

Abstract
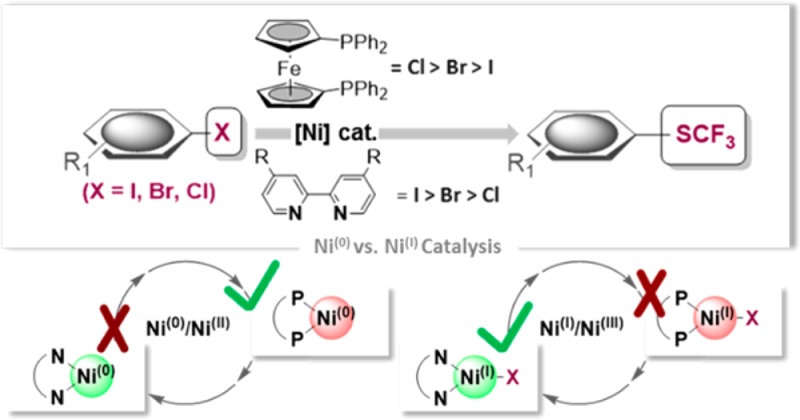
The direct introduction of the valuable SCF3 moiety into organic molecules has received considerable attention. While it can be achieved successfully for aryl chlorides under catalysis with Ni0(cod)2 and dppf, this report investigates the Ni-catalyzed functionalization of the seemingly more reactive aryl halides ArI and ArBr. Counterintuitively, the observed conversion triggered by dppf/Ni0 is ArCl > ArBr > ArI, at odds with bond strength preferences. By a combined computational and experimental approach, the origin of this was identified to be due to the formation of (dppf)NiI, which favors β-F elimination as a competing pathway over the productive cross-coupling, ultimately generating the inactive complex (dppf)Ni(SCF2) as a catalysis dead end. The complexes (dppf)NiI–Br and (dppf)NiI–I were isolated and resolved by X-ray crystallography. Their formation was found to be consistent with a ligand-exchange-induced comproportionation mechanism. In stark contrast to these phosphine-derived Ni complexes, the corresponding nitrogen-ligand-derived species were found to be likely competent catalysts in oxidation state I. Our computational studies of N-ligand derived NiI complexes fully support productive NiI/NiIII catalysis, as the competing β-F elimination is disfavored. Moreover, N-derived NiI complexes are predicted to be more reactive than their Ni0 counterparts in catalysis. These data showcase fundamentally different roles of NiI in carbon–heteroatom bond formation depending on the ligand sphere.
Keywords: nickel, cross-coupling, DFT, fluorine, mechanisms, ligand
Introduction
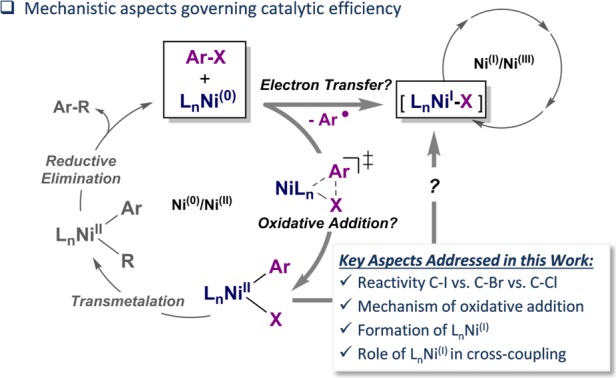
The past decade has seen numerous impressive advances in the area of homogeneous nickel catalysis.1 The limits of the oxidative addition step have continuously been pushed back to some of the least activated bonds, such as the recent breakthrough of aryl ether functionalizations, for example.2,3 In addition, nickel catalysis features a rich mechanistic portfolio, which ranges from the ability to more readily interchange among oxidation states 0, I, II, and III to the possibility for electron transfer processes (Figure 1) in cross-coupling.4 While capitalization on these diverse mechanistic possibilities has allowed the development of rich and novel synthetic organic chemistry in recent years, the very same mechanistic diversity also poses challenges in achieving the desired key features of modern and sustainable catalytic transformation: e.g. efficiency, low catalyst loading, recyclability, catalyst robustness, generality in substrate, and high functional group tolerance. To achieve high efficiency and generality, catalyst deactivation processes and side reactions will need to be overcome. This in turn requires a fundamental understanding of their origins. In this context, the role and potential catalytic competence of the odd oxidation state I has been questioned.5,6
Figure 1.
Key features and challenges of Ni catalysis.
Interestingly, while NiI species derived from N ligands have been postulated as catalytically competent intermediates in alkyl–alkyl couplings, e.g. in recent photoredox applications as well as in cross-coupling reactions of challenging electrophiles,6 for phosphine-ligand-derived NiI complexes, there are limited mechanistic data available. The latter complexes have been observed in reactions that employed Ni0 as catalyst but suggested to be catalytically inactive7,8 or reported to be less active than Ni0.9 On the other hand, Martin recently presented detailed mechanistic data supporting NiI as an active species in the activation of C–OMe bonds.10 Matsubara11a and Louie11b observed activities in Kumada and Suzuki couplings with NHC-bound NiI complexes. To shine a light on these contrasting observations, we undertook a combined computational and experimental study of the nickel-catalyzed trifluoromethylthiolation reactions of aryl halides as a case study.
Access to ArSCF3 compounds is of pharmaceutical and agrochemical significance due to their associated advantageous lipophilicity properties.12 Direct catalytic access is of particular interest.13,14 Aryl iodides and bromides can be converted to ArSCF3 via Pd0 15 and PdI–PdI catalysis with (Me4N)SCF3 or alternative nucleophilic SCF3 sources.16 In the context of Ni catalysis, Vicic has shown that a Ni(cod)2/bipyridine17 system allows for functionalization of aryl iodides and selected bromides, but not aryl chlorides. These in turn can be transformed with a phosphine-based catalyst system, Ni(cod)2/dppf, that forms [(dppf)Ni0(cod)] in situ.7,18
In this report, we will show that [(dppf)Ni0(cod)], in contrast to a bipyridine-derived Ni catalyst, counterintuitively leads to much lower conversions for those aryl halides that have weaker bonds: i.e., aryl iodides and bromides. We will unravel this reactivity behavior herein, unambiguously assigning the role of NiI for P- vs N-derived ligands in trifluoromethylthiolation, and uncover the pathways of their origins.
Results and Discussion
We started our investigations with the systematic comparison of the efficiency of C–SCF3 bond formation of 4-methoxy-substituted aryl halides in toluene at 45 °C with (Me4N)SCF3 under Ni(cod)2/dppf (10 mol %) catalysis conditions. While 4-chloroanisole was converted to the corresponding ArSCF3 in 52% yield, the generally more reactive aryl bromide gave only a 24% yield and the corresponding iodide as little as 16% of the trifluoromethylthiolated product (see Figure 2). These reactivities are at odds with the expected intrinsic ease of the aryl halides toward oxidative addition by a [Ni0] catalyst, as reinforced by the computed activation barriers of oxidative addition (see Figure 2). Our calculations at the M06L level of theory19 showed that the barrier to oxidatively add an aryl iodide to [(dppf)Ni0(cod)] is 7 kcal/mol lower in energy than that of the seemingly more reactive aryl chloride.
Figure 2.
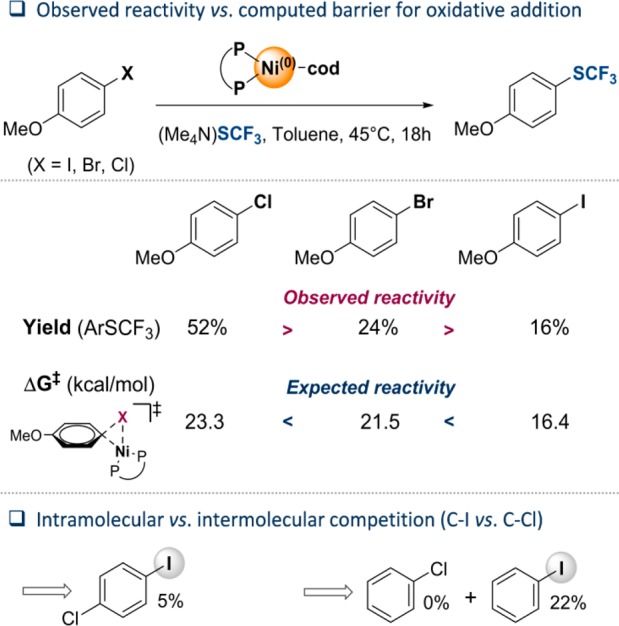
Observed reactivity order (C–Cl > C–Br > C–I) in the dppf/Ni0(cod)2-catalyzed trifluoromethylthiolation, at odds with the computed barriers.
To assess the inherent preference for C–SCF3 bond formation in greater detail, we subsequently undertook intra- and intermolecular competition experiments (C–I vs C–Cl), with Figure 2 presenting the results. While the selectivity followed the expected ease of oxidative addition, showing exclusive functionalization of the C–I site, the overall conversion to product was low (5% for intramolecular and 22% for intermolecular competition). Thus, the lower yields obtained in the reactions with the weaker C–halogen bond substrates do not correlate with the intrinsic reactivities toward oxidative addition but instead must arise from alternative factors that render the catalysis nonproductive.
To gain deeper insight, we performed 19F and 31P NMR spectroscopic analyses of the reaction mixtures after 15 h. These indicated that the characteristic signals of [(dppf)Ni0(cod)] had disappeared, and instead a new species had formed that gives two triplets in the 31P NMR (resonating at 30.8 ppm (J = 23.0 Hz) and at 22.1 ppm (J = 37.6 Hz)) and a dd in the 19F NMR spectrum (at −44.8 ppm (J = 37.6, 23.0 Hz)). While we had observed this species also in our previous studies,20 we had not previously been able to assign its structure or explain its origin. However, we now succeeded in the isolation and characterization of single crystals, unambiguously confirming that the thiocarbonyl-bound [Ni0] complex 1 was formed (Figure 3). Attempts to react ArI, ArBr, and ArCl with (Me4N)SCF3 and complex 1 showed no reaction, suggesting that 1 was catalytically inactive and therefore a product of catalyst deactivation.20 Computational analysis further indicates that the S=CF2 ligand is very strongly bound to the Ni center—ligand exchange with 1,5-cyclooctadiene is predicted to be endothermic by 21.3 kcal/mol.19
Figure 3.
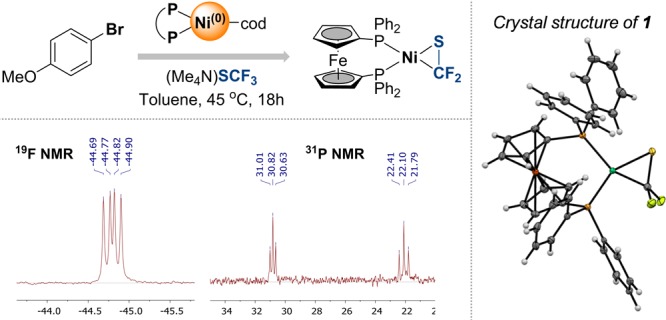
Deactivation of Ni0 to 1 occurring under catalytic conditions.
Species 1 could in principle arise from the trapping of a potential decomposition side product (F2C=S) of the employed (Me4N)SCF3 reagent by the [Ni0] catalyst.21 However, our separate subjection of (Me4N)SCF3 to Ni(cod)2/dppf at 45 °C indicated that this would be unlikely, as 1 was not formed. Moreover, there was no indication of the formation of F2C=S in solution, as judged by 19F NMR analysis. Instead, 1 is likely derived directly from an alternative Ni intermediate.
Given the formal loss of a fluorine atom, a β-fluoride elimination step is mechanistically required. Literature precedence suggests that β-fluoride elimination from [NiII] intermediates would be feasible.22 Thus, to gain insight whether the likely origin of 1 is a [NiII] or an alternative Ni species, we computationally investigated the ease of β-F elimination from [(dppf)NiII(SCF3)(Ph)] relative to the productive reductive elimination pathway (Figure 4). Our data, obtained at the CPCM(toluene)M06/def2TZVP//ωB97XD/6-31G(d)(SDD) level of theory suggests β-F elimination is disfavored by ΔΔG⧧ = 7.5 kcal/mol.23
Figure 4.
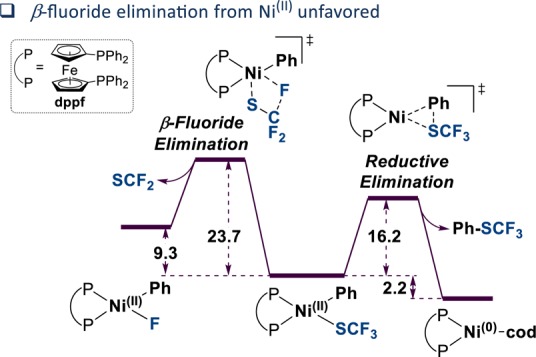
Computational comparison of β-F elimination and reductive elimination pathways from NiII. Shown are the ΔΔG⧧ values in kcal/mol, calculated at the CPCM(toluene)M06/def2TZVP//ωB97XD/6-31G(d)(SDD) level of theory.23
In line with these calculations are the following experimental observations: while subjection of the product ArSCF3 to Ni(cod)2/dppf will eventually give rise to the deactivation complex 1, this process occurs on a slower time scale in comparison to that observed for the forward reactions.24 Moreover, given the higher propensity for subsequent oxidative addition of the liberated [Ni0] to either ArI or ArBr (in comparison to ArCl) upon reductive elimination of ArSCF3 under catalysis conditions, the unproductive reverse reaction with the product, ArSCF3, can also be ruled out as the predominant cause of the catalysis dead end 1 for the weaker versus stronger C–halogen bond electrophiles.25
We speculated that the initial oxidative addition step may be the cause of the divergent reactivities. Thus, we subsequently monitored the oxidative addition of [(dppf)Ni0(cod)] to ArI, ArBr, and ArCl in the absence of (Me4N)SCF3. We observed that in all cases a paramagnetic species was formed, which we could unambiguously assign as [NiI] upon X-ray crystallographic analyses. While the oxidative addition to ArCl7 and ArBr give the tricoordinate monomer [(dppf)NiI-X], interestingly, for the iodide, a dimer is the favored species in the solid state (see Figure 4).26,27 The observed relative ease of the formation of [NiI] follows the order ArI > ArBr > ArCl.
These data are in line with the pioneering and seminal studies by Kochi on the oxidative addition of Ni0(PEt3)4 to aryl halides.28 Kochi proposed an electron transfer mechanism as origin of [NiI]. However, we detected the formation of biaryl products in all cases, suggesting that a different mechanism for the formation of [NiI] may be operative. As an alternative for the electron transfer mechanism there have been reports on [NiI] formation via ligand exchanges on a [NiII] intermediate.11
As illustrated in Figure 5, following the oxidative addition of [Ni0] to ArX, ligand exchange from [(dppf)NiII(X)(Ph)] to [(dppf)NiII(X)2] and [(dppf)NiII(Ph)2] may likely occur, followed by reductive elimination of biaryl from [NiII] and subsequent comproportionation of the resulting [Ni0] with [NiII] (see Figure 5).29 The formation of [(dppf)NiIIX2] as an intermediate was also unambiguously confirmed through its isolation (in addition to NiI) and characterization (via 1H NMR and X-ray crystallography) from the reaction of Ni0 and PhI in benzene.30 Our computational data indicate that the ligand exchange step from [(dppf)NiII(X)(Ph)] to [(dppf)NiIIX2] and [(dppf)NiII(Ph)2] is favorable, being exergonic for all halides (ΔG = −7.5, −5.9, and −3.3 kcal/mol for X = Cl, Br, I, respectively; see Figure 5). Subsequent reductive elimination of biphenyl and formation of [(dppf)Ni0(cod)] is also thermodynamically favored (by 17.1 kcal/mol). Finally, comproportionation of [(dppf)NiIIX2] and [(dppf)Ni0(cod)] is also exergonic for all halides, with ΔG = −1.5, −2.2, and −1.5 kcal/mol for X = Cl, Br, I, respectively (relative to the NiII and Ni0 complexes). As such, the steps leading from [(dppf)NiII(Ph)(X)] to [NiI] are thermodynamically favored (see Figure 5).
Figure 5.
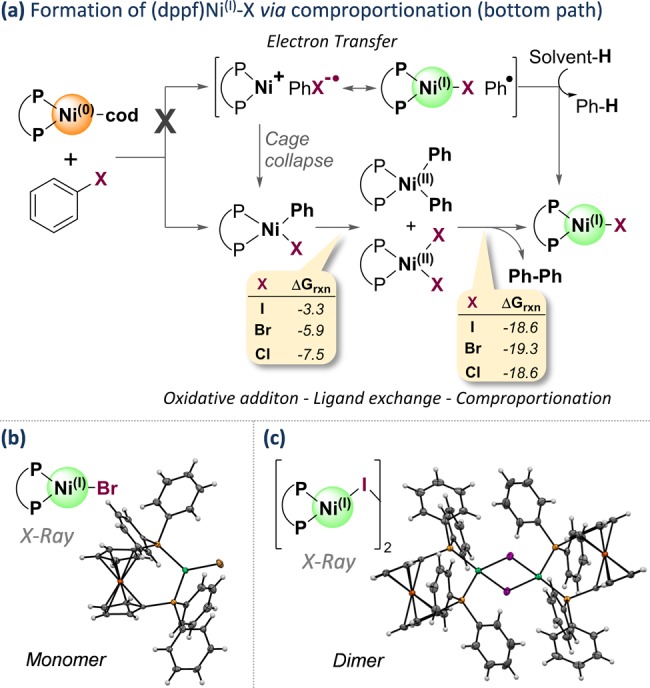
Likely mechanism of [NiI] formation and calculated free energy changes (in kcal/mol) of the ligand exchange and reductive elimination + comproportionation steps (a) and crystal structures of (dppf)NiI bromide (b) and iodide (c).19f,27
Would the [NiI] species that we observed ultimately generate complex 1, leading to a catalysis dead end, or perhaps remain a competent species for the generation of ArSCF3? To test this, we initially subjected (Me4N)SCF3 to [(dppf)NiI(Cl)]. This led to rapid formation of the deactivated thiocarbonyl-bound [Ni0] complex 1, as judged by 19F NMR analysis, suggesting that if any [(dppf)NiI(SCF3)] species were to be generated upon halogen to SCF3 exchange, facile β-fluoride elimination would take place (Figure 6). In line with this, our calculations of the β-F elimination from [(dppf)NiI(SCF3)] predicted that this process is relatively facile (ΔG⧧ = 20.6 kcal/mol). The thereby generated [(dppf)NiI(F)] complex could subsequently undergo disproportionation to generate [(dppf)NiII(F)2] and [Ni0]. The latter would ultimately trap S=CF2 to give 1 (Figure 6). Consistent with this mechanism, our quantitative 31P NMR spectroscopic analysis of the reaction mixture of (Me4N)SCF3 with [(dppf)NiI(Cl)] indicated that after 1 h ∼50% of the employed [(dppf)NiI(Cl)] was converted to [(dppf)Ni0(SCF2)] 1 (and ∼50% free dppf was also formed31).
Figure 6.
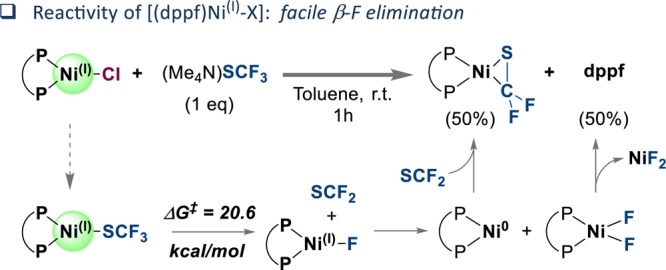
Facile reactivity of [NiI] to form 1 (P-P = dppf).
The transition states of β-fluoride elimination from NiI (panel (a)) and NiII (panel (b)) complexes are illustrated in Figure 7. The TS derived from NiI is slightly later, as expressed by the longer C–F distance (2.12 Å for NiI and 1.88 Å for NiII). This suggests a greater stabilization in the TS derived from NiI, which is reflected also in the shorter Ni–S distance in case of NiI (3.08 Å for NiI vs 3.31 Å for NiII) as well as the presence of a free coordination site.23
Figure 7.
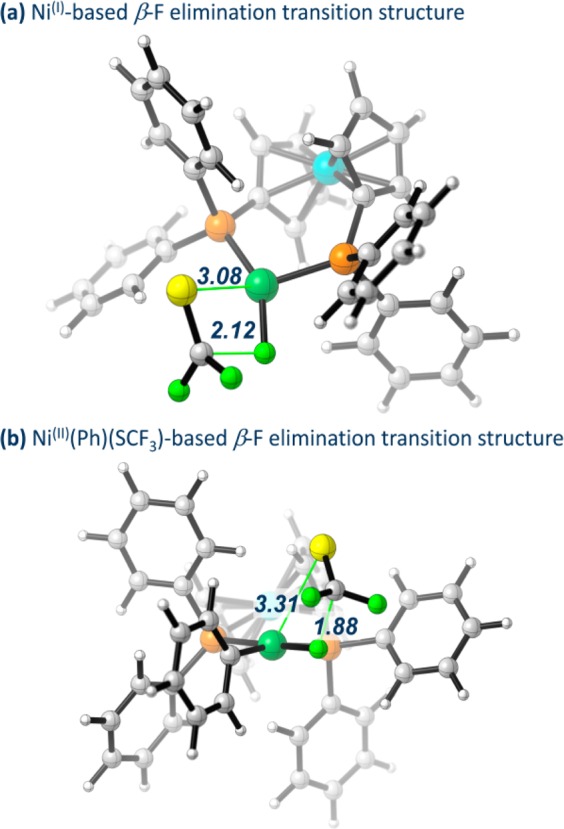
Calculated β-F elimination transition state structures from NiI (a) and NiII (b), shown with the Ni–S and C–F distances (in Å).
These results are a clear indicator that [(dppf)NiISCF3] is a competent and very potent source of the catalytically inactive complex 1.32 The generation of [NiI] therefore is detrimental to trifluoromethylthiolation. The origin of lower conversions of aryl iodides and aryl bromides in comparison to the generally less reactive aryl chlorides can be unambiguously correlated to their relative propensity to form [NiI]. The key to productive catalysis in (dppf)Ni0-derived catalysis is therefore a low concentration of the [(dppf)NiII(X)(Ar)] intermediate that is formed upon oxidative addition to prevent ligand exchanges, as well as a rapid transmetalation and follow-up reaction. Transmetalation generally follows MII–Cl > MII–Br > MII–I (for M = Pd, Ni),33 paralleling the observed efficiencies of C–SCF3 bond formation.
Many powerful Ni-catalyzed synthetic methods employ N-derived ligands, such as pyridine derivatives, instead of the otherwise more donating P-based ligands. In line with this, Vicic and co-workers elegantly showed that Ni(cod)2 along with dmbpy (=4,4′-dimethoxybipyridine) as a ligand results in the trifluoromethylthiolation of aryl iodides and certain bromides, but not chlorides.17 In light of our above observations with P-derived Ni0, there hence appear to be fundamental reactivity differences between N- and P-coordinated Ni catalysts. Given that N-derived ligands led to the opposite reactivity (X = I, efficient; X = Cl, no conversion), this implies that for N-based ligands either the formation of NiI is suppressed or the competing β-F elimination is no longer favorable, therefore avoiding catalyst deactivation products.
We thus also computationally assessed the dmbpy/Ni(cod)2-catalyzed trifluoromethylthiolation of aryl iodides.34 The obtained data suggest that, in stark contrast to Ni0/dppf, the corresponding Ni0/dmbpy system does not proceed via Ni0/NiII catalysis but instead by NiI/NiIII (see Figure 8). Interestingly, the Ni0/NiII catalytic cycle is disfavored primarily due to a high-barrier reductive elimination step of ArSCF3 from [(dmbpy)NiII(SCF3)(Ph)] (ΔG⧧ = 33.1 kcal/mol). In contrast, a NiI/NiIII pathway is characterized by much lower activation free energy barriers of 12.9 kcal/mol for oxidative addition and 16.1 kcal/mol for reductive elimination (Figure 8b). These data also agree with previous studies highlighting facile oxidative addition to (N-N)NiI complexes.6h,35
Figure 8.
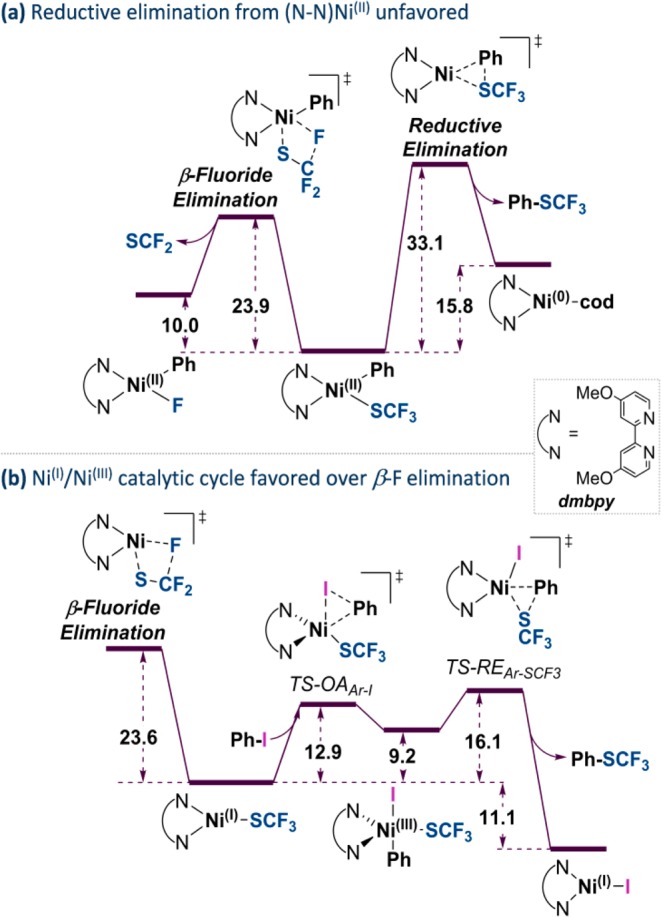
(a) Computational comparison of β-F-elimination and reductive elimination pathways with dmbpy-ligated NiII. (b) NiI/NiIII catalytic cycle being favored over β-F elimination.
Moreover, the corresponding β-F elimination from [(dmbpy)NiI(SCF3)] is significantly less favored (ΔG⧧ = 23.6 kcal/mol) than the productive pathway: oxidative addition of [(dmbpy)NiISCF3] to ArI occurs preferentially (ΔΔG⧧=10.7 kcal/mol). While the β-F eliminations are comparable in magnitude for N- and P-derived NiI species, the N-based system is overall more effective, as the barriers for the productive NiI/NiIII pathway are significantly lower. In comparison, the direct oxidative addition of iodobenzene to [(dppf)NiI(SCF3)] is calculated to proceed with a barrier of 32.8 kcal/mol, being significantly greater (ΔΔG⧧ = 12.2 kcal/mol) than the barrier of the competing β-F elimination pathway. The observed trends are likely related to the different steric properties of the dmbpy and dppf ligands. The smaller bipyridine ligand would still allow facile oxidative addition to the tricoordinate NiI-SCF3, while the lack of steric bulk would stabilize the NiII species and thus not allow facile reductive elimination. With dppf, on the other hand, the steric bulk of the phenyl groups readily allows reductive elimination from NiII but hampers oxidative addition to NiI.
Overall, these data strongly indicate that (dppf)NiI primarily is a less competent catalyst than (dppf)Ni0, as it suffers from a relatively high oxidative addition barrier to ArX, which renders the competing β-F elimination favored, ultimately giving the catalytically inactive complex 1. In stark contrast, bipyridine-ligand-derived NiI shows much lower barriers for productive catalysis, rendering the competing processes disfavored. The β-F elimination therefore has served as a mechanistic probe to differentiate between the divergent reactivities of the various plausible oxidation states (0, I, and II) as a function of ligand, using a combination of experiments and computation. Such unambiguous differentiations are otherwise challenging to accomplish.
Conclusions
In summary, using a combination of computational and experimental studies, we examined the key factors for efficiency in C–SCF3 bond formation, catalyzed by phosphine- and nitrogen-based nickel complexes. Our data show that, for dppf, [NiI] species may readily form with the relative ease ArI > ArBr > ArCl under typical [Ni0] catalysis conditions. This will be detrimental for the agrochemically and pharmaceutically relevant C–SCF3 bond formation, as the corresponding [(dppf)NiI-SCF3] undergoes facile β-fluoride elimination more readily over productive catalysis, leading to [(dppf)Ni0(SCF2)] complex 1, which is catalytically incompetent and a catalysis dead end. Our mechanistic data support that [(dppf)NiI] is derived from NiII precursors via a comproportionation mechanism under concomitant formation of biaryl, not through reductive electron transfer pathways. The reverse was observed for the nitrogen-based Ni/dmbpy system: the corresponding [NiI] species promotes efficient NiI/NiIII catalysis, rendering unproductive β-F elimination from NiI disfavored. In contrast, a Ni0/NiII cycle suffers from high activation barriers at the elementary steps (oxidative addition and reductive elimination) with bipyridine as ligand. These data highlight the prerequisites for selective Ni-catalyzed couplings of aryl halides and showcase the potential and reactivity of NiI as a catalyst for different ligands. Our future efforts are directed at exploring the full potential of catalysis at NiI.
Acknowledgments
We thank the RWTH Aachen, the MIWF NRW, and the European Research Council (ERC-637993) for funding. Calculations were performed with computing resources granted by JARA-HPC from RWTH Aachen University under project “jara0091”.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.6b03344.
Detailed experimental procedures, spectroscopic data, computational details, and energies of computed structures (PDF)
Cartesian coordinates of computed structures (XYZ)
Crystallographic data for 1 (CIF)
Crystallographic data for [(dppf)NiIBr] (CIF)
Crystallographic data for [(dppf)NiII]2 (CIF)
Crystallographic data for [(dppf)NiIII2] (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Johansson Seechurn C. C. C.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]; b Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]; c Tamaru Y., Introductory Guide to Organonickel Chemistry. In Modern Organonickel Chemistry; Wiley-VCH: Weinheim, Germany, 2005; pp 1–40. [Google Scholar]
- For reviews, see:; a Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Chem. Rev. 2011, 111, 1346–1416. 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yamaguchi J.; Muto K.; Itami K. Eur. J. Org. Chem. 2013, 2013, 19–30. 10.1002/ejoc.201200914. [DOI] [Google Scholar]; c Milburn R. R.; Snieckus V. Angew. Chem., Int. Ed. 2004, 43, 888–891. 10.1002/anie.200352633. [DOI] [PubMed] [Google Scholar]; d Gavryushin A.; Kofink C.; Manolikakes G.; Knochel P. Org. Lett. 2005, 7, 4871–4874. 10.1021/ol051615+. [DOI] [PubMed] [Google Scholar]; e Guan B.-T.; Wang Y.; Li B.-J.; Yu D.-G.; Shi Z.-J. J. Am. Chem. Soc. 2008, 130, 14468–14470. 10.1021/ja8056503. [DOI] [PubMed] [Google Scholar]; f Quasdorf K. W.; Tian X.; Garg N. K. J. Am. Chem. Soc. 2008, 130, 14422–14423. 10.1021/ja806244b. [DOI] [PubMed] [Google Scholar]; g Ehle A. R.; Zhou Q.; Watson M. P. Org. Lett. 2012, 14, 1202–1205. 10.1021/ol203322v. [DOI] [PubMed] [Google Scholar]; h Ge S.; Hartwig J. F. Angew. Chem., Int. Ed. 2012, 51, 12837–12841. 10.1002/anie.201207428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For Ni-catalyzed functionalization of C–O electrophiles, see:; a Yu D.-G.; Li B.-J.; Shi Z.-J. Acc. Chem. Res. 2010, 43, 1486–1495. 10.1021/ar100082d. [DOI] [PubMed] [Google Scholar]; b Muto K.; Yamaguchi J.; Lei A.; Itami K. J. Am. Chem. Soc. 2013, 135, 16384–16387. 10.1021/ja409803x. [DOI] [PubMed] [Google Scholar]; c Cornella J.; Zarate C.; Martin R. Chem. Soc. Rev. 2014, 43, 8081–8097. 10.1039/C4CS00206G. [DOI] [PubMed] [Google Scholar]; d Tobisu M.; Chatani N. Acc. Chem. Res. 2015, 48, 1717–1726. 10.1021/acs.accounts.5b00051. [DOI] [PubMed] [Google Scholar]; e Dankwardt J. W. Angew. Chem., Int. Ed. 2004, 43, 2428–2432. 10.1002/anie.200453765. [DOI] [PubMed] [Google Scholar]; f Tobisu M.; Shimasaki T.; Chatani N. Angew. Chem., Int. Ed. 2008, 47, 4866–4869. 10.1002/anie.200801447. [DOI] [PubMed] [Google Scholar]; g Liu X.; Hsiao C.-C.; Kalvet I.; Leiendecker M.; Guo L.; Schoenebeck F.; Rueping M. Angew. Chem., Int. Ed. 2016, 55, 6093–6098. 10.1002/anie.201510497. [DOI] [PubMed] [Google Scholar]
- a Montgomery J., Organonickel Chemistry. In Organometallics in Synthesis; Wiley: Hoboken, NJ, 2013; pp 319–428. [Google Scholar]; b Tasker S. Z.; Standley E. A.; Jamison T. F. Nature 2014, 509, 299–309. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Colon I.; Kelsey D. R. J. Org. Chem. 1986, 51, 2627–2637. 10.1021/jo00364a002. [DOI] [Google Scholar]
- For phosphine ligands, Ni0/NiII catalysis is generally assumed. The role of NiI is unclear.
- For N-type ligands and alkyl–alkyl and alkyl–aryl couplings, NiI is assumed to be a catalytic intermediate:; a Bakac A.; Espenson J. H. J. Am. Chem. Soc. 1986, 108, 719–723. 10.1021/ja00264a024. [DOI] [PubMed] [Google Scholar]; b Jones G. D.; Martin J. L.; McFarland C.; Allen O. R.; Hall R. E.; Haley A. D.; Brandon R. J.; Konovalova T.; Desrochers P. J.; Pulay P.; Vicic D. A. J. Am. Chem. Soc. 2006, 128, 13175. 10.1021/ja063334i. [DOI] [PubMed] [Google Scholar]; c Li Z.; Jiang Y.-Y.; Fu Y. Chem. - Eur. J. 2012, 18, 4345–4357. 10.1002/chem.201103882. [DOI] [PubMed] [Google Scholar]; d Powell D. A.; Maki T.; Fu G. C. J. Am. Chem. Soc. 2005, 127, 510–511. 10.1021/ja0436300. [DOI] [PubMed] [Google Scholar]; e Phapale V. B.; Buñuel E.; García-Iglesias M.; Cárdenas D. J. Angew. Chem., Int. Ed. 2007, 46, 8790–8795. 10.1002/anie.200702528. [DOI] [PubMed] [Google Scholar]; f Zuo Z.; Ahneman D. T.; Chu L.; Terrett J. A.; Doyle A. G.; MacMillan D. W. C. Science 2014, 345, 437–440. 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Weix D. J. Acc. Chem. Res. 2015, 48, 1767–1775. 10.1021/acs.accounts.5b00057. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. J. Am. Chem. Soc. 2015, 137, 4896–4899. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G.; Kalvet I.; Englert U.; Schoenebeck F. J. Am. Chem. Soc. 2015, 137, 4164–4172. 10.1021/jacs.5b00538. [DOI] [PubMed] [Google Scholar]
- Ge S.; Green R. A.; Hartwig J. F. J. Am. Chem. Soc. 2014, 136, 1617–1627. 10.1021/ja411911s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guard L. M.; MohadjerBeromi M.; Brudvig G. W.; Hazari N.; Vinyard D. J. Angew. Chem., Int. Ed. 2015, 54, 13352–13356. 10.1002/anie.201505699. [DOI] [PubMed] [Google Scholar]
- Cornella J.; Gómez-Bengoa E.; Martin R. J. Am. Chem. Soc. 2013, 135, 1997–2009. 10.1021/ja311940s. [DOI] [PubMed] [Google Scholar]
- Formation of NiI from NiII:; a Miyazaki S.; Koga Y.; Matsumoto T.; Matsubara K. Chem. Commun. 2010, 46, 1932–1934. 10.1039/b924716e. [DOI] [PubMed] [Google Scholar]; b Zhang K.; Conda-Sheridan M.; Cooke S. R.; Louie J. Organometallics 2011, 30, 2546–2552. 10.1021/om200090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Leo A.; Jow P. Y. C.; Silipo C.; Hansch C. J. Med. Chem. 1975, 18, 865–868. 10.1021/jm00243a001. [DOI] [PubMed] [Google Scholar]; b Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- For reviews of recent advances, see:; a Xu X.-H.; Matsuzaki K.; Shibata N. Chem. Rev. 2015, 115, 731–764. 10.1021/cr500193b. [DOI] [PubMed] [Google Scholar]; b Zheng H.; Huang Y.; Weng Z. Tetrahedron Lett. 2016, 57, 1397–1409. 10.1016/j.tetlet.2016.02.073. [DOI] [Google Scholar]
- For selected examples of alternative indirect, electrophilic, or metal-free approaches or examples involving alternative coupling partners (C–H, C–N2 ,or C–B(OH)2), see:; a Pooput C.; Medebielle M.; Dolbier W. R. Org. Lett. 2004, 6, 301–303. 10.1021/ol036303q. [DOI] [PubMed] [Google Scholar]; b Kieltsch I.; Eisenberger P.; Togni A. Angew. Chem., Int. Ed. 2007, 46, 754–757. 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]; c Pooput C.; Dolbier W. R.; Médebielle M. J. Org. Chem. 2006, 71, 3564–3568. 10.1021/jo060250j. [DOI] [PubMed] [Google Scholar]; d Chen C.; Xie Y.; Chu L.; Wang R.-W.; Zhang X.; Qing F.-L. Angew. Chem., Int. Ed. 2012, 51, 2492–2495. 10.1002/anie.201108663. [DOI] [PubMed] [Google Scholar]; e Tran L. D.; Popov I.; Daugulis O. J. Am. Chem. Soc. 2012, 134, 18237–18240. 10.1021/ja3092278. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Shao X.; Wang X.; Yang T.; Lu L.; Shen Q. Angew. Chem., Int. Ed. 2013, 52, 3457–3460. 10.1002/anie.201209817. [DOI] [PubMed] [Google Scholar]; g Pluta R.; Nikolaienko P.; Rueping M. Angew. Chem., Int. Ed. 2014, 53, 1650–1653. 10.1002/anie.201307484. [DOI] [PubMed] [Google Scholar]; h Danoun G.; Bayarmagnai B.; Gruenberg M. F.; Goossen L. J. Chem. Sci. 2014, 5, 1312–1316. 10.1039/c3sc53076k. [DOI] [Google Scholar]; i Xu J.; Mu X.; Chen P.; Ye J.; Liu G. Org. Lett. 2014, 16, 3942–3945. 10.1021/ol501742a. [DOI] [PubMed] [Google Scholar]; j Honeker R.; Ernst J. B.; Glorius F. Chem. - Eur. J. 2015, 21, 8047–8051. 10.1002/chem.201500957. [DOI] [PubMed] [Google Scholar]
- a Teverovskiy G.; Surry D. S.; Buchwald S. L. Angew. Chem., Int. Ed. 2011, 50, 7312–7314. 10.1002/anie.201102543. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yin G.; Kalvet I.; Schoenebeck F. Angew. Chem., Int. Ed. 2015, 54, 6809–6813. 10.1002/anie.201501617. [DOI] [PubMed] [Google Scholar]; c Xu C.; Shen Q. Org. Lett. 2014, 16, 2046–2049. 10.1021/ol5006533. [DOI] [PubMed] [Google Scholar]
- Alternatively, for a method with stoichiometric use of Cu, see:Weng Z.; He W.; Chen C.; Lee R.; Tan D.; Lai Z.; Kong D.; Yuan Y.; Huang K.-W. Angew. Chem., Int. Ed. 2013, 52, 1548–1552. 10.1002/anie.201208432. [DOI] [PubMed] [Google Scholar]
- Zhang C.-P.; Vicic D. A. J. Am. Chem. Soc. 2012, 134, 183–185. 10.1021/ja210364r. [DOI] [PubMed] [Google Scholar]
- During the preparation of this paper a Ni-catalyzed directing-group-based approach for the coupling of aryl chlorides was reported:Nguyen T.; Chiu W.; Wang X.; Sattler M. O.; Love J. A. Org. Lett. 2016, 18, 5492–5495. 10.1021/acs.orglett.6b02689. [DOI] [PubMed] [Google Scholar]
- a Calculations performed at the CPCM(toluene)M06L/def2TZVP//ωB97XD/6-31G(d)(SDD) level of theory.; b Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian, Inc., Wallingford, CT, 2013.; For the appropriateness of the chosen computational method, see:; c Sperger T.; Sanhueza I. A.; Kalvet I.; Schoenebeck F. Chem. Rev. 2015, 115, 9532–9586. 10.1021/acs.chemrev.5b00163. [DOI] [PubMed] [Google Scholar]; For synergistic studies employing computation and experiment, see:; d Tsang A. S. K.; Sanhueza I. A.; Schoenebeck F. Chem. - Eur. J. 2014, 20, 16432–16441. 10.1002/chem.201404725. [DOI] [PubMed] [Google Scholar]; e Sperger T.; Sanhueza I. A.; Schoenebeck F. Acc. Chem. Res. 2016, 49, 1311–1319. 10.1021/acs.accounts.6b00068. [DOI] [PubMed] [Google Scholar]; f We also performed comparisons between M06L and other methods for energy evaluations, such as M06, PBE0-D3, and B3LYP-D3, and found all to give qualitatively the same trends in reactivities and preferred reaction pathways. For optimizations, we found the geometries resulting from ωB97XD to reproduce the crystal structures well.
- Dürr A. B.; Yin G.; Kalvet I.; Napoly F.; Schoenebeck F. Chem. Sci. 2016, 7, 1076–1081. 10.1039/C5SC03359D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tavener S. J.; Adams D. J.; Clark J. H. J. Fluorine Chem. 1999, 95, 171–176. 10.1016/S0022-1139(99)00063-9. [DOI] [Google Scholar]; b Liu J.-B.; Xu X.-H.; Chen Z.-H.; Qing F.-L. Angew. Chem., Int. Ed. 2015, 54, 897–900. 10.1002/anie.201409983. [DOI] [PubMed] [Google Scholar]; c Baert F.; Colomb J.; Billard T. Angew. Chem., Int. Ed. 2012, 51, 10382–10385. 10.1002/anie.201205156. [DOI] [PubMed] [Google Scholar]
- For β-fluoride elimination from NiII, see:; a Ichitsuka T.; Fujita T.; Arita T.; Ichikawa J. Angew. Chem., Int. Ed. 2014, 53, 7564–7568. 10.1002/anie.201402695. [DOI] [PubMed] [Google Scholar]; b Ichitsuka T.; Fujita T.; Ichikawa J. ACS Catal. 2015, 5, 5947–5950. 10.1021/acscatal.5b01463. [DOI] [Google Scholar]; c Fujita T.; Arita T.; Ichitsuka T.; Ichikawa J. Dalton Trans. 2015, 44, 19460–19463. 10.1039/C5DT02160J. [DOI] [PubMed] [Google Scholar]; For DFT studies on β-fluoride elimination with Pd, see:; d Zhao H.; Ariafard A.; Lin Z. Organometallics 2006, 25, 812–819. 10.1021/om050703v. [DOI] [Google Scholar]
- We also investigated the potential of alternative mechanisms being at play, such as a phosphine participation in the C–F activation, in analogy to:Nova A.; Reinhold M.; Perutz R. N.; Macgregor S. A.; McGrady J. E. Organometallics 2010, 29, 1824–1831. 10.1021/om100064z. [DOI] [Google Scholar]; During the optimization of this TS, however, it collapsed back to our reported TS geometry, where the F atom is transferred directly to the Ni atom.
- Similarly, our attempts to “transmetalate” (Cl/SCF3 exchange) [(dppf)NiII(Cl)(o-tolyl)] to [(dppf)NiII(SCF3)(o-tolyl)] also eventually formed the thiocarbonyl-bound species 1 (see Figure 3).
- The [(dppf)NiII(SCF3)(Ar)] intermediate could also undergo ligand exchange to produce [(dppf)NiII(SCF3)2] and [(dppf)NiII(Ar)2]. Upon reductive elimination of Ar-Ar, comproportionation of Ni0 and NiII could take place to form a “NiI-SCF3” species which may then undergo the chemistry described in the article to give 1. While this alternative explains the formation of 1 from ArSCF3, it does not explain the different reactivities observed, ArI < ArBr < ArCl, which is the focus of the discussion.
- The complexes were recrystallized from different solvents ([NiI-Br] from benzene and [NiI-I] from THF).
- Our calculations of the spin density in the (dppf)NiIBr complex indicate that the unpaired electron is localized predominantly on the Ni center, suggesting that it is a NiI complex. Furthermore, we also compared the C–Fe distances in the ferrocene moieties of the obtained crystal structures with the corresponding distances in FeII (CCDC 1154857) and FeIII (CCDC 194434, 170258, 138350, and 1194362) ferrocenes. The data indicate that our NiI complexes are more similar to FeII ferrocene, indicating that there is no substantial electronic exchange between the ferrocene and the Ni center. See the Supporting Information for further information.
- Morrell D. G.; Kochi J. K. J. Am. Chem. Soc. 1975, 97, 7262–7270. 10.1021/ja00858a011. [DOI] [Google Scholar]
- For feasibility of comproportionation of Ni0 and NiII, see:; a Velian A.; Lin S.; Miller A. J. M.; Day M. W.; Agapie T. J. Am. Chem. Soc. 2010, 132, 6296–6297. 10.1021/ja101699a. [DOI] [PubMed] [Google Scholar]; b Beck R.; Shoshani M.; Krasinkiewicz J.; Hatnean J. A.; Johnson S. A. Dalton Trans. 2013, 42, 1461–1475. 10.1039/C2DT32008H. [DOI] [PubMed] [Google Scholar]
- It is likely that NiII precipitated from the reaction mixture due to its poor solubility in benzene. A single crystal grown from benzene/hexane yielded the NiII species, while from THF/hexane crystals of the NiI species were obtained.
- The formation of 1 was initially accompanied by the appearance of a dark red precipitate, which ultimately (after ∼1 h) turned bright green, and free dppf could then be detected by 31P NMR spectroscopic analysis. The free dppf may originate from the initially formed (dppf)NiF2, since our attempts to independently prepare (dppf)NiF2 from dppf and NiF2 turned out to be unsuccessful, as indicated by the lack of new signals in paramagnetic 1H NMR and quantitative recovery of free ligand, on the basis of 31P qNMR analysis.
- In comparison, it takes more than 24 h at 45 °C to fully convert PhSCF3 + [Ni0] to [Ni(SCF2)].
- a Casado A. L.; Espinet P. J. Am. Chem. Soc. 1998, 120, 8978–8985. 10.1021/ja9742388. [DOI] [Google Scholar]; b Ariafard A.; Lin Z.; Fairlamb I. J. S. Organometallics 2006, 25, 5788–5794. 10.1021/om0607705. [DOI] [Google Scholar]
- Calculations were performed at the CPCM(THF)M06/def2TZVP//ωB97XD/6-31G(d)(SDD) level of theory.
- Phapale V. B.; Guisán-Ceinos M.; Buñuel E.; Cárdenas D. J. Chem. - Eur. J. 2009, 15, 12681–12688. 10.1002/chem.200901913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.