

Abstract
The etiology of chronic bile duct injury and fibrosis in patients with autoimmune cholestatic liver diseases is complex, and likely involves immune cells such as lymphocytes. However, most models of biliary fibrosis are not autoimmune in nature. Biliary fibrosis can be induced experimentally by prolonged exposure of mice to the bile duct toxicant alpha-naphthylisothiocyanate (ANIT). We determined whether lymphocytes contributed to ANIT-mediated biliary hyperplasia and fibrosis in mice. Hepatic accumulation of T-lymphocytes and increased serum levels of anti-nuclear-auto-antibodies were evident in wild-type mice exposed to ANIT (0.05% ANIT in chow). This occurred alongside bile duct hyperplasia and biliary fibrosis. To assess the role of lymphocytes in ANIT-induced biliary fibrosis, we utilized RAG1−/− mice, which lack T- and B-lymphocytes. ANIT-induced bile duct injury, indicated by increased serum alkaline phosphatase activity, was reduced in ANIT-exposed RAG1−/− mice compared to ANIT-exposed wild-type mice. Despite this reduction in biliary injury, ANIT-induced bile duct hyperplasia was similar in wild-type and RAG1−/− mice. However, hepatic induction of profibrogenic genes including COL1A1, ITGβ6 and TGFβ2 was markedly attenuated in ANIT-exposed RAG1−/− mice compared to ANIT-exposed wild-type mice. Peribiliary collagen deposition was also reduced in ANIT-exposed RAG1−/− mice. The results indicate that lymphocytes exacerbate bile duct injury and fibrosis in ANIT-exposed mice without impacting bile duct hyperplasia.
Keywords: liver, fibrosis, lymphocytes, hepatocellular necrosis, bile duct, biliary hyperplasia, ANIT, alpha-naphthylisothiocyanate, RAG1, B cells, T cells
1. Introduction
Among the key functions of the liver is production and delivery of bile to the gall bladder (McCuskey and Sipes 2010). The synthesis of bile is complex and involves not only the synthesis of bile acids by hepatocytes, but also regulation of bile composition and flow by bile duct epithelial cells (BDECs; cholangiocytes) (Chen et al. 2008; Morell et al. 2013; O’Hara et al. 2013). BDECs line intrahepatic bile ducts, forming a conduit separating bile, which is both toxic and proinflammatory, from the liver parenchyma (Kanz 2010; Perez and Briz 2009; Sipka and Bruckner 2014). BDEC injury is one trigger of cholestatic liver disease, wherein bile flow out of the liver is disturbed, leading to elevated plasma levels of bile acids (Chen et al. 2008; Li and Crawford 2004; Morell et al. 2013; O’Hara et al. 2013). Although BDEC injury is a well-appreciated etiology of several cholestatic liver diseases in humans, the mechanisms responsible for biliary injury are not completely understood, and vary from increased pressure (i.e., obstructive cholestasis) to immune-mediated events in conditions such as primary biliary and primary sclerosing cholangitis (PBC and PSC) (Hirschfield et al. 2013; Lazaridis and LaRusso 2015; Li and Crawford 2004; Lindor et al. 2009; Lindor et al. 2015; Trauner et al. 1998).
Experimental models serve as an important platform to trace the mechanisms of chronic liver injury and fibrosis triggered by injury to intrahepatic BDECs (Kopec et al. 2016). Although obstructive cholestasis (i.e., bile duct ligation) and genetic models (Mdr2−/− mice) are utilized, BDECs can also be chronically injured by administration of certain xenobiotics. For example, owing to its unique metabolism and transport, the xenobiotic alpha-naphthylisothiocyanate (ANIT) is selectively toxic to BDECs (Becker and Plaa 1965; Jean et al. 1995; Plaa and Priestly 1976). In contrast to acute ANIT exposure in mice (Hill et al. 1999), chronic exposure of mice to ANIT elicits bile duct injury, hyperplasia and fibrosis alongside induction of mixed lymphocytic hepatic inflammation (Golbar et al. 2013; Joshi et al. 2014; Sullivan et al. 2010; Tjandra et al. 2000; Xu et al. 2004). As an experimental tool, ANIT-mediated BDEC cytotoxicity is currently perceived as the primary stimulus for liver fibrosis, with hepatic lymphocytic inflammation occurring as a consequence of biliary injury.
We tested the hypothesis that ANIT-mediated biliary hyperplasia and fibrosis in mice are lymphocyte-independent. Utilizing an established experimental setting of chronic ANIT exposure in mice, we quantified hepatic T- and B-lymphocyte accumulation and evaluated autoantibody production in ANIT-exposed mice. To directly address the role of lymphocytes in this model, we utilized RAG1−/− mice, which lack both T- and B-lymphocytes (Mombaerts et al. 1992).
2. Materials and Methods
2.1 Mice and ANIT exposure model
Male, wild-type mice (C57BL/6J) and RAG-1−/− mice (Mombaerts et al. 1992) were obtained from the Jackson Laboratory and studies were initiated when the mice were 10 weeks of age. Mice were housed at an ambient temperature of approximately 22°C with alternating 12 hour light/dark cycles and provided water and standard rodent chow ad libitum prior to study initiation. Custom diets were prepared by Dyets, Inc. (Bethlehem, PA). The ANIT diet was formulated in a standard rodent chow (Teklad 8940) containing 0.05% ANIT (Sigma-Aldrich, St. Louis, MO), as we have described previously (Joshi et al. 2016b). Groups of mice of each genotype were fed chow or chow containing ANIT (0.05%) for 4 weeks. At the end of the study, mice were anesthetized with isoflurane; citrate-anticoagulated blood and non-anticoagulated blood were collected from the caudal vena cava for collection of plasma and serum, respectively. The entire left lateral liver lobe was fixed in 10% neutral buffered formalin, the gall bladder removed, and the remaining liver snap frozen in liquid nitrogen. Mice were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited facility at Michigan State University. All animal procedures were approved by Michigan State University Institutional Animal Care and Use Committee.
2.2 Serum enzyme and autoantibody levels
Serum alanine aminotransferase (ALT) and alkaline phosphatase (ALP) activities were determined using commercial reagents (Thermo Fisher, Waltham, MA; Pointe Scientific, Canton, MI). Serum auto-antibody levels were determined using an auto-antigen microarray panel by the Genomics and Microarray Core Facility at The University of Texas Southwestern Medical Center. True fluorescent intensities for specific auto-antibodies were considered only if signal-to-noise ratios were >3.
2.3 Immunohistochemistry
Formalin-fixed, paraffin-embedded livers were cut at 5 microns and stained with picrosirius red, and immunohistochemically for cytokeratin-19 (CK-19), CD3 (T-cells), and CD45R (B-cells) by the Investigative Histopathology Laboratory at Michigan State University as described previously (Joshi et al. 2015; Joshi et al. 2016a). Primary antibodies utilized were polyclonal rabbit antibodies (Abcam, Cambridge, MA) and detected by HRP-conjugated polymer detection systems. Images comprising the entire left lateral lobe (>500 images) were captured using a Virtual Slide System VS110 (Olympus, Hicksville, NY) with a 20× objective. The area of positive sirius red and CK-19 staining was determined in an automated and unbiased fashion using a batch macro and the color de-convolution tool in ImageJ. Positive area for each stain was expressed individually and as a ratio of sirius red/CK-19. Similarly, the number of CD3 and CD45R-positive cells per image was determined using a batch macro, the color de-convolution tool, and particle analysis function of ImageJ.
2.4 RNA isolation, cDNA synthesis, and quantitative real-time PCR
Total RNA was isolated from approximately 20 mg of snap-frozen liver using TRI Reagent according to the manufacturer’s protocol (Molecular Research Center, Cincinnati, OH). 1 μg of total RNA was utilized for the synthesis of cDNA, accomplished using a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA) and a C1000 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA). SYBR Green quantitative real-time PCR (qPCR) amplification was performed using a CFX Connect thermal cycler (Bio-Rad) with primers purchased from IDT (Coralville, IA). The expression of each gene was normalized to the housekeeper gene GAPDH and the relative levels of each gene were evaluated using the ΔΔCt method. Mouse GAPDH primer sequences were 5′-GTGGACCTCATGGCCTACAT-3′ (forward primer), 5′-TGTGAGGGAGATGCTCAGTG-3′ (reverse primer). Mouse COL1A1 primer sequences were 5′-GAGCGGAGAGTACTGGATCG-3′ (forward primer), 5′-GCTTCTTTTCCTTGGGGTTC-3′ (reverse primer). Mouse ITGβ6 primer sequences were 5′-CTCACGGGTACAGTAACGCA-3′ (forward primer), 5′-AAATGAGCTCTCAGGCAGGC-3′ (reverse primer). Mouse TGFβ2 primer sequences were 5′-CCCCGGAGGTGATTTCCATC-3′ (forward primer), 5′-GATGGCATTTTCGGAGGGGA-3′ (reverse primer).
2.5 Statistics
Comparison of two groups was made using Student’s t-test. Comparison of three or more groups was made by two-way analysis of variance with Student-Newman-Keuls test used for post-hoc comparisons. Data not fitting a normal distribution were log-transformed. The criterion for statistical significance was P<0.05.
3. Results
3.1 Hepatic lymphocyte accumulation and autoantibody production accompanies ANIT-induced biliary hyperplasia and fibrosis in mice
Compared to mice fed control chow, bile duct hyperplasia increased significantly in ANIT-exposed mice, denoted by increased area stained by CK-19 (Fig. 1A–B). Likewise, biliary fibrosis increased in ANIT-exposed mice compared to mice fed control chow, as indicated by sirius red staining (Fig. 1C–D). In agreement with a previous study (Joshi et al. 2015), compared to mice fed control chow, a significant increase in CD3+ lymphocytes (T-cells) was evident in livers of ANIT-exposed mice, largely near the portal tracts (Fig. 1E–F). There was tendency for the CD3+ T-cells to localize to inflamed portal tracts (Fig. 1E). Very modest CD45R+ (B220+) B-cell accumulation was also evident in livers of ANIT-exposed mice (Fig. 1G–H). These data indicate that lymphocytic inflammation accompanies ANIT-induced biliary pathology in mice.
Figure 1. Hepatic lymphocyte accumulation, biliary hyperplasia and liver fibrosis in wild-type mice after chronic ANIT exposure.
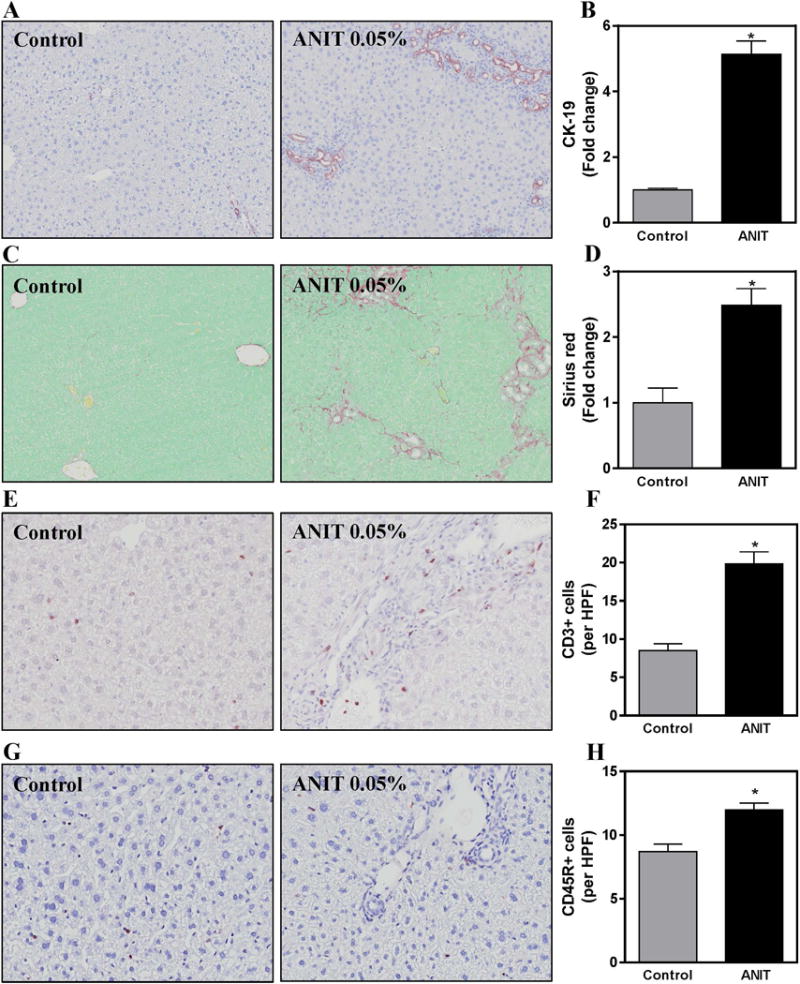
Wild-type mice were fed standard control rodent chow or diet containing 0.05% ANIT for 4 weeks. Representative photomicrographs (100×) show liver sections stained for (A) Cytokeratin-19 (CK-19, brown), (C) Sirius red (red), (E) CD3 T-lymphocytes (brown) and (G) CD45R B-lymphocytes (brown). For E and G, arrows denote positive staining for each cell type. Staining for (B) CK-19, (D) Sirius red, (F) CD3 T-lymphocytes and (H) CD45R B-lymphocytes per high power field (HPF, 200×) were quantified as described in Materials and Methods. Data are expressed as mean + SEM; n=5–10 mice per group. *p<0.05 vs. control diet.
Although their precise role as biomarkers and/or mediators of disease is debated, patients with cholestatic liver disease often display increased antibodies directed at various self-antigens. To determine whether the levels of specific auto-antibodies increase in ANIT-induced biliary fibrosis, we utilized an autoantigen microarray super panel that tests for levels of IgM and IgG antibodies directed against approximately 130 antigens typically targeted in autoimmune diseases. Compared to mice fed control chow, anti-nuclear (polymeraseβ (POLB), histone H4, dsDNA) antibodies were significantly increased in ANIT-exposed wild-type mice, as were antibodies against fibronectin, complement factor I and prothrombin, proteins primarily synthesized by liver (Table 1). These results suggest that ANIT-induced biliary fibrosis is accompanied by modest increases in serum levels of select auto-antibodies.
Table 1. Serum auto-antibody levels determined using an auto-antigen microarray panel.
Wild-type and RAG1−/− mice were fed standard control rodent chow or diet containing 0.05% ANIT for 4 weeks. Serum auto-antibody levels were determined using an auto-antigen microarray panel. Fluorescent intensities for specific auto-antibodies were considered only if signal-to-noise ratios were >3. Data are expressed as fold change. n=5 mice per group.
| Relative fluorescence (fold change) | |||
|---|---|---|---|
| Control | ANIT | Isotype | |
| POLB | 1.0+0.2 | 1.7+0.1 | IgG |
| Fibronectin | 1.0+0.1 | 1.5+0.2 | IgG |
| Complement Factor I | 1.0+0.1 | 1.5+0.2 | IgG |
| Prothrombin | 1.0+0.1 | 1.4+0.2 | IgG |
| Histone H4 | 1.0+0.1 | 1.6+0.2 | IgG |
| dsDNA | 1.0+0.2 | 3.0+0.9 | IgM |
3.2 Lymphocytes contribute to biliary injury but not hyperplasia in ANIT-exposed mice
To assess the role of lymphocytes in ANIT-induced bile duct injury we utilized RAG1−/− mice, which do not harbor mature T-cells or B-cells (Mombaerts et al. 1992). We first assessed biliary injury by measuring the activity of ALP, which is synthesized by HPCS and transported into bile canaliculi. Compared to mice fed control chow, serum ALP activity was significantly increased in ANIT-exposed wild-type mice and this was significantly reduced in RAG1−/− mice exposed to ANIT (Fig. 2A). Notably, the ANIT-mediated increase in serum ALT activity, a marker of hepatocyte necrosis, was unaffected by RAG1-deficiency (Fig. 2B). Interestingly, bile duct hyperplasia as indicated by CK-19 staining, was similarly increased in wild-type and in RAG1−/− mice exposed to ANIT (Fig. 2C–D). Collectively, these results suggest that a lymphocyte-mediated mechanism is partially responsible for biliary injury, but not the hyperplastic response of BDECs, in ANIT-exposed mice. Notably, this observation disconnects changes in biliary hyperplasia from biomarkers of BDEC injury.
Figure 2. Lymphocytes contribute to biliary injury but not hyperplasia in ANIT-exposed mice.
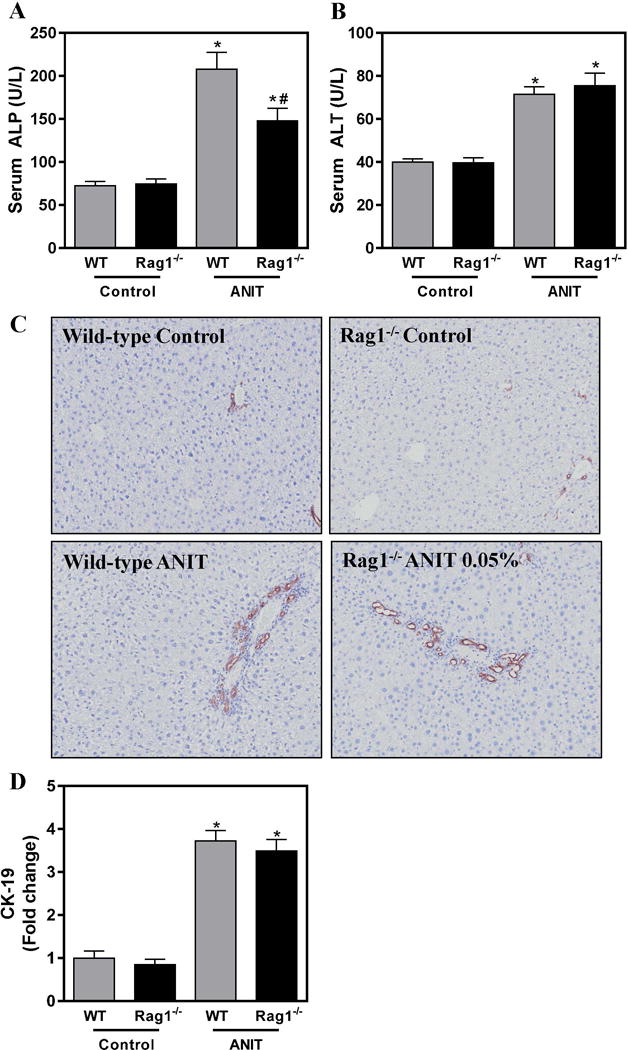
Wild-type (WT) and RAG1−/− mice were fed standard control rodent chow or diet containing 0.05% ANIT for 4 weeks. (A) Serum ALP and (B) Serum ALT activity were determined as described in Materials and Methods. (C) Representative photomicrographs (100×) show liver sections stained for CK-19 (brown). (D) CK-19 staining was quantified as described in Materials and Methods. Data are expressed as mean + SEM; n=5–10 mice per group. *p<0.05 vs. control diet. #p<0.05 vs. ANIT-exposed WT mice.
3.3 Lymphocytes contribute to biliary fibrosis in ANIT-exposed mice
Hepatic expression of profibrogenic COL1A1, ITGβ6 and TGFβ2 mRNAs was increased significantly in livers of ANIT-exposed mice compared to control chow exposed mice (Fig. 3A–C). In our previous studies, we noted that changes in the severity of biliary fibrosis were coupled to quantitative changes in the area occupied by BDECs (i.e. biliary hyperplasia). However, despite similar bile duct area (Fig. 2D), profibrogenic gene induction was significantly attenuated in ANIT-exposed RAG1−/− mice compared to ANIT-exposed wild-type mice (Fig. 3A–C). In agreement, hepatic collagen protein deposition increased significantly in ANIT-exposed wild-type mice, but was reduced by approximately 25% (P=0.07) in ANIT-exposed RAG1−/− mice (Fig.4A–C). Comparison of liver sections with qualitatively similar bile duct hyperplasia revealed a marked reduction in peribiliary collagen deposition in picrosirius red-stained serial sections (Fig. 4A–B). Consistent with this observation, a significant reduction (P<0.05) in peribiliary fibrosis was detected when sirius red-stained area was normalized to the CK-19-positive labeling in the same tissue section (Fig. 4D).
Figure 3. Lymphocytes contribute to induction of profibrogenic gene induction in ANIT-exposed mice.
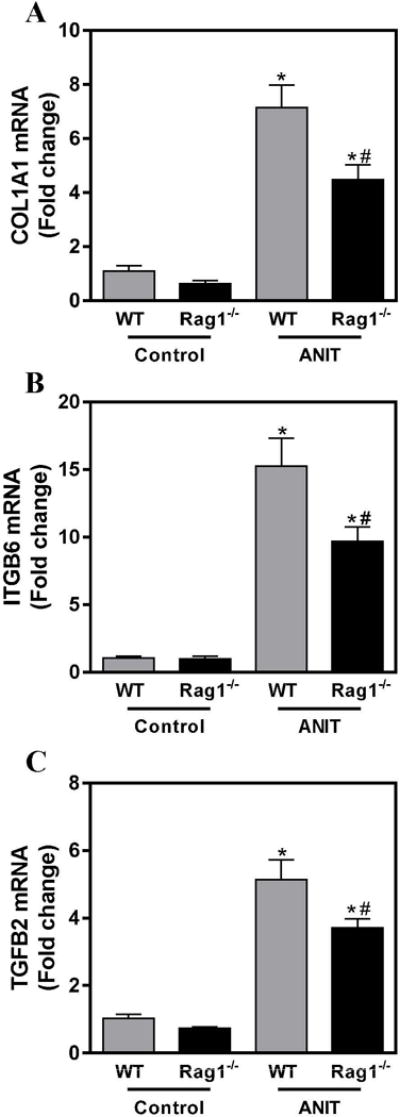
Wild-type (WT) and RAG1−/− mice were fed standard control rodent chow or diet containing 0.05% ANIT for 4 weeks. Hepatic expression of mRNAs encoding the profibrogenic genes (A) COL1A1, (B) ITGβ6 AND (C) TGFβ2 was determined by real-time qPCR. Data are expressed as mean + SEM; n=5–10 mice per group. *p<0.05 vs. control diet. #p<0.05 vs. 0.05% ANIT-exposed wild-type mice.
Figure 4. Lymphocytes contribute to biliary fibrosis in ANIT-exposed mice.
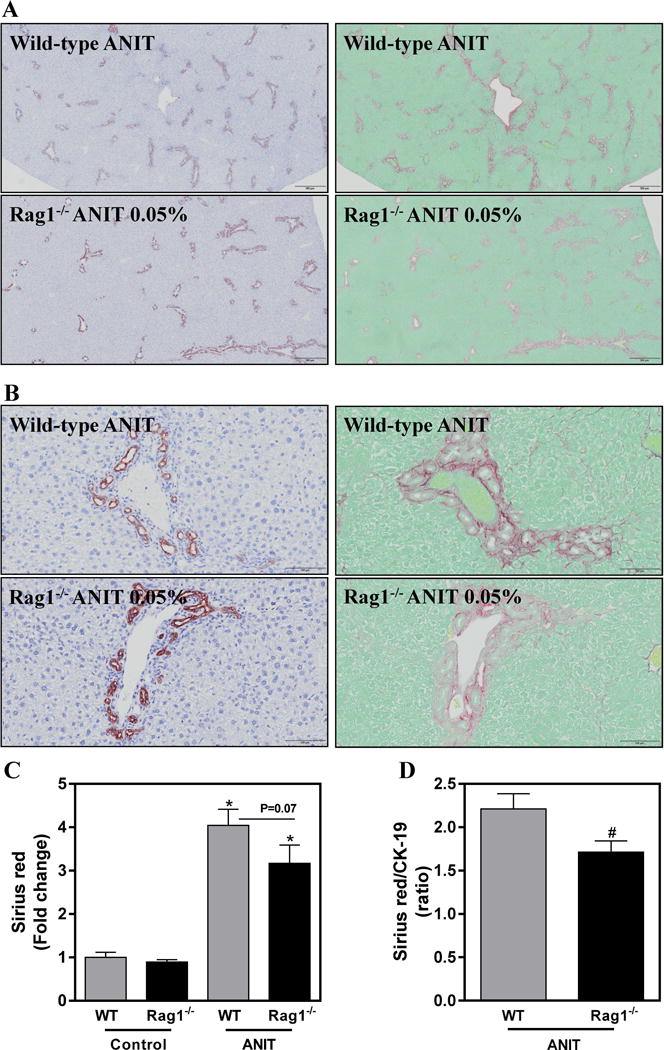
Wild-type (WT) and RAG1−/− mice were fed standard control rodent chow or diet containing 0.05% ANIT for 4 weeks. Representative photomicrographs show liver sections stained for CK-19 (brown) and Sirius red (red) at (A) 100× (scale bar 500 microns) and (B) 200× (scale bar 100 microns). (C) Sirius red staining was quantified as described in Materials and Methods. (D) Area of sirius red-staining expressed as a ratio of CK-19-positive staining. Data are expressed as mean + SEM; n=5–10 mice per group. *p<0.05 vs. control diet. #p<0.05 vs. ANIT-exposed WT mice.
4. Discussion
Strong evidence supports lymphocyte involvement in BDEC injury in cholestatic liver disease such as PBC and pediatric biliary atresia (Bird et al. 1988; Bjorkland et al. 1991; Guo et al. 2012). However, the role of lymphocytes in widely-utilized experimental models of cholestasis-associated biliary fibrosis is not entirely clear. In the case of ANIT exposure, a prototypical model of chemical-induced bile duct injury, the cytotoxic effects of ANIT are perceived as the primary trigger of chronic BDEC injury (Dahm et al. 2010; Jean et al. 1995; Joshi et al. 2016a; Plaa and Priestly 1976). Our hypothesis has been that the use of a model xenobiotic to chronically injure BDECs likely bypasses the requirement for immune cell involvement in long-term biliary injury. However, to our surprise, the present studies suggest that lymphocytes contribute, in part, to chronic bile duct injury and biliary fibrosis in mice exposed to ANIT. Overall, the results suggest that hepatic lymphocyte accumulation in livers of ANIT-exposed mice is not simply a reaction to tissue injury, and is instead an important determinant of the severity of biliary injury and fibrosis.
There exists an important dichotomy in the pathologies produced by an acute single oral gavage of ANIT and the chronic exposure of mice to ANIT in the chow. Acute administration of ANIT is well-appreciated to cause cholestatic liver damage in rodents (Becker and Plaa 1965; Plaa and Priestly 1976). Hepatocellular necrosis in rodents given an acute dose of ANIT requires ANIT transport into the bile, as Mrp2 deficient TR−/− rats are protected from ANIT-induced cholestatic liver injury (Dietrich et al. 2001). Enterohepatic recirculation enables ANIT to achieve toxic levels in the biliary tree, injuring BDECs, and triggering exaggerated neutrophil activation and hepatocellular injury (Dietrich et al. 2001; Jean et al. 1995; Jean and Roth 1995). In this paradigm of acute ANIT administration, lymphocytes do not appear to contribute to hepatocellular necrosis (Dahm et al. 1991). This is most easily accounted for by the dominant role neutrophils play in this particular form of liver injury. However, it also seems plausible that the high levels of ANIT achieved in the bile essentially circumvent the requirement for lymphocytes in producing biliary injury.
Contrasting acute ANIT administration, prolonged exposure of wild-type mice to ANIT is not associated with marked focal hepatocellular necrosis (Chang et al. 2005; Luyendyk et al. 2011; Sullivan et al. 2012; Sullivan et al. 2010). Rather, the mice develop bile duct hyperplasia and biliary fibrosis, reminiscent of primary sclerosing cholangitis (Golbar et al. 2013; Hirschfield et al. 2013; Joshi et al. 2015; Joshi et al. 2016a; Lindor et al. 2015; Sullivan et al. 2012; Sullivan et al. 2010). Mechanistically, this form of peribiliary fibrosis occurs by mechanisms congruent with multiple other experimental models of bile duct fibrosis (e.g. bile duct ligation, Mdr2−/− mice, biliary atresia) (Fickert et al. 2004; Popov et al. 2005; Popov et al. 2008; Sullivan et al. 2010). Consistent with previous studies, even in the context of chronic ANIT exposure, lymphocyte deficiency did not affect mild hepatocellular injury, as indicated by similar serum ALT activity. However, the reduction in serum ALP activity in ANIT-exposed RAG1−/− mice implies that a role for lymphocytes in biliary injury emerges in mice exposed chronically to ANIT. The mechanism whereby lymphocytes contribute to bile duct injury in ANIT-exposed mice is not known. Notably, we observed hepatic accumulation of CD3+ lymphocytes, but not CD45R B220+ B-cells in livers of wild-type mice after ANIT exposure. The extent of hepatic lymphocytic infiltration after ANIT exposure is considerably less than what is observed in other hepatic autoimmunity models. However, the accumulation of T-cells in ANIT-exposed mice near portal tracts suggests a potential role for these cells in biliary pathology. Additional studies are required to define the precise lymphocyte subtype contributing to biliary injury in ANIT-exposed mice.
Biliary hyperplasia is evident in livers of mice exposed to ANIT (Joshi et al. 2015; Joshi et al. 2016a). The mechanisms responsible for this hyperplasia are not entirely understood. Interestingly, despite the reduction of serum ALP activity in ANIT-exposed RAG1−/− mice, expansion of CK-19-positive bile ducts increased similar to wild-type mice. This disconnect is intriguing, as expansion and remodeling of the biliary tree after ANIT exposure is perceived as a compensatory response to bile duct injury, among other factors (Kossor et al. 1995). It is possible that ANIT triggers bile duct proliferation independent of cellular injury. Interestingly, we found that at non-cytotoxic concentrations, ANIT increased proliferation of MMNK-1 cells, a human cholangiocyte cell line, as indicated by MTT assay (unpublished results). Overall, the results from the current study imply that bile duct hyperplasia is independent of lymphocyte involvement in ANIT-exposed mice.
Quantitative increases in hepatic collagen deposits typically correspond to the relative increase in biliary mass in ANIT-exposed mice, largely because bile ducts themselves express mediators responsible for portal fibroblast activation and peribiliary fibrosis (Munger et al. 1999; Patsenker et al. 2008; Popov et al. 2008; Sullivan et al. 2010). Thus, there is typically a close connection between increased biliary hyperplasia and induction of profibrogenic mediators expressed by BDECs (Munger et al. 1999; Patsenker et al. 2008; Popov et al. 2008; Sullivan et al. 2010; Wang et al. 2007). Thus, we were surprised to find reduced expression of multiple profibrogenic genes in ANIT-exposed RAG1−/− mice, where biliary hyperplasia was unaffected. This associated with reduced peribiliary collagen protein deposition in ANIT-exposed RAG1−/− mice. This result indicates that lymphocytes contribute to peribiliary fibrosis in this model, but through a mechanism independent of bile duct hyperplasia.
The exact mechanism whereby lymphocytes contribute to fibrosis in this model is not known. Indeed, B-cell deficiency was demonstrated to confer protection from hepatic fibrosis in mice exposed to carbon tetrachloride (Bhogal and Bona 2005). Thus, we were surprised that the reduction in liver fibrosis after ANIT exposure was not larger in RAG-1−/− mice. One possibility is that lymphocytes direct the production of profibrogenic genes by BDECS, such as alphaVbeta6 integrin (αVβ6), a heterodimeric integrin that activates latent transforming growth factor β (TGFβ) (Hahm et al. 2007; Munger et al. 1999; Patsenker et al. 2008; Sullivan et al. 2010). Alternatively, lymphocytes may direct expression of profibrogenic cytokines by other cell types, such as macrophages (Parsons et al. 2007; Wynn 2008). Indeed, we found that TGFβ1 mRNA levels were significantly reduced in livers of ANIT-exposed RAG1−/− mice compared to ANIT-exposed wild-type mice (not shown). Additional studies are required to determine the precise mechanism whereby lymphocytes contribute to biliary fibrosis in this model.
5. Conclusion
In summary, we report that bile duct injury and biliary fibrosis are reduced in ANIT-exposed RAG1−/− mice, which lack T- and B-lymphocytes. Despite evidence of auto-antibodies against nuclear proteins, the results do not imply that ANIT-induced biliary fibrosis is a model of autoimmune liver disease. However, the novel observation that lymphocytes contribute to the profibrogenic response independent of bile duct remodeling sets the stage for utilizing chronic ANIT exposure as another potential experimental setting to decipher the role of lymphocytes in liver fibrosis.
Acknowledgments
Funding information: This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [R01 ES017537]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institutes of Health.
Abbreviations
- ANIT
alpha-naphthylisothiocyanate
- CK-19
cytokeratin-19
- ALT
alanine aminotransferase
- ALP
alkaline phosphatase
- TGFβ
transforming growth factor beta
- COL1A1
type I collagen
- ITGβ6
integrin beta 6
- αVβ6
alphaVbeta6 integrin
- PBC
primary biliary cirrhosis
- PSC
primary sclerosing cholangitis
- BDEC
bile duct epithelial cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authorship contributions
Participated in concept and research design: Nikita Joshi, Anna K. Kopec, James P. Luyendyk
Conducted experiments: Nikita Joshi, Anna K. Kopec, Holly Cline-Fedewa, James. P. Luyendyk
Performed data analysis and interpreted data: Nikita Joshi, Anna K. Kopec, James. P. Luyendyk
Wrote or contributed to the writing of the manuscript: Nikita Joshi, Anna K. Kopec, Holly Cline-Fedewa, James P. Luyendyk
Final approval of the version to be published: Nikita Joshi, Anna K. Kopec, Holly Cline-Fedewa, James P. Luyendyk
Disclosures of Conflicts of Interest: The authors have no conflicts to disclose
References
- Becker BA, Plaa GL. The nature of alpha-naphthylisothiocyanate-induced cholestasis. Toxicol Appl Pharmacol. 1965;7:680–685. doi: 10.1016/0041-008x(65)90125-0. [DOI] [PubMed] [Google Scholar]
- Bhogal RK, Bona CA. B cells: no longer bystanders in liver fibrosis. J Clin Invest. 2005;115:2962–2965. doi: 10.1172/JCI26845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird P, Calvert JE, Mitchison H, Ling NR, Bassendine M, James OF. Lymphocytes from patients with primary biliary cirrhosis spontaneously secrete high levels of IgG3 in culture. Clin Exp Immunol. 1988;71:475–480. [PMC free article] [PubMed] [Google Scholar]
- Bjorkland A, Festin R, Mendel-Hartvig I, Nyberg A, Loof L, Totterman TH. Blood and liver-infiltrating lymphocytes in primary biliary cirrhosis: increase in activated T and natural killer cells and recruitment of primed memory T cells. Hepatology. 1991;13:1106–1111. doi: 10.1002/hep.1840130617. [DOI] [PubMed] [Google Scholar]
- Chang ML, Yeh CT, Chang PY, Chen JC. Comparison of murine cirrhosis models induced by hepatotoxin administration and common bile duct ligation. World J Gastroenterol. 2005;11:4167–4172. doi: 10.3748/wjg.v11.i27.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XM, O’Hara SP, LaRusso NF. The immunobiology of cholangiocytes. Immunol Cell Biol. 2008;86:497–505. doi: 10.1038/icb.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahm L, Ganey P, Roth R. Alpha-Naphthylisothiocyanate. Elsevier; 2010. [Google Scholar]
- Dahm LJ, Schultze AE, Roth RA. An antibody to neutrophils attenuates alpha-naphthylisothiocyanate-induced liver injury. J Pharmacol Exp Ther. 1991;256:412–420. [PubMed] [Google Scholar]
- Dietrich CG, Ottenhoff R, de Waart DR, Oude Elferink RP. Role of MRP2 and GSH in intrahepatic cycling of toxins. Toxicology. 2001;167:73–81. doi: 10.1016/s0300-483x(01)00459-0. [DOI] [PubMed] [Google Scholar]
- Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, Krause R, Lammert F, Langner C, Zatloukal K, Marschall HU, Denk H, Trauner M. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127:261–274. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Golbar HM, Izawa T, Ichikawa C, Tanaka M, Juniantito V, Sawamoto O, Kuwamura M, Yamate J. Slowly progressive cholangiofibrosis induced in rats by alpha-naphthylisothiocyanate (ANIT), with particular references to characteristics of macrophages and myofibroblasts. Exp Toxicol Pathol. 2013;65:825–835. doi: 10.1016/j.etp.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Guo C, Zhu J, Pu CL, Deng YH, Zhang MM. Combinatory effects of hepatic CD8+ and NK lymphocytes in bile duct injury from biliary atresia. Pediatr Res. 2012;71:638–644. doi: 10.1038/pr.2012.17. [DOI] [PubMed] [Google Scholar]
- Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, Simon KJ, Chun Wang L, Leone DR, Lobb RR, McCrann DJ, Allaire NE, Horan GS, Fogo A, Kalluri R, Shield CF, 3rd, Sheppard D, Gardner HA, Violette SM. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170:110–125. doi: 10.2353/ajpath.2007.060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Jean PA, Roth RA. Bile duct epithelial cells exposed to alpha-naphthylisothiocyanate produce a factor that causes neutrophil-dependent hepatocellular injury in vitro. Toxicol Sci. 1999;47:118–125. doi: 10.1093/toxsci/47.1.118. [DOI] [PubMed] [Google Scholar]
- Hirschfield GM, Karlsen TH, Lindor KD, Adams DH. Primary sclerosing cholangitis. Lancet. 2013;382:1587–1599. doi: 10.1016/S0140-6736(13)60096-3. [DOI] [PubMed] [Google Scholar]
- Jean PA, Bailie MB, Roth RA. 1-naphthylisothiocyanate-induced elevation of biliary glutathione. Biochem Pharmacol. 1995;49:197–202. doi: 10.1016/0006-2952(94)00469-2. [DOI] [PubMed] [Google Scholar]
- Jean PA, Roth RA. Naphthylisothiocyanate disposition in bile and its relationship to liver glutathione and toxicity. Biochem Pharmacol. 1995;50:1469–1474. doi: 10.1016/0006-2952(95)02051-9. [DOI] [PubMed] [Google Scholar]
- Joshi N, Kopec AK, O’Brien KM, Towery KL, Cline-Fedewa H, Williams KJ, Copple BL, Flick MJ, Luyendyk JP. Coagulation-driven platelet activation reduces cholestatic liver injury and fibrosis in mice. J Thromb Haemost. 2015;13:57–71. doi: 10.1111/jth.12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Kopec AK, Ray JL, Cline-Fedewa H, Nawabi A, Schmitt T, Nault R, Zacharewski TR, Rockwell CE, Flick MJ, Luyendyk JP. Fibrin deposition following bile duct injury limits fibrosis through an alphaMbeta2-dependent mechanism. Blood. 2016a;127:2751–2762. doi: 10.1182/blood-2015-09-670703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Kopec AK, Towery K, Williams KJ, Luyendyk JP. The antifibrinolytic drug tranexamic acid reduces liver injury and fibrosis in a mouse model of chronic bile duct injury. J Pharmacol Exp Ther. 2014;349:383–392. doi: 10.1124/jpet.113.210880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N, Ray JL, Kopec AK, Luyendyk JP. Dose-dependent effects of alpha-naphthylisothiocyanate disconnect biliary fibrosis from hepatocellular necrosis. J Biochem Mol Toxicol. 2016b doi: 10.1002/jbt.21834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanz M. Anatomy and physiology of the biliary epithelium. Elsevier; 2010. [Google Scholar]
- Kopec AK, Joshi N, Luyendyk JP. Role of hemostatic factors in hepatic injury and disease: animal models de-liver. J Thromb Haemost. 2016;14:1337–1349. doi: 10.1111/jth.13327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kossor DC, Goldstein RS, Ngo W, DeNicola DB, Leonard TB, Dulik DM, Meunier PC. Biliary epithelial cell proliferation following alpha-naphthylisothiocyanate (ANIT) treatment: relationship to bile duct obstruction. Fundam Appl Toxicol. 1995;26:51–62. doi: 10.1006/faat.1995.1074. [DOI] [PubMed] [Google Scholar]
- Lazaridis KN, LaRusso NF. The Cholangiopathies. Mayo Clin Proc. 2015;90:791–800. doi: 10.1016/j.mayocp.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MK, Crawford JM. The pathology of cholestasis. Semin Liver Dis. 2004;24:21–42. doi: 10.1055/s-2004-823099. [DOI] [PubMed] [Google Scholar]
- Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, American Association for Study of Liver, D Primary biliary cirrhosis. Hepatology. 2009;50:291–308. doi: 10.1002/hep.22906. [DOI] [PubMed] [Google Scholar]
- Lindor KD, Kowdley KV, Harrison ME, American College of G ACG Clinical Guideline: Primary Sclerosing Cholangitis. Am J Gastroenterol. 2015;110:646–659. doi: 10.1038/ajg.2015.112. quiz 660. [DOI] [PubMed] [Google Scholar]
- Luyendyk JP, Kassel KM, Allen K, Guo GL, Li G, Cantor GH, Copple BL. Fibrinogen deficiency increases liver injury and early growth response-1 (Egr-1) expression in a model of chronic xenobiotic-induced cholestasis. Am J Pathol. 2011;178:1117–1125. doi: 10.1016/j.ajpath.2010.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCuskey R, Sipes I. Introduction to the Liver and its Response to Toxicants. Elsevier; 2010. [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Morell CM, Fabris L, Strazzabosco M. Vascular biology of the biliary epithelium. J Gastroenterol Hepatol. 2013;28(Suppl 1):26–32. doi: 10.1111/jgh.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- O’Hara SP, Tabibian JH, Splinter PL, LaRusso NF. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol. 2013;58:575–582. doi: 10.1016/j.jhep.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CJ, Takashima M, Rippe RA. Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol. 2007;22(Suppl 1):S79–84. doi: 10.1111/j.1440-1746.2006.04659.x. [DOI] [PubMed] [Google Scholar]
- Patsenker E, Popov Y, Stickel F, Jonczyk A, Goodman SL, Schuppan D. Inhibition of integrin alphavbeta6 on cholangiocytes blocks transforming growth factor-beta activation and retards biliary fibrosis progression. Gastroenterology. 2008;135:660–670. doi: 10.1053/j.gastro.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez MJ, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–1689. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaa GL, Priestly BG. Intrahepatic cholestasis induced by drugs and chemicals. Pharmacol Rev. 1976;28:207–273. [PubMed] [Google Scholar]
- Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43:1045–1054. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- Popov Y, Patsenker E, Stickel F, Zaks J, Bhaskar KR, Niedobitek G, Kolb A, Friess H, Schuppan D. Integrin alphavbeta6 is a marker of the progression of biliary and portal liver fibrosis and a novel target for antifibrotic therapies. J Hepatol. 2008;48:453–464. doi: 10.1016/j.jhep.2007.11.021. [DOI] [PubMed] [Google Scholar]
- Sipka S, Bruckner G. The immunomodulatory role of bile acids. Int Arch Allergy Immunol. 2014;165:1–8. doi: 10.1159/000366100. [DOI] [PubMed] [Google Scholar]
- Sullivan BP, Cui W, Copple BL, Luyendyk JP. Early growth response factor-1 limits biliary fibrosis in a model of xenobiotic-induced cholestasis in mice. Toxicol Sci. 2012;126:267–274. doi: 10.1093/toxsci/kfr311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BP, Weinreb PH, Violette SM, Luyendyk JP. The coagulation system contributes to alphaVbeta6 integrin expression and liver fibrosis induced by cholestasis. Am J Pathol. 2010;177:2837–2849. doi: 10.2353/ajpath.2010.100425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjandra K, Sharkey KA, Swain MG. Progressive development of a Th1-type hepatic cytokine profile in rats with experimental cholangitis. Hepatology. 2000;31:280–290. doi: 10.1002/hep.510310204. [DOI] [PubMed] [Google Scholar]
- Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med. 1998;339:1217–1227. doi: 10.1056/NEJM199810223391707. [DOI] [PubMed] [Google Scholar]
- Wang B, Dolinski BM, Kikuchi N, Leone DR, Peters MG, Weinreb PH, Violette SM, Bissell DM. Role of alphavbeta6 integrin in acute biliary fibrosis. Hepatology. 2007;46:1404–1412. doi: 10.1002/hep.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lee G, Wang H, Vierling JM, Maher JJ. Limited role for CXC chemokines in the pathogenesis of alpha-naphthylisothiocyanate-induced liver injury. Am J Physiol Gastrointest Liver Physiol. 2004;287:G734–741. doi: 10.1152/ajpgi.00300.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]