

Abstract
(−)-Lomaiviticin A (1) is a C2-symmetric cytotoxin that contains two diazofluorene functional groups and which induces double-strand breaks (DSBs) in DNA. Evidence suggests DNA cleavage is initiated by hydrogen atom abstraction from the deoxyribose backbone. Here we demonstrate the formation of the vinyl radicals 1· and 2· from 1 by 1,7-addition of thiols to the diazofluorenes. These radicals can affect hydrogen atom abstraction from methanol and acetone. The first addition of thiol to 1 proceeds at a much greater rate than the second. The diazosulfide 5 formed en route to 1· has been detected at −50 °C and undergoes decomposition to 1· with a half-life of 110 min at −20 °C under air. These data, which constitute the first direct evidence for the generation of 1· and 2· from 1, provide insights into the mechanism of DNA cleavage by 1.
The C2-symmetric bacterial metabolite (−)-lomaiviticin A (1, Scheme 1) induces double-strand breaks (DSBs) in DNA1 and is undergoing preclinical evaluation as a combination2 and monotherapy1e for the treatment of DNA DSB repair-deficient tumors.3 1 binds DNA by a mode of association involving penetration of both diazotetrahydrobenzo[b]fluorene (diazofluorene) residues into the duplex.4 The related metabolite (−)-lomaiviticin C (2)1b,c contains only one diazo substituent and does not induce DNA DSBs.1d In vitro reactivity studies of synthetic diazofluorene analogs5 led to the hypothesis that carbon-centered radical intermediates form from the diazofluorene. It was later proposed that 1 is transformed to the sp2-radicals 1· and 2· in tissue culture and that these affect strand cleavage6 by hydrogen atom abstraction from the deoxyribose backbone.1d A single hydrodediazotization of 1 generates 2; 2-fold reaction of 1 forms the double hydrodediazotization product 3.
Scheme 1.

Proposed Pathway for the Conversion of (−)-Lomaiviticin A (1) to (−)-Lomaiviticin C (2) and the Double Hydrodediazotization Product 3 via the Vinyl Radical Intermediates 1· and 2·
Here we provide the first direct experimental evidence for the formation of 1· and 2· from 1 by 1,7-addition of thiol-based nucleophiles to the diazofluorene. Our studies lead to the unexpected observation that the rate of the first thiol addition (1→2) vastly exceeds that of the second (2→3). The radicals 1· and 2· affect hydrogen atom abstraction from methanol and acetone (BDE = 95 and 93.9 kcal/mol, respectively).7
We first studied the reactivity of 1 toward N-acetyl-l-cysteine methyl ester (NACME) in methanol-d4 and acetone-d6. Each experiment was conducted under air and argon. In the absence of base, mixtures of (−)-lomaiviticin A (1) bis(trifluoroacetate) (1.22 mM) and NACME (40 equiv) remained unchanged after at least 8 h at 25 °C.8 When triethylamine (TEA, 40 equiv) was added to a solution of 1 (1.22 mM) and NACME (40 equiv) in methanol-d4 at 25 °C under air, an instantaneous color change from vivid red to dark-brown red was observed. Immediate (<5 min) analysis by 1H NMR spectroscopy revealed the formation of 2-d (94%, entry 1, Table 1). Upon aging, the solution of 2-d transformed to 3-d2, with a half-life of 49 min (83%). Under argon, the yield of 2-d was 97%, and the rate of conversion to 3-d2 was faster (t1/2 = 5 h at 5 °C, entry 2). (−)-Lomaiviticin A (1) was also instantaneously converted to 2-d in acetone-d6 (84% and 96% yield under air and argon, entries 3, and 4, respectively) but 3-d2 was not observed. Instead, 2-d slowly transformed to unidentified decomposition products. This sequence was readily followed by 1H NMR spectroscopy as transformation of the C2-symmetric structure 1 to the C1-symmetric structure 2-d results in doubling of most signals (Figure 1). Loss of the remaining diazo substituent restores C2-symmetry (as 3-d2), leading to simplification of the spectra. These experiments reveal that the rate of 1,7-addition to the first diazofluorene of 1 is faster than the remaining diazofluorene in 2.
Table 1.
Hydrodediazotization Studies of 1
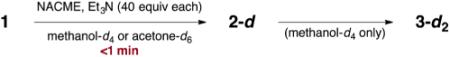
| ||||
|---|---|---|---|---|
|
| ||||
| entry | atmos. | solvent | yield 2-d2a | yield 3-d2a (t1/2, T) |
| 1 | air | CD3OD | 94% | 83% (49 min, 25 °C) |
| 2 | argon | CD3OD | 97% | 87% (5 h, 5 °C) |
| 3 | air | acetone-d6 | 84% | n/db |
| 4 | argon | acetone-d6 | 96% | n/db |
Yields were determined by 1H NMR spectroscopy using 1,4-dicyanobenzene as an internal standard and are based on 1.
n/d = not detected.
Figure 1.
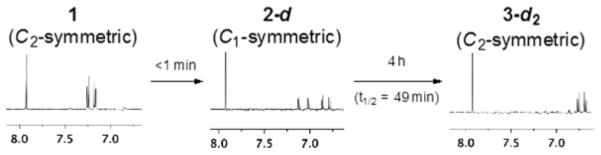
Aryl region of the 1H NMR spectra of 1, 2-d, and 3-d2. Conditions: 1 (1.22 mM), TEA (40 equiv), NACME (40 equiv), methanol-d4, air, 25 °C.
The source of deuterium at the vinylic position of 2-d was elucidated by repeating the experiments separately in methanol and methanol-d4, and analyzing the products by LC/HRMS. (−)-Lomaiviticin C (2) ionizes by ejection of the aminosugar residue proximal to the hydroxyfulvene, leading to a prominent daughter ion corresponding to 4 upon MS analysis (Figure 2A).1b,9 LC/HRMS analysis of the first hydrodediazotization of 1 in methanol indicated formation of 4 ([M]+ = C60H66N3O21+: calculated, =1164.4183; observed = 1164.4169; error = 1.20 ppm), whereas the same experiment conducted in methanol-d4 provided 4-d ([M]+ = C60DH65N3O21+: calculated = 1165.4246; observed = 1165.4220; error = 2.23 ppm, Figure 2B). To identify the site of bond cleavage in methanol (e.g., C–H/D or O–H/D), and to remove any potential complications arising from O–H/D exchange, we conducted additional experiments in CH3OD and CD3OH. Mass spectral analysis of the hydrodediazotization of 1 in CH3OD revealed generation of 4 (observed = 1164.4162; error = 1.80 ppm), and analysis of the same experiment in CD3OH indicated generation of 4-d (observed = 1165.4222; error = 2.06 ppm). Strictly analogous results were obtained when the 2-fold hydrodediazotization of 1 was conducted in CH3OD or CD3OH. Reaction in the former solvent formed the protiated product 3 whereas reaction in the latter solvent generated 3-d2 (Figure S1). These results indicate that the newly formed C–H/D bonds in 2 and 3 derive from C–H/D bond cleavage in methanol and provide compelling evidence for the intermediacy of the sp2 radicals 1· and 2·.
Figure 2.

(A) Ionization of 2 or 2-d leads to ejection of the aminosugar residue and observation of the elimination products 4 or 4-d by HRMS analysis.1b,9 (B) 1, 2: Selected region of the HRMS spectrum of 4, generated by hydrodediazotization of 1 in CH3OH or CH3OD, respectively. 3, 4: Selected region of the HRMS spectrum of 4-d, generated by hydrodediazotization of 1 in CD3OH or CD3OD, respectively. 5, 6: Predicted isotope distribution of 4 and 4-d, respectively.
The requirement for base in the conversion of 1 to 2 and 3 is consistent with 1,7-addition of thiolate to generate a diazosulfide intermediate, followed by loss of dinitrogen and thiyl radical (Scheme 2). To probe this, we monitored the reactivity of 1 toward benzylthiol in the presence of triethylamine at low temperature.10 Addition of benzylthiol (10 equiv) and triethylamine (10 equiv) to a solution of 1 (1.22 mM) in acetone-d6 at −50 °C under air instantaneously formed the diazosulfide 5 (81%, Scheme 3). The diazosulfide 5 was generated as a ~1:1 mixture of E:Z isomers that converted to a 3:1 mixture (presumably, E:Z) after standing for 1 h at −50 °C. The 3:1 mixture of diazosulfides 5 was stable for at least 12 h at −50 °C and was characterized by 1H, HSQC, and HMBC NMR analysis. The protons α to sulfur in the major isomer of 5 appeared as two distinct doublets (J = 14.0 Hz) centered at 4.94 and 4.75 ppm. These were correlated to the same carbon atom (36.5 ppm; 28.4 ppm in free benzylthiol) in the HSQC spectrum and to the quaternary carbon of the phenyl ring in 5 (130.5 ppm; 128.8 in free benzylthiol) in the HMBC spectrum. Warming to −20 °C induced transformation of 5 to 2-d, with a half-life of 110 min (79% yield from 1 at 98% conversion of 5). Under argon, the diazosulfide 5 was formed in quantitative yield and transformed to 2-d with a half-life of 49 min (>99%). No intermediates were detected when the conversion of 2-d to 3-d2 was monitored carefully by NMR spectroscopy, suggesting decomposition of the putative diazosulfide derived from 2-d is faster than its formation.
Scheme 2.

Postulated Pathway for the Formation of 1· and 2· via Nucleophilic Addition of Thiol
Scheme 3.

Generation and Decomposition of the Diazosulfide 5.a
aSpectroscopic data shown corresponds to the major diazosulfide isomer. Reaction was run under air. 1H NMR data were acquired at −20 °C.
DFT calculations were employed to gain insight into the relative rates of addition to 1 and 2.1d The optimized structure of 1 using the B3LYP 6-31G(d) level of theory and an aqueous solvent model is shown in Figure 3 and indicates that the distance from the diazo carbon to the opposing diazofluorene is 3.8 Å. We propose that the developing anionic charge in the transition state for addition to 1 is stabilized by a through-space interaction with the adjacent electron-deficient diazofluorene. The transformation of 1 to 2 converts a diazofluorene to an electron-rich hydroxyfulvene, and the transition state for the second addition may not benefit from the same stabilization. It is also possible that the hydroxyfulvene in 2 is strongly hydrogen-bound (or deprotonated) under these conditions, which would further decrease electrophilicity. The structure shown in Figure 3 parallels of 1 bound to DNA,4 suggesting these transannular interactions are relevant in tissue culture.
Figure 3.
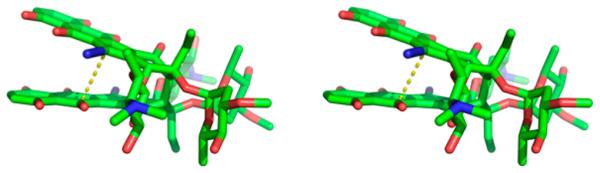
Stereoview of DFT-minimized structure of 1 in water [B3LYP 6-31G(d)]. Hydrogen atoms are omitted for clarity.
These data provide several insights into the mechanism of DNA cleavage by 1. First, these experiments show that the radicals 1· and 2· can be formed from 1 by nucleophilic addition, and that these are competent to cleave relatively strong C–H bonds, providing support for DSB induction by a hydrogen atom abstraction mechanism. Second, the faster rate of nucleophilic addition to 1 than 2 may provide an explanation for the greater proportion of single-strand breaks than DSBs produced by 1 (in an in vitro plasmid cleavage assay, this ratio was ~5:11d): dissociation of 2 from the duplex may be competitive with the formation of 2•. Finally, the facile conversion of 1 to 2 suggests that natural 2 derives from hydrodediazotization of 1 during bacterial growth. As 2 is several orders of magnitude less potent,1b this reactivity may constitute a fortuitous detoxification pathway for the producing strain. A question currently unresolved is the nature of the nucleophile in the presence of DNA. Given the short lifetimes of sp2 radicals, we hypothesize that 1· and 2· are generated after binding, potentially by direct addition of a nucleotide11 to the coordinated metabolite.
Supplementary Material
ACKNOWLEDGMENTS
Financial support from the National Institutes of Health (NIGMS R01-GM090000) and Yale University is gratefully acknowledged. We thank Dr. Eric Paulson for NMR assistance, Dr. Wei Shen for assistance with bacterial cultures, and Dr. Seewon Joung, Dr. Alan Healy, and Mr. Herman Nikolayevskiy for helpful comments on the paper.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b09657.
Detailed experimental procedures and complete spectroscopic data for all new compounds (PDF)
Notes
The authors declare no competing financial interest.
ORCID
Seth B. Herzon: 0000-0001-5940-9853
REFERENCES
- (1).(a) He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J. Am. Chem. Soc. 2001;123:5362. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]; (b) Woo CM, Beizer NE, Janso JE, Herzon SB. J. Am. Chem. Soc. 2012;134:15285. doi: 10.1021/ja3074984. [DOI] [PubMed] [Google Scholar]; (c) Kersten RD, Lane AL, Nett M, Richter TKS, Duggan BM, Dorrestein PC, Moore BS. ChemBioChem. 2013;14:955. doi: 10.1002/cbic.201300147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Colis LC, Woo CM, Hegan DC, Li Z, Glazer PM, Herzon SB. Nat. Chem. 2014;6:504. doi: 10.1038/nchem.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Colis LC, Hegan DC, Kaneko M, Glazer PM, Herzon SB. J. Am. Chem. Soc. 2015;137:5741. doi: 10.1021/ja513117p. For a review, see. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Herzon SB, Woo CM. Nat. Prod. Rep. 2012;29:87. doi: 10.1039/c1np00052g. [DOI] [PubMed] [Google Scholar]
- (2).Colis LC, Herzon SB. Bioorg. Med. Chem. Lett. 2016;26:3122. doi: 10.1016/j.bmcl.2016.04.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Rajski SR, Williams RM. Chem. Rev. 1998;98:2723. doi: 10.1021/cr9800199. For reviews of DNA-damaging natural products, see: [DOI] [PubMed] [Google Scholar]; (b) Wolkenberg SE, Boger DL. Chem. Rev. 2002;102:2477. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]; (c) Tse WC, Boger DL. Chem. Biol. 2004;11:1607. doi: 10.1016/j.chembiol.2003.08.012. [DOI] [PubMed] [Google Scholar]
- (4).Woo CM, Li Z, Paulson EK, Herzon SB. Proc. Natl. Acad. Sci. U. S. A. 2016;113:2851. doi: 10.1073/pnas.1519846113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Laufer RS, Dmitrienko GI. J. Am. Chem. Soc. 2002;124:1854. doi: 10.1021/ja0167809. For the first proposals for the generation of sp2 radicals from the diazofluorene, see: [DOI] [PubMed] [Google Scholar]; (b) Feldman KS, Eastman KJ. J. Am. Chem. Soc. 2006;128:12562. doi: 10.1021/ja0642616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Burrows CJ, Muller JG. Chem. Rev. 1998;98:1109. doi: 10.1021/cr960421s. For a review of DNA cleavage by hydrogen atom abstraction, see: For a review of natural products that affect hydrogen atom abstraction from DNA, see: [DOI] [PubMed] [Google Scholar]; (b) Murphy JA, Griffiths J. Nat. Prod. Rep. 1993;10:551. doi: 10.1039/np9931000551. [DOI] [PubMed] [Google Scholar]
- (7).(a) Bordwell FG, Cheng JP, Harrelson JA. J. Am. Chem. Soc. 1988;110:1229. [Google Scholar]; (b) Comprehensive Handbook of Chemical Bond Energies. CRC Press; Boca Raton: 2007. BDEs of C–H bonds; pp. 19–145. [Google Scholar]
- (8). The addition of TEA alone to solutions of 1 in methanol-d4 or acetone-d6 led to rapid precipitation of the metabolite, see Supporting Information.
- (9).Mulcahy SP, Woo CM, Ding WD, Ellestad GA, Herzon SB. Chem. Sci. 2012;3:1070. [Google Scholar]
- (10). Attempts to characterize a diazosulfide formed by the addition of NACME to 1 were complicated by line broadening.
- (11).(a) Chin A, Hung M-H, Stock LM. J. Org. Chem. 1981;46:2203. For the addition of nucleotides to arenediazonium salts, see: [Google Scholar]; (b) Hung MH, Stock LM. J. Org. Chem. 1982;47:448. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.