

Abstract
RNA editing in mammals is a form of post-transcriptional modification in which adenosine is converted to inosine by the adenosine deaminases acting on RNA (ADAR) family of enzymes. Based on evidence of altered ADAR expression in epithelial ovarian cancers (EOC), we hypothesized that single nucleotide polymorphisms (SNPs) in ADAR genes modify EOC susceptibility, potentially by altering ovarian tissue gene expression. Using directly genotyped and imputed data from 10,891 invasive EOC cases and 21,693 controls, we evaluated the associations of 5,303 SNPs in ADAD1, ADAR, ADAR2, ADAR3, and SND1. Unconditional logistic regression was used to estimate odds ratios (OR) and 95% confidence intervals (CI), with adjustment for European ancestry. We conducted gene-level analyses using the Admixture Maximum Likelihood (AML) test and the Sequence-Kernel Association test for common and rare variants (SKAT-CR). Association analysis revealed top risk-associated SNP rs77027562 (OR (95% CI)= 1.39 (1.17-1.64), P=1.0×10−4) in ADAR3 and rs185455523 in SND1 (OR (95% CI)= 0.68 (0.56-0.83), P=2.0×10−4). When restricting to serous histology (n=6,500), the magnitude of association strengthened for rs185455523 (OR=0.60, P=1.0×10−4). Gene-level analyses revealed that variation in ADAR was associated (P<0.05) with EOC susceptibility, with PAML=0.022 and PSKAT-CR=0.020. Expression quantitative trait locus analysis in EOC tissue revealed significant associations (P<0.05) with ADAR expression for several SNPs in ADAR, including rs1127313 (G/A), a SNP in the 3′ untranslated region. In summary, germline variation involving RNA editing genes may influence EOC susceptibility, warranting further investigation of inherited and acquired alterations affecting RNA editing.
Keywords: polymorphisms, RNA editing, ovarian cancer risk
INTRODUCTION
Over the past decade it has been recognized that the complexity of higher organisms is related to the information stored in non-protein-coding regions of the genome. Such complexity may be attributed to a range of processing events and post-transcriptional modifications that affect the fate of RNA, including alternative splicing, 5′ capping, 3′ polyadenylation, and RNA editing [1-3]. The most common type of RNA editing in eukaryotes is site-selective hydrolytic deamination of adenosine into inosine (A-to-I) within double-stranded RNAs, and recent bioinformatic analyses and high-throughput sequencing efforts have revealed that A-to-I editing is widespread and alters non-coding and protein-coding sequences throughout the genome [4].
A-to-I editing is mediated by a family of adenosine deaminases acting on RNA (ADARs), and this process modulates expression of genes and biological pathways via several mechanisms [4]. Indeed, altered expression and/or activity of ADAR enzymes has been linked to a variety of conditions, including cardiovascular and neurological diseases and cancers [4]. Epithelial ovarian cancer (EOC) is the fifth leading cause of cancer death among women in the United States [5], and ADAR expression levels have been reported to be significantly higher in serum and peritoneal fluid from patients with EOCs compared with benign ovarian tumors [6, 7], suggesting ADARs may be useful biomarkers for the diagnosis and management of EOC.
We hypothesized that germline single nucleotide polymorphisms (SNPs) involving ADAR-related/RNA editing genes may contribute to EOC risk. The main purpose of this investigation was to determine whether SNPs in five ADAR genes (ADAD1, ADAR, ADAR2, ADAR3, and SND1) were associated with EOC susceptibility. We used data available from a large-scale genotyping collaboration involving 10,891 EOC cases and 21,693 controls from the international Ovarian Cancer Association Consortium (OCAC) [8]. We also sought to evaluate the overall contribution of each gene on EOC susceptibility and to determine whether candidate SNPs associated with altered expression of corresponding genes in EOC tumor tissue.
RESULTS
Study population
The study sample included 10,891 invasive EOC patients and 21,693 controls of European ancestry (Supplementary Table 1). Selected subject characteristics are shown in Table 1. The mean age at diagnosis for cases was 58.1 years, the mean age at interview for controls was 56.1 years. Cases were more likely than controls to be nulliparous and to have never used oral contraceptives. Most cases had serous histology (59.7%), distant stage (63.0%), and high-grade disease (58.9%).
Table 1. Characteristics of study participants (N = 32,584).
| Variable | Cases (n= 10,891) | Controls (n= 21,693) |
|---|---|---|
| Age at diagnosis/interview(y), mean (SD) | 58.1 (11.4) | 56.1 (24.9) |
| History of pregnancy | ||
| Yes | 6021 (80.4) | 15190 (87.9) |
| No | 1318 (17.6) | 1868 (10.8) |
| Unknown | 149 (2.0) | 217 (1.3) |
| Oral contraceptive use | ||
| Ever | 4017 (57.4) | 10572 (63.3) |
| Never | 2864 (41.0) | 5900 (35.3) |
| Unknown | 112 (1.6) | 243 (1.5) |
| Histology | ||
| Serous | 6500 (59.7) | NA |
| Mucinous | 696 (6.4) | |
| Endometrioid | 1439 (13.2) | |
| Clear Cell | 660 (6.1) | |
| Mixed Cell | 369 (3.4) | |
| Other or unknown epithelial type | 1227(11.3) | |
| Stage | ||
| Localized | 1425 (15.7) | NA |
| Regional | 1838 (20.2) | |
| Distant | 5721 (63.0) | |
| Unknown | 103 (1.1) | |
| Grade | ||
| I/II | 2882 (32.8) | NA |
| III/IV | 5174 (58.9) | |
| Other/Unknown | 729 (8.3) |
Variant-level association analysis and overlap with regulatory domains
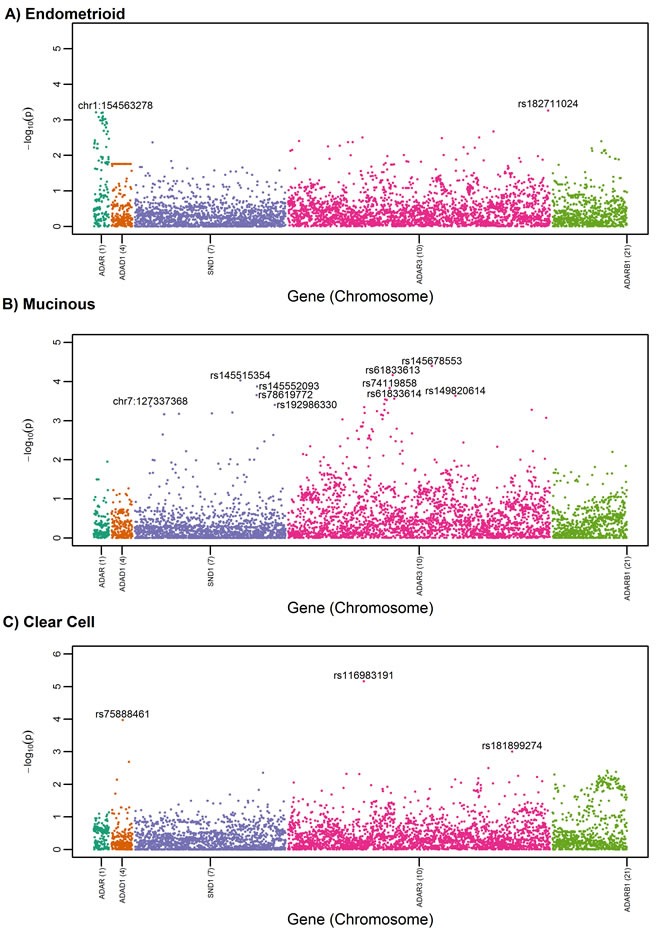
SNP-level association analysis revealed top-ranked SNPs (defined as the top 5% of SNPs having the most statistically significant P values) in ADAR, ADAR3, and SND1 in the all-histologies and serous-only analyses (Figure 1A and 1B). Table 2 summarizes association results for the most statistically significant SNPs overall or by serous histology (P < 4.0×10−3); associations were not significant after correction for multiple testing (FDR > 0.15). Most of the top-ranked variants were imputed, rare or low frequency (MAF < 0.05), and not part of a shared haplotype. rs77027562 (A>G; MAF = 0.009), the top risk-associated variant among all histologies (OR (95% CI) = 1.39 (1.17-1.64, P = 1.0 x10−4)), resides in an intron of ADAR3. ADAR3 SNP rs77027562 and its proxies (r2>0.80) reside in genomic regions that overlap with regulatory domains, particularly enhancers in blood and brain (Table 3). The next top-ranked variant, SND1 rs185455523 (T>A), was associated with a decreased EOC risk (OR (95% CI) = 0.68 (0.56-0.83), P = 1.5 x10−4), but this SNP and its proxies do not appear to overlap with regulatory domains. When analysis was restricted to the 6,500 patients with invasive serous adenocarcinomas, the magnitude of association was slightly attenuated for ADAR3 rs77027562 (OR = 1.33, P = 6.1×10−3) and slightly stronger for SND1 rs185455523 (OR = 0.60, P = 1×10−4). Exploratory analysis for the less common histologic subtypes (endometrioid (n = 1,439), mucinous (n = 696), and clear cell (n = 660)) revealed several SNP-level associations unique to each sub-type (Figure 2A-2C). For example, rs145678553-C in ADAR3 is a rare variant (MAF = 0.0047) associated with an increased risk for mucinous EOC (OR (95%CI) = 3.46 (1.91-6.26), P = 3.99x 10−5), and rs116983191-A in ADAR3 is a low-frequency variant (MAF = 0.044) associated with clear cell carcinoma (OR (95%CI) = 1.86 (1.42-2.43, P = 6.91 × 10−6). rs145678553-C was not represented in Haploreg. rs116983191-A is located in promoter and enhancer regions, but not in tissues relevant to ovarian cancer.
Figure 1. Manhattan plot for candidate RNA editing SNPs among a) all invasive cases (n = 10,891) versus controls (n = 21,693) and b) serous cases (n = 6,500) versus controls.
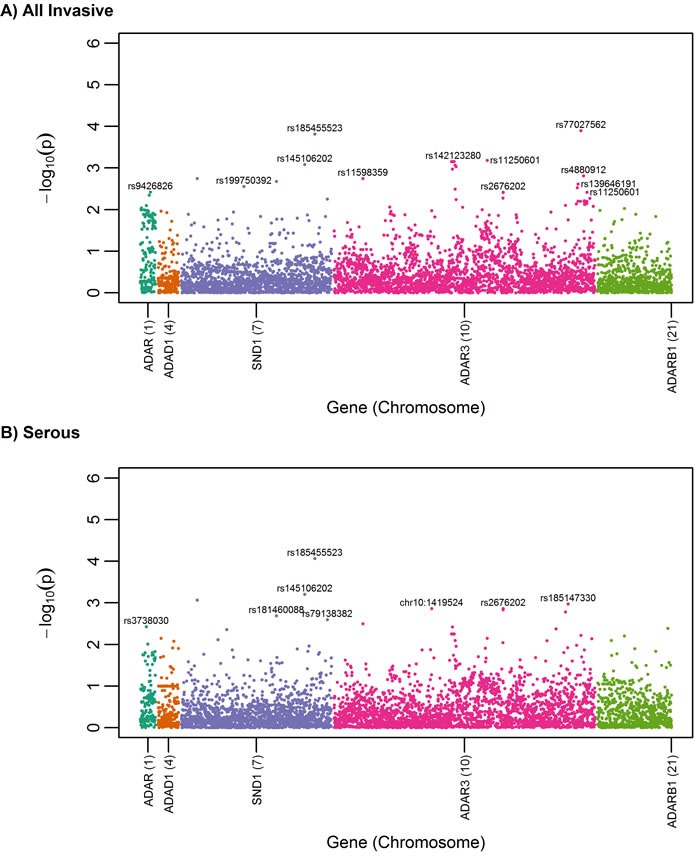
Table 2. Top-ranked RNA editing SNP-EOC risk associations among all histologies (N = 10,891) or serous histology (N = 6,500) versus controls (N = 21,693), sorted by gene and p-value.
| Gene | SNP | Alleles | MAF | Imputation accuracy R2 | All histologies OR (95% CI) | P | FDR | Serous OR (95% CI) | P | FDR |
|---|---|---|---|---|---|---|---|---|---|---|
| ADAR | rs9426826 | C>G | 0.481 | 0.86 | 1.05 (1.02-1.09) | 0.0038 | 0.76 | 1.04 (1-1.08) | 0.0759 | 0.9996 |
| rs3738030 | A>C | 0.116 | 0.79 | 0.93 (0.89-0.98) | 0.0080 | 0.83 | 0.91 (0.86-0.97) | 0.0038 | 0.9996 | |
| ADAR3 | rs77027562a | A>G | 0.009 | 0.41 | 1.39 (1.17-1.64) | 0.0001 | 0.34 | 1.33 (1.08-1.62) | 0.0061 | 0.9996 |
| rs11250601 | C>T | 0.070 | 0.62 | 0.89 (0.83-0.95) | 0.0007 | 0.34 | 0.9 (0.83-0.97) | 0.0071 | 0.9996 | |
| rs142123280b | A>G | 0.001 | 0.48 | 2.08 (1.36-3.17) | 0.0007 | 0.34 | 2.01 (1.23-3.29) | 0.0056 | 0.9996 | |
| rs4880912 | T>C | 0.200 | 0.82 | 1.07 (1.03-1.11) | 0.0015 | 0.51 | 1.06 (1.01-1.11) | 0.0267 | 0.9996 | |
| rs11598359 | C>T | 0.005 | 0.45 | 0.68 (0.54-0.87) | 0.0018 | 0.53 | 0.63 (0.47-0.86) | 0.0032 | 0.9996 | |
| rs6560760 | C>T | 0.025 | 0.59 | 1.17 (1.06-1.3) | 0.0024 | 0.65 | 1.07 (0.95-1.22) | 0.2688 | 0.9996 | |
| rs2676202c | C>T | 0.122 | 0.66 | 0.93 (0.88-0.98) | 0.0038 | 0.76 | 0.9 (0.85-0.96) | 0.0014 | 0.9996 | |
| rs139646191 | TAGAA>T | 0.062 | 0.66 | 1.11 (1.03-1.18) | 0.0038 | 0.76 | 1.07 (0.98-1.16) | 0.1242 | 0.9996 | |
| rs139812582 | G>A | 0.002 | 0.47 | 1.71 (1.15-2.54) | 0.0078 | 0.83 | 2.03 (1.31-3.14) | 0.0017 | 0.9996 | |
| chr10:1419524 | T>TGG | 0.009 | 0.60 | 0.79 (0.65-0.95) | 0.0106 | 0.83 | 0.68 (0.54-0.86) | 0.0014 | 0.9996 | |
| rs185147330 | C>T | 0.005 | 0.45 | 1.32 (1.05-1.67) | 0.0176 | 0.86 | 1.54 (1.19-2) | 0.0011 | 0.9996 | |
| SND1 | rs185455523 | T>A | 0.008 | 0.56 | 0.68 (0.56-0.83) | 0.0002 | 0.34 | 0.6 (0.46-0.77) | 0.0001 | 0.45 |
| rs145106202d | G>C | 0.009 | 0.87 | 0.73 (0.61-0.88) | 0.0008 | 0.35 | 0.67 (0.53-0.84) | 0.0006 | 0.9996 | |
| rs181460088 | C>T | 0.008 | 0.80 | 0.73 (0.6-0.89) | 0.0021 | 0.58 | 0.67 (0.52-0.86) | 0.0020 | 0.9996 | |
| rs199750392 | G>GT | 0.036 | 0.61 | 1.14 (1.05-1.25) | 0.0028 | 0.71 | 1.11 (1-1.23) | 0.0550 | 0.9996 | |
| rs79138382 | C>T | 0.007 | 0.72 | 1.32 (1.09-1.61) | 0.0056 | 0.83 | 1.42 (1.13-1.77) | 0.0025 | 0.9996 |
Significant SNPs (P < 4.0×10−3) are listed and SNPs in LD (r2>0.60) with more significant SNP are not reported:
7 SNPs in LD not reported
12 SNPs in LD not reported
1 SNP in LD not reported
1 SNP in LD not reported
Table 3. HaploReg results for top-ranked ADAR3 SNP rs77027562 and its proxies from univariate analyses.
| Position (hg38) | LD (r2) | SNP (Ref>Alt) | MAF in EUR | Functional Annotation | CR | Promoter histone marks | Enhancer histone marks | DNAse site | Proteins bound | eQTL | Motifs Changed |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr10:1688744 | -- | rs77027562 (A>G) | 0.02 | Intronic | No | BRN | BLD | ERalpha-a, RXRA, Zfp281 | |||
| Chr10:1675149 | 0.94 | rs12258319 (G>T) | 0.98 | Intronic | No | BLD | Pax-5 | ||||
| Chr10:1675875 | 0.94 | rs7077743 (C>T) | 0.98 | Intronic | No | BLD | BDP1, CAC-binding-protein, HNF4, p300 | ||||
| Chr10:1676470 | 0.94 | rs6560758 (T>C) | 0.98 | Intronic | No | ||||||
| Chr10:1678882 | 0.94 | rs10751814 (A>G) | 0.98 | Intronic | No | GR, Rad21 | |||||
| Chr10:1680294 | 0.94 | rs7089727 (A>G) | 0.98 | Intronic | No | ESDR, IPSC, BLD, LNG | ESC, BLD | CTCF | |||
| Chr10:1681695 | 0.94 | rs6560759 (T>C) | 0.98 | Intronic | No | ESDR, BLD | Myc | ||||
| Chr10:1687566 | 0.94 | rs79784382 (T>A) | 0.02 | Intronic | No | BRN | Cphx, Duxl, HNF6, Hmx, Hoxa13, Pbx-1, Pbx3 |
Abbreviations: LD, linkage disequilibrium; MAF, minor allele frequency; EUR, European; CR, conserved region; eQTL, expression quantitative trait loci. Tissue groups: BRN, brain cells; BLD, blood and T-cells; ESDR, embryonic stem cell derived cells; IPSC, induced pluripotent stem cells; LNG, lung cell; ESC, embryonic stem cells. Proxies were defined as variants in LD (r2>0.8) with the index SNP rs77027562 (bolded) in 1000 genomes project Phase 1 data for Europeans. All data was accessed using HaploReg v4.1 available at: http://www.broadinstitute.org/mammals/haploreg/documentation_v4.1.html. Both conservation prediction algorithms, GERP and SiPhy-omega, were used. Only eQTLs for ADAR genes (5 genes) are given.
Figure 2. Manhattan plot for candidate RNA editing SNPs among a) endometrioid cases (n = 1,439) versus controls (n = 21,693), b) mucinous cases (n = 696) versus controls, and c) clear cell cases (n = 660) versus controls.
Gene-level analyses
Gene-level analyses based on AML and SKAT-CR revealed that variation in ADAR was nominally associated (P < 0.05) with susceptibility to all invasive EOC, with P = 0.02 using both methods (Table 4). Histology-specific analyses revealed that ADAR variation was associated with endometrioid EOC susceptibility (PSKAT-CR = 0.005/PAML = 0.008). When using a Bonferroni threshold of 0.0025, only ADAR3 variation was significantly associated with mucinous histology (PSKAT-CR = 0.0016/PAML = 0.031).
Table 4. Association between RNA editing genes and EOC susceptibility.
| Gene | Total N Markers (N Tested) | N Rare Markers (MAF<0.01) | N Common Markers (MAF≥0.01) | All Invasive | Serous | Endometrioid | Mucinous | Clear cell | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P. SKAT-CR | P.AML Trend | P. SKAT-CR | P.AML Trend | P. SKAT-CR | P.AML Trend | P. SKAT-CR | P.AML Trend | P. SKAT-CR | P.AML Trend | ||||
| ADAD1 | 210 (210) | 98 | 112 | 0.698 | 0.749 | 0.300 | 0.433 | 0.054 | 0.179 | 0.857 | 0.804 | 0.696 | 0.635 |
| ADAR1 | 155 (155) | 50 | 105 | 0.020 | 0.022 | 0.110 | 0.101 | 0.005 | 0.008 | 0.943 | 0.841 | 0.780 | 0.411 |
| ADAR2 | 754 (754) | 301 | 4563 | 0.861 | 0.894 | 0.623 | 0.720 | 0.134 | 0.496 | 0.338 | 0.208 | 0.105 | 0.041 |
| ADAR3 | 2656 (2654) | 787 | 1867 | 0.216 | 0.266 | 0.334 | 0.541 | 0.587 | 0.470 | 0.002 | 0.031 | 0.234 | 0.502 |
| SND1 | 1528 (1527) | 764 | 763 | 0.630 | 0.809 | 0.703 | 0.376 | 0.919 | 0.895 | 0.632 | 0.204 | 0.773 | 0.535 |
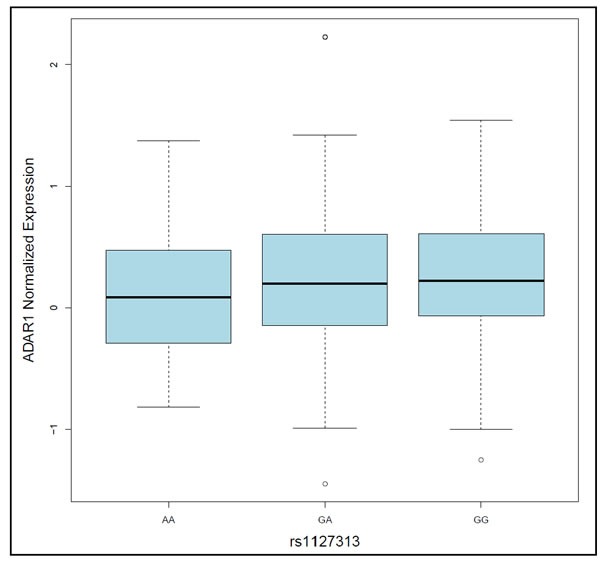
To examine associations between genotype and gene expression for the 5 candidate RNA editing genes, expression quantitative trait locus (eQTL) analysis was performed using matched genotype and tissue expression data from The Cancer Genome Atlas (TCGA) high-grade serous adenocarcinoma tumors (https://tcga-data.nci.nih.gov/tcga/). eQTL analysis revealed statistically significant associations (P < 0.05) with ADAR expression for several SNPs in ADAR, including rs1127313 (G/A), a SNP in the 3′UTR within a putative miRNA binding site that was associated with susceptibility in all histologies (OR = 1.05, P = 0.009). rs1127313 is also in high LD (r2 = 0.86) with top ADAR risk SNP rs9426826 (see Table 2). ADAR tumor tissue expression was slightly higher among G allele carriers of rs1127313 compared to A allele carriers (P = 0.027; Figure 3). rs1127313 is also an eQTL for ADAR in whole blood (Supplementary Table 2), and lies in a genomic region with enhancer features and DNase I hypersensitivity site in several tissues, including ovary. Statistically-significant cis-eQTLs were not detected for SNPs in other candidate RNA editing genes.
Figure 3. Box-plot showing that ADAR1 tumor tissue expression differed, albeit only slightly, by rs1127313 genotype (p = 0.027).
DISCUSSION
An emerging body of data suggest that defects in RNA editing may contribute to a range of human diseases, including cancer [2-4, 9-11]. The current large-scale collaboration represents the first comprehensive association study of germline variants involving RNA editing genes and susceptibility to epithelial ovarian cancer. At the SNP- level, the strongest associations were observed for SNPs in RNA editing genes ADAR3 and SND1, but no associations reached genome-wide levels of statistical significance. Gene-level analyses highlighted ADAR and ADAR3 as potential contributors to EOC susceptibility within the set of ADAR-related genes. Finally, positive eQTLs were also observed between ADAR genotype and ADAR expression in EOC tumor tissue.
Focused evaluations of RNA editing SNP-disease associations are limited [12], especially with cancer as an outcome, so it is not possible to compare our SNP findings to those of other studies of cancer risk. We are, however, unaware of GWAS hits in or near these genes. Several recent studies [2, 3] have evaluated the genomic landscape and clinical relevance of RNA editing in numerous human tissue types. These analyses used RNA-sequencing data from both tumor and normal samples profiled as part of TCGA Project. Striking differences in RNA-editing patterns were observed in tumors relative to matched normal tissues for 12 cancer types [2]. Further analyses revealed that altered RNA editing patterns in tumors correlated with ADAR expression, and that non-random, clinically-relevant RNA editing events (frequently located in noncoding RNAs, nonsynonymous sites, intronic regions, and non-Alu elements) correlated with tumor classification and patient survival and with increased cell survival and altered drug sensitivity [2, 3]. Interestingly, gene amplification-associated overexpression of ADAR was recently shown to enhance lung tumorigenesis and contribute to poor outcomes by affecting downstream RNA editing patterns [10]. As mentioned previously, ADAR expression levels have been reported to be significantly higher in serum and/or peritoneal fluid from patients with EOCs compared with benign ovarian tumors [6, 7]. Although high-grade serous EOCs from TCGA were not profiled as part of the aforementioned genomic investigations [18, 19], Haploreg 4.1 effectively integrates GTEX eQTL results for normal ovary.
Taken together with several lines of investigation from ovarian [6, 7] and other cancers [2, 3, 10] the current study suggests that ADARs (and ADAR in particular) may be useful biomarkers for the diagnosis and management of EOC. Thus, with replication, ADAR genotype status and/or expression level may serve as a risk factor for EOC. Indeed, we find that our top risk SNP in ADAR, rs9426826, has several proxy variants (r2>0.8, Supplementary Table 2) that are strongly associated with expression of this gene in blood (rs1127313: 7.23×10−14) and to a lesser extent, expression in high-grade serous EOC tumors (rs1127313: P = 0.027). Based on growing data which demonstrate the inhibition of tumor growth in the presence of ADAR inhibitors [13] and other therapeutic agents such as the IGFR-1R inhibitor BMS536924 and the MEK inhibitors CI1040 and trametinib [2], ADAR genotype and/or expression may help identify women whose tumors may respond to new combinations of therapies.
Strengths of the current study include the large sample size that primarily enabled detection of small effects for common variants, the relatively homogeneous population of EOC cases, and the multi-tiered genomic evaluation. However, this study was underpowered to detect the rare variants that were identified and is burdened by the low imputation quality. Additionally, the study is limited in that eQTL analysis did not permit adjustment for somatic copy number changes and DNA methylation status, factors that can influence transcript abundance and confound associations between germline polymorphisms and gene expression [14-16]. Moreover, it is possible that the top-ranked SNPs could potentially affect genes other than the RNA editing genes that drive candidate selection. Efforts to replicate these findings are needed; data will be available soon from a large, independent cohort of EOC cases genotyped by OCAC for this purpose (Amos et al, The OncoArray Consortium: a Network for Understanding the Genetic Architecture of Common Cancers (provisionally accepted, CEBP). Mechanistic studies to reveal how ADAR polymorphisms may affect oncogenic phenotypes will also be required, as will systematic investigations of the genomic landscape and clinical relevance of RNA editing in EOC using data from TCGA or other sources.
In summary, this study provides data to support the hypothesis that germline polymorphisms in ADAR related genes may influence gene expression and susceptibility to EOC. Further investigations are needed to determine whether inherited and acquired alterations affecting RNA editing serve as biological mechanisms to promote the development of EOC.
MATERIALS AND METHODS
Study population
A total of 41 studies (32 case-control and 9 case-only) from OCAC contributed to this investigation (Supplementary Table 1). Briefly, cases were women diagnosed with histologically confirmed primary invasive EOC (95%), fallopian tube cancer (1%), or primary peritoneal cancer (4%). Controls were women without cancer and with at least one intact ovary on the reference date. Individual studies were grouped into 26 case-control strata. All studies provided data on disease status, age at diagnosis/interview, self-reported racial group, and histologic subtype.
Genotyping, quality control (QC), and imputation
Peripheral blood was the primary source of germline DNA and was collected in the course of clinical care or research at each of the participating sites. The candidate SNPs selected for the current investigation were genotyped using a custom Illumina Infinium iSelect Array as part of the international Collaborative Oncological Gene-environment Study (iCOGS), an effort to evaluate 211,155 genetic variants for association with cancer risk [17].
Briefly, OCAC genotyping was conducted at McGill University and Génome Québec Innovation Centre (Montréal, Canada) and Mayo Clinic Medical Genomics Facility. Each 96-well plate well contained 250ng genomic DNA (or 500 ng whole genome-amplified DNA). Raw intensity data files were sent to the COGS data coordination center at the University of Cambridge for genotype calling and QC using the GenCall algorithm. Sample and SNP quality control procedures have been described previously; in brief, samples were excluded with call rates < 95%, >1% discordance, < 80% European ancestry, or ambiguous gender, and SNPs were excluded with call rates < 95% or monomorphism [18, 19].
To improve genomic coverage and power [14], we imputed genotypes based on data from the 1000 Genomes Project (1KGP); we used IMPUTE2 version 2 after pre-phasing with SHAPEIT [20]. All 14 populations in the 1KGP were used as the reference. Before imputation, we excluded poorly performing SNPs according to the genotyping success rates, deviation from Hardy-Weinberg equilibrium (HWE) (P < 1×10−7), and replicate errors. To ensure the quality of the imputed genotypes, maximum likelihood genotype imputation was carried out and an estimate of the squared correlation between the imputed and true genotypes was calculated. Imputation quality is significantly decreased for low and rare frequency variants [21]. To be more inclusive of rare variants, we considered imputed SNPs with an r2> 0.40 as well-imputed [22] and included them in our analyses. The average imputation quality for included variants is detailed in Supplementary Table 4, overall and by MAF categories.
Gene and SNP selection
Five candidate genes were chosen for this study based on published literature which directly showed or suggested roles in the regulation of A-to-I RNA editing [1, 4, 23]. The genes included adenosine deaminase domain containing 1 (ADAD1), adenosine deaminase, RNA-specific (ADAR/ADAR1), adenosine deaminase, RNA-specific, B1 (ADARB1/ADAR2), adenosine deaminase, RNA-specific, B2 (ADAR3/ADARB2), and staphylococcal nuclease and Tudor domain containing 1 (SND1). In total, 5,303 SNPs in the 5 genes, 77 genotyped directly and 5,226 imputed, were available for statistical analysis.
Population stratification
HapMap DNA samples from European (CEU, n = 60), African (YRI, n = 53) and Asian (JPT+CHB, n = 88) populations were also genotyped as part of the same custom Illumina iSelect Array. The program LAMP [24] was used to estimate intercontinental ancestry based on the HapMap (release no. 23) genotype frequency data for these three populations. Eligible subjects with greater than 90 percent European ancestry were defined as European (n = 39,773). We then used a set of 37,000 unlinked autosomal markers to perform principal components analysis within each major population subgroup. To enable this analysis on very large sample sizes we used an in-house program written in C++ using the Intel MKL libraries for eigenvectors (available at http://ccge.medschl.cam.ac.uk/software/).
Statistical analysis
Descriptive statistics were calculated in terms of means and standard deviations for continuous variables and frequencies and percents for categorical variables. The primary association analysis focused on individuals of European ancestry. Unconditional logistic regression was used to estimate odds ratios (OR) and their 95% confidence intervals (CI) between genotype and case status under a log-additive genetic model, with adjustment for the first five principal components representing sub-European ancestry. Due to the heterogeneous nature of EOC, subgroup analyses were conducted to estimate genotype-specific odds ratios by histologic subtype: serous, endometrioid, mucinous, and clear cell carcinomas. False discovery rates (FDR) [25] were used to adjust for multiple comparisons, and FDR of 15% was used to declare significance.
Two methods of gene-level evaluations were also conducted to combine association evidence from SNPs within each gene evaluated: the Admixture Maximum Likelihood (AML) Test [26] and the Sequence-Kernel Association test for the combined effect of common and rare variants (SKAT-CR) [27]. AML is an approach that simultaneously examines the global null hypothesis (of no SNP-outcome associations) and estimates the proportion of underlying false hypotheses. The AML uses univariate SNP-level results to calculate the AML Cochran-Armitage Trend test. Compared to other methods, AML has been shown to have similar or higher statistical power to detect associations except under the unlikely scenario that greater than 20% of all variants are associated with the outcome [26]. SKAT-CR evaluates the cumulative effect of rare and common variants, but does not consider low-frequency variants. These gene-level approaches were undertaken to complement SNP-level findings, and aimed to reduce the degrees of freedom, avoid model-fitting issues due to multicollinearity from LD, and to improve statistical power. The Bonferroni method was used to account for multiple comparisons.
Expression quantitative trait locus (eQTL) analysis was performed to examine for association between genotype (n = 5,303, imputed as above in n = 5 genes) and corresponding gene expression for the 5 candidate RNA editing genes. Matched genotype and gene expression profiling data were obtained for 402 high-grade serous EOC samples evaluated in the Cancer Genome Atlas (TCGA) Project using previously described methods [19]. Briefly, germline genotypes and matched tumor gene expression data were downloaded from the TCGA data portal. To conduct the eQTL analysis, we used germline genotypes of SNPs/proxies as independent variables and expression levels as traits. Expression levels between minor allele carriers versus non-carriers were compared using the Wilcoxon rank sum statistic. Haploreg v4.1http://www.broadinstitute.org/mammals/haploreg/haploreg.php) [28] was used to evaluate the putative function of candidate SNPs.
SUPPLEMENTARY MATERIALS TABLES
Footnotes
CONFLICTS OF INTEREST
None
GRANT SUPPORT
Funding for this study was supported in part by the US National Institute of Health (R01-CA-11343 and R01-CA114343-S1) and the Genetic Associations and Mechanisms in Oncology (GAME-ON), a NCI Cancer Post-GWAS Initiative (U19-CA148112). In addition, we acknowledge the following: AUS: U.S. Army Medical Research and Materiel Command (DAMD17-01-1-0729), National Health & Medical Research Council of Australia, Cancer Councils of New South Wales, Victoria, Queensland, South Australia and Tasmania, Cancer Foundation of Western Australia; National Health and Medical Research Council of Australia (199600 and 400281). The Australian Ovarian Cancer Study Management Group (D. Bowtell, G. Chenevix-Trench, A. deFazio, D. Gertig, A. Green, P. Webb) and ACS Investigators (A. Green, P. Parsons, N. Hayward, P. Webb, D. Whiteman) thank all the clinical and scientific collaborators (see http://www.aocstudy.org/) and the women for their contribution. GCT & PW are supported by Fellowships from NHMRC; AP is funded by a Medical Research Council studentship; BAV: ELAN funds of the University of Erlangen-Nuremberg. BEL: Nationaal Kankerplan, we would like to thank Gilian Peuteman, Thomas Van Brussel, Annick Van den Broeck and Joke De Roover for technical assistance; DOV: U.S. National Institutes of Health R01-CA112523 and R01-CA87538; GER: German Federal Ministry of Education and Research, Programme of Clinical Biomedical Research (01 GB 9401) and the German Cancer Research Center (DKFZ). GRR: Roswell Park Cancer Institute Alliance Foundation, P30 CA016056. HAW: U.S. National Institutes of Health (R01-CA58598, N01-CN-55424 and N01-PC-67001); HJO and HMO: Intramural funding; Rudolf-Bartling Foundation; HOC: Helsinki University Research Fund; HOP: DOD DAMD17-02-1-0669 and NCI K07-CA080668, R01-CA95023, P50-CA159981; NIH/National Center for Research Resources/General Clinical Research Center grant M01-RR000056; R01-CA126841; LAX: American Cancer Society Early Detection Professorship (SIOP-06-258-01-COUN) and the National Center for Advancing Translational Sciences (NCATS), Grant UL1TR000124; MAL: Funding for this study was provided by research grant R01- CA61107 from the National Cancer Institute, Bethesda, MD; research grant 94 222 52 from the Danish Cancer Society, Copenhagen, Denmark; and the Mermaid I project.; MAY: National Institutes of Health (R01-CA122443, P30-CA15083, P50-CA136393), Mayo Foundation; Minnesota Ovarian Cancer Alliance; Fred C. and Katherine B. Andersen Foundation; MCC: MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database;; MDA: DOD Ovarian Cancer Research Program (W81XWH-07-0449); NEC: National Institutes of Health R01-CA54419 and P50-CA105009 and Department of Defense W81XWH-10-1-02802; NHS: National Institute of Health (UM1-CA176726, and R01-CA67262). The NHS would like to thank the participants and staff for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY; NJO: National Cancer Institute (NIH-K07 CA095666, R01-CA83918, NIH-K22-CA138563, and P30-CA072720) and the Cancer Institute of New Jersey, and NCI CCSG award (P30-CA008748); NOR: Helse Vest, The Norwegian Cancer Society, The Research Council of Norway; NTH: Radboud University Medical Centre; ORE: OHSU Foundation; OVA: This work was supported by Canadian Institutes of Health Research grant (MOP-86727) and by NIH/NCI 1 R01CA160669-01A1; POC: Pomeranian Medical University; POL: Intramural Research Program of the NCI; PVD: Herlev Hospitals Forskningsrad, Direktor Jacob Madsens og Hustru Olga Madsens fond, Arvid Nilssons fond, Gangsted fonden, Herlev Hospitals Forskningsrad and Danish Cancer Society; RMH: Cancer Research UK (no grant number is available); SEA: Cancer Research UK (C490/A10119 C490/A10124); UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge, SEARCH team, Craig Luccarini, Caroline Baynes, Don Conroy; SRO: Cancer Research UK (C536/A13086, C536/A6689) and Imperial Experimental Cancer Research Centre (C1312/A15589) and to thank all members of Scottish Gynaecological Clinical Trials group and COTROC1 investigators; STA: US National Institutes of Health U01-CA71966, R01-CA16056, K07-CA143047, and U01-CA69417 for recruitment of controls by the Cancer Prevention Institute of California; TOR: NIH grants R01 CA063678, CA063682, and CA149429; UCI: NIH R01-CA058860, and the Lon V Smith Foundation grant VLS-39420; UKO: The UKOPS study was funded by The Eve Appeal (The Oak Foundation) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. We particularly thank I. Jacobs, M. Widschwendter, E. Wozniak, A. Ryan, J. Ford and N. Balogun for their contribution to the study; UKR: Cancer Research UK (C490/A6187); UK National Institute for Health Research Biomedical Research Centres at the University of Cambridge; USC: P01-CA17054, P30-CA14089, R01-CA61132, N01-PC67010, R03-CA113148, R03-CA115195, N01-CN025403, and California Cancer Research Program (00-01389V-20170, 2II0200). WOC: National Science Centren (N N301 5645 40) The Maria Sklodowska-Curie Memorial Cancer Center and Institute of Oncology, Warsaw, Poland. This study was supported in part by the Biostatistics and Cancer Informatics Core Facilities at the H. Lee Moffitt Cancer Center & Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292).
REFERENCES
- 1.Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G. Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis. 2011;32(11):1569–1577. doi: 10.1093/carcin/bgr124. [DOI] [PubMed] [Google Scholar]
- 2.Han L, Diao L, Yu S, Xu X, Li J, Zhang R, Yang Y, Werner HM, Eterovic AK, Yuan Y, Li J, Nair N, Minelli R, et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer cell. 2015;28(4):515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, Eisenberg E, Levanon EY. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell reports. 2015;13(2):267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- 4.Galeano F, Tomaselli S, Locatelli F, Gallo A. A-to-I RNA editing: the “ADAR” side of human cancer. Semin Cell Dev Biol. 2012;23(3):244–250. doi: 10.1016/j.semcdb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 5.American Cancer Society. Cancer Facts and Figures 2016. Atlanta: American Cancer Society. 2016.
- 6.Urunsak IF, Gulec UK, Paydas S, Seydaoglu G, Guzel AB, Vardar MA. Adenosine deaminase activity in patients with ovarian neoplasms. Arch Gynecol Obstet. 2012;286(1):155–159. doi: 10.1007/s00404-012-2279-5. [DOI] [PubMed] [Google Scholar]
- 7.Pragathi P, Bharath Kumar PV, Amar Kumar P, Ramakanth Reddy M, Sravani V, Neeraja J, Reeba Mary E, Gopalakrishna K. Evaluation of serum adenosine deaminase and 5′-nucleotidase activities as probable markers in ovarian cancer. Indian journal of clinical biochemistry : IJCB. 2005;20(2):195–197. doi: 10.1007/BF02867428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fasching PA, Gayther S, Pearce L, Schildkraut JM, Goode E, Thiel F, Chenevix-Trench G, Chang-Claude J, Wang-Gohrke S, Ramus S, Pharoah P, Berchuck A. Role of genetic polymorphisms and ovarian cancer susceptibility. Mol Oncol. 2009;3(2):171–181. doi: 10.1016/j.molonc.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aberrant RNA Editing Is a Potential Driver in Cancer. Cancer discovery. 2015;5(12):OF12. [Google Scholar]
- 10.Anadon C, Guil S, Simo-Riudalbas L, Moutinho C, Setien F, Martinez-Cardus A, Moran S, Villanueva A, Calaf M, Vidal A, Lazo PA, Zondervan I, Savola S, et al. Gene amplification-associated overexpression of the RNA editing enzyme ADAR1 enhances human lung tumorigenesis. Oncogene. 2015. [DOI] [PMC free article] [PubMed]
- 11.Zipeto MA, Jiang Q, Melese E, Jamieson CH. RNA rewriting, recoding, and rewiring in human disease. Trends in molecular medicine. 2015;21(9):549–559. doi: 10.1016/j.molmed.2015.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Oguro R, Kamide K, Katsuya T, Akasaka H, Sugimoto K, Congrains A, Arai Y, Hirose N, Saitoh S, Ohishi M, Miura T, Rakugi H. A single nucleotide polymorphism of the adenosine deaminase, RNA-specific gene is associated with the serum triglyceride level, abdominal circumference, and serum adiponectin concentration. Exp Gerontol. 2012;47(2):183–187. doi: 10.1016/j.exger.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Barry CP, Lind SE. Adenosine-mediated killing of cultured epithelial cancer cells. Cancer Res. 2000;60(7):1887–1894. [PubMed] [Google Scholar]
- 14.Li Q, Seo JH, Stranger B, McKenna A, Pe'er I, Laframboise T, Brown M, Tyekucheva S, Freedman ML. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell. 2013;152(3):633–641. doi: 10.1016/j.cell.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrenson K, Li Q, Kar S, Seo JH, Tyrer J, Spindler TJ, Lee J, Chen Y, Karst A, Drapkin R, Aben KK, Anton-Culver H, Antonenkova N, et al. Cis-eQTL analysis and functional validation of candidate susceptibility genes for high-grade serous ovarian cancer. Nature communications. 2015;6:8234. doi: 10.1038/ncomms9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Q, Stram A, Chen C, Kar S, Gayther S, Pharoah P, Haiman C, Stranger B, Kraft P, Freedman ML. Expression QTL-based analyses reveal candidate causal genes and loci across five tumor types. Hum Mol Genet. 2014;23(19):5294–5302. doi: 10.1093/hmg/ddu228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pharoah PD, Tsai YY, Ramus SJ, Phelan CM, Goode EL, Lawrenson K, Buckley M, Fridley BL, Tyrer JP, Shen H, Weber R, Karevan R, Larson MC, et al. GWAS meta-analysis and replication identifies three new susceptibility loci for ovarian cancer. Nat Genet. 2013;45(4):362–370. doi: 10.1038/ng.2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Permuth-Wey J, Chen YA, Tsai YY, Chen Z, Qu X, Lancaster JM, Stockwell H, Dagne G, Iversen E, Risch H, Barnholtz-Sloan J, Cunningham JM, Vierkant RA, et al. Inherited Variants in Mitochondrial Biogenesis Genes May Influence Epithelial Ovarian Cancer Risk. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2011. [DOI] [PMC free article] [PubMed]
- 19.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44(8):955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nature reviews Genetics. 2010;11(7):499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 22.Zheng HF, Rong JJ, Liu M, Han F, Zhang XW, Richards JB, Wang L. Performance of genotype imputation for low frequency and rare variants from the 1000 genomes. PLoS One. 2015;10(1):e0116487. doi: 10.1371/journal.pone.0116487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daniel C, Lagergren J, Ohman M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie. 2015;117:22–7. doi: 10.1016/j.biochi.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 24.Sankararaman S, Sridhar S, Kimmel G, Halperin E. Estimating local ancestry in admixed populations. American journal of human genetics. 2008;82(2):290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyrer JP, Guo Q, Easton DF, Pharoah PD. The admixture maximum likelihood test to test for association between rare variants and disease phenotypes. BMC Bioinformatics. 2013;14:177. doi: 10.1186/1471-2105-14-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. American journal of human genetics. 2013;92(6):841–853. doi: 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic acids research. 2012;40(Database issue):D930–934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.