a T cell-dependent manner, although those data have not been replicated (43).
LAG3 is broadly expressed on several additional hematopoietic cell types, including CD11clow B220+ PDCA-1+ plasmacytoid dendritic cells (pDCs) that constitutively express LAG3 at a greater level than any other subset (44). Resting pDCs have a ~70-fold increase of Lag3 mRNA as compared with resting T cells. Interestingly, while T cell LAG3 mRNA levels increase approximately 10 fold upon activation, CpG activation of pDCs did not enhance Lag3 mRNA expression. LAG3 mRNA has been detected in the red pulp of the spleen, which is a localization site for pDCs (27, 45). Notably, LAG3 is not expressed on any myeloid or lymphoid DC subset. LAG3 is also expressed on NK cells (~10%) and invariant NKT cells (27), although the significance of such expression is unclear.
Lag3 mRNA has also been detected in the thymic medulla and at the base of the cerebellum (27). As discussed above, LAG3 has recently been shown to bind α-synuclein fibrils, triggering endocytosis into neurons (37). This is significant as the pathogenesis of Parkinson’s disease may result from cell-to-cell transmission of misfolded preformed fibrils of α-synuclein, thus it is possible that LAG3 blockade may have a role in the treatment of Parkinson’s disease.
LAG3 Signaling
The LAG3 signal transduction mechanism is unknown. However, LAG3 possesses a distinct cytoplasmic domain not shared by other IRs suggesting that it may possess a unique mode of action. The LAG3 cytoplasmic domain has an unusual motif consisting of glutamic acid and proline di-peptide repeats (“EP motif”) (23) (Figure 2). Early studies suggested that LAG3-associated protein (LAP), identified in a yeast two-hybrid screen, may bind to the EP motif (46). Alternatively, LAP may facilitate LAG3 co-localization with CD3, CD4 and/or CD8 within glycosphingolipid-enriched microdomains (lipid rafts) (47). Partitioning of co-stimulatory molecules into lipid rafts, followed by the formation of the immune synapse, concentrates signaling molecules to enhance TCR signaling. Thus, compartmentalization with IRs, including LAG3, may serve to prevent overt activation. Although LAP was initially hypothesized to be important for LAG3 signaling, LAG3 mutants lacking the EP motif retain activity - suggesting that it may not be essential for LAG3 function (22).
One motif that does appear to be essential for LAG3 activity is the KIEELE motif that is conserved between all primates, mice and rats. A single lysine residue (Lys468 in mouse) in this sequence is indispensable and is conserved across all species sequenced to date (22) (Figure 2). As this motif does not appear to occur in any other protein, it may recruit or mediate an as yet unidentified signaling molecule or mechanism. Therefore, elucidation of potential LAG3 intracellular binding partners and signaling mechanism remains a top priority.
LAG3 may also mediate bidirectional signaling into the interacting APCs. MHC class II binding to LAG3-expressing Tregs has been shown to inhibit DC activation, thereby suppressing their maturation (48). CD86 upregulation was inhibited along with reduced IL-12 secretion mediated by an ITAM inhibitory signaling pathway involving FcγRγ and ERK-mediated recruitment of SHP-1. This is truly a reverse signaling mechanism as a LAG3 mutant without the cytoplasmic tail was sufficient to suppress DCs function. LAG3-expressing Tregs may utilize this mechanism to enforce tolerance by indirectly inhibiting DC function. A similar reverse signaling mechanism for LAG3 was also evident in interaction between DC and melanoma cells. When exposed to LAG3-transfected cells, MHC class II-expressing, but not MHC class II-negative, melanoma cells were resistant to Fas-mediated apoptosis via activation of MAPK/ERK and PI3K/Akt survival pathways (31).
Regulation of LAG3 Expression and Function
Although LAG3 clearly plays a negative regulatory role in controlling T cell activation and function, a second layer of control is mediated by modulation of LAG3 expression at the transcriptional level and also by cell surface shedding, which releases sLAG3 (Figure 3). This additional layer of control over LAG3 expression may help to ensure optimal immunoregulation. LAG3 cleavage is mediated by the metalloproteinases ADAM10 and ADAM17 (A Disintegrin And Metalloproteinase domain-containing protein), which also cleave a wide range of transmembrane proteins including TNFα, CD62L and TIM3 (49).
Figure 3. LAG3 cell surface shedding mediated by ADAM10/17 metalloproteinases.
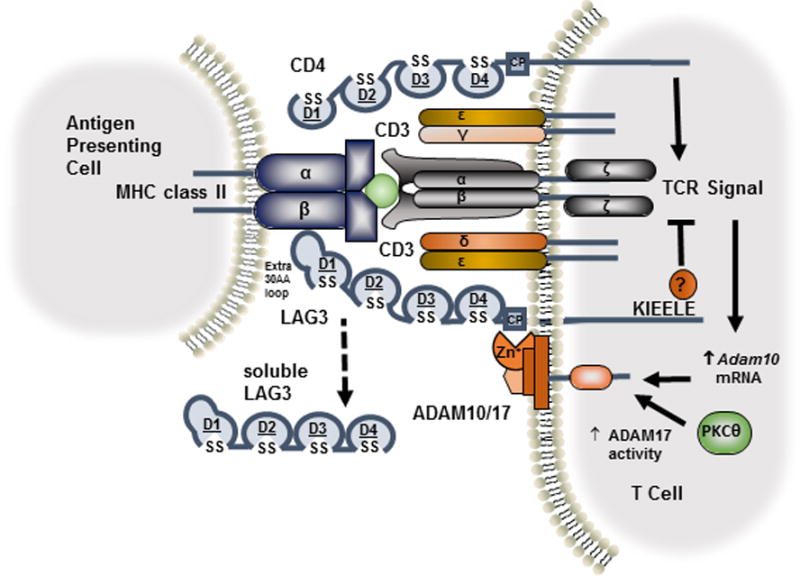
Upon TCR activation, Adam10 mRNA increases and ADAM17 enzymatic activity is enhanced by protein kinase C-θ-dependent phosphorylation. ADAM10/17 cleaves LAG3 within the connecting peptide (CP) between the membrane-proximal D4 domain and the transmembrane domain, releasing soluble LAG3.
ADAM10 constitutively cleaves LAG3 on resting LAG3+ T cells. Following T cell activation, Adam10 mRNA increases ~12-fold 24 hours after activation, further enhancing shedding (50). Unlike ADAM10, LAG3 shedding by ADAM17 only occurs following activation and its activity is controlled by serine phosphorylation in a TCR- and PKCθ-dependent manner. ADAM10 and ADAM17 cleave LAG3 within the connecting peptide between the membrane-proximal D4 domain and the transmembrane domain, releasing sLAG3 (18) (Figure 3). While no biological function for sLAG3 has been found, cleavage of LAG3 is required for optimal T cell function. Prevention of LAG3 shedding by generation of non-cleavable LAG3 mutants resulted in reduced proliferation and attenuated IL-2 and IFNγ production due to enhanced inhibitory activity (50). Likewise, ADAM10 knockdown using a shRNA retroviral vector significantly inhibited proliferation and IFNγ release in a LAG3-dependent manner. Taken together, these results suggest that ADAM10/17-mediated LAG3 shedding acts in a negative feedback loop to moderate its inhibitory function.
The function of sLAG3 generated by LAG3 shedding has been controversial, with some studies considering this to be a “waste product” with no obvious biological function in vitro or in vivo (50). However, one group has suggested that sLAG3 impairs the differentiation of monocytes into macrophages and DCs, which in turn have diminished immunostimulatory capacities (51). From a clinical perspective, sLAG3 may serve as a prognostic biomarker as serum levels in patients with active tuberculosis correlated with a more favorable prognosis (52). Further, it has been suggested that sLAG3 could serve as an early biomarker for type 1 diabetes onset, based on studies in non-obese diabetic (NOD) mice (53). sLAG3 therefore may be a clinically relevant indicator of enhanced LAG3 expression within inflammatory sites - providing a rationale for studying sLAG3 in the serum of patients receiving immunotherapy. Indeed, in a cohort of 246 patients with metastatic hormone-receptor positive breast cancer, those with detectable serum levels of sLAG3 at the time of diagnosis demonstrated an advantage in both disease-free and overall survival compared with patients with undetectable levels of sLAG3. These correlative data support further studies to determine if sLAG3 could serve as a prognostic factor in human cancer and a predictive biomarker for the use of LAG3-targeted or other immunotherapies (18, 54).
In addition to LAG3 shedding, another mechanism that regulates expression is its intracellular storage in lysosomal compartments, which may serve to facilitate rapid LAG3 cell surface expression following T cell activation (55, 56). In resting T cells that have been previously activated, LAG3 is localized near the microtubule-organizing center (MTOC) in Rab11b+ vesicles, which mediate endosomal recycling of transferrin receptors to the plasma membrane (56). While LAG3 can be degraded in lysosomal compartments, it can also be rapidly translocated to the surface upon TCR stimulation to control the T cell response. The cytoplasmic domain of LAG3 was shown to be essential for its trafficking to the cell surface, mediated by protein kinase C signaling, although the two serine phosphorylation sites in the LAG3 cytoplasmic domain did not appear to be involved (55). Lysosomal degradation is a major limiting step in the translocation of LAG3 to the cell surface, as blockade of lysosomal enzyme activity enhances surface expression.
Physiological Role of LAG3
Early studies suggested that LAG3 was a negative regulator of T cell activation and function since blockade of LAG3 on human CD4 clones resulted in enhanced proliferation with elevated production of IL-2, IL-4, IFN-γ and TNFα (57). Likewise, LAG3-deficient (Lag3–/–) CD4+ OT-II T cells also secreted more of these cytokines following in vitro stimulation, although with reduced expansion attributed to increased cell death (58). Normal T cell function was restored following ectopic expression of LAG3, but not mutants that lacked either the entire cytoplasmic domain or the KIEELE motif. This study demonstrated that the largely conserved KIEELE motif mediates a cell intrinsic signal that is essential for the negative regulatory function of LAG3 on T cells.
Initial studies utilizing Lag3−/− mice found no defects in the T cell compartment; here young (~5-week-old) mice were used for analysis (27). However, further studies using aged mice (~16-week-old) revealed that the number of T cells had doubled compared with wild-type mice suggesting a role for LAG3 in regulating T cell homeostasis (28). Lag3–/– mice also have more Mac-1+ macrophages, Gr-1+ granulocytes and CD11chi DCs, populations that do not express LAG3, suggesting a cell extrinsic impact mediated by an enhanced T cell compartment (28). This suggests that there may be cell extrinsic homeostatic roles for LAG3 on other populations that have yet to be explored. To confirm whether the uncontrolled expansion of T cells was a result of LAG3 deficiency, Lag3–/– or Lag3+/+ mice were injected with the superantigen, staphylococcal enterotoxin B (SEB) which activates Vβ7/8+ T cells in an antigen non-specific manner. Following injection, a significant increase of SEB-reactive T cells was reported in the absence of LAG3 (59). Moreover, there was notable splenomegaly in Lag3–/– mice on day 2 post SEB injection, which was not present in control wild-type mice. Further analysis showed that this increase resulted from a deficiency in cell cycle arrest and cell death. Similarly, an increased expansion of Lag3–/– OT-II T cells following in vivo stimulation with OVA was also demonstrated providing further evidence that LAG3 negatively controls T cell homeostasis. Early studies further suggested that LAG3 expression may distinguish Th1 from Th2 cells; as in vitro studies showed that IL-12 stimulated LAG3 expression was abrogated by IFNγ blockade (59). Subsequent work with human T cells suggested that LAG3 expression was not a reliable discriminatory marker for these subsets (60).
A role of LAG3 in the control of memory T cell expansion was also shown following infection of Lag3–/– mice with Sendai virus, a murine parainfluenza virus in which large populations of memory T cells persist in secondary lymphoid organs and peripheral tissues (59). In this acute viral infection model, there was a significant increase in IFNγ-producing Sendai-specific CD4+ (HN419–433/Ab) and CD8+ (NP324-332/Kb) T cells in Lag3–/– mice 30 days post-infection, as compared to wild-type mice. This suggested that LAG3 may control the size of the memory T cell pool. In a chronic viral infection model utilizing murine gammaherpesvirus 68 (MHV-68), a more limited role for LAG3 was shown with the number of viral specific CD4+ T cells peaking later at day 36 post-infection in Lag3–/– mice, compared with day 21 in wild-type mice (59). Unlike the acute viral model, there were no differences in the CD8+ T cell response in this model of chronic infection.
A more specific role for LAG3 on CD8+ T cells was demonstrated in a model of self-tolerance. These studies used animals that expresses influenza HA as a self-antigen; HA-specific T cells (Clone 4) were adoptively transferred to HA-expressing hosts (61). In this model, LAG3 blockade enhanced the accumulation of HA-specific CD8+ T cells. Likewise, adoptively transferred LAG3–/– HA-specific CD8+ T cells expanded and produced large amounts of IFNγ, suggesting that LAG3 limits self-tolerance. In addition, a second model utilizing ProHA × TRAMP mice that express HA as a prostate-specific, tumor-associated antigen resulting in a spontaneous tumor-tolerizing environment, expansion of HA-specific clone 4 CD8+ T cell effectors was observed following LAG3 blockade and Vaccinia virus-HA vaccination (61). Moreover, these CD8+ T cells regained effector function, as shown with an increased frequency of IFNγ producing cells. The effect elicited by blocking LAG3 was shown to be a CD8+ T cell intrinsic effect, and was not dependent on CD4+ T cells. Further studies using this model demonstrate that distinct CD8+ T cell sub-populations develop rapidly after antigen encounter under these tolerizing conditions, with differential LAG3 and PD1 expression levels (62). These subsets have different cytolytic function, with the LAG3negPD1hi subset functionally inferior to the LAG3+PD1int subset with respect to IFNγ release.
LAG3 expression on Tregs has also been shown to be required for maximal suppressive activity, as blockade abrogates Treg function in in vitro proliferation assays (38). In addition, anti-LAG3 blocked protection mediated by antigen-specific CD4+ Tregs in an in vivo model of pulmonary vasculitis (38). These findings suggested that LAG3 was essential for Treg function in this model, which would otherwise suppress the infiltrating effector T cells that mediate disease pathogenesis. Additionally, transfection of LAG3 in non-Treg CD4+ T cells resulted in the acquisition of a regulatory phenotype, with reduced proliferation of co-cultured responder T cells. This gain of immunosuppressive function occurred with transfection of full-length LAG3 but not a truncated mutant lacking the cytoplasmic tail. More recent studies have shown that LAG3 promotes Treg differentiation whilst LAG3 blockade inhibits Treg induction (63). This study also showed that blockade or genetic deletion of LAG3 skewed CD4+ T cells into a Th1 phenotype, with LAG3 limiting IL-2 and STAT5 signaling that modulates the ability to be suppressed by Tregs. Other studies also support a role for STAT5 in LAG3 signaling; T cells in which STAT5 was specifically knocked out did show enhanced proliferation in response to LAG3 blockade in a well-controlled homeostatic proliferation model (63).
LAG3 is constitutively expressed at a much greater level on pDCs than any other cell type, yet its functional role on these cells is not well understood. Lag3–/– pDCs show enhanced in vivo expansion following CpG stimulation when compared with wild-type pDCs, but do not have an altered expression profile of activation markers, including MHC class II and CD80/86, or differential cytokine production, including expression of IFNα (44). Interestingly, there appears to be a reciprocal homeostatic interplay between Lag3–/– pDCs and T cells, which proliferate less in their presence as compared to conditions in which they are co-cultured with wild-type pDCs. This provides an additional extrinsic mechanism for the negative regulatory role of LAG3 on T cell homeostasis. In humans, LAG3+ pDCs were found to infiltrate the melanoma environment and interact with HLA-DR-expressing tumor cells in vivo. In vitro it was also shown that MHC class II-expressing melanoma cells could stimulate LAG3+ pDCs to mature and produce IL-6 (64). This was confirmed in vivo with LAG3+ pDCs showing elevated IL-6 production and an activated phenotype in close proximity to melanoma cells. In a separate study, it was shown that increased IL-6 drives the release of CCL2 by monocytes in vitro, which then may recruit myeloid-derived suppressor cells (MDSCs) (65). Thus, it was hypothesized that LAG3+ pDCs may indirectly drive MDSC-mediated immunosuppression through engagement of MHC class II+ melanoma cells.
The role of LAG3 on NK cells and iNKT cells is also not well understood. Proliferation of activated NKT cells is reduced as a result of LAG3 signaling, resulting in cell cycle arrest in the S phase (66). Moreover, elevated expression of LAG3 was associated with impaired iNKT cytokine production (IFNγ) during chronic HIV infection, but this was not noted on other T cell subsets (67). Interestingly, PD1 expression levels were not affected. The functional significance of LAG3 on iNKT cells in this scenario is unclear, although a significant percentage (~40%) of this cell type expressed LAG3 in this cohort, which inversely correlated with IFNγ production. Finally, the role of LAG3 on B cells is controversial as analysis has been limited and expression was only reported in a single study (43).
Role of LAG3 in controlling Autoimmunity
The expression of IRs is necessary to control immune responses, thereby preventing exacerbated T cell activation and the onset of autoimmunity. Indeed, as described above, LAG3 has an important role as a negative regulator and thus LAG3 insufficiency results in the exacerbation of disease in a number of autoimmunity models.
For example, Lag3–/– NOD mice exhibit a highly accelerated diabetes onset with 100% incidence at a timepoint when wild-type mice were just starting to develop hypoglycemia (68). Enhanced antigen-specific T cell infiltration into the islets is also evident in Lag3–/– NOD mice. LAG3 blockade at 7 weeks of age, prior to diabetes onset, also accelerated disease onset. In an in vivo model of colitis, Lag3–/– T cells co-transferred with wild-type Tregs were relatively resistant to suppression, unlike wild-type conventional T cells, thus resulting in a more severe form of colitis demonstrated by histology and enhanced reduction in body weight of the animals (63). Finally in a model of mercury-induced autoimmunity, LAG3 blockade or genetic ablation resulted in an increased susceptibility to disease as a failure to maintain tolerance (69).
Although Lag3–/– C57BL/6 mice do not develop spontaneous disease, Lag3–/–Pdcd1–/– mice succumb to a lethal systemic autoimmunity, which was also not evident in Pdcd1–/– mice (70). This phenotype provides evidence for a synergistic cooperation between LAG3 and PD1 in maintaining immune homeostasis and preventing autoimmunity.
Role of LAG3 in mediating T cell exhaustion
Although LAG3 limits autoimmunity, sustained co-expression with other IRs (e.g. PD1, TIGIT, TIM3, 2B4, CD160) in tolerogenic environments can result in a state of functional exhaustion, exemplified by lack of proliferation, cytokine secretion and cytolytic activity (10–12). Initial studies that assessed the impact of IRs on T cell exhaustion were performed in a lymphocytic choriomeningitis virus (LCMV) clone 13 chronic infection model. During the course of disease, transcriptional analysis defined a distinct subset of exhausted T cells with an elevated expression of IRs (71). Exhausted CD8+ T cells had a severe defect in cytokine production, which increased over time. LAG3 was found to strongly correlate with the severity of infection, with PD1 co-expression observed on gp33 virus protein-specific CD8+ T cells (72). Although blockade of LAG3 alone had little effect on the resolution of LCMV, dual LAG3/PDL1 blockade synergized to reduce viral load by reversing exhaustion, thus improving anti-viral CD8+ T cell responses (29). Elucidating the role of LAG3 during LCMV infection is relevant here because tumors share several common features with chronic infections; i.e. chronic antigenic exposure and the consequent development of dysfunctional, exhausted T cells.
In mice, LAG3 co-expresses with PD1 on tumor-infiltrating CD4+ and CD8+ T cells in melanoma (B16-F10), colon adenocarcinoma (MC38) and fibrosarcoma (Sa1N) tumors (70). CD8+ T cells expressing both LAG3 and PD1 are also the dominant tumor-infiltrating lymphocyte (TIL) population in CT26, a colon carcinoma in which LAG3 was shown to control T cell proliferation/cell cycle progression - resulting in a state of hypofunction (73).
LAG3 monotherapy in mice with MC38 and Sa1N tumors was largely ineffective with a small reduction of tumor growth and very limited tumor clearance. Interestingly, dual LAG3/PD1 co-blockade synergistically limited the growth of MC38 and resulted in tumor clearance in 80% of mice (70). This compared with 40% remission in mice receiving anti-PD1 alone. Likewise, in the Sa1N tumor model, LAG3/PD1 blockade resulted in 70% tumor-free animals, compared to 20% survival with anti-PD1 alone. Increased survival was a result of augmented CD8+ T cell infiltration with enhanced IFNγ production. This profound synergistic cooperativity between LAG3 and PD1 was also evident in Lag3–/–Pdcd1–/– mice, which were able to clear large tumor burdens, whereas only delayed tumor growth was evident in the respective single knockout mice (70).
Synergy between LAG3 and PD1 has also been reported in other tumor models, demonstrating that dual immunotherapeutic efficacy may extend to multiple tumor subtypes. Combinatorial blockade of LAG3 and PD1 synergistically enhanced antitumor immunity in an ovarian tumor derivative of the ID8 model (74). Dual targeting increased CD4+ and CD8+ T cell infiltration, as well as increasing the frequency of single and double-producing IFNγ+/TNFα+ CD8+ T cells in this model. In the EG7 lymphoma model, 100% of mice treated with a combination of anti-LAG3 and anti-PD1 cleared their tumors compared with 50% of mice becoming tumor-free with anti-PD1 alone (74). Mice only exhibited delayed tumor growth with LAG3 blockade as a monotherapy. In a B16-F10 model of recurrent melanoma, dual targeting of LAG3 and PD1 at the time of relapse resulted in significant tumor regression (75). Synergistic anti-tumor efficacy was also demonstrated in a murine multiple myeloma model (5T33) following a lymphodepleting dose of whole body irradiation (76). The rationale for dual blockade in this model was that expression of LAG3 is augmented on CD4+ and CD8+ T cells following whole body irradiation and anti-PDL1 treatment. Here, the combination of anti-PDL1 and anti-LAG3 mAbs resulted in long-term survival of 80% of mice compared with 40% of mice receiving anti-PDL1 alone. As above, the frequency of myeloma-reactive CD8+ and CD4+ T cells was also enhanced following combinatorial therapy. Finally, in an immunotherapeutic model that utilizes a MVA-BN-HER2 poxvirus, which is a DNA virus with an engineered HER2 tumor-associated antigen vector, additional administration of anti-LAG3 and anti-PDL1 mAbs led to complete regression of CT26-HER2 tumors, unlike a combination of MVA-BN-HER2 and anti-PDL1 alone (77). As in other models, LAG3 expression was upregulated with anti-PDL1 therapy, possibly as a compensatory measure, providing a rationale for combinatorial blockade.
Overall these promising pre-clinical data suggesting a clear synergistic interplay between LAG3 and PD1 has prompted the analysis of human tumor samples for expression of these IRs and their impact on intratumoral T cell function, and have paved the way for clinical trials of combinatorial immunotherapeutic regimens.
Translational Relevance
In many human patient tumor samples, LAG3 co-expression with PD1 correlates with a state of T cell dysfunction. For example, in ovarian cancer, tumor-infiltrating NY-ESO-1 specific CD8+ T cells expressed high levels of PD1, with some co-expressing LAG3 (78). These cells displayed an exhausted phenotype with a reduced capacity to produce IFNγ and TNFα. In vitro dual blockade of both IRs improved proliferation and cytokine production (IFNγ) of these tumor-specific CD8+ T cells, which was not augmented by blocking LAG3 or PD1 alone. Likewise, antigen-specific T cells (Melan-A/MART-1) isolated from melanoma patients’ metastases expressed elevated levels of LAG3 amongst other IRs (CTLA4, TIM3) as compared to expression on peripheral blood lymphocytes (79). PD-L1+ melanomas also co-expressed LAG3+ TIL, providing strong evidence that up-regulation of this IR could mediate an escape mechanism from PD1 therapy, in which resistance might possibly be overcome with the addition of LAG3 blockade (80). Compared to peripheral blood, hepatitis B virus (HBV)-specific CD8+ TIL isolated from hepatocellular carcinoma (HCC) patients exhibit a significant up-regulation of LAG3, with consequent functional deficiencies, such as reduced IFNγ production (81). In colon cancer, LAG3 was also expressed at higher levels in microsatellite instability (MSI) tumors, compared with microsatellite stability (MSS) tumors, which are more amenable to checkpoint blockade (82).
LAG3 is also highly expressed on Tregs found in peripheral blood, tumor-involved lymph nodes and within tumor tissue isolated from patients with advanced (stage III and IV) melanoma and colorectal cancer (83). Moreover, a correlation was shown between LAG3+ Tregs and production of immunosuppressive cytokines (IL-10, TGF-β1) at these sites, versus LAG3-negative cells (83). These LAG3+ Tregs display a terminal-effector (CD45RA+CCR7–) phenotype, and proliferate less than their LAG3-negative counterparts (83).
In head and neck squamous cell carcinoma (HNSCC) patients as well as NSCLC patients, LAG3 is also preferentially expressed on tumor-infiltrating Tregs (84, 85). Another study with a colorectal cancer cohort showed that there was a prevalent intratumoral population of CD4+ Foxp3– cells that expressed LAG3, likely human Tr1 cells, which produced high levels of IL-10 and TGF-β and were in significantly more suppressive than the corresponding CD4+ Foxp3+ Tregs (86). Interestingly, these LAG3+ IL-10+ CD49b+ Foxp3– Tr1 cells have been associated with progression of colorectal cancer (87). Therefore, LAG3 contributes towards immunosuppression within the tumor microenvironment, and this suppression is mediated by LAG3+ Tregs and also by non-Foxp3 regulatory populations. In hematological malignancies, Lag3 mRNA has been demonstrated in chronic lymphocytic leukemia (CLL) patients to be a prognostic marker, possibly relating to pathogenesis, but only in patients with unmutated IGHV (Immunoglobulin Variable Heavy Chain Region) (88, 89). Taken together, these multiple analyses of human tumor samples suggest that LAG3 may be a viable candidate for targeted monotherapy, and has significant promise in combinatorial therapeutic approaches along with anti-PD1.
Clinical Development of LAG3 Targeted Immunotherapy
There are currently four LAG3 modulating agents that have entered the clinic as anti-cancer therapeutics, with several more in preclinical development. Initial LAG3-driven clinical trials focused on a first-in-class agent, IMP321 (Prima BioMed/Immutep) designed as an APC activator. Three different LAG3-specific mAbs have been developed for the treatment of cancer; BMS-986016 (Bristol-Myers Squibb, fully human IgG4), LAG525 (Novartis, humanized IgG4) and MK-4280 (Merck). Table 1 summarizes current clinical studies involving LAG3-targeted immunotherapy that have either been completed, in progress or are currently recruiting participants (ClinicalTrials.gov). In addition, it should be noted that a fourth anti-LAG3 antagonistic mAb (GSK2831781) developed by GlaxoSmithKline was designed to deplete LAG3+ expressing cells in patients with autoimmune diseases; this agent is in clinical trials for patients with plaque psoriasis (NCT02195349). While this agent will not be further discussed here, it serves to highlight that LAG3 modulating therapeutics could have applications beyond cancer.
Table 1.
Clinical Studies of LAG-3 targeted immunotherapy. Table represents a list of clinical trials listed on ClinicalTrials.gov (current as of October 2016 – search terms: “LAG-3 and cancer”, “BMS-936558 and cancer”, “LAG525 and cancer”, “MK-4280 and cancer” and “IMP321 and cancer”). Studies were excluded if administration of LAG-3 targeted therapy did not include treatment of patients with cancer.
|
ClinicalTrials.gov ID / Sponsor (year opened) |
Trial | Patient Population Phase (estimated enrollment) Primary endpoint |
Description | Status/Outcomes |
|---|---|---|---|---|
| IMP321 – LAG-3Ig fusion protein (Immutep S.A.; Prima BioMed) | ||||
|
NCT00351949 Immutep S.A. (2006) |
IMP321 Phase I Trial in Metastatic Renal Cell Carcinoma (mRCC) |
|
|
|
|
NCT00349934 Immutep S.A (2006) |
IMP321 Plus First-line Paclitaxel in Metastatic Breast Carcinoma |
|
|
|
|
NCT00365937 Cliniques universitaires Saint-Luc-Université Catholique de Louvain (2006) |
Immunization of Disease-Free Melanoma Patients With Different HLA-A2 Peptides |
|
|
|
|
NCT00324623 Centre Hospitalier Universitaire Vaudois (2006) |
Cyclophosphamide and Fludarabine Followed by Cellular Adoptive Immunotherapy and Vaccine Therapy in Patients with Metastatic Melanoma |
|
|
|
|
NCT00732082 Washington University School of Medicine (2008) |
Lag-3 and Gemcitabine for Treatment of Advanced Pancreas Cancer |
|
|
|
|
NCT01308294 Centre Hospitalier Universitaire Vaudois (2010) |
Immunotherapy of HLA-A2 Positive Stage II–IV Melanoma Patients (LAG-3/IMP321) |
|
|
|
|
NCT02614833 Immutep S.A (2015) |
IMP321 as Adjunctive to Standard Chemotherapy Paclitaxel Metastatic Breast Carcinoma |
|
|
|
|
NCT02676869 Prima BioMed Ltd (2016) |
Phase I study of IMP321 Adjuvant to Anti-PD-1 Therapy in Unresectable or Metastatic Melanoma |
|
|
|
| BMS-986016 (Bristol-Myers Squibb) | ||||
|
NCT01968109 Bristol-Myers Squibb (2013) |
Safety Study of Anti-LAG-3 With and Without Anti-PD-1 in the Treatment of Solid Tumors |
|
|
|
|
NCT02061761 Bristol-Myers Squibb (2014) |
Safety Study of Anti-LAG-3 in Relapsed or Refractory Hematologic Malignancies |
|
|
|
|
NCT02750514 Bristol-Myers Squibb (2016) |
A Study to Test Combination Treatment in People with Advanced Non-Small Cell Lung Cancer |
|
|
|
|
NCT02658981 Sidney Kimmel Comprehensive Cancer Center (2016) |
Anti-LAG-3 or Urelumab Alone and in Combination with Nivolumab in Treating Patients with Glioblastoma |
|
|
|
|
NCT02935634 Bristol-Myers Squibb (2016) |
A Study to Test Combination Treatments in People with Advanced Gastric Cancer (FRACTION-GC) |
|
|
|
| LAG525 (Novartis) | ||||
|
NCT02460224 Novartis Pharmaceuticals (2015) |
Safety and Efficacy of LAG525 Single Agent and in Combination With PDR001 in Patients with Advanced Malignancies |
|
|
|
| MK-4280-001 (Merck) | ||||
|
NCT02720068 Merck Sharp & Dohme Corp. (2016) |
Study of MK-4280 as Monotherapy and in Combination with Pembrolizumab (MK-3475) in Adults with Advanced Solid Tumors (MK-4280-001) |
|
|
|
Subcutaneous, SQ; injection, inj; dose-limiting toxicity, DLT; day, D; week, wk; month, mo; adverse events, AE; antigen presenting cells, APCs; natural killer, NK; objective response rate, ORR; metastatic renal cell carcinoma, mRCC; progression free-survivial, PFS; cytolytic T lymphocyte, CTL; peripheral blood mononuclear cells, PBMCs; T-cell receptor, TCR; MTD, maximum tolerated dose; overall survival, OS; progression of disease, POD; radiation therapy, RT.
Early clinical studies with IMP321
Although current clinical work focuses on the development of antagonistic mAbs, initial LAG3-driven clinical trials centered on IMP321, a soluble dimeric recombinant protein consisting of four LAG3 extracellular domains fused to the Fc portion of human IgG1 (LAG3-Ig). This fusion protein was intended as a LAG3 antagonist but its clinical development has more recently been re-focused on its use as an immune adjuvant to activate APCs. This re-focusing was based on the observation that LAG3-Ig interaction with MHC class II on human immature DCs induced the up-regulation of CD80/CD86, secretion of IL-12 and TNFα, and promoted morphological changes such as the formation of dendritic projections (90, 91). LAG3-Ig stimulation of DCs also induced a distinct pattern of chemokines (CCL22, CCL17) allowing migration to secondary lymphoid organs for priming of naïve CD4+ and CD8+ T cells (92). Additionally, in this study, cross-linking with anti-MHC class II antibodies did not result equivalent maturation, suggesting that LAG3-Ig results in a distinct downstream effect. In addition, LAG3-Ig induced CCR7 surface expression and consequent chemotaxis studies indicated that this would direct migration to draining lymph nodes (92). Clinical grade LAG3-Ig (IMP321) binding to 10% of MHC class II+ human PBMCs stimulated myeloid cells to produce TNFα and CCL4 (93). In turn, a minority of CD8+ T cells (~1%) produced IFNγ and/or TNFα as a result of DC activation. Therefore, it was hypothesized that IMP321 may act as an immunopotentiator to activate CD8+ T cells via maturation of DCs and could be used as a viable cancer immunotherapeutic approach.
The first-in-man phase I dose-escalation of IMP321 as a monotherapy was performed in patients with advanced metastatic RCC (NCT00351949) (94). In this study, 21 patients were treated with IMP321, administered biweekly as a subcutaneous dose ranging from 0.05mg to 30mg for a total of six injections. IMP321 monotherapy was safe, well-tolerated and demonstrated significant induction of effector-memory T-cells expressing CD28. Although no objective responses were reported, 7 out of 8 patients did experience stable disease with higher doses of IMP321 (>6mg) as compared to only 3 out of 11 patients with the lower dose group (<6mg), which associated with significantly less tumor growth. Taken together, these tolerability data, and the modest yet favorable signal for the activity of IMP321 monotherapy provided a rationale to combine the agent with first-line chemotherapy or other immunotherapies, such as cancer vaccines, in order to enhance overall anti-tumor activity. Indeed, based on these findings, two chemo-immunotherapy trials were initiated in advanced pancreatic cancer (NCT00732082) and metastatic breast cancer (NCT00349934).
The first clinical trial of IMP321 in the United States, as a single-center phase I dose-escalation study at Washington University, evaluated the safety of IMP321 combined with gemcitabine (100mg/m2) as front-line therapy in a cohort of 18 patients with advanced pancreatic adenocarcinoma (95). Chemotherapy can induce tumor cell apoptosis releasing antigens, and since IMP321 stimulates the maturation of DCs, this combination was hypothesized to enhance CD8+ T cell expansion and recognition of tumors. IMP321 was administered via subcutaneous injection on day 2 and 16 of a four-week cycle for a total of 6 months. As with the monotherapy, the combination of gemcitabine and IMP321 was considered to be well-tolerated at all dose-levels and no severe adverse events were attributed to IMP321. However, there was no significant immunomodulation of CD11b+CD14+ monocytes, CD11b+CD11c+ conventional DCs, or CD8+ or CD4+ T cell subsets (including CD4+CD25+Foxp3+ Tregs) when comparing pre- and post-treatment parameters by flow cytometry. The lack of dose-effect from a toxicity and immunological standpoint suggested that IMP321 dosing was suboptimal and the authors recommended that future studies explore higher dose levels.
The combination of IMP321 and Paclitaxel as first-line chemo-immunotherapy was assessed in 33 patients with metastatic breast cancer (NCT00349934). In this Phase I, non-randomized fixed-dose escalation study, patients were treated with a combination of paclitaxel (80mg/m2, day 1, 8, 15 every 28 days) and soluble IMP321 delivered every two weeks the day following chemotherapy (day 2 and 16 every 28 days) for a total of six cycles (46). Three cohorts of patients were treated, each with an increasing dose of IMP321 administered as 0.25mg, 1.25mg and 6.25mg via subcutaneous injection. Among the 30 patients who received all six cycles of IMP321, there were no significant local or systemic IMP321-related adverse events. Further, there were significant and durable immunological effects observed with this regimen, which appeared to correlate with favorable clinical outcomes, compared with a historical control group (Paclitaxel alone). Indeed, patient sera collected at pre-treatment and post-treatment (day 1, 85 and 170) demonstrated phenotypic changes in both primary and secondary target cells. There was an absolute and proportional increase in MHC class II-expressing APCs (both monocytes and DCs) and monocytes were significantly more activated at higher IMP321 doses for at least three months (day 85, compared to day 1). Furthermore, as regards to secondary target cells, there was an absolute increase in the NK cell and CD8+ T cell populations; among the CD8+ subset there was a proportional enhancement of cells with a terminally differentiated effector memory phenotype (CD62L−CD45RA+). There was a 50% objective tumor response at the end of IMP321 treatment and decreased tumor size correlated with an increase in the absolute number of monocytic cells. Finally, after six months, 90% of patients demonstrated clinical benefit, overall – again comparing favorably with historical controls. Given these results, a phase II multi-center double-blinded randomized trial was opened in 2015 with a target enrollment of 211 patients with metastatic hormone receptor-positive breast cancer (NCT0261483). The 1:1 randomization will compare paclitaxel in combination with IMP321 versus paclitaxel plus placebo with a primary endpoint of progression free survival (PFS). Immunologic correlative studies include assessment of tumor-infiltrating immune cell activation status and tumor cell molecular profiling obtained from archival tumor tissue.
There are a number of clinical trials which are actively exploring the role of IMP321 as an immunologic adjuvant in advanced melanoma. IMP321 has also been developed as a vaccine adjuvant, combining Montanide ISA51 VG (mannide monooleate surfactant and mineral oil) with various tumor-specific peptides, including Melan-A and NY-ESO-1 to activate tumor-specific CD8+ T cells, as well as Mage-A3 to elicit a helper CD4+ T cell response (96). A four-armed phase I/II trial initiated in 2006 set out to determine the cytolytic T-lymphocyte (CTL) response and toxicity profile of several HLA-A2 peptides administered alone or in combination with immunologic adjuvants, IMP321 or Montanide (NCT00365937). A CTL response was defined as a 10-fold increase in the frequency of circulating CTLs. The HLA-A2 peptide cocktail consisted of eight peptides injected intradermally or subcutaneously in two sites every three weeks on five occasions: MAGE-1.A2, MAGE-3A.2, MAGE-4.A2, MAGE-10.A2, MAGE-C2.A2, NA17.A2, Tyrosinase.A2 and NY-ESO-1.A2. There was a target enrollment of 28 patients with seven patients per treatment arm; however, this study was terminated early after enrolling 19 patients due to new regulations initiated by the pharmaceutical company associated with the HLA-A2 melanoma-associated peptides.
In a similar cancer vaccine approach, advanced (stage IV) melanoma patients were treated in a Phase I trial with MART-1 peptide vaccination, with or without IMP321 to investigate potential synergy of adoptive T-cell transfer and immunomodulation (97). Eligible patients were HLA-A2 positive with tumor expression of MART-1 and Melan-A who had progressive disease following treatment with melan-A peptide vaccination. In this study, 12 HLA-A2 positive patients with measurable pre-treatment endogenous anti-MART1 CD8+ T-cell responses underwent leukopheresis prior to lymphodepletion with cyclophosphamide and fludarabine. Subsequently, these patients underwent autologous adoptive transfer with reinfusion of harvested PBMCs delivered in combination with MART-1 peptide vaccination in the presence or absence of IMP321. The investigators hypothesized that adjuvant IMP321 would elicit more robust anti-tumor immunity and result in expansion of MART-1-specific CD8+ T cells. Indeed, among the six patients treated with IMP321, there was significant enhancement of the effector phenotype of MART-1-specific CD8+ T cells, which further correlated with increased functionality as measured by effector cytokine production. Furthermore, analysis of MART-1 specific CD8+ T cells among patients treated with IMP321 demonstrated decreased expression of exhaustion markers, including: PD1, LAG3, TIM3, 2B4 and CD160. In addition to superior antigen-specific CTL responses and functionality, immunization with IMP321 was noted to selectively restrain expansion of Tregs suggesting the relative increase in the CD8+ effector to Treg ratio may in part explain the favorable immunological responses observed with IMP321. Unfortunately, despite the induction of seemingly potent anti-tumor immunity, only one of six patients in this study achieved a transient partial response - underscoring the need for further optimization.
Across trials, clinical and immunological outcomes of IMP321 treatment have been variable and interpretation of these results is complicated by slow accrual in several trials. While some of these differences are attributable to trial design and IMP321 dosing/administration, it is also plausible that important differences in biology and tumor histology underlie this heterogeneity in clinical outcomes. Indeed, in patients with metastatic breast cancer, IMP321 appeared to have some evidence of activity, which is surprising in light of the notion that breast cancer is a weakly immunogenic tumor type.
The recently opened phase I TACTI-mel (TwoACTive Immunotheraputics in melanoma) trial highlights the concept of targeting different mechanisms of action by using IMP321 as an “APC activator” in combination with immune checkpoint inhibition to release the putative “brakes” on T cell function in order to augment anti-tumor immune responses. This trial, which was initiated in 2016, is specifically evaluating the combination of IMP321 with Pembrolizumab in patients with unresectable or metastatic melanoma (NCT02676869). This study will primarily assess the safety profile of this combination, but it is anticipated that the addition of IMP321 may improve the objective response rates as compared with PD1 blockade alone.
Taken together, IMP321 has demonstrated minimal activity as a monotherapy and there has been modest success with IMP321 when combined with cytotoxic chemotherapies and vaccine-based strategies. Ultimately, larger, well-controlled randomized studies exploring IMP321 in combination with cytotoxic chemotherapy or with immune checkpoint inhibitors are needed to validate this strategy. Alternatively, the advent of mAb-based immune checkpoint blockade has transformed the treatment landscape in several malignancies. As previously discussed, substantial preclinical data suggest that blockade of LAG3 enhances anti-tumor immune responses and there is evidence of synergy when combined with PD1 blockade. These studies have provided ample rationale to translate this approach in humans and mAbs targeting LAG3 are now actively being explored clinically.
Targeting LAG3 with antagonistic monoclonal antibodies
While objective clinical responses have been observed with PD1/PDL1-targeted therapies across a variety of tumor subtypes, the majority of patients fail to respond and only a small proportion of patients achieve durable responses and long-term survival in most tumor types. As such, dual immune checkpoint blockade and combinatorial immunotherapy are being extensively explored in order to improve response rates, PFS and overall survival (OS). While experience with metastatic melanoma has demonstrated improved efficacy with the combination of CTLA4 and PD1 blockade, these clinical benefits have come at the expense of increased toxicity with a corresponding increase in the proportion of patients that exhibit serious adverse events (98). As such, there is growing emphasis on testing new strategies and evaluating novel combinations of immune checkpoint inhibitors that increase efficacy but without substantially increasing toxicity, with considerable interest in PD1/LAG3 combinations (Table 1).
BMS-986016 was the first anti-LAG3 mAb to be developed, and it is currently being evaluated in several phase I and phase II trials in a variety of solid and hematological malignancies. The initial phase I/IIa trial launched by BMS in 2013 aimed to evaluate the efficacy of LAG3 blockade as a monotherapy or in combination with Nivolumab among patients with advanced malignancies (cervical, ovarian, bladder, colorectal, HPV-positive HNSCC, gastric, hepatocellular, RCC) who were naïve to immune-oncology agents (NCT01968109). NSCLC patients were allowed to be enrolled and treated per protocol as a first-line treatment or if they had progressed while on anti-PD1/PDL1 or anti-CTLA4 therapy. The primary endpoint of this study was safety and tolerability and to determine the maximum tolerated dose in order to inform dosing for the phase IIa cohort-expansion portion of this trial. The investigators aim to enroll 360 patients with an estimated completion date in 2018. A nearly identical phase I/IIa trial will explore the safety and tolerability of LAG3 blockade with BMS-986016 with or without Nivolumab in the setting of refractory or recurrent B-cell malignancies including non-Hodgkin lymphoma, CLL, Hodgkin lymphoma and multiple myeloma (NCT02061761). Given that PD1 blockade is rapidly becoming a standard of care (SOC) in the NSCLC anti-cancer armamentarium, the FRACTION-Lung study has set out to identify new agents that synergize with Nivolumab to improve objective response rates and PFS (NCT02750514). This phase II trial, with a target enrollment of 504 patients, will randomize patients to either Nivolumab monotherapy, Nivolumab in combination with BMS-986016 or Nivolumab in combination with desatinib. Finally, while immune checkpoint inhibition is in its infancy for primary central nervous system tumors, a recently initiated phase I trial is testing BMS-986016 as a monotherapy or in combination with Nivolumab among patients with progressive or recurrent glioblastoma/gliosarcoma following chemoradiation plus temozolomide or following re-resection with measurable residual disease post-operatively (NCT02658981).
A second humanized IgG4 anti-LAG3 mAb has been developed by Novartis (LAG525) and is also currently in Phase I/II clinical development to determine its safety and pharmacokinetic profile. This trial administers this agent as a monotherapy or in combination with a novel anti-PD1 inhibitor (PDR001) (NCT02460224). Initially, a dose-limiting toxicity study of LAG525 will be performed in patients with advanced/metastatic solid tumors (NSCLC, RCC, melanoma) to determine the maximum tolerated dose. A second Phase II trial will involve a dose expansion phase of LAG525 or the LAG525/PDR001 combination to assess the overall response rate in these patients. As part of this study, biomarker analysis, including correlation of PDL1 expression with clinical outcomes, and mRNA profiling of IFNγ-related genes will be performed. Further, evaluation of humoral immune responses including the potential emergence of anti-LAG525 antibodies will be assessed as secondary outcomes.
Merck has also recently entered phase I clinical testing of their mAb against LAG3 (MK-4280). Similar to several clinical trials discussed above, the investigators aim to evaluate the safety and tolerability of this agent as a monotherapy or combination with PD1 blockade (Pembrolizumab) in a dose-escalation cohort of 70 patients with metastatic solid tumors (NCT02720068).
Other mAbs and novel reagents targeting LAG3 are also at various stages of pre-clinical development. Tesaro, in collaboration with AnaptysBio, has developed an anti-LAG3 monospecific antagonist mAb (TSR-033), which will be entering Phase I clinical trials shortly. To capitalize on the enhanced efficiency of dual LAG3 and PD1 blockade in pre-clinical models, a number of bispecific anti-LAG3/PD1 antagonistic mAbs are being developed. One approach by Tesaro, partnered with AnaptysBio, utilizes a somatic hypermutation/mammalian cell system platform (SHM-XEL) that couples antibody libraries with in vitro somatic hypermutation in mammalian cells to generate high affinity antibodies (99). Similarly, MacroGenics has developed a bispecific agent that simultaneously binds LAG3 and PD1 (MGD013), generated using their Dual-Affinity Re-Targeting (DART) platform, this agent is currently under clinical testing. This technology, previously used for the generation of a CD19/CD3 DART protein designed to redirect T cells to eliminate CD19-expressing cells in hematological malignancies, covalently links two polypeptide chains between the variable domains of the two antibodies by a disulphide bridge, with a short linker connecting the binding domains to promote heterodimerization (100). Several other companies are developing their own novel reagents for targeting LAG3, attesting to the broad interest in the LAG3 pathway.
In summary, clinical trial development of antagonistic LAG3 mAbs was initially cautious but has expanded considerably recently, based in part on preclinical evidence supporting promising synergy with PD1 blockade (70). Thus it is likely that over a thousand patients with a variety of solid and hematological malignancies will be enrolled in clinical protocols exploring LAG3-based immune checkpoint blockade in the coming years.
Key Questions and Future Directions.
Since its discovery in 1990, we have gained considerable insight into the function and therapeutic potential of LAG3. There are now at least four LAG3-targeted therapeutics in the clinic with many more on the way. However, there are many important questions that remain to be addressed that will impact our understanding of LAG3 biology and mechanism of action, and its targeting in the clinic.
How does LAG3 work? This fundamentally important question remains largely unknown and elusive. While we know that LAG3 signaling impacts TCR signaling and function, we do not know how this is mediated. Given the unusual motifs present in the LAG3 cytoplasmic domain (e.g. EP and KIEELE), one might predict that its mode of action is unique and distinct from other IRs. Further elucidation of LAG3 function may have unforeseen implications for therapeutic development and may also highlight novel combinatorial therapeutic approaches.
What are the key ligands for LAG3? While MHC class II is the canonical LAG3 ligand, controversy remains. A full elucidation of all LAG3 ligands, when and where they are expressed and how they are utilized by LAG3 will facilitate mechanistic understanding and clinical development. LAG3 ligands may also serve as important biomarkers that may predict efficacy.
What is the mechanistic basis that underlies LAG3 synergy with PD1 and do synergies exist with other IRs? Addressing this key question will provide important insight into LAG3 biology and may facilitate optimization of LAG3 targeted therapies. A more complete evaluation of the impact of targeting LAG3 in combination with other IRs and alternate immunotherapeutic modalities is warranted. Clearly, the greatest focus will be on the therapeutic impact of combinatorial PD1/LAG3 immunotherapy.
What is the impact of LAG3 on different cell populations? While the role of LAG3 in CD4+ and CD8+ effector T cells has been studied extensively, its role on other cell types remains obscure. For instance, the role of LAG3, and other IRs, on Tregs is controversial as LAG3 has been suggested to mediate their regulatory activity but could also limit their intrinsic activity in a manner analogous to effector cells. This question is important as it may underlie the differential impact of IR-targeted immunotherapies. The role of LAG3 on NK cells, NKT cells and pDCs also remains largely unclear.
Could sLAG3 serve as a biomarker? Evidence exists for sLAG3 as a potential clinical biomarker in identifying cancer patients with improved prognostic outcomes. Whether sLAG3 sera levels in patients predict responsiveness to LAG3 targeted therapies, or correlates with clinical outcome in response to LAG3-based immunotherapy remains to be determined. And, while sLAG3 does not appear to have any physiological role, more analysis of this issue is warranted.
Given the success of CTLA4- and PD1/PDL1-targeted therapeutics in cancer, there is considerable interest in the outcome of the growing number of clinical trials with LAG3 therapeutics. As LAG3 is essentially the third IR to be targeted in the clinic, the outcome of these trials could significantly increase or dampen enthusiasm for subsequent targets in the pipeline (TIM3, TIGIT, etc.). Only time will tell if LAG3 continues to lag behind.
Acknowledgments
We thank Creg Workman and Kate Vignali for critical reading of the manuscript. This work was supported by the National Institutes of Health (P01 AI108545, P50 CA097190 to D.A.A.V.), NCI Comprehensive Cancer Center Support CORE grant (P30 CA047904, to D.A.A.V.) NCI Comprehensive Cancer Center Support CORE grant (P30 CA006973, to C.G.D), National Institutes of Health (R01 CA154555 to C.G.D), a PCF/Movember Challenge Award (to C.G.D) and the Melanoma Research Association (C.G.D). C.G.D. and D.A.A.V. declares competing financial interests. They have submitted patents that are pending or granted on LAG3, and are entitled to a share in net income generated from licensing of these patent rights for commercial development.
References
- 1.Zarour HM. Reversing T-cell Dysfunction and Exhaustion in Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:1856–1864. doi: 10.1158/1078-0432.CCR-15-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009;138:30–50. doi: 10.1016/j.cell.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 3.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature medicine. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 4.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. International journal of cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 5.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 6.Gravitz L. Cancer immunotherapy. Nature. 2013;504:S1. doi: 10.1038/504S1a. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gettinger S, Rizvi NA, Chow LQ, Borghaei H, Brahmer J, Ready N, Gerber DE, et al. Nivolumab Monotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2016;34:2980–2987. doi: 10.1200/JCO.2016.66.9929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turnis ME, Andrews LP, Vignali DA. Inhibitory receptors as targets for cancer immunotherapy. European journal of immunology. 2015;45:1892–1905. doi: 10.1002/eji.201344413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annual review of immunology. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 13.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, Hercend T. LAG-3, a novel lymphocyte activation gene closely related to CD4. The Journal of experimental medicine. 1990;171:1393–1405. doi: 10.1084/jem.171.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huard B, Mastrangeli R, Prigent P, Bruniquel D, Donini S, El-Tayar N, Maigret B, et al. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:5744–5749. doi: 10.1073/pnas.94.11.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang JH, Meijers R, Xiong Y, Liu JH, Sakihama T, Zhang R, Joachimiak A, et al. Crystal structure of the human CD4 N-terminal two-domain fragment complexed to a class II MHC molecule. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10799–10804. doi: 10.1073/pnas.191124098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moebius U, Pallai P, Harrison SC, Reinherz EL. Delineation of an extended surface contact area on human CD4 involved in class II major histocompatibility complex binding. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:8259–8263. doi: 10.1073/pnas.90.17.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li N, Workman CJ, Martin SM, Vignali DA. Biochemical analysis of the regulatory T cell protein lymphocyte activation gene-3 (LAG-3; CD223) Journal of immunology. 2004;173:6806–6812. doi: 10.4049/jimmunol.173.11.6806. [DOI] [PubMed] [Google Scholar]
- 19.Annunziato F, Manetti R, Tomasévic I, Guidizi MG, Biagiotti R, Giannò V, Germano P, et al. Expression and release of LAG-3-encoded protein by human CD4+ T cells are associated with IFN-gamma production. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1996;10:769–776. doi: 10.1096/fasebj.10.7.8635694. [DOI] [PubMed] [Google Scholar]
- 20.Hannier S, Triebel F. The MHC class II ligand lymphocyte activation gene-3 is co-distributed with CD8 and CD3-TCR molecules after their engagement by mAb or peptide-MHC class I complexes. International immunology. 1999;11:1745–1752. doi: 10.1093/intimm/11.11.1745. [DOI] [PubMed] [Google Scholar]
- 21.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. Journal of immunology. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 22.Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. Journal of immunology. 2002;169:5392–5395. doi: 10.4049/jimmunol.169.10.5392. [DOI] [PubMed] [Google Scholar]
- 23.Mastrangeli R, Micangeli E, Donini S. Cloning of murine LAG-3 by magnetic bead bound homologous probes and PCR (gene-capture PCR) Analytical biochemistry. 1996;241:93–102. doi: 10.1006/abio.1996.0382. [DOI] [PubMed] [Google Scholar]
- 24.Baixeras E, Huard B, Miossec C, Jitsukawa S, Martin M, Hercend T, Auffray C, et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. The Journal of experimental medicine. 1992;176:327–337. doi: 10.1084/jem.176.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. European journal of immunology. 1995;25:2718–2721. doi: 10.1002/eji.1830250949. [DOI] [PubMed] [Google Scholar]
- 26.Weber S, Karjalainen K. Mouse CD4 binds MHC class II with extremely low affinity. International immunology. 1993;5:695–698. doi: 10.1093/intimm/5.6.695. [DOI] [PubMed] [Google Scholar]
- 27.Workman CJ, Rice DS, Dugger KJ, Kurschner C, Vignali DA. Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3) Eur J Immunol. 2002;32:2255–2263. doi: 10.1002/1521-4141(200208)32:8<2255::AID-IMMU2255>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 28.Workman CJ, Vignali DA. Negative regulation of T cell homeostasis by lymphocyte activation gene-3 (CD223) Journal of immunology. 2005;174:688–695. doi: 10.4049/jimmunol.174.2.688. [DOI] [PubMed] [Google Scholar]
- 29.Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nature immunology. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruiter DJ, Mattijssen V, Broecker EB, Ferrone S. MHC antigens in human melanomas. Seminars in cancer biology. 1991;2:35–45. [PubMed] [Google Scholar]
- 31.Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, Triebel F, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. Journal of immunology. 2011;186:5173–5183. doi: 10.4049/jimmunol.1002050. [DOI] [PubMed] [Google Scholar]
- 32.Donia M, Andersen R, Kjeldsen JW, Fagone P, Munir S, Nicoletti F, Andersen MH, et al. Aberrant Expression of MHC Class II in Melanoma Attracts Inflammatory Tumor-Specific CD4+ T- Cells, Which Dampen CD8+ T-cell Antitumor Reactivity. Cancer research. 2015;75:3747–3759. doi: 10.1158/0008-5472.CAN-14-2956. [DOI] [PubMed] [Google Scholar]
- 33.Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res. 2015;3:412–423. doi: 10.1158/2326-6066.CIR-14-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dumic J, Dabelic S, Flogel M. Galectin-3: an open-ended story. Biochimica et biophysica acta. 2006;1760:616–635. doi: 10.1016/j.bbagen.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Tang L, Zhang G, Wei H, Cui Y, Guo L, Gou Z, et al. Characterization of a novel C-type lectin-like gene, LSECtin: demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. The Journal of biological chemistry. 2004;279:18748–18758. doi: 10.1074/jbc.M311227200. [DOI] [PubMed] [Google Scholar]
- 36.Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, Du X, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer research. 2014;74:3418–3428. doi: 10.1158/0008-5472.CAN-13-2690. [DOI] [PubMed] [Google Scholar]
- 37.Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, Ge P, et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science. 2016;353 doi: 10.1126/science.aah3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, et al. Role of LAG-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 39.Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, Guo B, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nature medicine. 2013;19:739–746. doi: 10.1038/nm.3179. [DOI] [PubMed] [Google Scholar]
- 40.White AM, Wraith DC. Tr1-Like T Cells - An Enigmatic Regulatory T Cell Lineage. Frontiers in immunology. 2016;7:355. doi: 10.3389/fimmu.2016.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fahrer AM, Konigshofer Y, Kerr EM, Ghandour G, Mack DH, Davis MM, Chien YH. Attributes of gammadelta intraepithelial lymphocytes as suggested by their transcriptional profile. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10261–10266. doi: 10.1073/pnas.171320798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denning TL, Granger SW, Mucida D, Graddy R, Leclercg G, Zhang W, Honey K, et al. Mouse TCRalphabeta+CD8alphaalpha intraepithelial lymphocytes express genes that down-regulate their antigen reactivity and suppress immune responses. Journal of immunology. 2007;178:4230–4239. doi: 10.4049/jimmunol.178.7.4230. [DOI] [PubMed] [Google Scholar]
- 43.Kisielow M, Kisielow J, Capoferri-Sollami G, Karjalainen K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. European journal of immunology. 2005;35:2081–2088. doi: 10.1002/eji.200526090. [DOI] [PubMed] [Google Scholar]
- 44.Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, Vignali DA. LAG-3 regulates plasmacytoid dendritic cell homeostasis. Journal of immunology. 2009;182:1885–1891. doi: 10.4049/jimmunol.0800185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nature immunology. 2004;5:1219–1226. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 46.Iouzalen N, Andreae S, Hannier S, Triebel F. LAP, a lymphocyte activation gene-3 (LAG-3)-associated protein that binds to a repeated EP motif in the intracellular region of LAG-3, may participate in the down-regulation of the CD3/TCR activation pathway. European journal of immunology. 2001;31:2885–2891. doi: 10.1002/1521-4141(2001010)31:10<2885::aid-immu2885>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 47.Macon-Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology. 2005;115:170–178. doi: 10.1111/j.1365-2567.2005.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, Flores M, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. Journal of immunology. 2008;180:5916–5926. doi: 10.4049/jimmunol.180.9.5916. [DOI] [PubMed] [Google Scholar]
- 49.Clayton KL, Douglas-Vail MB, Nur-ur Rahman AK, Medcalf KE, Xie IY, Chew GM, Tandon R, et al. Soluble T cell immunoglobulin mucin domain 3 is shed from CD8+ T cells by the sheddase ADAM10, is increased in plasma during untreated HIV infection, and correlates with HIV disease progression. Journal of virology. 2015;89:3723–3736. doi: 10.1128/JVI.00006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, Hartmann D, et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. The EMBO journal. 2007;26:494–504. doi: 10.1038/sj.emboj.7601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buisson S, Triebel F. LAG-3 (CD223) reduces macrophage and dendritic cell differentiation from monocyte precursors. Immunology. 2005;114:369–374. doi: 10.1111/j.1365-2567.2004.02087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lienhardt C, Azzurri A, Amedei A, Fielding K, Sillah J, Sow OY, Bah B, et al. Active tuberculosis in Africa is associated with reduced Th1 and increased Th2 activity in vivo. European journal of immunology. 2002;32:1605–1613. doi: 10.1002/1521-4141(200206)32:6<1605::AID-IMMU1605>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 53.Delmastro MM, Styche AJ, Trucco MM, Workman CJ, Vignali DA, Piganelli JD. Modulation of redox balance leaves murine diabetogenic TH1 T cells “LAG-3-ing” behind. Diabetes. 2012;61:1760–1768. doi: 10.2337/db11-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Triebel F, Hacene K, Pichon MF. A soluble lymphocyte activation gene-3 (sLAG-3) protein as a prognostic factor in human breast cancer expressing estrogen or progesterone receptors. Cancer Lett. 2006;235:147–153. doi: 10.1016/j.canlet.2005.04.015. [DOI] [PubMed] [Google Scholar]
- 55.Bae J, Lee SJ, Park CG, Lee YS, Chun T. Trafficking of LAG-3 to the surface on activated T cells via its cytoplasmic domain and protein kinase C signaling. Journal of immunology. 2014;193:3101–3112. doi: 10.4049/jimmunol.1401025. [DOI] [PubMed] [Google Scholar]
- 56.Woo SR, Li N, Bruno TC, Forbes K, Brown S, Workman C, Drake CG, et al. Differential subcellular localization of the regulatory T-cell protein LAG-3 and the coreceptor CD4. European journal of immunology. 2010;40:1768–1777. doi: 10.1002/eji.200939874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huard B, Tournier M, Hercend T, Triebel F, Faure F. Lymphocyte-activation gene 3/major histocompatibility complex class II interaction modulates the antigenic response of CD4+ T lymphocytes. European journal of immunology. 1994;24:3216–3221. doi: 10.1002/eji.1830241246. [DOI] [PubMed] [Google Scholar]
- 58.Workman CJ, Vignali DA. The CD4-related molecule, LAG-3 (CD223), regulates the expansion of activated T cells. European journal of immunology. 2003;33:970–979. doi: 10.1002/eji.200323382. [DOI] [PubMed] [Google Scholar]
- 59.Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. Journal of immunology. 2004;172:5450–5455. doi: 10.4049/jimmunol.172.9.5450. [DOI] [PubMed] [Google Scholar]
- 60.Rogala B, Gluck J, Mazur B. Do the molecules CD26 and lymphocytes activation gene-3 differentiate between type 1 and 2 T cell response? Journal of investigational allergology & clinical immunology. 2002;12:198–203. [PubMed] [Google Scholar]
- 61.Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, Anders R, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. The Journal of clinical investigation. 2007;117:3383–3392. doi: 10.1172/JCI31184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, Hipkiss E, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. Journal of immunology. 2009;182:6659–6669. doi: 10.4049/jimmunol.0804211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Durham NM, Nirschi CJ, Jackson CM, Elias J, Kochel CM, Anders RA, Drake CG. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PloS one. 2014;9:e109080. doi: 10.1371/journal.pone.0109080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E, Santinami M, et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. The Journal of investigative dermatology. 2014;134:1893–1902. doi: 10.1038/jid.2014.29. [DOI] [PubMed] [Google Scholar]
- 65.Huang B, Lei Z, Zhao J, Gong W, Liu J, Chen Z, Liu Y, et al. CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer letters. 2007;252:86–92. doi: 10.1016/j.canlet.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 66.Byun HJ, Jung WW, Lee DS, Kim S, Kim SJ, Park CG, Chung HY, et al. Proliferation of activated CD1d-restricted NKT cells is down-modulated by lymphocyte activation gene-3 signaling via cell cycle arrest in S phase. Cell biology international. 2007;31:257–262. doi: 10.1016/j.cellbi.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 67.Juno JA, Stalker AT, Waruk JL, Oyugi J, Kimani M, Plummer FA, Kimani J, et al. Elevated expression of LAG-3, but not PD-1, is associated with impaired iNKT cytokine production during chronic HIV-1 infection and treatment. Retrovirology. 2015;12:17. doi: 10.1186/s12977-015-0142-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, Pan X, Drake CG, et al. Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. Journal of immunology. 2011;187:3493–3498. doi: 10.4049/jimmunol.1100714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jha V, Workman CJ, McGaha TL, Li L, Vas J, Vignali DA, Monestier M. Lymphocyte Activation Gene-3 (LAG-3) negatively regulates environmentally-induced autoimmunity. PloS one. 2014;9:e104484. doi: 10.1371/journal.pone.0104484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschi CJ, Bettini ML, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer research. 2012;72:917–927. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 72.Richter K, Agnellini P, Oxenius A. On the role of the inhibitory receptor LAG-3 in acute and chronic LCMV infection. International immunology. 2010;22:13–23. doi: 10.1093/intimm/dxp107. [DOI] [PubMed] [Google Scholar]
- 73.Waugh KA, Leach SM, Moore BL, Bruno TC, Buhrman JD, Slansky JE. Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model. Journal of immunology. 2016;197:1477–1488. doi: 10.4049/jimmunol.1600589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang RY, Eppolito C, Lele S, Shrikant P, Matsuzaki J, Odunsi K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget. 2015;6:27359–27377. doi: 10.18632/oncotarget.4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, Fulton A, et al. Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. Journal of immunology. 2013;190:4899–4909. doi: 10.4049/jimmunol.1300271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jing W, Gershan JA, Weber J, Tlomak D, McOlash L, Sabatos-Peyton C, Johnson BD. Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. Journal for immunotherapy of cancer. 2015;3:2. doi: 10.1186/s40425-014-0043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Foy SP, Sennino B, dela Cruz T, Cote JJ, Gordon EJ, Kemp F, Xavier V, et al. Poxvirus-Based Active Immunotherapy with PD-1 and LAG-3 Dual Immune Checkpoint Inhibition Overcomes Compensatory Immune Regulation, Yielding Complete Tumor Regression in Mice. PloS one. 2016;11:e0150084. doi: 10.1371/journal.pone.0150084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:7875–7880. doi: 10.1073/pnas.1003345107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, Wieckowski S, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. The Journal of clinical investigation. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taube JM, Young GD, McMiller TL, Chen S, Salas JT, Pritchard TS, Xu H, et al. Differential Expression of Immune-Regulatory Genes Associated with PD-L1 Display in Melanoma: Implications for PD-1 Pathway Blockade. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:3969–3976. doi: 10.1158/1078-0432.CCR-15-0244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li FJ, Zhang Y, Jin GX, Yao L, Wu DQ. Expression of LAG-3 is coincident with the impaired effector function of HBV-specific CD8(+) T cell in HCC patients. Immunology letters. 2013;150:116–122. doi: 10.1016/j.imlet.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 82.Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer discovery. 2015;5:43–51. doi: 10.1158/2159-8290.CD-14-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, Parmiani G, et al. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J Immunol. 2010;184:6545–6551. doi: 10.4049/jimmunol.0903879. [DOI] [PubMed] [Google Scholar]
- 84.Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, Ferris RL. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;109:2629–2635. doi: 10.1038/bjc.2013.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei T, Zhang J, Qin Y, Wu Y, Zhu L, Lu L, Tang G, et al. Increased expression of immunosuppressive molecules on intratumoral and circulating regulatory T cells in non-small-cell lung cancer patients. American journal of cancer research. 2015;5:2190–2201. [PMC free article] [PubMed] [Google Scholar]
- 86.Scurr M, Ladell K, Besneux M, Christian A, Hockey T, Smart K, Bridgeman H, et al. Highly prevalent colorectal cancer-infiltrating LAP(+) Foxp3(−) T cells exhibit more potent immunosuppressive activity than Foxp3(+) regulatory T cells. Mucosal immunology. 2014;7:428–439. doi: 10.1038/mi.2013.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen J, Chen Z. The effect of immune microenvironment on the progression and prognosis of colorectal cancer. Medical oncology. 2014;31:82. doi: 10.1007/s12032-014-0082-9. [DOI] [PubMed] [Google Scholar]
- 88.Kotaskova J, Tichy B, Trbusek M, Francova HS, Kabathova J, Malcikova J, Doubek M, et al. High expression of lymphocyte-activation gene 3 (LAG3) in chronic lymphocytic leukemia cells is associated with unmutated immunoglobulin variable heavy chain region (IGHV) gene and reduced treatment-free survival. The Journal of molecular diagnostics: JMD. 2010;12:328–334. doi: 10.2353/jmoldx.2010.090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang J, Xiang Y, Ding L, Keen-Circle K, Borlawsky TB, Ozer HG, Jin R, et al. Using gene co-expression network analysis to predict biomarkers for chronic lymphocytic leukemia. BMC bioinformatics. 2010;11(Suppl 9):S5. doi: 10.1186/1471-2105-11-S9-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andreae S, Piras F, Burdin N, Triebel F. Maturation and activation of dendritic cells induced by lymphocyte activation gene-3 (CD223) Journal of immunology. 2002;168:3874–3880. doi: 10.4049/jimmunol.168.8.3874. [DOI] [PubMed] [Google Scholar]
- 91.Avice MN, Sarfati M, Triebel F, Delespesse G, Demeure CE. Lymphocyte activation gene-3, a MHC class II ligand expressed on activated T cells, stimulates TNF-alpha and IL-12 production by monocytes and dendritic cells. Journal of immunology. 1999;162:2748–2753. [PubMed] [Google Scholar]
- 92.Buisson S, Triebel F. MHC class II engagement by its ligand LAG-3 (CD223) leads to a distinct pattern of chemokine and chemokine receptor expression by human dendritic cells. Vaccine. 2003;21:862–868. doi: 10.1016/s0264-410x(02)00533-9. [DOI] [PubMed] [Google Scholar]
- 93.Brignone C, Grygar C, Marcu M, Schakel K, Triebel F. A soluble form of lymphocyte activation gene-3 (IMP321) induces activation of a large range of human effector cytotoxic cells. Journal of immunology. 2007;179:4202–4211. doi: 10.4049/jimmunol.179.6.4202. [DOI] [PubMed] [Google Scholar]
- 94.Brignone C, Escudier B, Grygar C, Marcu M, Triebel F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2009;15:6225–6231. doi: 10.1158/1078-0432.CCR-09-0068. [DOI] [PubMed] [Google Scholar]
- 95.Wang-Gillam A, Plambeck-Suess S, Goedegebuure P, Simon PO, Mitchem JB, Hornick JR, Sorscher S, et al. A phase I study of IMP321 and gemcitabine as the front-line therapy in patients with advanced pancreatic adenocarcinoma. Investigational new drugs. 2013;31:707–713. doi: 10.1007/s10637-012-9866-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Legat A, Maby-El Hajjami H, Baumgaertner P, Cagnon L, Abed Maillard S, Geldhof C, Iancu EM, et al. Vaccination with LAG-3Ig (IMP321) and Peptides Induces Specific CD4 and CD8 T-Cell Responses in Metastatic Melanoma Patients–Report of a Phase I/IIa Clinical Trial. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:1330–1340. doi: 10.1158/1078-0432.CCR-15-1212. [DOI] [PubMed] [Google Scholar]
- 97.Romano E, Michielin O, Voelter V, Laurent J, Bichat H, Stravodimou A, Romero P, et al. MART-1 peptide vaccination plus IMP321 (LAG-3Ig fusion protein) in patients receiving autologous PBMCs after lymphodepletion: results of a Phase I trial. Journal of translational medicine. 2014;12:97. doi: 10.1186/1479-5876-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette CP, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. The New England journal of medicine. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bowers PM, Horlick RA, Kehry MR, Neben TY, Tomlinson GL, Altobell L, Zhang X, et al. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods. 2014;65:44–56. doi: 10.1016/j.ymeth.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 100.Liu L, Lam CK, Long V, Widjaja L, Yang Y, Li H, Jin L, et al. MGD011, a CD19 × CD3 Dual Affinity Re-Targeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-cell Malignancies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-16-0666. [DOI] [PubMed] [Google Scholar]