

SUMMARY
We previously reported a genetic analysis of heart failure traits in a population of inbred mouse strains treated with isoproterenol to mimic catecholamine-driven cardiac hypertrophy. Here, we apply a co-expression network algorithm, wMICA, to perform a systems-level analysis of left ventricular transcriptomes from these mice. We describe the features of the overall network but focus on a module identified in treated hearts that is strongly related to cardiac hypertrophy and pathological remodeling. Using the causal modeling algorithm NEO, we identified the gene Adamts2 as a putative regulator of this module and validated the predictive value of NEO using small interfering RNA-mediated knockdown in neonatal rat ventricular myocytes. Adamts2 silencing regulated the expression of the genes residing within the module and impaired isoproterenol-induced cellular hypertrophy. Our results provide a view of higher order interactions in heart failure with potential for diagnostic and therapeutic insights.
Graphical abstract
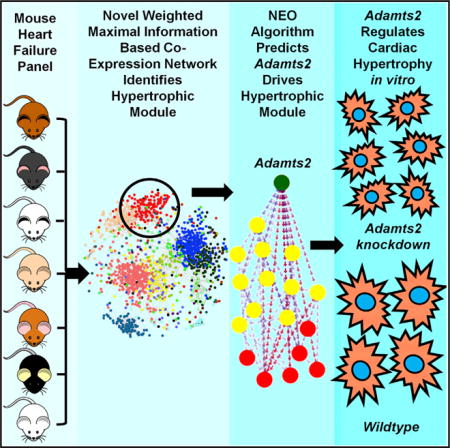
INTRODUCTION
Heart failure (HF) is a common disorder characterized by impaired heart function, cardiac hypertrophy, and chamber remodeling (Frangogiannis, 2012). Despite reports of significant genetic heritability, genome-wide association studies (GWAS) involving tens of thousands of patients have had only modest success, likely due to the complex, heterogeneous nature of the disease (reviewed in Rau et al., 2015a). These complexities can be minimized in genetic studies of model organisms such as mice, and classical quantitative trait locus (QTL) linkage analyses in mice have identified a number of novel HF-related genes (McNally et al., 2015; Wheeler et al., 2009).
In previous work, we have shown that a GWAS approach can be applied to populations of common inbred strains of mice if associations are corrected for population structure (Bennett et al., 2010). We studied a population of over 100 commercially available inbred strains of mice selected for diversity, constituting a resource that we termed the Hybrid Mouse Diversity Panel (HMDP). The mapping resolution of this approach is at least an order of magnitude better than traditional QTL analyses involving genetic crosses and has led to the identification of novel genes for a number of traits (reviewed in Lusis et al., 2016). We recently applied this approach to identify loci and genes that contribute to HF traits in an isoproterenol (ISO) model, which mimics the chronic β-adrenergic stimulation that often occurs in human HF. Association analyses identified both known and novel genes contributing to hypertrophy, cardiac fibrosis, and echocardiographic traits (Rau et al., 2015b; Wang et al., 2016).
We now report an extension of this study in which we seek to understand genes and pathways that contribute to HF through the modeling of biological networks. We apply an improved version of the Maximal Information Component Analysis (MICA) algorithm (Rau et al., 2013), with increased versatility and power, to left ventricular transcriptomes of the HMDP population before and after treatment with ISO to form modules of functionally related genes. Several modules that showed significant association to HF-related phenotypes were identified. We focused our analysis on a module based on treated expression data as it exhibited striking correlations with a number of HF traits and contained several genes previously implicated in HF, such as Nppa and Timp1. We then applied the NEO (Near Edge Orientation) algorithm (Aten et al., 2008) to develop a directed network with predicted causal interactions among the module genes. The results suggested that Adamts2, a metalloproteinase not previously associated with HF, plays a key role in modulating the expression of other genes in the module in response to ISO stimulation. Using an in vitro model, we validated several of these causal links and demonstrated that Adamts2 expression affected several proxy measurements of cardiac hypertrophy.
RESULTS
Gene Network Analysis Using Weighted MICA
Prior research (Farber, 2013) using the HMDP benefited from the use of systems-level transcriptomics to generate mRNA co-expression networks. We previously reported an unbiased gene network construction algorithm, termed MICA, which has several conceptual improvements over traditional co-expression methods in that it captures both linear and nonlinear interactions within the data and allows genes to be spread proportionally across multiple modules (Rau et al., 2013). Previous research on gene networks (Langfelder and Horvath, 2008) has shown that weighted network construction algorithms, in which all edges are included in the analysis, have greater versatility and power than unweighted algorithms, in which edges are included or excluded based on a hard threshold. Therefore, we have improved upon our original algorithm (STAR Methods) and developed a modified, weighted form of MICA, which we term wMICA. We describe here the first application of wMICA to the analysis of HF, using gene expression data across inbred strains of mice from the HMDP HF study.
Left ventricular tissue from the HMDP was processed using Illumina Mouse Ref 2.0 gene expression arrays. Probes were filtered for transcripts that were significantly expressed in at least 25% of samples and had a coefficient of variation of at least 5%. This resulted in a final set of 8,126 probes, representing 31.6% of the total probes on the array. Three gene networks, with 20 modules each, were generated from these data: one based only on transcripts from the untreated hearts, one based only on the treated hearts, and a third based on the change in gene expression between these two conditions (Data S1).
Two measures were used for the preliminary analyses of these networks. We calculated significant Gene Ontology (GO) enrichments within each of these modules at several module membership cutoffs, using the Database for Annotation, Visualization and Integrated Discovery (DAVID). Significant enrichment for one or more GO terms suggests that the module represents a collection of genes that are biologically related to one another and are less likely to be an artifact of the module identification process. We also used principal-component analysis (PCA) to identify the first principal component (often called the “eigengene”) of each module. This eigengene can be correlated to HF-related phenotypes to identify modules that, as a whole, are most likely related to specific features of cardiac pathogenesis. As wMICA allows genes to reside within multiple modules, we used a weighted PCA algorithm to calculate the first weighted principal component of each module based on each gene’s module membership. The weighted eigengene was then correlated to a number of HF-related phenotypes from the HMDP panel. They include seven organ weights, eight echocardiographic parameters, and five plasma traits that, in conjunction with either an organ weights or functional parameters, suggest a change in metabolism status associated with HF.
Heart Failure Co-expression Networks
MICA Modules Generated from Untreated Left Ventricular Tissue
Three MICA-generated modules were observed to have very strong (DAVID score > 7) enrichments, while five other modules showed significant (score > 3) enrichments. Only a single module (not one of the eight with strong enrichments to a GO category) had even suggestive (p < 0.01) correlations to an HF-related phenotype. Therefore, the co-expression gene modules identified in the pre-treated hearts appear to have limited correlation with susceptibility to HF.
MICA Modules Generated from Treated Left Ventricular Tissue
Thirteen of the modules of the MICA network constructed from treated hearts had significant DAVID enrichments, including three modules (3, 4, and 5) with enrichment scores greater than 10 (Figure 1A; Table S1). Three modules (1, 5, and 19) contained at least one significant (p < 10−4) correlation between the eigengene and either an organ weight or echocardiographic parameter (Figure 1B; Figure S1). Four additional modules (4, 12, 15, and 18) contained at least two suggestive (p < 0.01) correlations between a module and a HF-related trait.
Figure 1. Visualization, Functional Enrichment, and Trait Correlations of the Treated Left Ventricular Gene Co-expression Network.
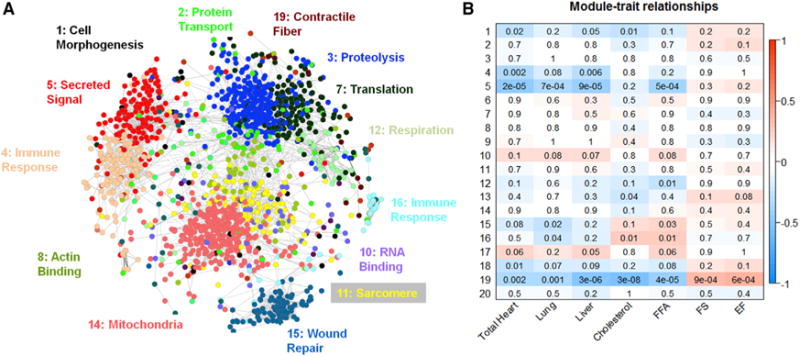
(A) 8126 transcripts were arranged into 20 distinct modules by the network construction algorithm MICA. For the ease of visualization, only probes with a module membership of greater than 50% in a module (4,392) were included, and probes with a module membership of over 50% in module 9 (2,783) were removed, as they tended to mask the relationships between the genes in the smaller modules. Circles (nodes) represent transcripts, and lines (edges) represent significant (MI score > 0.35) relationships between nodes. Probes are arranged by correlation strength where shorter distances indicate high correlation. Node color indicates maximal module membership. The groups of transcripts corresponding to each module were analyzed using DAVID to identify any significant enrichment for biological processes. The most significant biological processes for that particular module has been labeled in the color corresponding to the module.
(B) A weighted PCA algorithm was used to calculate the first weighted principal component of each module. These weighted first principal components were then correlated to the HF-related phenotypes. Strength of correlation is indicated by color, while the p value of the correlation is printed within each box. EF, ejection fraction; FFA, free fatty acid; FS, fractional shortening.
Module 5 has 309 genes that have maximal module membership within the module. It showed strong correlations to 7 of 20 measured HF traits: total heart weight, left and right ventricle weights, left ventricular internal dimension, lung weight, liver weight, and plasma free fatty acids, suggesting a strong relationship between this module and cardiac hypertrophy and HF (Figure 1B). Module 5 also showed highly significant GO enrichments for several biological processes: extracellular matrix (p < 10−18), secreted signaling (p < 10−16), and cell adhesion (p < 10−9). Of the 47 probes (42 genes) that possess greater than 70% module membership (Figure 2A), 20 genes (47.7%) have previously been described as involved in heart hypertrophy or cardiac remodeling based on transgenic studies in animal models or mendelian forms of HF. These genes include Nppa, a well-known marker of cardiac hypertrophy; Timp1, an extracellular matrix regulator (Barton et al., 2003); and the collagen genes Col12a1, Col14a1, and Col6a2. We further observed three genes (Pcolce, Sprx, and Sprx2) that we had previously identified via GWAS as candidate genes for cardiac phenotypes in this panel of mice (Rau et al., 2015b). These features led us to characterize module 5 in greater detail.
Figure 2. Visualization and Causal Modeling of Module 5.
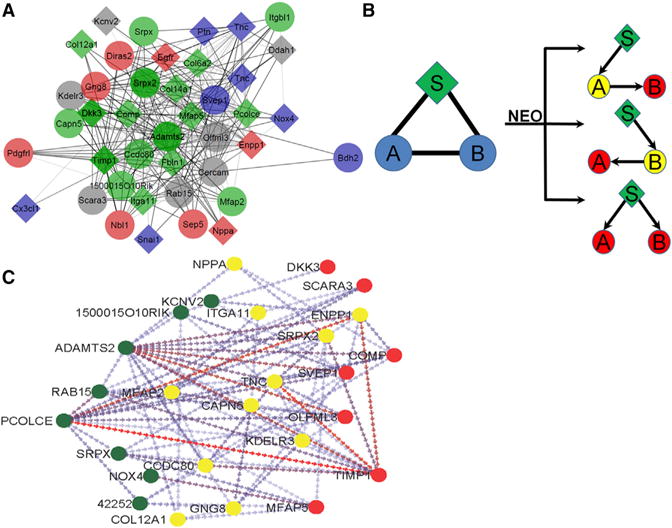
(A) 309 probes contain a maximal membership in module 5. To examine the most important genes in the module, only those 47 probes (42 genes) that possess a module membership of 70% or greater (the “core” module) are shown. Diamonds indicate genes that have been previously implicated in heart disease. The color of the diamonds and circles indicate GO category membership, with blue indicating glycoprotein, red indicating signaling, green indicating extracellular region, and gray indicating no significant GO category membership.
(B) A schematic depicting the way in which the NEO algorithm uses genetic markers (indicated with an S) as anchors to infer directionality between two genes (indicated with an A and B).
(C) Module 5 after NEO. 44 probes of the module, representing 93.6% of the core module, showed at least one edge with significant inferred directionality. Circles indicate the nodes, while arrows indicate the inferred directionality by NEO between two nodes. Node color indicates the percentage of edges with inferred directions to or from the node, with green indicating 75% or greater inferred edges originating from the node, yellow indicating a roughly equal percentage of edges originating from and traveling to the node, and red indicating that 75% or greater of the edges originating from another node.
MICA Modules Generated from the Change in Gene Expression between Control and Treated Mice: Delta Network
We observed the strongest associations between gene modules and GO terms in the MICA network generated from the changes in gene expression induced by ISO and subsequent remodeling, with 11 modules showing significant enrichments. Four of these modules had DAVID scores greater than 10, and two had scores greater than 15. Despite strong enrichments for GO terms, no eigengene/trait correlations for any organ weight or echocardiographic parameter had a p value less than 0.01.
Causality Modeling of Modules Using the NEO Algorithm
Using co-expression networks based on common genetic variation in populations, it is possible to model causal interactions and orient edges. The NEO (Aten et al., 2008) algorithm uses SNPs as anchors to infer directionality between SNP/gene/gene triads based on several possible models (Figure 2B). We applied the NEO algorithm to the largest connected components of the core genes of modules 5.
Of the 47 probes in the core network of module 5, 44 had at least one edge with significant directionality (Figure 2C). Genes with directed edges were classified into three categories (reactive, intermediary, and driver) based upon the number of directed edges traveling from the gene of interest to other genes in the module and the number of directed edges traveling from other genes in the module to the gene of interest (Table S2). We observed 12 “reactive” genes in which 75% or more of their directed edges travel to the gene from other genes in the module. The most reactive gene is Timp1, an important marker of left ventricular remodeling and HF (Barton et al., 2003), which is predicted to be affected by 18 other genes in the module. Similarly, we classified 12 genes as drivers based on the observation that, at minimum, 75% of their directed edges originate from the gene and travel to other genes in the module. Of the 12 drivers, 3 are notable. The first, Pcolce, a previously reported GWAS candidate gene (Rau et al., 2015b), has 11 outputs representing 26% of the genes in the core of module 5. The second, Nox4, which has a single output to Mfap5, has been previously established to be a key contributor to oxidative stress during pressure overload (Kuroda et al., 2010). The third driver, the metalloproteinase Adamts2, has not been previously implicated in HF or muscle development and has the largest number of directed edges within the module, with 23 (56% of module 5 genes) causal and 2 reactive (to the exocytosis regulator Rab15 and the extracellular matrix organizer Ccdc80) edges. Furthermore, Adamts2 showed significant correlations to lung weight (R = 0.46 · p = 4.6 · 10−6), heart weight (R = 0.43 · p = 1.5 · 10−5), and free fatty acids (R = 0.43, p = 2.0 · 10−5) after ISO stimulation, and it has both the highest degree (30) and third highest betweenness centrality of any gene within the module. Examination of sequence variation in and near the Adamts2 gene revealed a significant cis-eQTL (expression QTL) but no evidence of alternative splicing or deleterious missense mutations (see Table S4 for a sequence-level analysis of each “driver” gene from this module). Taken together, these results suggest that Adamts2 is a previously unidentified regulator of cardiac pathology.
Validation of the Driver Role of Adamts2 in Module 5 Using Cultured Cardiomyocytes
To test the NEO-predicted regulation of gene expression by Adamts2, we measured the expression of a set of seven predicted downstream targets residing in module 5, as well as a gene that was not predicted to be a target, the calcium transporter Atp2a2 (Figure 3; Figure S4). These seven genes (Col12a1, Kcnv2, Mfap2, Nppa, Pcolce, Timp1, and Tnc) were selected based on three criteria: strength of predicted relationship to Adamts2, number of directed edges in module 5, and previous associations with cardiac function and/or cardiovascular disease (Table S3). Using small interfering RNA (siRNA) to silence Adamts2, we achieved a 28%–45% decrease in Adamts2 expression when compared to transfection control (Figure 3A). We did not observe any consistent changes in Atp2a2 expression upon siRNA knockdown (Figure S2), consistent with the NEO prediction that it is not a target of Adamts2 (Figure 2). In contrast, we observed significant changes in four genes predicted to be targets (Kcnv2, Mfap2, Nppa, and Tnc) (Figure 3), suggesting that Adamts2 acts to regulate their expression under ISO-treated conditions in cardiomyocytes. The failure to observe the predicted responses for the other genes is likely due either to insufficient knockdown of Adamts2 or incorrect prediction by the NEO algorithm.
Figure 3. Adamts2 Alters the Expression of Module-5 Genes in Cardiomyocytes.
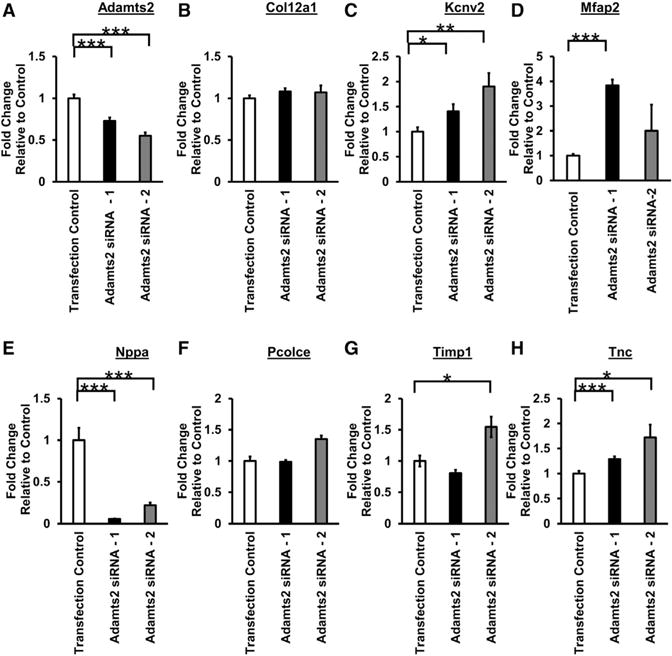
(A–H) NRVMs were transfected with either control siRNA or Adamts2 siRNA. Following overnight transfection, NRVMs were treated with isoproterenol (60 μM)-containing media for 48 hr. mRNA levels were quantified using qRT-PCR. Plots show average ± SEM. n = 9. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
Adamts2 Is a Regulator of β-Adrenergic-Induced Cardiomyocyte Hypertrophy In Vitro
As module 5 is significantly correlated with changes in numerous HF-related traits, we hypothesized that changes in the expression in Adamts2, a predicted driver of module 5, would alter cardiomyocyte size and/or viability in response to ISO. Indeed, following treatment with ISO, cells transfected with the control siRNA nearly doubled in cellular cross-sectional area (Figures 4A and 4B). Cells that expressed less Adamts2 due to siRNA transfection were smaller than scramble-transfected cells following treatment with ISO, being about the same size as the control cells without ISO stimulation (Figures 4A and 4B).
Figure 4. Adamts2 Regulates Cardiac Hypertrophy In Vitro.
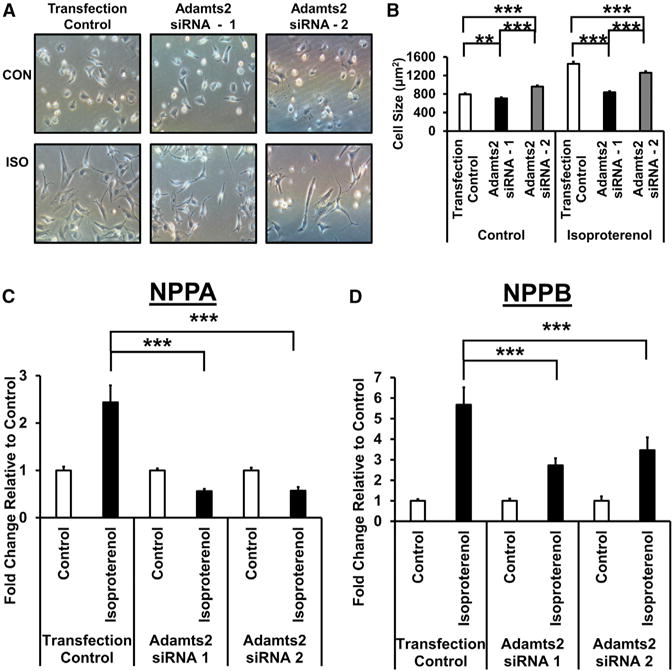
NRVMs were transfected with either control siRNA or Adamts2 siRNA. Following overnight transfection, NRVMs were treated with either control or isoproterenol (60 μM)-containing media for 48 hr.
(A) Representative images of cardiomyocytes transfected with either control or Adamts2 siRNA following 48 hr treatment with control or isoproterenol-containing media.
(B) Quantification of cardiomyocyte cell size following transfection with either control or Adamts2 siRNA and a 48-hr treatment with control or isoproterenol-containing media. Plots show average ± SEM. n = 6.
(C and D) mRNA expression levels of Nppa and Nppb were quantified using qRT-PCR. Plots show average ± SEM. n = 8–9. *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
At the molecular level, treatment with ISO induced the expression of the hypertrophic markers Nppa and Nppb, which rose 2.4-fold and 5.7-fold, respectively, in cells transfected with the control siRNA. Knockdown of Adamts2 strongly impaired these inductions, with Nppa induction reduced by approximately 75% and Nppb expression reduced by approximately 65% (Figures 4C and 4D). The significant response of the hypertrophic markers to relatively modest changes in Adamts2 expression under an adrenergic stimulus suggests a non-linear relationship between these genes. As such, these findings indicate that Adamts2 acts as a regulator of β-adrenergic-induced cardiac hypertrophy in cardiomyocytes.
DISCUSSION
We report network modeling of the molecular pathways contributing to HF in an ISO-treated mouse model. Prior efforts to analyze transcriptome networks underlying HF and related cardiomyopathies in human studies have met with limited successes, likely due in part to the high degree of heterogeneity of HF etiology and progression in humans (Drozdov et al., 2013; Moreno-Moral et al., 2013). Additionally, human heart samples are generally available only from extremely late-stage HF, masking pathways involved in the initial stages in favor of the reactive pathways. In contrast, by using the HMDP mice, we were able to model networks with minimal environmental heterogeneity. In addition to the conclusions reported here, our weighted module membership tables and network graphs (Data S1) should be useful in exploring how genes or pathways of interest intersect with other genes and pathways involved in HF.
Although we constructed MICA networks from three separate sets of gene expression data, we found that the treated network returned the most relevant modules. The control network had generally weaker GO enrichments and only a single module with even suggestive (p < 0.01) correlations with organ weights or echocardiographic parameters. The delta network had GO enrichments on par with the treated modules but no significant correlations with the HF phenotypes. Hierarchical clustering analysis revealed that 11 of the 20 modules from the delta network were highly correlated (r > 0.8) to one another, suggesting that the identified modules in the network are, in fact, largely reflecting a single “mega-module” of genes that have reacted strongly to ISO stimulation but whose underlying genetic variability has been masked to such a degree that they cannot be cleanly differentiated into phenotypically relevant modules. The network based on ISO-treated gene expression, on the other hand, contained well-defined modules strongly enriched for GO categories and very significantly associated with HF traits.
We focused on module 5 of the ISO-treated network in detail since it was significantly correlated with the traits of total heart weight, lung weight, liver weight, and plasma free fatty acids, as well as several functional parameters. Additionally, a total of 15 module-5 genes have been implicated in the development of HF and HF-related traits. Of these, six genes are predicted to be cardioprotective, as deficiencies promote cardiac remodeling (Col14a1 [Tao et al., 2012], Dkk3 [Bao et al., 2015], and Timp1 [Ikonomidis et al., 2005]) or decrease angiogenesis and neovascularization (Olfml3 [Miljkovic-Licina et al., 2012], Srpx2 [Miljkovic-Licina et al., 2009], and Ptn [Li et al., 2007]). In contrast, six genes are probably maladaptive, as their overexpression leads to increases in cardiac remodeling (Col6a2 [Grossman et al., 2011], Cx3cl1 [Xuan et al., 2011], Egfr [Messaoudi et al., 2012], Pcolce [Kessler-Icekson et al., 2006], and Tnc [Nishioka et al., 2010]), oxidative stress (Nox4), or fibrosis (Itga11 [Talior-Volodarsky et al., 2012]) during HF. The remaining four genes have been implicated in the development of vascular disorders such as generalized arterial calcification of infancy (Enpp1 [Hofmann Bowman and McNally, 2012]) or thoracic abdominal aneurysm (Mfap5 [Barbier et al., 2014]). Two genes have been implicated in cardiovascular development through the regulation of ventricular morphogenesis (Fbln1 [Cooley et al., 2012]) or cardiac valve formation (Snai1 [Tao et al., 2011]).
Despite the fact that ISO activates the neurohormonal signaling cascade, many of the genes residing within module 5, with the exception of Nppa, are not directly related to canonical β-adrenergic signaling. Rather, approximately one third (29%) have been previously implicated in inflammatory signaling, integrin signaling, or transforming growth factor β (TGF-β) signaling. Four genes (Cx3cl1, Capn5, Cercam, and Ccdc80) are related to the activation of inflammatory signaling. In particular, an inhibitor of Cx3cl1, known as fractalkine, suppresses progression of HF in both MI (myocardial infarction) and TAC (tranverse aortic constriction) models (Xuan et al., 2011). Five module-5 genes (Comp, Fbln1, Itga11, Svep1, and Tnc) either have direct interactions with integrins expressed on the cardiomyocyte surface or are integrin proteins. In addition, several module-5 genes interact with proteins known to contribute to integrin signaling, including fibrillin 1 (Mfap2 and Mfap5; Hanssen et al., 2004), decorin (Col14a1; Ehnis et al., 1997), fibronectin (Col6a2; Tillet et al., 1994), and FAK (Srpx2; Tanaka et al., 2009). Thus, it seems likely that integrin-mediated signaling plays a role in ISO-induced stress responses and subsequent pathology.
TGF-β signaling plays an important role in the pathogenesis of cardiac remodeling through its effects on cardiomyocytes, immune cells, and mesenchymal cells (Dobaczewski et al., 2011), and 13 of the module-5 genes are directly involved in TGF-β signaling, with four genes, including Pcocle (Moali et al., 2005), which activated or enhanced signaling; and two, including Dkk3, which antagonize signaling. They also include downstream targets of TGF-β signaling such as: Enpp1 (Goding et al., 2003), Fbln1 (Chen et al., 2013), Nox4 (Yan et al., 2014), Timp1 (Hall et al., 2003), and Tnc (Jinnin et al., 2004).
Using NEO causal modeling, we identified Adamts2 as a key driver of module 5. Adamts2 is a member of the “disintegrin and metalloproteinase with thrombospondin motifs” family of metalloproteases (Tortorella et al., 2009). Mutations in Adamts2 can cause the disorder Ehler Danlos Type VIIC, characterized by severe skin fragility and joint laxity (hyperextensibility) (Porter et al., 2005). Using siRNA knockdown of Adamts2 in cardiomyocytes, we validated five novel targets of Adamts2, including Kcnv2, Mfap2, Tnc, Nppa, and Nppb. Of these, Tnc, Nppa, and Nppb are currently associated with the development of HF, while Kcnv2 and Mfap2 are two novel genes not previously associated with HF.
While the direct molecular mechanism by which Adamts2 expression acts to regulate these five target genes is unknown, recent work has suggested that Adamts2 may act as a upstream regulator of TGF-β signaling through direct cleavage of the proteins TGF-β RIII and DKK3 (Bekhouche et al., 2016). Dkk3, a known cardioprotective molecule in HF also resides within module 5. Using siRNA knockdown of Adamts2 in neonatal rat ventricular cardiomyocytes (NRVMs), we showed that expression of Adamts2 is crucial to ISO-induced cardiac hypertrophy in vitro, possibly due to its regulation of Dkk3 cleavage.
In summary, network modeling of expression data across a diverse population of mice exposed to ISO identified a co-expressed group of genes strongly associated with HF traits. Furthermore, causal modeling revealed Adamts2 to be a key regulator of the module and of cardiac hypertrophy. It is likely that the ISO-induced cardiac pathology reflects only some aspects of the complex spectrum of human heart disease, and it will be of interest to compare our results with those generated using other stressors such as the α-adrenergic agonist angiotensin or pressure overload by trans-aortic constriction. It will also be of interest to compare networks generated in animal models with those observed in human studies.
STAR+METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| SYBRFast Master Mix | KAPA | KK4611 |
| Lipofectamine RNAimax | ThermoFisher Scientific | 13778150 |
| DMEM with pyruvate | Fisher | MT-10-013-CV |
| FBS PREMIUM CELLGRO 500ML | Fisher | MT35015CV |
| Penicillin/Streptomycin | Fisher | MT-30-002-CI |
| ITS | Fisher | CB-40351 |
| Isoproterenol | Sigma | 16504 |
| QIAzol | QIAGEN | 79306 |
| Isopropyl Alcohol | Sigma | 19516 |
| Ethanol | Sigma | E7023 |
| Osmotic Micropumps | Alzet | 1004 |
| Critical Commercial Assays | ||
| Reverse Transcription Kit | Applied Biosystems | 4374967 |
| miRNAeasy RNA extraction Kit | QIAGEN | 217004 |
| Microarray | Illumina | Mouse Reference 8 v 2.0 |
| Deposited Data | ||
| HMDP HF Data | Gene Expression Omnibus | GSE48760 |
| HMDP Mouse Diversity Array Genotypes | http://cgd.jax.org/datasets/diversityarray.shtml | N/A |
| Experimental Models: Cell Lines | ||
| Neonatal Rat Ventricular Myocytes | Primary Cell Culture | N/A |
| Experimental Models: Organisms/Strains | ||
| HMDP Strains | Jackson Labs | See Table S1 |
| Rats for NRVM extraction | UCLA | Sprague-Dawley |
| Recombinant DNA | ||
| None | N/A | N/A |
| Sequence-Based Reagents | ||
| Adamts2 DsiRNA #1 | IDT Tech | RNC.RNAI.N001137622.12.1 |
| Adamts2 DsiRNA #2 | IDT Tech | siRNA-2 #RNC.RNAI.N001137622.12.5 |
| Negative Scramble Control | IDT Tech | DS NC1 |
| Software and Algorithms | ||
| Weighted Maximal Information Component Analysis v0.9 | https://github.com/ChristophRau/wMICA | N/A |
| Database for Annotation, Visualization and Integrated Discovery v 6.7 | https://david.ncifcrf.gov/ | N/A |
| Near Edge Orientation | https://labs.genetics.ucla.edu/horvath/aten/NEO/ | N/A |
| NEQC Normalization | CRAN | Limma package |
| comBAT | Bioconductor | SVA package |
| Other | ||
| qPCR Machine | Roche | LightCycler 480 |
| RNA quality control | Agilent | Bioanalyzer |
CONTACT FOR REAGENT AND RESOURCE SHARING
Please contact Dr. Aldons J. Lusis, Department of Human Genetics, UCLA for reagents and resources at jlusis@mednet.ucla.edu
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
A total of 98 HMDP mouse strains (see Table S6) were obtained from The Jackson Laboratory and then bred at UCLA. Progeny were then studied for HF traits following ISO treatment as previously described (Rau et al., 2015b; Wang et al., 2016). Briefly, Isoproterenol (30 mg per kg body weight per day) was administered for 21 days in 8–10 week old female mice using ALZET osmotic mini-pumps, which were surgically implanted intraperitoneally. All animal experiments were conducted following guidelines established and approved by the University of California, Los Angeles Institutional Animal Care and Use Committee (IACUC) and housed in an IACUC-approved vivarium with daily monitoring by vivarium personnel. Neonatal Rat Ventricular Cardiomyocytes (NRVM) were isolated as previously described (Brown et al., 2005) in accordance with UCLA IACUC guidelines. Briefly, 2–4 day old Sprague-Dawley rats were killed by decapitation, rinsed briefly in ethanol and hearts excised. Ventricles were isolated, and minced in a buffer solution. The buffer solution was removed and replaced with a collagenase-pancreatin solution and incubated at 37°C for 30 min. Myocytes were then separated using a Percoll density gradient.
METHOD DETAILS
RNA Isolation and Microarray Processing
Following homogenization of left ventricular tissue samples in QIAzol, RNA was extracted using the QIAGEN miRNAeasy extraction kit, and verified as having a RIN > 7 by Agilent Bioanalyzer. Two RNA samples were pooled for each strain/experimental condition and arrayed on Illumina Mouse Reference 8 version 2.0 chips. Analysis was conducted using the Neqc algorithm included in the limma R package (Smyth, 2005) and batch effects addressed using COMbat (Johnson et al., 2007). In designing our study, we were cautious and distributed the treated and control conditions evenly across our three batches as well as endeavoring to include a diverse set of genetic backgrounds in each batch. Thus, we do not believe that our data suffer from the potential batch artifacts as reported in Nygaard and Rødland, 2016. To check our results, however, we used wMICA on data from a single batch of our data and recovered a module similar to module 5 (significance of overlap = 5.1E-26) and with similar GO enrichments.
wMICA
We used a modified form of Maximal Information Component Analysis (Rau et al., 2013) to form gene networks on the LV transcriptomes generated from the ISO-treated mice. Probes were limited to the 8126 probes which were both expressed in at least 25% of either the untreated or treated data and whose coefficient of variation was greater than 5%. The MICA algorithm consists of two parts. For the first step, relationships between genes are determined using the MINE algorithm (Reshef et al., 2011). For this, we used the MINERVA R package (Albanese et al., 2013) instead of the original Java implementation due to MINERVA’s significant improvement in run-time. The second step utilizes the ICMg algorithm (Parkkinen and Kaski, 2010) to determine the proportional module membership for each gene.
Analyses were performed as described (Rau et al., 2013), except that the ICMg algorithm was modified as follows to allow for weighted edges. ICMg is an iterative process in which each edge is independently interrogated for each iteration utilizing Gibbs sampling with the following equation:
where {L}′ is the set of all links excluding the one being interrogated, {z}′ is the set of module assignments for the links excluding the link being interrogated, nz is the count of links assigned to component z, i and j represent the genes linked by edge z0 and qzi counts the module-node co-occurrences between module z and node i. C is the total number of modules, and M is the total number of nodes. α and β are control parameters that modify the overall distribution of module sizes and the average module membership per gene per module. To allow for weighted edges, we altered how the matrix q was updated. Previously, and qzj were incremented by 1 each time an edge was placed into a module in an iteration. Instead, we increased these entries in the q matrix by the MIC score of the edge (Lij), and instead of dividing q by the number of iterations in order to get the proportional module memberships for each gene within each module, we divided each element of q by its respective column sum.
Weighted principle components (“eigengenes”) of each module were determined through the use of the dudi.pca function from the ade4 R package (Chessel et al., 2004) and compared to HF-related phenotypes using the heatmap function of WGCNA (Langfelder and Horvath, 2008). For all control variables, the standard values were used.
eQTL Analysis
All strains in this study have been previously genotyped using the Mouse Diversity Array (Rau et al., 2015c), which contains over 200,000 high-quality, informative SNPs. eQTLs were calculated for 13,155 expressed genes using the Efficient Mixed Model Association algorithm (Kang et al., 2008) which accounts for the population structure among the strains using the following model:
where m is the mean, b is the allele effect of the SNP, x is the (nx1) vector of observed genotypes of the SNP (using additive coding of 0,0.5,1), u is the random effects due to genetic relatedness with var , and e is the random noise with var . K denotes the identity-by-state kinship matrix estimated from all the SNPs, I denotes the (n×n) identity matrix, and 1n is the (n×1) vector of ones. Both u and e follow normal distributions. We estimated and using restricted maximum likelihood (REML) and computed p values using the standard F test to test the null hypothesis b = 0. Spot checking of 100 probe QQ plots was performed and all appeared normal.
NEO
The Network Edge Orienting (NEO) R software package (Aten et al., 2008) uses transcriptome and genotype information to infer a causal link between two genes. We applied this analysis to the genes of several modules identified as being significantly associated with phenotypes of interest. For each edge contained within the module, NEO was performed using those two genes and any SNP for which either gene had an eQTL (cis or trans) with a p value of less than 1E-4. Default parameters for NEO was used as originally described (Aten et al., 2008) in which NEO estimates the likelyhoods of all local structural equation models and returns a Local Edge Orientation (LEO) likelyhood score between nodes(genes) A and B., and we kept any SNP/gene/gene combination which yielded a LEO_NB.AtoB or LEO_NB.BtoA score greater than 0.75 and for which mlogp.M.AtoB or mlogp.M.BtoA was less than 0.05.
Prior work (Kang et al., 2008) has demonstrated that for the HMDP, 4.1E-6 is the correct significance threshold for a single genome-wide trait, and this threshold was used for trans-eQTLs. For cis-eQTLS, our significance threshold of 3.6E-4 was calculated using a FDR of 5% for all SNPs which lay within 1 Mb of a probe used in this study (roughly 100 SNPs/probe), using standard permutation analysis methods (total of 100 permutations of all data).
To make a final determination of edge orientation, we examined all SNP/gene/gene combinations kept in the above step. For each edge, we examined only combinations that included that edge. If all of the combinations were either “forward” (gene A affects gene B) or “reverse” (gene B affects gene A), then the edge was classified as either “forward” or “reverse,” respectively. Otherwise, the difference between the sum of the LEO_NB.AtoB scores (for the “forward” combinations) and the sum of the LEO_NB.BtoA scores (for the “reverse” combinations) was determined. If this difference was larger than 1, then the edge was classified as either “forward” or “reverse” (depending on which sum was larger). Otherwise, the directionality of the edge could not be determined and it was classified as “undirected.”
Cell Culture and Treatments
Following isolation, NRVM were plated in DMEM containing 10% Fetal bovine serum (FBS) and 1% antibiotics overnight. For the rest of the culture period the cells were maintained in DMEM containing 1% ITS (Fisher – CB-40351) and 1% antibiotics. For some studies cells were treated with 60uM Isoproterenol (Sigma – I6504) for 48 hr.
Adamts2 knockdown experiments were performed using IDT DsiRNA (Adamts2 siRNA –1 # RNC.RNAI.N001137622.12.1, Adamts2 siRNA-2 #RNC.RNAI.N001137622.12.5, Negative Control – DS NC1) at the indicated concentrations. All transfections were performed with Invitrogen Lipofectamine RNAimax reagent.
cDNA Synthesis and qRT-PCR
Total RNA isolation from NRVMs was performed by phenol-chloroform extraction using QIAzol lysis reagent (QIAGEN – 79306). RNA was quantified using a Nanodrop UV-Vis Spectrophotometer prior to cDNA synthesis. mRNA reverse transcription was performed using the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit (Thermo-Fisher Scientific – 4374967). Quantitative PCR was performed using the KAPA SYBRFast Master Mix in a Roche LightCycler 480 instrument. Analysis was performed using the Lightcycler 480 software, with standard curves and product melt curves performed for every set of gene primers. Primers used for qRT-PCR are provided in Table S5.
QUANTIFICATION AND STATISTICAL ANALYSIS
Combat
The significance of overlap between the module 5 constructed from all data as opposed to data from a single batch was calculated using a hypergeometric test with a threshold of significance of p < 0.05.
wMICA Module Correlation to Traits
Correlation and Significance of correlation between the wMICA module eigengenes (the first principle components of the genes within the module) were calculated using the Heatmap function of the WGCNA package(Langfelder and Horvath, 2008), which uses Pearson correlation for its calculations.
GO Enrichment with DAVID
All Gene Ontology enrichments were performed using DAVID (Huang et al., 2009). DAVID’s enrichments are performed using the EASE algorithm, which is a slightly more conservative form of the Fisher’s exact test, in which the upper left quadrant of the 2×2 Fisher’s exact contingency table is reduced by 1. See manuscript text and Tables S1, S2 and S4 for values.
eQTL Analysis
Prior work (Kang et al., 2008) has demonstrated that for the HMDP, 4.1E-6 is the correct significance threshold for a single genome-wide trait, and this threshold was used for trans-eQTLs. For cis-eQTLS, our significance threshold of 3.6E-4 was calculated using a FDR of 5% for all SNPs which lay within 1 Mb of a probe used in this study (roughly 100 SNPs/probe), using standard permutation analysis methods (total of 100 permutations of all data). We have previously observed that cis-eQTL at this level are highly conserved in the HMDP (Hasin-Brumshtein et al., 2014).
NRVMs and qPCR
See figure legends for n and manuscript text for significance. Significance values were obtained using Student’s t test after testing data for normality.
DATA AND SOFTWARE AVAILABILITY
Online Database
Data S1, containing the co-expression networks and module membership assignments may be accessed on Mendeley Data. doi: http://dx.doi.org/10.17632/ntd85jnwxd.1.
Microarray data may be accessed at the Gene Expression Omnibus using accession ID: GSE48760.
Phenotypic data may be found on Mendeley Data. doi: http://dx.doi.org/10.17632/y8tdm4s7nh.1.
All phenotypic, expression and eQTL data may also be accessed at https://systems.genetics.ucla.edu/data/hmdp_hypertrophy_heart_failure.
wMICA Software Package
The latest version of the wMICA algorithm may be found at https://github.com/ChristophRau/wMICA.
Supplementary Material
Highlights.
Co-expression networks were created from heart transcriptomes of 100 mouse strains
A module was identified that strongly correlated to heartfailure-related traits
The NEO algorithm identified Adamts2 as an important driver of the module
In vitro studies demonstrated that Adamts2 is a regulator of cardiac hypertrophy
Acknowledgments
This work was supported in part by NIH grants HL110667, HL123295, HL114437, and HL28481. C.D.R. was supported by NIH training grant T32HL69766, J.W. was supported by NIH training grant HL007895, and M.C.R. was supported by Ruth L. Kirschstein National Research Service award T32GM718539. We would like to thank Doug Chapski, Mario Deng, Adriana Huertas-Vazquez, Hrayr Karaguesian, Jonathan Hoffman, Jim O’Hearn, Tom Vondriska, and Nick Wisniewski for fruitful discussions regarding this manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes two figures, and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.cels.2016.10.016.
AUTHOR CONTRIBUTIONS
Conceptualization, C.D.R., Y.W., and A.J.L.; Methodology, C.D.R., M.S., A.K., J.N.W., Y.W., and A.J.L.; Software, C.D.R.; Formal Analysis, C.D.R. and M.C.R.; Investigation, C.D.R., M.C.R., M.T., J.J.-C.W., and S.R.; Writing – Original Draft, C.D.R. and M.C.R.; Writing – Reviewing & Editing, C.D.R., M.C.R., J.N.W., Y.W., and A.J.L.; Supervision, Y.W. and A.J.L.; Funding Acquisition, A.K., J.N.W., Y.W., and A.J.L.
References
- Albanese D, Filosi M, Visintainer R, Riccadonna S, Jurman G, Furlanello C. Minerva and minepy: a C engine for the MINE suite and its R, Python and MATLAB wrappers. Bioinformatics. 2013;29:407–408. doi: 10.1093/bioinformatics/bts707. [DOI] [PubMed] [Google Scholar]
- Aten JE, Fuller TF, Lusis AJ, Horvath S. Using genetic markers to orient the edges in quantitative trait networks: the NEO software. BMC Syst Biol. 2008;2:34. doi: 10.1186/1752-0509-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao MW, Cai Z, Zhang XJ, Li L, Liu X, Wan N, Hu G, Wan F, Zhang R, Zhu X, et al. Dickkopf-3 protects against cardiac dysfunction and ventricular remodelling following myocardial infarction. Basic Res Cardiol. 2015;110:25. doi: 10.1007/s00395-015-0481-x. [DOI] [PubMed] [Google Scholar]
- Barbier M, Gross MS, Aubart M, Hanna N, Kessler K, Guo DC, Tosolini L, Ho-Tin-Noe B, Regalado E, Varret M, et al. MFAP5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet. 2014;95:736–743. doi: 10.1016/j.ajhg.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton PJR, Birks EJ, Felkin LE, Cullen ME, Koban MU, Yacoub MH. Increased expression of extracellular matrix regulators TIMP1 and MMP1 in deteriorating heart failure. J Heart Lung Transplant. 2003;22:738–744. doi: 10.1016/s1053-2498(02)00557-0. [DOI] [PubMed] [Google Scholar]
- Bekhouche M, Leduc C, Dupont L, Janssen L, Delolme F, Vadon-Le Goff S, Smargiasso N, Baiwir D, Mazzucchelli G, Zanella-Cleon I, et al. Determination of the substrate repertoire of ADAMTS2, 3, and 14 significantly broadens their functions and identifies extracellular matrix organization and TGF-β signaling as primary targets. FASEB J. 2016;30:1741–1756. doi: 10.1096/fj.15-279869. [DOI] [PubMed] [Google Scholar]
- Bennett B, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, Neubauer M, Neuhaus I, Yordanova R, Guan B, et al. A high resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010;20:281–290. doi: 10.1101/gr.099234.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Beygui RE, MacLellan WR, Laks H, Dunn JC, Wu BM. Modulation of gene expression in neonatal rat cardiomyocytes by surface modification of polylactide-co-glycolide substrates. J Biomed Mater Res A. 2005;74:419–429. doi: 10.1002/jbm.a.30344. [DOI] [PubMed] [Google Scholar]
- Chen L, Ge Q, Black JL, Deng L, Burgess JK, Oliver BGG. Differential regulation of extracellular matrix and soluble fibulin-1 levels by TGF-β in airway smooth muscle cells. PLoS ONE. 2013;8:e65544. doi: 10.1371/journal.pone.0065544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessel D, Dufour AB, Thioulouse J. The ade4 package – I: One-table methods. R News. 2004;4:5–10. [Google Scholar]
- Cooley MA, Fresco VM, Dorlon ME, Twal WO, Lee NV, Barth JL, Kern CB, Iruela-Arispe ML, Argraves WS. Fibulin-1 is required during cardiac ventricular morphogenesis for versican cleavage, suppression of ErbB2 and Erk1/2 activation, and to attenuate trabecular cardiomyocyte proliferation. Dev Dyn. 2012;241:303–314. doi: 10.1002/dvdy.23716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobaczewski M, Chen W, Frangogiannis NG. Transforming growth factor (TGF)-β signaling in cardiac remodeling. J Mol Cell Cardiol. 2011;51:600–606. doi: 10.1016/j.yjmcc.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdov I, Didangelos A, Yin X, Zampetaki A, Abonnenc M, Murdoch C, Zhang M, Ouzounis CA, Mayr M, Tsoka S, Shah AM. Gene network and proteomic analyses of cardiac responses to pathological and physiological stress. Circ Cardiovasc Genet. 2013;6:588–597. doi: 10.1161/CIRCGENETICS.113.000063. [DOI] [PubMed] [Google Scholar]
- Ehnis T, Dieterich W, Bauer M, Kresse H, Schuppan D. Localization of a binding site for the proteoglycan decorin on collagen XIV (undulin) J Biol Chem. 1997;272:20414–20419. doi: 10.1074/jbc.272.33.20414. [DOI] [PubMed] [Google Scholar]
- Farber CR. Systems-level analysis of genome-wide association data. G3 (Bethesda) 2013;3:119–129. doi: 10.1534/g3.112.004788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110:159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goding JW, Grobben B, Slegers H. Physiological and pathophysiological functions of the ecto-nucleotide pyrophosphatase/phosphodiesterase family. Biochim Biophys Acta. 2003;1638:1–19. doi: 10.1016/s0925-4439(03)00058-9. [DOI] [PubMed] [Google Scholar]
- Grossman TR, Gamliel A, Wessells RJ, Taghli-Lamallem O, Jepsen K, Ocorr K, Korenberg JR, Peterson KL, Rosenfeld MG, Bodmer R, Bier E. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet. 2011;7:e1002344. doi: 10.1371/journal.pgen.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MC, Young DA, Waters JG, Rowan AD, Chantry A, Edwards DR, Clark IM. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-beta 1. J Biol Chem. 2003;278:10304–10313. doi: 10.1074/jbc.M212334200. [DOI] [PubMed] [Google Scholar]
- Hanssen E, Hew FH, Moore E, Gibson MA. MAGP-2 has multiple binding regions on fibrillins and has covalent periodic association with fibrillin-containing microfibrils. J Biol Chem. 2004;279:29185–29194. doi: 10.1074/jbc.M313672200. [DOI] [PubMed] [Google Scholar]
- Hasin-Brumshtein Y, Hormozdiari F, Martin L, van Nas A, Eskin E, Lusis AJ, Drake TA. Allele-specific expression and eQTL analysis in mouse adipose tissue. BMC Genomics. 2014;15:471. doi: 10.1186/1471-2164-15-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann Bowman MA, McNally EM. Genetic pathways of vascular calcification. Trends Cardiovasc Med. 2012;22:93–98. doi: 10.1016/j.tcm.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomidis JS, Hendrick JW, Parkhurst AM, Herron AR, Escobar PG, Dowdy KB, Stroud RE, Hapke E, Zile MR, Spinale FG. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol. 2005;288:H149–H158. doi: 10.1152/ajpheart.00370.2004. [DOI] [PubMed] [Google Scholar]
- Jinnin M, Ihn H, Asano Y, Yamane K, Trojanowska M, Tamaki K. Tenascin-C upregulation by transforming growth factor-beta in human dermal fibroblasts involves Smad3, Sp1, and Ets1. Oncogene. 2004;23:1656–1667. doi: 10.1038/sj.onc.1207064. [DOI] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- Kang HM, Zaitlen NA, Wade CM, Kirby A, Heckerman D, Daly MJ, Eskin E. Efficient control of population structure in model organism association mapping. Genetics. 2008;178:1709–1723. doi: 10.1534/genetics.107.080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler-Icekson G, Schlesinger H, Freimann S, Kessler E. Expression of procollagen C-proteinase enhancer-1 in the remodeling rat heart is stimulated by aldosterone. Int J Biochem Cell Biol. 2006;38:358–365. doi: 10.1016/j.biocel.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci USA. 2010;107:15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wei H, Chesley A, Moon C, Krawczyk M, Volkova M, Ziman B, Margulies KB, Talan M, Crow MT, Boheler KR. The proangiogenic cytokine pleiotrophin potentiates cardiomyocyte apoptosis through inhibition of endogenous AKT/PKB activity. J Biol Chem. 2007;282:34984–34993. doi: 10.1074/jbc.M703513200. [DOI] [PubMed] [Google Scholar]
- Lusis AJ, Seldin MM, Allayee H, Bennett BJ, Civelek M, Davis RC, Eskin E, Farber CR, Hui S, Mehrabian M, et al. The Hybrid Mouse Diversity Panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. J Lipid Res. 2016;57:925–942. doi: 10.1194/jlr.R066944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015;21:174–182. doi: 10.1016/j.cmet.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messaoudi S, Zhang AD, Griol-Charhbili V, Escoubet B, Sadoshima J, Farman N, Jaisser F. The epidermal growth factor receptor is involved in angiotensin II but not aldosterone/salt-induced cardiac remodelling. PLoS ONE. 2012;7:e30156. doi: 10.1371/journal.pone.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljkovic-Licina M, Hammel P, Garrido-Urbani S, Bradfield PF, Szepetowski P, Imhof BA. Sushi repeat protein X-linked 2, a novel mediator of angiogenesis. FASEB J. 2009;23:4105–4116. doi: 10.1096/fj.09-135202. [DOI] [PubMed] [Google Scholar]
- Miljkovic-Licina M, Hammel P, Garrido-Urbani S, Lee BPL, Meguenani M, Chaabane C, Bochaton-Piallat ML, Imhof BA. Targeting olfactomedin-like 3 inhibits tumor growth by impairing angiogenesis and pericyte coverage. Mol Cancer Ther. 2012;11:2588–2599. doi: 10.1158/1535-7163.MCT-12-0245. [DOI] [PubMed] [Google Scholar]
- Moali C, Font B, Ruggiero F, Eichenberger D, Rousselle P, François V, Oldberg A, Bruckner-Tuderman L, Hulmes DJS. Substrate-specific modulation of a multisubstrate proteinase. C-terminal processing of fibrillar procollagens is the only BMP-1-dependent activity to be enhanced by PCPE-1. J Biol Chem. 2005;280:24188–24194. doi: 10.1074/jbc.M501486200. [DOI] [PubMed] [Google Scholar]
- Moreno-Moral A, Mancini M, D’Amati G, Camici P, Petretto E. Transcriptional network analysis for the regulation of left ventricular hypertrophy and microvascular remodeling. J Cardiovasc Transl Res. 2013;6:931–944. doi: 10.1007/s12265-013-9504-x. [DOI] [PubMed] [Google Scholar]
- Nishioka T, Onishi K, Shimojo N, Nagano Y, Matsusaka H, Ikeuchi M, Ide T, Tsutsui H, Hiroe M, Yoshida T, Imanaka-Yoshida K. Tenascin-C may aggravate left ventricular remodeling and function after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2010;298:H1072–H1078. doi: 10.1152/ajpheart.00255.2009. [DOI] [PubMed] [Google Scholar]
- Nygaard V, Rødland EA. Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses. Biostatistics. 2016;17:29–39. doi: 10.1093/biostatistics/kxv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkkinen JA, Kaski S. Searching for functional gene modules with interaction component models. BMC Syst Biol. 2010;4:4. doi: 10.1186/1752-0509-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau CD, Wisniewski N, Orozco LD, Bennett B, Weiss J, Lusis AJ. Maximal information component analysis: a novel non-linear network analysis method. Front Genet. 2013;4:28. doi: 10.3389/fgene.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau CD, Lusis AJ, Wang Y. Genetics of common forms of heart failure: challenges and potential solutions. Curr Opin Cardiol. 2015a;30:222–227. doi: 10.1097/HCO.0000000000000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau CD, Wang J, Avetisyan R, Romay MC, Martin L, Ren S, Wang Y, Lusis AJ. Mapping genetic contributions to cardiac pathology induced by beta-adrenergic stimulation in mice. Circ Cardiovasc Genet. 2015b;8:40–49. doi: 10.1161/CIRCGENETICS.113.000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau CD, Parks B, Wang Y, Eskin E, Simecek P, Churchill GA, Lusis AJ. High-density genotypes of inbred mouse strains: improved power and precision of association mapping. G3 (Bethesda) 2015c;5:2021–2026. doi: 10.1534/g3.115.020784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reshef DN, Reshef YA, Finucane HK, Grossman SR, McVean G, Turnbaugh PJ, Lander ES, Mitzenmacher M, Sabeti PC. Detecting novel associations in large data sets. Science. 2011;334:1518–1524. doi: 10.1126/science.1205438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. limma: linear models for microarray data. In: Gentleman R, Irizarry W, Huber W, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Talior-Volodarsky I, Connelly KA, Arora PD, Gullberg D, McCulloch CA. α11 integrin stimulates myofibroblast differentiation in diabetic cardiomyopathy. Cardiovasc Res. 2012;96:265–275. doi: 10.1093/cvr/cvs259. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Arao T, Maegawa M, Matsumoto K, Kaneda H, Kudo K, Fujita Y, Yokote H, Yanagihara K, Yamada Y, et al. SRPX2 is overexpressed in gastric cancer and promotes cellular migration and adhesion. Int J Cancer. 2009;124:1072–1080. doi: 10.1002/ijc.24065. [DOI] [PubMed] [Google Scholar]
- Tao G, Levay AK, Gridley T, Lincoln J. Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev Biol. 2011;359:209–221. doi: 10.1016/j.ydbio.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao G, Levay AK, Peacock JD, Huk DJ, Both SN, Purcell NH, Pinto JR, Galantowicz ML, Koch M, Lucchesi PA, et al. Collagen XIV is important for growth and structural integrity of the myocardium. J Mol Cell Cardiol. 2012;53:626–638. doi: 10.1016/j.yjmcc.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillet E, Wiedemann H, Golbik R, Pan TC, Zhang RZ, Mann K, Chu ML, Timpl R. Recombinant expression and structural and binding properties of alpha 1(VI) and alpha 2(VI) chains of human collagen type VI. Eur J Biochem. 1994;221:177–185. doi: 10.1111/j.1432-1033.1994.tb18727.x. [DOI] [PubMed] [Google Scholar]
- Tortorella MD, Malfait F, Barve RA, Shieh HS, Malfait AM. A review of the ADAMTS family, pharmaceutical targets of the future. Curr Pharm Des. 2009;15:2359–2374. doi: 10.2174/138161209788682433. [DOI] [PubMed] [Google Scholar]
- Wang JJC, Rau C, Avetisyan R, Ren S, Romay MC, Stolin G, Gong KW, Wang Y, Lusis AJ. Genetic dissection of cardiac remodeling in an isoproterenol-induced heart failure mouse model. PLoS Genet. 2016;12:e1006038. doi: 10.1371/journal.pgen.1006038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler FC, Tang H, Marks OA, Hadnott TN, Chu PL, Mao L, Rockman HA, Marchuk DA. Tnni3k modifies disease progression in murine models of cardiomyopathy. PLoS Genet. 2009;5:e1000647. doi: 10.1371/journal.pgen.1000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xuan W, Liao Y, Chen B, Huang Q, Xu D, Liu Y, Bin J, Kitakaze M. Detrimental effect of fractalkine on myocardial ischaemia and heart failure. Cardiovasc Res. 2011;92:385–393. doi: 10.1093/cvr/cvr221. [DOI] [PubMed] [Google Scholar]
- Yan F, Wang Y, Wu X, Peshavariya HM, Dusting GJ, Zhang M, Jiang F. Nox4 and redox signaling mediate TGF-β-induced endothelial cell apoptosis and phenotypic switch. Cell Death Dis. 2014;5:e1010. doi: 10.1038/cddis.2013.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.