

Abstract
Sonic hedgehog (Shh), both as a mitogen and as a morphogen, plays an important role in cell proliferation and differentiation during early development. Here, we show that Shh inhibits glutamate transporter activities in neurons, rapidly enhances extracellular glutamate levels, and affects the development of epilepsy. Shh is quickly released in response to epileptic, but not physiological, stimuli. Inhibition of neuronal glutamate transporters by Shh depends on heterotrimeric G protein subunit Gαi and enhances extracellular glutamate levels. Inhibiting Shh signaling greatly reduces epileptiform activities in both cell cultures and hippocampal slices. Moreover, pharmacological or genetic inhibition of Shh signaling markedly suppresses epileptic phenotypes in kindling or pilocarpine models. Our results suggest that Shh contributes to the development of epilepsy and suppression of its signaling prevents the development of the disease. Thus, Shh can act as a modulator of neuronal activity, rapidly regulating glutamate levels and promoting epilepsy.
Keywords: Epilepsy, extracellular glutamate, glutamate transporter, neuronal activity, sonic hedgehog
Subject Categories: Neuroscience, Signal Transduction
Introduction
The hedgehog family of secreted proteins controls a wide variety of processes in embryonic development, stem cell maintenance, and cancer cell proliferation 1. Shh is one of the three members in the family and has important roles in early neuronal development, including axon guidance 2, neuronal differentiation 3, and cortical microcircuit formation 4. When Shh is released by the secretion cells, it binds to Patched (Ptch1) to relieve its inhibition on Smoothened (Smo), a transmembrane protein homologous with members of G‐protein‐coupled receptors (GPCRs), and thus trigger responses, leading to enhanced Gli or Ci (in Drosophila) expression and Gli/Ci‐dependent transcription 5. As most of the effects of Shh are mediated by Gli transcription, its expression levels are always used as an indicator of the activation of this signaling 5, 6. It has been reported that in mouse embryonic stem cells and developing spinal neurons 3, 7, Shh can induce an intracellular Ca2+ elevation. Shh and its signaling molecules are also expressed in mature central nervous system (CNS) though their functions are not clear 8, 9, 10.
Epilepsy is one of the most common chronic neurological disorders with the characteristic of recurrent unprovoked seizures. About one percent of population worldwide has been diagnosed with epilepsy 11. Individuals who got the first unprovoked seizure have a high risk to expect a recurrence within 2 years of the initial seizure. Understanding of the mechanism underlying epileptogenesis is critical for prevention and treatment of epilepsy. Generally, epileptic seizures result from an imbalance between neuronal excitation and inhibition. As the principal excitatory neurotransmitter in the mammalian brain, glutamate depolarizes neurons, which inevitably plays a role in the initiation and spreading of seizure activity, even when the primary defect is not of glutamatergic origin 11, 12. Therefore, it is of vital importance that the extracellular glutamate level is kept low 13. Since there seem to be no extracellular enzymes which can efficiently metabolize glutamate, the only rapid way to clear glutamate from the extracellular fluid is by cellular uptake 14, a task predominantly operated by the glutamate transporters. It has been known for decades that glutamate uptake activity is not constant, but subject to regulation 14. Down‐regulation of glutamate transporters in mice was found to cause spontaneous seizures 15, 16, 17.
Five members of excitatory amino acid transporter proteins (EAATs) have been identified and named as EAAT1‐5 in mammals 14, 18. Among them, EAAT1 (GLAST), EAAT2 (GLT‐1), and EAAT3 (EAAC1) are expressed in hippocampus and cortex throughout the developing and adult stage 14, 18, 19. The EAAC1 is localized to neurons, whereas GLAST and GLT‐1 predominantly to glia 14, 18, 19. When a glutamate is taken into the cell by the glutamate transporters, 3Na+ and 1H+ influx and 1K+ efflux are associated to generate a depolarization current 14, 19. The expression of these transporters can be regulated by growth factors to affect the homeostasis of synaptic glutamate levels, though the regulation of their activities remains controversial 14, 20.
It has been reported that Shh expression in temporal lobe epileptic foci of the patients is greatly increased, indicating that Shh may participate in the changes that occur during temporal lobe epileptic development 21. Here, we report that Shh can enhance the extracellular glutamate levels and control epileptogenesis.
Results and Discussion
Shh release and its pathway activation under epileptic stimulation
We initially studied the expression pattern of molecules associated with Shh pathway in rat hippocampus, including Ptch1, Smo, and Gli, and found that they were expressed from early stages to adulthood (Fig EV1A and B) and functional in cultures (Fig EV1C and D). We then examined the influence of limbic seizures on Shh pathway and found that a 2‐h status epilepticus (SE) induced by pilocarpine (Fig 1A–C) or one electrographic seizure induced by kindling stimuli (Fig 1D–F) all increased Gli1 protein levels in the cortex and hippocampus within 24 h (Figs 1A–F and EV1F–I). By contrast, Shh expression was not changed in the same period (Figs 1A–F and EV1E and G). These results suggest that Shh pathway is activated likely due to Shh release in epilepsy rather than enhanced expression level of Shh. To test whether Shh is indeed released under epileptic stimulation, we examined Shh levels in vivo from hippocampi and cortex in pilocarpine model. The levels of Shh detected by enzyme‐linked immunosorbent assay (ELISA) were significantly increased 0.5, 1, and 1.5 h after the seizure induction (Fig 1G). Also, Shh levels in the medium of slices or hippocampal neurons incubated in the medium with picrotoxin (Pic) or Mg2+‐free (0Mg), conditions known to induce epileptiform activities in slices or cells 22, 23, 24, were markedly enhanced within 1 h in hippocampal slices (Fig 1H) or 15 min in hippocampal neurons (Fig 1I). Thus, epileptic neuronal activity rapidly increases Shh release. Consistently, up‐regulation of Gli1 in neurons was found 4 h after the incubation in 0Mg for 30 min (Fig 1J), suggesting that Shh pathway was activated by the secreted Shh.
Figure EV1. Expression of molecules in Shh pathway and verification of antibodies against Shh or Gli1.
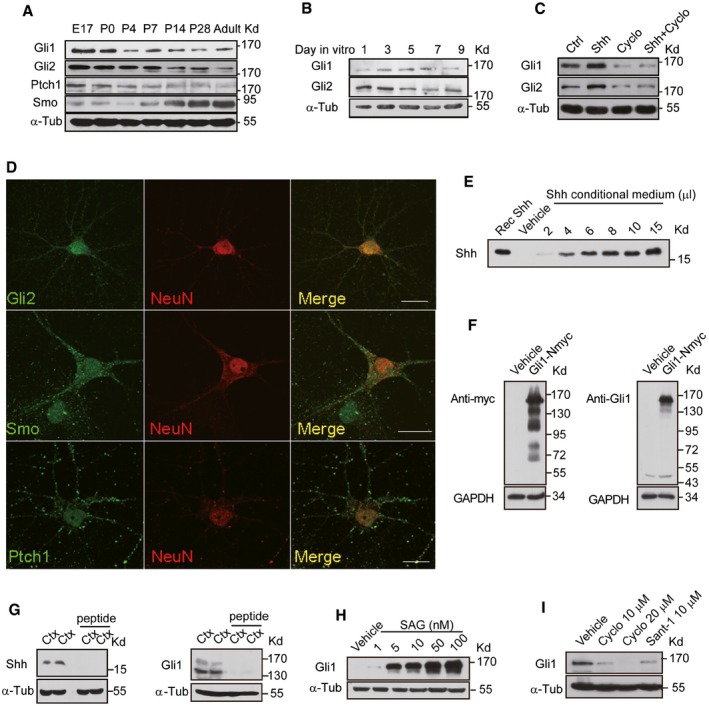
-
A–CWestern blots of the total lysates extracted from rat hippocampus at different developmental stages (A), from cultured hippocampal neurons at different days in vitro (B) and neurons treated with either vehicle (Ctrl), Shh, cyclopamine (Cyclo), or Shh plus Cyclo for 24 h (C) with the indicated antibodies.
-
DRepresentative immunostaining of cultured hippocampal neurons with the indicated antibodies. Scale bar: 10 μm.
-
EWestern blots of the medium from HEK293 cells transfected with empty vectors (Vehicle) or Shh construct by the anti‐Shh antibody. The numbers indicate different loading volume (μl) of conditional medium. Recombinant Shh (Rec Shh; Sigma) was used as a positive control.
-
FTotal lysates of HEK293 cells transfected with empty vectors (Vehicle) or myc‐tagged Gli1 construct (Gli1‐Nmyc) were Western‐blotted and analyzed with the anti‐myc antibody (left) or the anti‐Gli1 antibody (right).
-
GWestern blots of cortical extracts from mice using Shh (left) or Gli1 (right) antibodies with or without pre‐incubation of antigenic peptide.
-
H, IThe expression level of Gli1 detected by Western blots of lysates from NIH3T3 cells incubated with SAG (Smoothened agonist) at the indicated concentrations (H) or from hippocampal neurons incubated with Cyclo or Sant‐1 at the indicated concentrations (I).
Figure 1. Shh release and its pathway activation in response to epileptic stimulations.
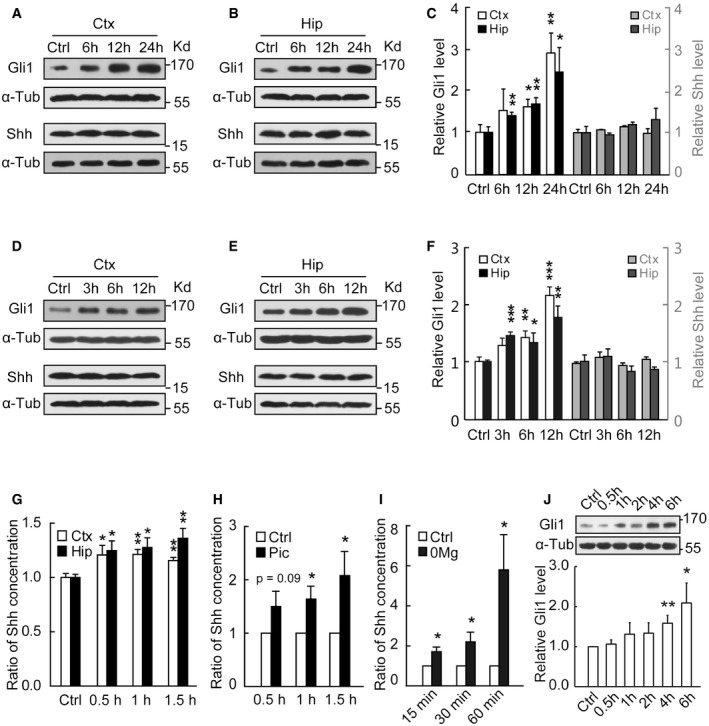
-
A–CRepresentative Western blots of the cortical (Ctx) or hippocampal (Hip) extracts from mice at the indicated time after seizure activity in pilocarpine model. (C) Quantification of Gli1 or Shh expression levels shown in (A, B). n = 8–14 mice.
-
D–FRepresentative Western blots of the cortical (Ctx) or hippocampal (Hip) extracts from mice at the indicated time after seizure activity in kindling model. Samples were obtained from mice evoked with a single kindling stimulation to induce seizure activity as evidenced in EEG. (F) Quantification of Gli1 or Shh expression levels shown in (D, E). n = 8–23 mice.
-
GShh levels assayed by ELISA from mouse cortex and hippocampus at the indicated time after the initiation of status epilepticus (SE) induced by pilocarpine (n = 7–10).
-
H, IShh levels assayed by ELISA in the medium of slices (H, n = 9) or hippocampal neurons (I, n = 6) incubated with picrotoxin (Pic) or Mg2+‐free (0Mg) for the indicated times.
-
JRepresentative Western blots and quantification of Gli1 expression levels from hippocampal neurons incubated with 0Mg for the indicated times (n = 13–19).
However, increase of Shh release was not found under physiological conditions, such as theta‐burst stimulation (TBS), 20‐Hz stimulation, or 50‐mM‐KCl stimulation (Fig EV2A–C). Moreover, inhibition of Shh signaling did not change neuronal transmission in physiological conditions, including basal synaptic transmission, AMPA receptor‐mediated excitatory postsynaptic current (AMPA‐EPSC), paired‐pulse facilitation, or long‐term potentiation (LTP) (Fig EV2D–G). These results suggest that Shh did not have any role in the physiological conditions. Therefore, compared with other factors (BDNF, NGF or NT‐3, etc.) 11, 25 whose secretion can be increased in response to both LTP and epileptic stimulations, Shh is unique in its release in epilepsy and in regulation of neural activity, but not synaptic efficacy.
Figure EV2. Inhibition of endogenous Shh pathway reduces epileptiform activities without affecting physiological synaptic transmission.
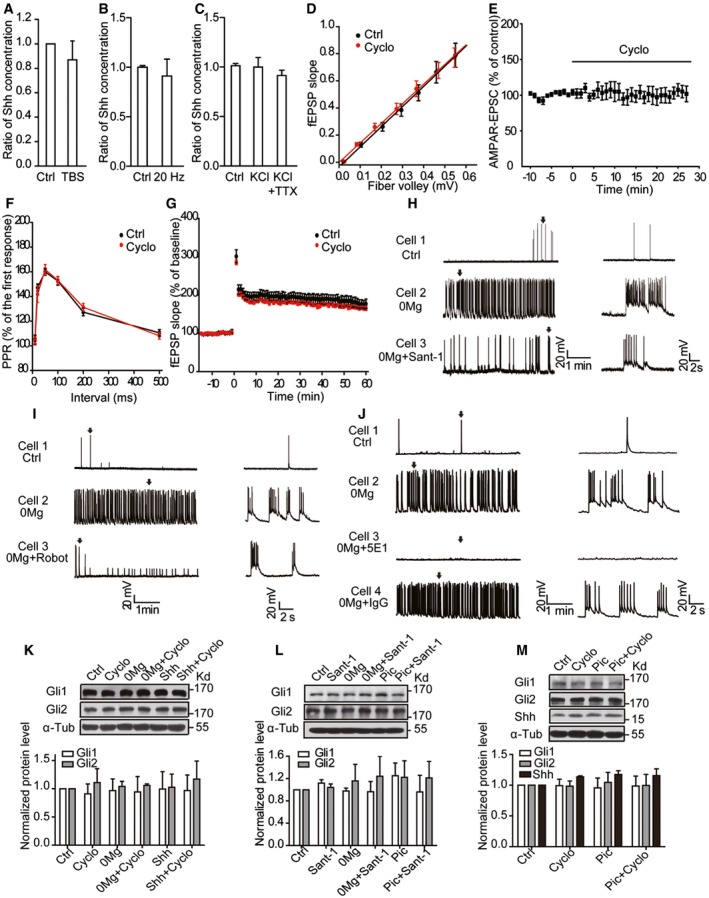
-
AShh levels determined by ELISA in the medium of hippocampal slices with or without (Ctrl) TBS stimuli. TBS: theta‐burst stimuli. n = 4–8.
-
B, CShh levels determined by ELISA in the medium of hippocampal neurons with or without 20‐Hz electrical stimulation for 30 min (B, n = 6–7) or incubated with the indicated treatments (C, n = 11). KCl, 50 mM; TTX, 1 μM.
-
DInput–output curves recorded from CA1 stratum radiatum of hippocampal slices treated with Cyclo or vehicle (Ctrl). fEPSP: field excitatory postsynaptic potential. n = 8.
-
ENormalized amplitude of AMPA receptor‐mediated current at −70 mV. Black line: perfusion of Cyclo. EPSC: excitatory postsynaptic current. n = 11.
-
FQuantification of the paired‐pulse ratio (PPR) of fEPSP in CA1 of hippocampal slices treated with Cyclo or vehicle (Ctrl). n = 9.
-
GTBS‐induced LTP in the presence of Cyclo or vehicle (Ctrl). The slope of fEPSP plotted as percent of baseline before TBS. n = 9.
-
H–JEffects of Sant‐1 (H), robotnikinin (Robot, I), or 5E1 (J) on the spontaneous epileptiform activity from hippocampal neurons incubated with 0Mg. Left: representative traces of whole‐cell recordings with the indicated treatments. Right: the expanded view of a single burst (arrow) from the left parallel panels.
-
K, LRepresentative Western blots and quantification of Gli1 and Gli2 expression levels from cultured hippocampal neurons under the indicated treatments for 30 min. n = 3.
-
MRepresentative Western blots and quantification of Gli1, Gli2, and Shh expression levels from hippocampal slices under the indicated treatments. n = 3.
Inhibiting Shh pathway reduces the epileptiform activity
We next investigated whether Shh is involved in the formation of epileptiform activities. Neurons incubated in 0Mg medium for 30 min showed the robust epileptiform activity with a train of high‐frequency action potentials overlaying on the plateau of large depolarization shifts with the frequency of 7.38 ± 0.56 or 6.52 ± 0.58 burst/min (Fig 2A–C, E left panel or Fig EV2H). In the presence of cyclopamine or Sant‐1, agents known to specifically inhibit Smo 26, 27, the frequency was 0.22 ± 0.09 or 0.64 ± 0.18 burst/min, respectively (Fig 2A–C and E left panel, and Fig EV2H). All neurons incubated with 0Mg medium showed epileptiform activities, whereas in the presence of cyclopamine or Sant‐1, 26.32% or 54.55% exhibited such activities (Fig 2C and D left panel). Moreover, robotnikinin, an agent targeting Shh 28, and 5E1, a Shh‐neutralizing antibody 29, 30, both reduced the epileptiform activities (middle and right panels of Fig 2D and E; Fig EV2I and J). Additionally, in the hippocampal slices the overall frequency of spontaneous epileptiform bursts induced by picrotoxin was reduced from 8.07 ± 0.52 bursts/min to 6.09 ± 0.55 by cyclopamine (Fig 2F–H). Meanwhile, the expression of Gli1 or Gli2 was not changed when neurons or slices were treated with Shh or the inhibitors in Mg2+‐free or Pic conditions in 30 min (Figs EV2K–M and 1J). Thus, inhibition of Shh pathway suppresses the formation of epileptiform activity independent of Gli transcription.
Figure 2. Inhibiting Shh pathway reduces the epileptiform activity.
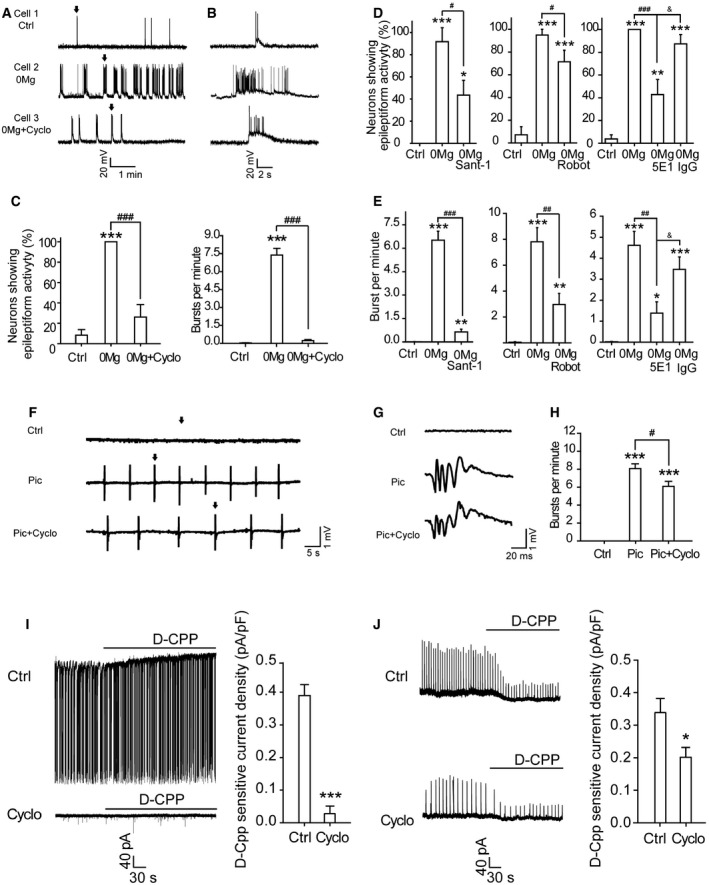
-
A–CWhole‐cell recordings of spontaneous epileptiform activity from cultured hippocampal neurons. (A) Representative traces showing the neuronal activity under the indicated treatments. Ctrl: extracellular solution. Cyclo: cyclopamine. (B) The expanded view of a single burst (arrow) from (A). (C) Quantification of the percentage of neurons showing epileptiform activity (left, n = 6–7) and the burst frequency (right, n = 18–19) shown in (A). Cells were incubated in the presence of Cyclo in extracellular solution for 30 min and then transferred to 0Mg plus Cyclo (0Mg + Cyclo) and incubated for another 30 min.
-
D, EThe effects of Sant‐1, robotnikinin (Robot), or 5E1 on the percentage of neurons showing epileptiform activity (D, n = 6–10) and the burst frequency (E, n = 15–22).
-
F–HExtracellular recordings of spontaneous epileptiform activity from CA1 stratum pyramidal of hippocampal slices. (F) Representative traces under the indicated treatments. Ctrl: artificial cerebrospinal fluid (aCSF). (G) The expanded view of a single burst (arrow) in (F). Slices were incubated in the presence of vehicle or Cyclo in aCSF for 30 min and then transferred to picrotoxin in aCSF (Pic) or with Cyclo (Pic + Cyclo) and recorded for 30 min. (H) Quantification of the spontaneous burst frequency of (F) from 10 slices (seven rats).
-
I, JLeft are representative traces showing the D‐CPP‐sensitive currents recorded from cultured hippocampal neurons (I) or slices (J) incubated in 0Mg or Pic, respectively, with or without Cyclo. Black lines: application of D‐CPP. On the right is shown quantification of D‐CPP‐sensitive current density from 18 to 26 cells (I) or 16–18 slices (15 rats) (J).
Shh increases the extracellular glutamate levels
To explore the mechanism by which Shh regulates epileptic activities, we assessed its effect on Ca2+ changes in hippocampal neurons. Shh induced a slow and sustained intracellular Ca2+ elevation in an NMDA receptor (NMDAR)‐dependent manner (Fig EV3A–D), but it did not influence NMDA‐ or AMPA‐induced currents, suggesting that Shh did not directly affect these receptors (Fig EV3E and F). Under Mg2+‐free or Pic conditions, whole‐cell recording can detect a large and slow NMDA receptor current, the tonic NMDA receptor current, an indication of ambient glutamate in the extracellular space 31. By application of D‐CPP, an antagonist to the NMDA receptors, the amplitude of the tonic NMDA receptor currents can be measured (Fig EV3G and H). Cyclopamine greatly reduced the tonic NMDA receptor currents in both hippocampal slices and neurons (Fig 2I and J). However, the NMDA‐induced currents were not affected by cyclopamine (Fig EV3I). Together, these results support an explanation that Shh increases extracellular glutamate to induce the NMDAR currents. Indeed, Shh markedly increased the glutamate levels in the culture medium in a cyclopamine‐sensitive manner (Fig 3A). In contrast, Shh did not affect the neuronal excitability (Fig EV3J–L). We then explored whether Shh increases glutamate release or inhibits its uptake. In the paired‐pulse facilitation experiments, the paired‐pulse ratio was not different between vehicle‐ and Shh‐incubated hippocampal slices (Fig 3B). Therefore, it is unlikely that Shh affects glutamate release. In contrast, Shh greatly inhibited 3H‐glutamate uptake in hippocampal neurons (Fig 3C). This inhibition was reversed by cyclopamine (Fig 3C). Together, these results suggest that Shh inhibits glutamate uptake to increase extracellular glutamate levels.
Figure EV3. Shh induces an APV‐sensitive [Ca2+]i elevation and Shh does not change the hippocampal neuron excitability.
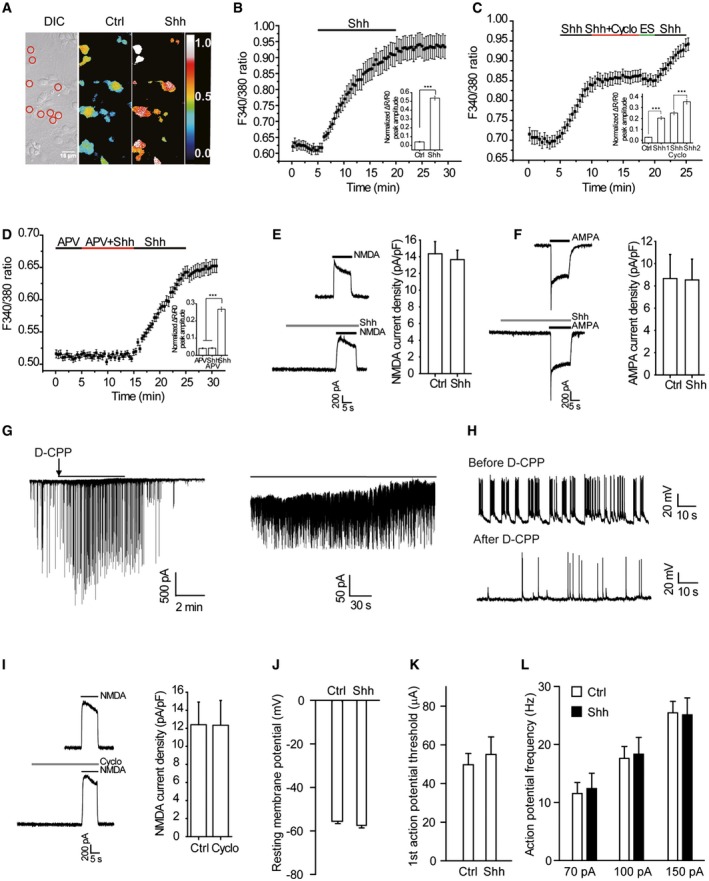
-
AAn example of Fura‐2 AM‐loaded neurons. Red circles in differential interference contrast (DIC) panel indicate the regions from where the F340/380 ratios were obtained. The pseudo color panels show changes in F340/380 ratios in the cells before (Ctrl) and after Shh perfusion. Scale bar, 10 μm.
-
B–DRepresentative traces showing F340/380 ratio of neurons after Shh perfusion. Effects of Cyclo (C) and APV (D) on Shh‐induced [Ca2+]i elevation. Insets: quantification of the peak amplitudes of ΔR/R0 values. ES, extracellular solution. APV: 50 μM. n = 45–80 cells in each group of at least three independent experiments.
-
E, FLeft: representative traces of NMDA or AMPA currents induced by 100 μM NMDA plus 1 μM glycine or 100 μM AMPA in the presence or absence of Shh. Shh was applied for 30 s and then applied with the indicated agonists. Membrane potential was clamped at +40 mV for NMDA‐induced currents, or at −70 mV for AMPA‐induced currents. Right: quantification of the effect of Shh on NMDA‐ or AMPA‐induced current density from 7 to 8 cells in each group.
-
GLeft: a representative trace showing the D‐CPP‐sensitive currents recorded from cultured hippocampal neurons incubated in 0Mg; right: the expanded view. Arrow: application of D‐CPP. Black lines: expanded regions.
-
HWhole‐cell recordings of spontaneous activities before and after application of D‐CPP from the neurons in 0Mg.
-
ILeft: representative traces of NMDA currents induced by 100 μM NMDA plus 1 μM glycine in the presence or absence of Cyclo. Cyclo was applied for 30 s and then applied with the indicated agonists. Membrane potential was clamped at +40 mV. Right: quantification of the effect of Cyclo on NMDA‐induced current density from 7 to 8 cells in each group.
-
JQuantification of resting membrane potentials of neuron treated with Ctrl or Shh for 30 min.
-
KAveraged current threshold of neurons treated with Ctrl or Shh.
-
LFiring frequency of neurons treated with Ctrl or Shh in response to 70‐, 100‐, and 150‐pA current injections for 500 ms.
Figure 3. Shh inhibits glutamate transporter activities and increases the extracellular glutamate levels.
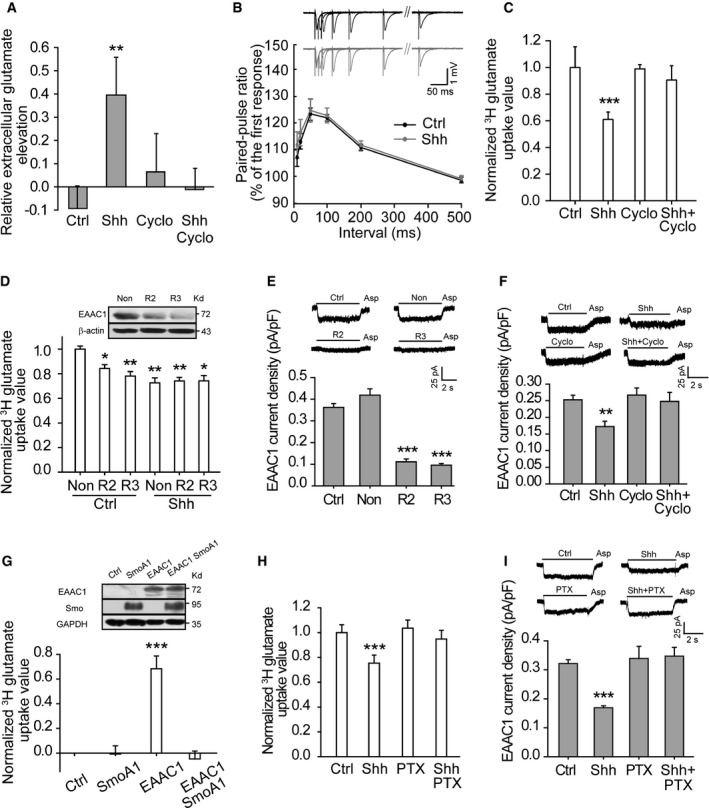
-
AGlutamate levels assayed by HPLC in the medium of neurons incubated with the indicated agents. n = 4–6.
-
BThe paired‐pulse ratio of field excitatory postsynaptic potentials recorded in CA1 of hippocampal slices treated with vehicle (Ctrl) or Shh (13–14 slices, six rats). Insets: representative traces recorded in response to paired‐pulse stimuli with different intervals.
-
C, DQuantification of 3H‐glutamate uptake by neurons treated with the indicated agents (C, n = 10) or transfected with two lentivirus‐based RNAi against EAAC1 (R2 and R3) or nonsense RNAi (Non) in response to vehicle (Ctrl) or Shh (D, n = 3–6). No difference between R2 or R3 in Ctrl and those in Shh. Inset of (D): representative Western blots for EAAC1.
-
E, FAspartate (Asp)‐evoked currents at −70 mV from hippocampal neurons transfected with RNAi or Non (E, n = 17–23) or treated with the indicated agents (F, n = 16–18).
-
GUpper panel: representative Western blots of EAAC1 and Smo from HEK293 cells transfected with empty vectors (Ctrl), constitutively active form of Smoothened (SmoA1), EAAC1, or EAAC1 plus SmoA1. Lower panel: 3H‐glutamate uptake by cells transfected with the indicated vectors. n = 9.
-
H, IShh effects on 3H‐glutamate uptake (H, n = 6) or Asp‐evoked currents (I, n = 17–23) of neurons with or without pertussis toxin (PTX) pretreatment.
Shh rapidly inhibits the activity of glutamate transporters through Gαi/o proteins
We then studied whether Shh regulates glutamate transporter activities. We found that cyclopamine no longer inhibited the epileptiform activity in the presence of TBOA, a non‐selective antagonist of glutamate transporters (Fig EV4A–C), suggesting that cyclopamine and TBOA might act on the similar pathway to regulate the epileptic activities. It has been reported that glial transporters are barely expressed in culture systems 32, 33. Consistently, the glutamate uptake in neuronal cultures was slightly inhibited by dihydrokainate (DHK), a selective inhibitor of GLT‐1 14 (Fig EV4D). In contrast, expressing EAAC1 RNAi, but not GLAST RNAi, inhibited the glutamate uptake (Figs 3D and EV4E–G), suggesting that EAAC1 is the main glutamate transporter in the cultured neurons. Moreover, Shh no longer suppressed the glutamate uptake when the EAAC1 RNAi was expressed (Fig 3D). Additionally, aspartate (Asp), a non‐selective agonist of glutamate transporters, induced an inward current in neurons, which was blocked by EAAC1 RNAi (Fig 3E). Inhibiting GLT‐1 and GLAST did not change the inward current (Fig EV4H). Therefore, EAAC1‐dependent current was the major component of Asp‐induced current in the cultures. Further, Shh reduced the Asp‐induced inward current density in a manner that was sensitive to cyclopamine (Figs 3F and EV4I). In the presence of TBOA, cyclopamine no longer affected Shh inhibition of EAAC1 current (Fig EV4J). In addition, Shh also increased extracellular glutamate and reduced the EAAC1 current in the cultures treated with ARA‐C (Fig EV4K and L), suggesting that neuronal EAAC1 was inhibited by Shh. To further confirm Shh inhibition of EAAC1, we studied the effect of SmoA1, a constitutively active form of Smo 34, on EAAC1 activities in HEK293 cells. Expressing SmoA1 eliminated the increase in 3H‐glutamate uptake caused by expressing EAAC1 (Fig 3G). Collectively, these results suggest that Shh inhibits EAAC1 activities to elevate the extracellular glutamate.
Figure EV4. Shh inhibits EAAC1 to enhance extracellular glutamate in a Gαi/o‐dependent manner.
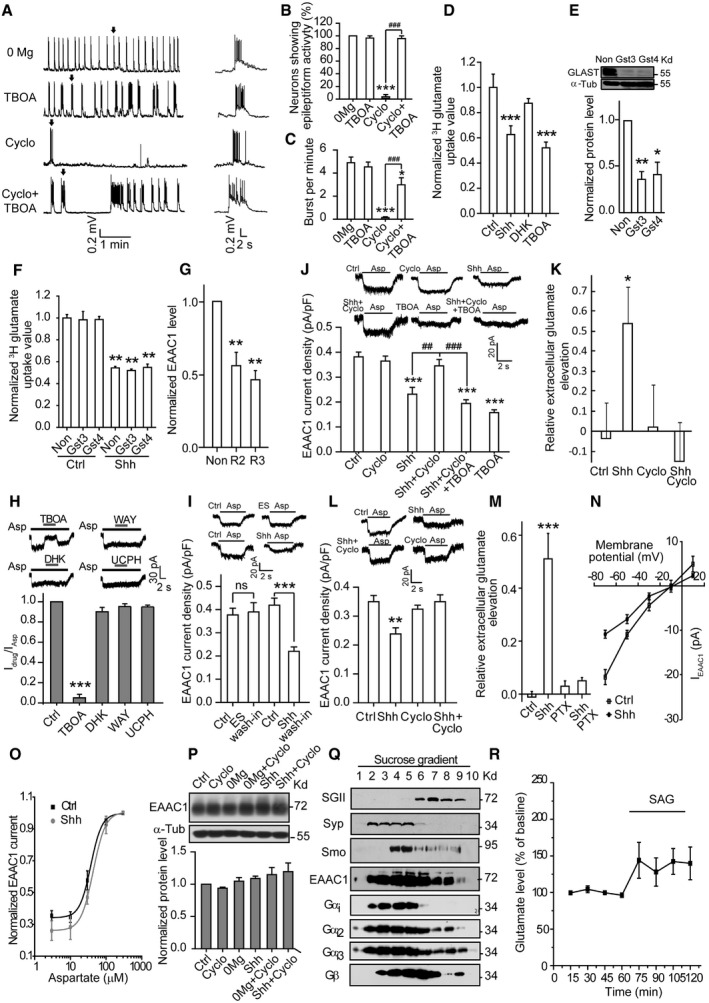
-
A–CWhole‐cell recordings of spontaneous epileptiform activity from cultured hippocampal neurons. (A) Left: representative traces showing the neuronal activity under the indicated treatments. Right: the expanded view of a single burst (arrow) from the left panel. (B) Quantification of the percentage of neurons showing epileptiform activity, n = 4–5. (C) The burst frequency shown in (A), n = 15–18.
-
DQuantification of 3H‐glutamate uptake by neurons in response to vehicle (Ctrl) or the indicated agents. n = 3–9.
-
EUpper panel: representative Western blots of the extracts from hippocampal neurons transfected with two lentivirus‐based RNAi against GLAST (Gst3 or Gst4) or nonsense RNAi (Non). Lower panel: quantification of normalized GLAST protein levels, n = 3.
-
FStatistics of 3H‐glutamate uptake by cultured hippocampal neurons transfected with the indicated RNAi in response to Ctrl or Shh. n = 3.
-
GQuantification of EAAC1 levels in cultured hippocampal neurons transfected with two lentivirus‐based RNAi against EAAC1 (R2 or R3) or nonsense RNAi (Non). n = 3.
-
HQuantification of the normalized current amplitude (13–22 cells in each group) of aspartate (Asp)‐evoked current in neurons treated with Ctrl, TBOA, DHK, WAY213623 (WAY, a selective inhibitor of GLT‐1), or UCPH‐101 (UCPH, a selective inhibitor of GLAST).
-
IThe effect of Shh on EAAC1 current density in the wash‐in method (n = 9). ES: extracellular solution. ns: not significant.
-
JQuantification of EAAC1 current density from hippocampal neurons with the indicated treatments (n = 15–17).
-
KGlutamate levels assayed by HPLC in the medium of neurons incubated with ARA‐C and treated with the indicated agents. n = 6–11.
-
LAsp‐evoked currents from hippocampal neurons incubated with ARA‐C and treated with the indicated agents. n = 14–15.
-
MGlutamate levels in the medium of neurons pretreated with Ctrl or Shh in the absence or presence of pertussis toxin (PTX). n = 8–9.
-
NThe current–voltage (I–V) relationship of Asp‐induced currents recorded from hippocampal neurons in response to vehicle (Ctrl) or Shh, from 10 to 24 cells in each group.
-
OThe dose–response curve of Asp‐evoked EAAC1 currents, obtained from hippocampal neurons at −70 mV. There was no significant difference between Ctrl and Shh in all concentrations from 12–13 cells. The EC50 of Ctrl or Shh was 37.33 ± 2.56 or 43.61 ± 0.63 μM, respectively. The dose–response curve was fitted by logistic equation: y = A 2 + (A 1 – A 2)/[1 + (x/x 0)p], where y is the response; A 1 and A 2 are the maximum and minimum response, respectively; x is the drug concentration; and p is the Hill coefficient.
-
PUpper panel: representative Western blots of the extracts of hippocampal neurons under the indicated treatments using the indicated antibodies. Lower panel: quantification of the normalized expression level of EAAC1. n = 3.
-
QRepresentative Western blots of fractionated lysates of rat hippocampus with antibodies against secretogranin II (SGII), synaptophysin (Syp), Smo, EAAC1, Gαi‐pan (Gαi), Gαi2, Gαi3, or Gβ‐pan (Gβ). SGII: a marker of large vesicles; Syp: a marker of small vesicles.
-
REffect of SAG on extracellular glutamate in mouse hippocampus. n = 11.
It has been reported that Smo can signal through Gαi/o, but not members of Gs, Gq, and G12 families, to regulate downstream effectors 35, 36, 37. Pretreatment of the neurons with pertussis toxin (PTX), an agent known to selectively inhibit Gαi/o 35, blocked the increase in extracellular glutamate levels, prevented Shh inhibition of glutamate uptake, and reversed the inhibition of EAAC1 current (Figs EV4M and 3H and I), suggesting that Gαi/o protein is necessary for Shh to inhibit EAAC1 activity. To detail Shh regulation of EAAC1 activity, we examined the I–V curve or Asp dose–response curve of EAAC1 (Fig EV4N and O) and found that neither of them was affected by Shh. Further, the expression level of EAAC1 was unchanged 30 min after treatment with Shh (Fig EV4P). Using the sucrose gradient centrifugation (0.3–2.0 M gradient) to isolate the subcellular fractions from hippocampal neurons, we found that Smo, Gαi1‐3, Gβ, and EAAC1 were mainly presented in the same fractions (Fig EV4Q). Together, these results point to a possibility that Shh inhibits EAAC1 activity by suppressing its surface expression to elevate extracellular glutamate in a manner that is dependent on Gαi.
Inhibiting Shh pathway suppresses epileptogenesis
We next explored the possible role of Shh in epilepsy using the mouse kindling model (Fig EV5A), which is commonly used to quantify the epilepsy development 11. Cyclopamine greatly delayed the development of epilepsy, as evidenced by slowed progression of behavioral seizure class and decreased prolongation of electrographic seizure duration (ESD) (Fig 4A and B). It took more stimulation to reach the fully kindled state in cyclopamine‐treated mice (16.5 ± 1.2 stimulations; Fig 4C) than its controls (9.07 ± 1.2 stimulations). Then, we inhibited endogenous Shh using its neutralizing antibody 5E1 to see whether Shh pathway was indeed involved in the development of kindling‐induced epilepsy. Similar to the effect of cyclopamine, the administration of 5E1 greatly reduced the severity of epilepsy with delayed behavioral seizure class and decreased prolongation of ESD (Fig 4D and E). More stimulation was needed to reach the fully kindled state in 5E1‐treated mice (18.7 ± 1.61 stimulations; Fig 4F) than in its controls (12.94 ± 1.3 stimulations; Fig 4F).
Figure EV5. Genetic inhibition of Smo suppresses epileptogenesis in mouse kindling model.
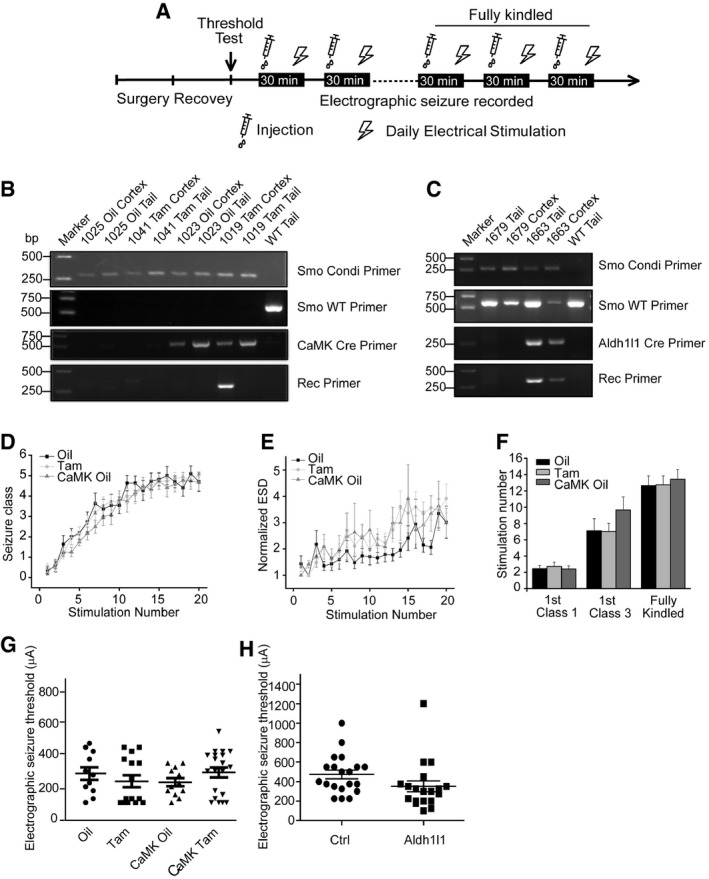
-
ASchematic of the kindling protocol.
-
B, CPCR analysis of lysates form the cortex or the tail tissue of wild‐type mice (WT) and mice with conditional knockout of Smo in CaMKIIα‐positive neurons (B) or Aldh1l1‐positive cells (C). The presence of the Smo +/fl or Smo fl/fl was revealed by the fragments amplified by Smo Conditional Primer (Smo Condi Primer) and Smo WT Primer (Smo fl/fl without a fragment). The presence of the Cre was revealed by the fragments amplified by Cre Primer (CaMK Cre Primer in B or Aldh1l1 Cre Primer in C). The ablation of Smo was revealed by the fragment amplified by recombinant primer (Rec Primer). Oil: injection of vehicle oil; Tam: injection of tamoxifen. Primers for genotyping were as published 50, 51.
-
D–FThe intensification of behavioral seizure class (D), evoked electrographic seizure duration (ESD) (E), and the number of stimulations required to reach equivalent seizure intensity (F) in the indicated control mice.
-
GMean electrographic seizure threshold in Oil, Tam, CaMK Oil, and CaMK Tam group littermates. Each data point represents a result from a single mouse.
-
HMean electrographic seizure threshold in Ctrl or Aldh1l1 group littermates. Each data point represents a result from a single mouse. Ctrl: Smo +/fl (n = 20); Aldh1l1: Smo +/fl Aldh1l1‐Cre (n = 19).
Figure 4. Inhibiting Shh pathway suppresses epileptogenesis in mouse epilepsy models.
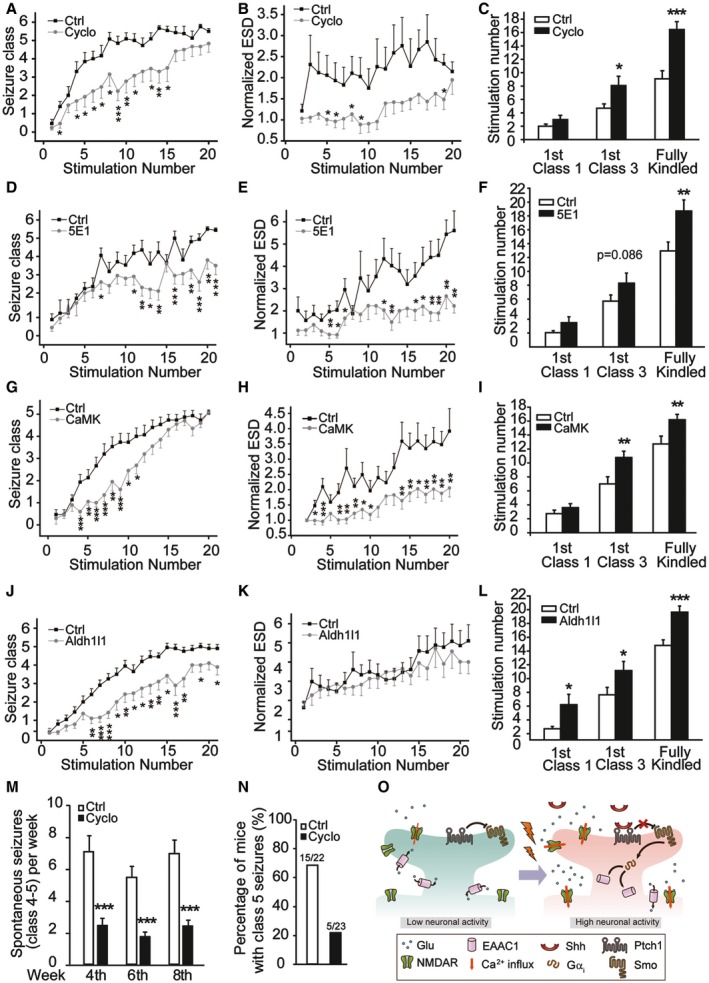
-
A–LEffects of Cyclo (A–C, n = 14), 5E1 (D–F, n = 16–20), or ablation of Smo in CaMKIIα‐positive neurons (G–I), or ablation of Smo in Aldh1l1‐positive astrocytes (J–L) on the progression of kindling including seizure class (A, D, G, J), evoked electrographic seizure duration (ESD) (B, E, H, K), and the number of stimulations required to reach equivalent seizure intensity (C, F, I, L). In (A–F), Ctrl are vehicle‐treated mice. In (G–I), Ctrl: Smo fl/fl induced by tamoxifen; CaMK: Smo fl/fl CaMKIIα‐Cre ERT2 induced by tamoxifen, n = 14‐20. In (J–L), Ctrl: Smo +/fl; Aldh1l1: Smo +/fl Aldh1l1‐Cre, n = 19–20.
-
MFrequency of spontaneous seizures (class 4–5) in mice administrated with Cyclo or vehicle (Ctrl) at 4th, 6th, and 8th week after pilocarpine SE induction. n = 22–23.
-
NPercentage of mice with class 5 seizures within 8 weeks after pilocarpine SE induction. n = 22–23.
-
OSchematic diagram depicting a working model for Shh regulation of epileptogenesis. Epileptic activity triggers sequential responses, including Shh release, inhibition of glutamate transporter activity, and increase in extracellular glutamate, leading to epilepsy development.
To clearly show the role of Shh signaling in epilepsy development, we generated Smo fl/fl CaMKIIα‐Cre ERT2 mice, which specifically lack Smo in CaMKIIα‐positive neurons after tamoxifen treatment (Fig EV5B). The epilepsy development in Smo fl/fl CaMKIIα‐Cre ERT2 mice was notably delayed (Figs 4G–I and EV5D–F), in a pattern reminiscent of that seen in cyclopamine‐treated mice. Since in addition to the neuronal glutamate transporter EAAC1, astrocytic transporter GLT‐1 is the main transporter to control glutamate uptake in hippocampus 14, 15, 38, we then generated Smo +/fl Aldh1l1‐Cre mice, in which Smo was specifically ablated in Aldh1l1‐positive astrocytes (Fig EV5C). Down‐regulation of Smo in astrocytes also suppressed the epilepsy development (Fig 4J–L). No difference in the basal electrographic seizure threshold was observed between genetically modified mice and their controls (Fig EV5G and H). Therefore, Smo in both neurons and astrocytes participates in epilepsy development in mouse kindling models. Since stimulation of Shh pathway increased extracellular glutamate in vivo (Fig EV4R) and glutamate transporter family displays a considerable homology (50–60% at the amino acid level) 39, it is likely that the activity of transporters, including GLT‐1 and GLAST, can be also regulated by Shh to contribute to the enhancement in extracellular glutamate.
It has been reported that elevation of glutamate levels plays a crucial role in spontaneous seizures induced by pilocarpine 40, 41. To further test the effects of Shh inhibition on epileptogenesis, we examined the frequency of spontaneous seizures in pilocarpine‐induced spontaneous seizures. The frequency of spontaneous seizures (class 4–5) was markedly inhibited in cyclopamine‐treated group (2.44 ± 0.47, 1.74 ± 0.27, or 2.39 ± 0.39 in the 4th, 6th, or 8th week, respectively) than in the control group (7.05 ± 1.02, 5.46 ± 0.67, or 6.91 ± 0.87 in the 4th, 6th, or 8th week, respectively) in every week of time (Fig 4M). Overall, 68.18% of control mice developed class 5 seizure within 8 weeks, whereas 21.74% of cyclopamine‐treated mice did (Fig 4N). Together, these results are consistent with an explanation that Shh pathway contributes to the development of epilepsy. Therefore, Smo could be a novel target for anti‐epileptogenesis therapy.
Here, we report a novel function of Shh in CNS. Our data show that Shh is specifically released under epileptic stimulations and regulates the extracellular glutamate levels independent of enhanced Gli expression in the hippocampal neurons. The current findings led us to propose a model that epileptic activity induces Shh release to activate Smo and trigger subsequent responses, including stimulation of Gαi proteins, inhibition of surface expression of the neuronal glutamate transporter EAAC1, increase in extracellular glutamate levels, and enhancement in neuronal activities, contributing to epileptogenesis (Fig 4O). Therefore, Shh–glutamate signaling likely initiates a positive feedback to amplify the network excitation to promote the development of epilepsy. Our findings thus provide evidence to explain the fact that people face much higher risk of permanent epilepsy after one experience of seizure. Thus, Smo can induce a long‐term effect through the Gli‐expression pathway and also induce a short‐term effect through the Gαi proteins in neurons. Furthermore, because of the close relationship between glutamate transporters and excitotoxicity, our work also suggests a novel candidate for further study of excitotoxicity‐related diseases.
In conclusion, our findings point to a previously unknown role of Shh in modulating extracellular glutamate and in contributing to epileptogenesis independent of Gli transcription and to the existence of Shh‐regulated Gi protein and EAAC1 activities that is essential for Shh‐controlled neuronal activity. The demonstration of Shh‐dependent epileptiform activity in cultures and slices and epileptogenesis in animal models further expands our understanding of the diverse functions of Shh.
Materials and Methods
Animals
The 129SV Smo fl/fl mice were from Jackson Lab, CaMKIIα‐Cre ERT2 mice from European Mouse Mutant Archive, and Aldh1l1‐Cre mice from Mutant Mouse Resource & Research Centers. We crossed these Cre mice with Smo fl/fl mice to generate Smo‐conditional knockout mice. Smo fl/fl CaMKIIα‐Cre ERT2 recombination was induced by tamoxifen (Tam, intraperitoneally, once a day for 7 consecutive days) at adulthood. Tam (10 mg/ml, Sigma, T‐5648) solution was prepared in corn oil containing 10% ethanol. Corn oil containing 10% ethanol (Oil) served as the control for Tam. We performed experiments 4 weeks after the induction. Oil‐treated Smo fl/fl CaMKIIα‐Cre ERT2 mice, oil‐treated Smo fl/fl mice, and tamoxifen‐treated Smo fl/fl mice served as controls for Tam‐treated Smo fl/fl CaMKIIα‐Cre ERT2 mice. Homozygous Smo fl/fl Aldh1l1‐Cre mice were lethal during the embryonic development, so Smo +/fl Aldh1l1‐Cre mice were used (Smo +/fl as controls). We used adult male C57BL/6 mice in pilocarpine or kindling epilepsy models. The C57BL/6 mice were from SLAK Laboratory Animal Shanghai China. For transgenic mice, both male and female mice were used in epilepsy experiments. For C57BL/6 mice, male mice were used in the experiments. All animal studies followed the animal welfare guidelines of Institute of Neuroscience, CAS.
Reagents and antibodies
Rabbit anti‐Gli1 (2534), anti‐Shh (2287), Gli1 blocking peptide (1641S), Shh blocking peptides (13937S), and anti‐Gαi1 antibodies were from Cell Signaling; rabbit anti‐Gli2, anti‐EAAC1, anti‐GLAST, and anti‐Gαi2 antibodies from Abcam; rabbit anti‐Gαi3 from Millipore; and mouse anti‐β‐actin, anti‐α‐tubulin, and anti‐GAPDH antibodies from Sigma‐Aldrich. Fura‐2 AM, Alexa Fluor 488‐conjugated goat anti‐rabbit and Texas‐Red‐conjugated goat anti‐mouse secondary antibodies were from Molecular Probes, and HRP‐conjugated goat anti‐rabbit and anti‐mouse secondary antibodies from Amersham. All other reagents were purchased from Sigma‐Aldrich.
Animal models for seizures and epilepsy
Pilocarpine model
Pilocarpine hydrochloride (Sigma‐Aldrich) dissolved in 0.9% (wt/vol) sterile saline was intraperitoneally (i.p.) administered to adult male mice (C57BL/6) at a dosage of 300 mg/kg (body weight). Scopolamine methyl nitrate (2 mg/kg, i.p.; Sigma‐Aldrich) was injected 30 min before pilocarpine to block peripheral side effects. Diazepam (4 mg/kg, i.p.; Sigma‐Aldrich) was used to terminate status epilepticus (SE, 2 h) of continuous seizures to standardize the duration of seizure activity.
To detect Gli1 and Shh levels, mice were killed at varying intervals (6, 12, or 24 h after the termination of SE) and their expression levels in cortex and hippocampi were analyzed. Vehicle (saline, 0.9% wt/vol sterile saline)‐injected mice served as controls.
To detect the spontaneous seizures, mice were divided into two groups after SE induction, vehicle (HBC, 45% wt/vol HBC (2‐hydroxypropyl‐β‐cyclodextrin, Sigma‐Aldrich) in PBS)‐ or cyclopamine (Cyclo, 10 mg/kg, Selleckchem, Sigma‐Aldrich, Abcam)‐treated group. A video monitoring system was used for recording the behaviors at the 4th, 6th, and 8th week (7 h/day, 5 days/week) after SE induction. HBC or Cyclo was administrated every other day from the first day after SE induction to the end of the 8th week.
To examine Shh release in vivo, mice were killed at varying intervals (0.5, 1, or 1.5 h after the initiation of SE) and their cortex and hippocampi were dissected and ground gently by grinding rod with heparin. The samples were centrifuged at 100 g at 4°C and Shh in the supernatant was determined by ELISA. Animals were divided into groups randomly.
Kindling model
Kindling model was established according to the method described previously with a little modification 42. We implanted a bipolar electrode used for stimulating and recording stereotactically in the left amygdala of adult mice under sodium pentobarbital anesthesia, at the following coordinates (with bregma as the reference): 1.2 mm posterior, 2.8 mm lateral, and 4.6 mm below dura. Four screws were also inserted into the skull through a drilled hole without piercing the dura. One of the screws served as a ground electrode. The electrodes and screws were fixed with a mixture of acrylic and dental cement. After a recovery period of 5–7 days, we determined the electrographic seizure threshold (EST) for each animal by applying a 1‐s train of 1‐ms biphasic rectangular pulses at 60 Hz beginning at 60 μA. More stimulation, increasing by 20 μA steps, were delivered at 10 min intervals until an electrographic seizure with a duration of no less than 3 s was detected by the electroencephalogram (EEG) recording from the bipolar electrode. Both EEG and behavioral seizures were recorded. We scored the behavioral progression of stimulation‐evoked seizures according to Racine's standard classification 43 with a little modification: 0, no behavioral change; 1, eye blinking and/or facial clonus; 2, head nodding; 3, unilateral forelimb clonus; 4, rearing with bilateral forelimb clonus; 5, generalized clonic convulsions with loss of hind limb control; and 6, severe whole body convulsions with continuous jumping behavior. We defined the fully kindled state as the occurrence of three consecutive class 5 or 6 seizures. We stimulated and recorded each mouse once a day.
To detect Gli1 and Shh expression, mice with an electrographic seizure detected were divided into groups randomly and killed at varying intervals (3, 6, or 12 h after the stimuli) and their protein levels in cortex and hippocampi were analyzed. Mice undergone surgery but without stimulation were used as controls.
To test the effect of inhibition of Shh pathway on kindling development, Cyclo (10 mg/kg) and its vehicle (HBC), or 5E1 (900 ng/mouse, DSHB) and its control IgG were delivered intraperitoneally (i.p.) or intracerebroventricularly (i.c.v.), respectively, 30 min before each stimulation. Animals were divided into groups randomly.
Cell culture and transfection
Primary hippocampal neurons were isolated and cultured as described previously 44. Briefly, the neurons were obtained by dissociating the hippocampus from SD rat brains of embryonic day 18 and then seeded at a density of 5 × 104/cm2 onto coverslips (No. 1 Glass, Warner Instruments, Connecticut, USA) which had been coated with 50 μg/ml poly‐D‐lysine (Sigma‐Aldrich). Cells were cultured in Neurobasal supplemented with B‐27 and 0.5 mM glutamax for 9–12 days before use. In ARA‐C (cytosine β‐D‐arabinofuranoside, Sigma C1768, 10 μM) experiments, the ARA‐C was added 24–48 h after plating.
The HEK293 cells cultured in DMEM supplemented with 10% FBS were transfected with control (1 μg pCIG and 3 μg pEGFP), pEGFP‐smoothened (1 μg pCIG and 3 μg pEGFP‐constitutively active smoothened), pCIG‐EAAC1 (1 μg pCIG‐EAAC1 and 3 μg pEGFP), or pCIG‐EAAC1 plus pEGFP‐smoothened (1 μg pCIG‐EAAC1 and 3 μg pEGFP‐constitutively active form of smoothened) using Lipofectamine 2000 (Invitrogen).
Electrophysiology
Whole‐cell recordings in cultured neurons
Whole‐cell patch‐clamp recordings were carried out at room temperature (22–25°C) at DIV (days in vitro) 9–12. Patch electrodes were pulled with a Flaming/Brown micropipette puller (P‐97, Sutter Instruments, USA) and fire‐polished. The recording electrodes had a resistance of 4–6 MΩ when filled with different internal solutions. The liquid junction potential was auto‐adjusted each time by pipette offset. After the formation of whole‐cell recording, access resistances were generally < 20 MΩ.
To record NMDA‐activated currents, the pipette solution A (in mM: CsCl 140, EGTA 10, Mg2ATP 0.3, CaCl2 0.3, and HEPES 10, pH adjusted to 7.3 with CsOH) and the external solution (ES) A (in mM: NaCl 140, KCl 5, CaCl2 1, MgCl2 1, glucose 10, and HEPES 10, pH adjusted to 7.4 with NaOH) were used. The membrane potential was held at +40 mV. To record AMPA‐activated currents, the pipette solution B (in mM: potassium gluconate 120, KCl 20, MgCl2 2, HEPES 10, EGTA 10, and Na2ATP 2, pH adjusted to 7.3 with KOH) and ES A were used. The membrane potential was held at −70 mV.
To record epileptiform discharge or tonic NMDA receptor‐mediated currents, the pipette solution B and the ES B (in mM: NaCl 145, KCl 3, CaCl2 2, MgCl2 2, glucose 10, and HEPES 10, pH adjusted to 7.4 with NaOH) were used. In recording of epileptiform discharge, Mg2+‐free ES (0Mg) was prepared from the external solution B, in which MgCl2 was omitted. ES was used as a control. Cells were incubated in various ES for 30 min before recordings. In 0Mg+Cyclo group, cells were incubated in ES with Cyclo (10 μM in DMSO, used in all in vitro experiments) for 30 min and then transferred to 0Mg plus Cyclo and incubated for another 30 min. Recordings were performed for 10 min. The epileptiform burst is defined as a large depolarization shift with ≥ 10 mV depolarization and ≥ 300 ms in duration and at least five action potentials as described in the previous study 45. Neurons with at least two epileptiform bursts during 10 min recording were defined as “neurons showing epileptiform activity” 45.
In recording of D‐CPP (50 μM)‐sensitive tonic NMDA currents, 0Mg was used as control. The cells were incubated in 0Mg for 30 min before recording. In 0Mg+Cyclo group, the cells were incubated in the presence of Cyclo for 30 min and then transferred to 0Mg plus Cyclo and incubated for another 30 min. The membrane potential was held at −70 mV.
To record aspartate (Asp, 100 μM)‐evoked EAAC1 currents, the pipette solution C (in mM: KNO3 140, MgCl2 2.5, HEPES 10, EGTA 11, Na2ATP 5, and HEPES 10, pH adjusted to 7.3 with KOH) and the ES C (in mM: NaCl 135, KCl 5.4, CaCl2 1.8, MgCl2 1.3, glucose 10, HEPES 10, D‐CPPene (NMDA receptor antagonist) 0.05, CNQX (AMPA/KA receptor antagonist) 0.02, and bicuculline (GABAA receptor antagonist) 0.02, pH adjusted to 7.4 with NaOH) were used. The membrane potential was held at −80 mV.
To test the effect of Shh on neuronal excitability, a series of depolarizing currents from 0 to 200 in 5 pA step increment was injected 46 and the resting membrane potential, the averaged current threshold to induce the first action potential, and frequencies of firing in response to 70‐, 100‐, and 150‐pA current injections with or without Shh were determined with pipette solution B and the ES B.
Drug solutions were prepared in external solutions and applied to neurons by pressure using the 8‐Channel Focal Perfusion System (ALA Scientific Instruments, USA). Neurons were bathed constantly in external solutions between drug applications. Drug solution exchange was accomplished by electronic control.
Recordings in hippocampal slices
Hippocampal slices were prepared from male SD rats (90–140 g). Animals were anesthetized with 1% pentobarbital sodium, and their brains were rapidly removed. Transverse slices were cut 400 μm thick on a vibration microtome (Leica VT1000S, Leica Microsystems, Wetzlar, Germany) in ice‐cold dissection buffer (in mM: sucrose 213.26, KCl 2.5, MgSO4 2, CaCl2 0.5, NaH2PO4 1.25, NaHCO3 26, and D‐glucose 10). Before use, the slices were equilibrated in artificial cerebrospinal fluid (aCSF, in mM: NaCl 124, KCl 2.5, MgSO4 1, CaCl2 2, NaH2PO4 1.25, NaHCO3 26, and D‐glucose 10) saturated with 95% O2/5% CO2 for at least 1 h. Recordings were in a submerge chamber at a flow rate of 1.5–2 ml/min with aCSF.
To record spontaneous epileptiform discharge, the aCSF containing 6.5 mM KCl and 100 μM picrotoxin (Pic) was used and recordings were performed at 30°C with aCSF as a control. Slices were pre‐incubated with DMSO or Cyclo for 30 min and then perfused with Pic or Pic plus Cyclo. Borosilicate glass microelectrodes with 1–3 MΩ resistance filled with aCSF were positioned at CA1 stratum pyramidal. Recordings were performed for 30 min after the frequency of discharge was stabilized.
In paired‐pulse experiments, microelectrodes were positioned at CA1 stratum radiatum to record field excitatory postsynaptic potential (fEPSP). Paired‐pulse stimuli were delivered at 10, 20, 50, 100, 200, and 500‐ms intervals every 20 s at 30–40% of maximal response. Paired‐pulse ratio was the percentage of the second stimulus‐evoked fEPSP amplitude divided by the first stimulus‐evoked fEPSP amplitude in a given paired‐pulse stimuli in individual slices.
In recording of AMPA‐EPSC, picrotoxin (100 μM) was added with a holding at −70 mV and pipette solution B was used. In LTP experiment, theta‐burst stimulation (TBS, five trains of stimuli which contain five burst (five stimuli at 100 Hz) at 5 Hz repeated at 0.1 Hz) was given by a tungsten electrode positioned at striatum radiatum of CA1.
To record D‐CPP‐sensitive tonic NMDA receptor currents, Pic was used as a control. Whole‐cell recordings were performed at CA1 pyramidal neurons with a holding at +40 mV. The inner solution (in mM: CsCl 140, EGTA 10, Mg2ATP 0.3, CaCl2 0.3, and HEPES 10) was used. The cells were incubated in Pic for 30 min before recording. In Pic+Cyclo group, the cells were incubated in the presence of Cyclo for 30 min and then transferred to Pic plus Cyclo and incubated for another 30 min.
Data were acquired using MultiClamp700A and 700B and Digidata1322AA and 1440A (Axon Instruments, California, USA), sampled at 10 kHz, and filtered at 2 kHz. Offline analysis was done by Clampfit 9.0 and 10.2 software (Axon Instruments, California, USA).
ELISA
Briefly, hippocampal neurons (2 × 106 per 3.5‐cm dish) were cultured for 10 days and replaced with the medium with 450 μl Mg2+‐free external solution (0Mg) (as in electrophysiology). After incubation for different times (15, 30, or 45 min), all supernatant was immediately collected and assayed in the plate coated with anti‐Shh antibody. In the TTX experiment, cells were treated with 50 mM KCl, CNQX, 10 μM; APV, 100 μM with or without TTX (1 μM) for 30 min. In the electro‐stimulation experiments, the cells were stimulated at 20 Hz for 30 min 47, 48. All cell experiments used ES as a control. For determination of Shh secretion from slices, acute hippocampal slices (400 μm thick) were transferred to 900 μl oxygen‐bubbled aCSF (as a control) or Pic (as in electrophysiology) in 3.5‐cm dish and incubated for different times (0.5, 1, or 1.5 h). After incubation, slices were rinsed with manual stir bars, and the supernatant was collected and centrifuged at 4°C for 10 min. To detect the Shh levels under physiological condition, TBS (as in electrophysiology) was given by a tungsten electrode positioned at striatum radiatum of CA1. All the supernatant was applied in one anti‐Shh‐coated well with a two‐step incubation. Shh levels were determined by the ELISA kit (R&D MSHH00) and calculated from the standard curve prepared for each plate, using Origin 7.5 software. The standard curves were linear within the range used (0–500 pg/ml Shh). The quantities of Shh in experimental samples were always within the linear range of the standard curve.
Cytosolic Ca2+ measurement
Changes in [Ca2+]i concentration were measured using Fura‐2 AM (Invitrogen). Briefly, a total of 1 × 105 primary cultured neurons were seeded on coverslips and incubated with 2 μM Fura‐2 AM at 37°C for 25 min. Cells were washed three times with normal external solution (in mM: NaCl 120, KCl 16, CaCl2 2, MgCl2 2, glucose 12, sucrose 12, and HEPES‐free acid 10, pH 7.4) and imaged using a Nikon Eclipse Ti microscope (Nikon, Japan) with dual excitation wavelengths for Fura‐2 AM at 340 and 380 nm and detection of fluorescent emission at 500 nm.
High performance liquid chromatography (HPLC) assay
After two washes with external solution (in mM: NaCl 145, KCl 3, CaCl2 2, MgCl2 2, glucose 10, and HEPES 10, pH adjusted to 7.4 with NaOH), cells were incubated with vehicle (bovine serum albumin in external solution, BSA) for 30 min. Then, the incubating solution was collected for HPLC assay as a baseline control. Immediately after removing the solution, cells were treated with either BSA or Shh (Sigma‐Aldrich, Selleckchem, 500 ng/ml used in all experiments) for another 30 min. The incubating solution was also collected for HPLC test.
The HPLC system (Agilent Jordax Eclipse Plus C18 2.1 mm I.D × 150 mm analytical column, 3.5/micron) and fluorescence detector (Agilent G1321A HPLC‐FLD) with excitation wavelength at 340 nm and emission wavelength at 450 nm were used. Column temperature was maintained at 36°C. The mobile phase was formed by methanol, acetonitrile, and water.
3H‐glutamate uptake assay
After two washes with the uptake buffer (in mM: glucose 6, KCl 4, NaCl 130, CaCl2 1.3, MgSO4 1.2, KH2PO4 1, and HEPES [pH 7.3] 25), cells were pre‐incubated with vehicle (BSA), Shh, or inhibitors for 10 min. Uptake assays were started by adding [3H] L‐glutamic acid at 10−6 M final concentration (specific activity 25 Ci/mmol, PerkinElmer) diluted in the uptake buffer. Incubation was at 37°C for 6 min. The reaction was stopped by rapidly adding 1 ml of cold uptake buffer and followed by two washes with the cold medium. Then, 1 N NaOH was added to cells and the radioactivity was assessed by liquid scintillation counting.
Microdialysis
The mice were implanted with CMA 7 guide cannula (CMA microdialysis AB, Kista, Sweden) with the following coordinates (to bregma): 2.5 mm posterior, 3.1 mm lateral, and 2.4 mm below dura. Seven days after implantation, the guide cannula were replaced with CAM 7 microdialysis probes (CMA microdialysis AB, Kista, Sweden), the tip of which was covered with a 1.0 mm length of permeable hollow fiber. The dialysis cannula was connected to a microinfusion pump (CMA/100 microdialysis pump, CAM Microdialysis) and continuously perfused with aCSF at 1.0 μl/min. Following a 2‐h stabilization period (the concentration of glutamate is between 2 and 5 μM), the dialysates were collected in tubes at 15‐min intervals. The baseline lasted for 60 min before administration of SAG (Smoothened agonist, 4 mM via the microdialysis probe). The HPLC system was used for analysis of glutamate.
RNAi constructs and lentiviral vectors
Specific sequences of short hairpin RNA (shRNA) targeting rat EAAC1, GLAST mRNA sequence and a nonsense shRNA were designed and constructed into the pLentiLox3.7 (pLL3.7) lentiviral vector, which has a GFP tag 49. The lentivirus was packaged and amplified in HEK293T cells. The cultured hippocampal neurons were infected at an MOI of 5, unless otherwise noted. The shRNA sequences are described below.
Nonsense shRNA forward:
T‐(Gttctccgaacgtgtcacg)‐(TTCAAGA)‐(gacgtgacacgttcggagaaC)‐TTTTTTC;
Nonsense shRNA reverse:
TCGA GAAAAAA (Gttctccgaacgtgtcacg)‐(TCTCTTGAA)‐(cgtgacacgttcggagaaC)‐A
Rat shEAAC1‐2 forward:
T‐(Gccgtggcagctgtgttca)‐(TTCAAGAGA)‐(tgaacacagctgccacggC)‐TTTTTTC
Rat shEAAC1‐2 reverse:
TCGAGAAAAAA (Gccgtggcagctgtgttca)‐(TCTCTTGAA)‐(tgaacacagctgccacggC)‐A
Rat shEAAC1‐3 forward:
T‐(Gtcaacattgtgaacccct)‐(TTCAAGAGA)‐(aggggttcacaatgttgaC)‐TTTTTTC
Rat shEAAC1‐3 reverse:
TCGA GAAAAAA (Gtcaacattgtgaacccct)‐(TCTCTTGAA)‐(aggggttcacaatgttgaC)‐A
Rat shGLAST‐3 forward:
T‐(Ggatgtgaagagctacctg)‐(TTCAAGAGA)‐(caggtagctcttcacatcC)‐TTTTTTC
Rat shGLAST‐3 reverse:
TCGA GAAAAAA (Ggatgtgaagagctacctg)‐(TCTCTTGAA)‐(caggtagctcttcacatcC)‐A
Rat shGLAST‐4 forward:
T‐(Gaagcctgctttaaacagt)‐(TTCAAGAGA)‐(actgtttaaagcaggcttC)‐TTTTTTC
Rat shGLAST‐4 reverse:
TCGA GAAAAAA (Gaagcctgctttaaacagt)‐(TCTCTTGAA)‐(actgtttaaagcaggcttC)‐A.
Statistical analysis
Data were expressed as mean ± SEM. Statistical analysis for Ca2+ imaging, electrophysiology, HPLC, 3H‐glutamate uptake and ELISA was evaluated using Student's t‐test. P‐values less than 0.05 were considered statistically significant. All statistical analysis was performed using Office Excel 2004 (Microsoft Corporation, Redmond, WA) or Origin 7.5 (OriginLab). The results of the percentage of neurons showing epileptiform activity and burst frequency in hippocampal neurons used one‐way ANOVA. Other results used Student's t‐test. The F‐test was done always before Student's t‐test.
Author contributions
SF and SM conducted experiments and wrote the manuscript. CJ did whole‐cell recording in cultures and epilepsy model experiments. YS and SY did ELISA work and epilepsy model experiments. KZ, DL, and LF did whole‐cell recording in slices. YL did whole‐cell recording in cultures. JC did Ca2+ imaging analysis. All authors discussed the results and commented on the manuscript. YW supervised the study and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
The work was supported by the grant (81130081) from NNSF of China. The authors thank Y. Zhao for Shh, Gli1, and SmoA1 constructs, ZJ. Fan for technical assistance, and YF. Li for diagram modification.
EMBO Reports (2016) 17: 682–694
Reference
- 1. Jiang J, Hui CC (2008) Hedgehog signaling in development and cancer. Dev Cell 15: 801–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Charron F, Stein E, Jeong J, McMahon AP, Tessier‐Lavigne M (2003) The morphogen sonic hedgehog is an axonal chemoattractant that collaborates with netrin‐1 in midline axon guidance. Cell 113: 11–23 [DOI] [PubMed] [Google Scholar]
- 3. Belgacem YH, Borodinsky LN (2011) Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc Natl Acad Sci U S A 108: 4482–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harwell CC, Parker PR, Gee SM, Okada A, McConnell SK, Kreitzer AC, Kriegstein AR (2012) Sonic hedgehog expression in corticofugal projection neurons directs cortical microcircuit formation. Neuron 73: 1116–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Varjosalo M, Taipale J (2008) Hedgehog: functions and mechanisms. Genes Dev 22: 2454–2472 [DOI] [PubMed] [Google Scholar]
- 6. Robbins DJ, Fei DL, Riobo NA (2012) The Hedgehog signal transduction network. Sci Signal 5: re6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heo JS, Lee MY, Han HJ (2007) Sonic hedgehog stimulates mouse embryonic stem cell proliferation by cooperation of Ca2+/protein kinase C and epidermal growth factor receptor as well as Gli1 activation. Stem Cells 25: 3069–3080 [DOI] [PubMed] [Google Scholar]
- 8. Traiffort E, Charytoniuk D, Watroba L, Faure H, Sales N, Ruat M (1999) Discrete localizations of hedgehog signalling components in the developing and adult rat nervous system. Eur J Neurosci 11: 3199–3214 [DOI] [PubMed] [Google Scholar]
- 9. Sasaki N, Kurisu J, Kengaku M (2010) Sonic hedgehog signaling regulates actin cytoskeleton via Tiam1‐Rac1 cascade during spine formation. Mol Cell Neurosci 45: 335–344 [DOI] [PubMed] [Google Scholar]
- 10. Ihrie RA, Shah JK, Harwell CC, Levine JH, Guinto CD, Lezameta M, Kriegstein AR, Alvarez‐Buylla A (2011) Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron 71: 250–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morimoto K, Fahnestock M, Racine RJ (2004) Kindling and status epilepticus models of epilepsy: rewiring the brain. Prog Neurobiol 73: 1–60 [DOI] [PubMed] [Google Scholar]
- 12. During MJ, Spencer DD (1993) Extracellular hippocampal glutamate and spontaneous seizure in the conscious human brain. Lancet 341: 1607–1610 [DOI] [PubMed] [Google Scholar]
- 13. Fonnum F (1984) Glutamate: a neurotransmitter in mammalian brain. J Neurochem 42: 1–11 [DOI] [PubMed] [Google Scholar]
- 14. Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1–105 [DOI] [PubMed] [Google Scholar]
- 15. Rothstein JD, Dykes‐Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP et al (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686 [DOI] [PubMed] [Google Scholar]
- 16. Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, Coulter DA, Rothstein JD (2002) A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J Neurosci 22: 6372–6379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T et al (1997) Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT‐1. Science 276: 1699–1702 [DOI] [PubMed] [Google Scholar]
- 18. Maragakis NJ, Rothstein JD (2004) Glutamate transporters: animal models to neurologic disease. Neurobiol Dis 15: 461–473 [DOI] [PubMed] [Google Scholar]
- 19. Storck T, Schulte S, Hofmann K, Stoffel W (1992) Structure, expression, and functional analysis of a Na(+)‐dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci U S A 89: 10955–10959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J (2003) Regulation of glial glutamate transporter expression by growth factors. Exp Neurol 183: 124–135 [DOI] [PubMed] [Google Scholar]
- 21. Fang M, Lu Y, Chen GJ, Shen L, Pan YM, Wang XF (2011) Increased expression of Sonic hedgehog in temporal lobe epileptic foci in humans and experimental rats. Neuroscience 182: 62–70 [DOI] [PubMed] [Google Scholar]
- 22. Cao HY, Jiang YW, Liu ZW, Wu XR (2003) Effect of recurrent epileptiform discharges induced by magnesium‐free treatment on developing cortical neurons in vitro. Brain Res Dev Brain Res 142: 1–6 [DOI] [PubMed] [Google Scholar]
- 23. DeLorenzo RJ, Pal S, Sombati S (1998) Prolonged activation of the N‐methyl‐D‐aspartate receptor‐Ca2+ transduction pathway causes spontaneous recurrent epileptiform discharges in hippocampal neurons in culture. Proc Natl Acad Sci U S A 95: 14482–14487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Salazar P, Tapia R, Rogawski MA (2003) Effects of neurosteroids on epileptiform activity induced by picrotoxin and 4‐aminopyridine in the rat hippocampal slice. Epilepsy Res 55: 71–82 [DOI] [PubMed] [Google Scholar]
- 25. Morimoto K, Sato K, Sato S, Yamada N, Hayabara T (1998) Time‐dependent changes in neurotrophic factor mRNA expression after kindling and long‐term potentiation in rats. Brain Res Bull 45: 599–605 [DOI] [PubMed] [Google Scholar]
- 26. Schmutz M, Portet C, Jeker A, Klebs K, Vassout A, Allgeier H, Heckendorn R, Fagg GE, Olpe HR, van Riezen H (1990) The competitive NMDA receptor antagonists CGP 37849 and CGP 39551 are potent, orally‐active anticonvulsants in rodents. Naunyn Schmiedebergs Arch Pharmacol 342: 61–66 [DOI] [PubMed] [Google Scholar]
- 27. Chen JK, Taipale J, Young KE, Maiti T, Beachy PA (2002) Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A 99: 14071–14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stanton BZ, Peng LF, Maloof N, Nakai K, Wang X, Duffner JL, Taveras KM, Hyman JM, Lee SW, Koehler AN et al (2009) A small molecule that binds Hedgehog and blocks its signaling in human cells. Nat Chem Biol 5: 154–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hall JM, Bell ML, Finger TE (2003) Disruption of sonic hedgehog signaling alters growth and patterning of lingual taste papillae. Dev Biol 255: 263–277 [DOI] [PubMed] [Google Scholar]
- 30. Parra LM, Zou Y (2010) Sonic hedgehog induces response of commissural axons to Semaphorin repulsion during midline crossing. Nat Neurosci 13: 29–35 [DOI] [PubMed] [Google Scholar]
- 31. Herman MA, Jahr CE (2007) Extracellular glutamate concentration in hippocampal slice. J Neurosci 27: 9736–9741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo Y, Wei Q, Huang Y, Xia W, Zhou Y, Wang S (2013) The effects of astrocytes on differentiation of neural stem cells are influenced by knock‐down of the glutamate transporter, GLT‐1. Neurochem Int 63: 498–506 [DOI] [PubMed] [Google Scholar]
- 33. Pita‐Almenar JD, Zou S, Colbert CM, Eskin A (2012) Relationship between increase in astrocytic GLT‐1 glutamate transport and late‐LTP. Learn Mem 19: 615–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA (2000) Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 406: 1005–1009 [DOI] [PubMed] [Google Scholar]
- 35. Riobo NA, Saucy B, Dilizio C, Manning DR (2006) Activation of heterotrimeric G proteins by Smoothened. Proc Natl Acad Sci U S A 103: 12607–12612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA (2011) Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J Biol Chem 286: 19589–19596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barzi M, Kostrz D, Menendez A, Pons S (2011) Sonic Hedgehog‐induced proliferation requires specific Galpha inhibitory proteins. J Biol Chem 286: 8067–8074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gaillet S, Plachez C, Malaval F, Bezine MF, Recasens M (2001) Transient increase in the high affinity [3H]‐L‐glutamate uptake activity during in vitro development of hippocampal neurons in culture. Neurochem Int 38: 293–301 [DOI] [PubMed] [Google Scholar]
- 39. Beart PM, O'Shea RD (2007) Transporters for L‐glutamate: an update on their molecular pharmacology and pathological involvement. Br J Pharmacol 150: 5–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smolders I, Khan GM, Manil J, Ebinger G, Michotte Y (1997) NMDA receptor‐mediated pilocarpine‐induced seizures: characterization in freely moving rats by microdialysis. Br J Pharmacol 121: 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Curia G, Longo D, Biagini G, Jones RS, Avoli M (2008) The pilocarpine model of temporal lobe epilepsy. J Neurosci Methods 172: 143–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. He XP, Minichiello L, Klein R, McNamara JO (2002) Immunohistochemical evidence of seizure‐induced activation of trkB receptors in the mossy fiber pathway of adult mouse hippocampus. J Neurosci 22: 7502–7508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Racine RJ (1972) Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32: 281–294 [DOI] [PubMed] [Google Scholar]
- 44. Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27‐supplemented Neurobasal, a new serum‐free medium combination. J Neurosci Res 35: 567–576 [DOI] [PubMed] [Google Scholar]
- 45. Sun Y, Wu Z, Kong S, Jiang D, Pitre A, Wang Y, Chen G (2013) Regulation of epileptiform activity by two distinct subtypes of extrasynaptic GABAA receptors. Mol Brain 6: 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cummins TR, Rush AM, Estacion M, Dib‐Hajj SD, Waxman SG (2009) Voltage‐clamp and current‐clamp recordings from mammalian DRG neurons. Nat Protoc 4: 1103–1112 [DOI] [PubMed] [Google Scholar]
- 47. Hirase H, Leinekugel X, Czurko A, Csicsvari J, Buzsaki G (2001) Firing rates of hippocampal neurons are preserved during subsequent sleep episodes and modified by novel awake experience. Proc Natl Acad Sci U S A 98: 9386–9390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Louie K, Wilson MA (2001) Temporally structured replay of awake hippocampal ensemble activity during rapid eye movement sleep. Neuron 29: 145–156 [DOI] [PubMed] [Google Scholar]
- 49. Rubinson DA, Dillon CP, Kwiatkowski AV, Sievers C, Yang L, Kopinja J, Rooney DL, Zhang M, Ihrig MM, McManus MT et al (2003) A lentivirus‐based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nat Genet 33: 401–406 [DOI] [PubMed] [Google Scholar]
- 50. Zhang XM, Ramalho‐Santos M, McMahon AP (2001) Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R symmetry by the mouse node. Cell 106: 781–792 [PubMed] [Google Scholar]
- 51. Long F, Zhang XM, Karp S, Yang Y, McMahon AP (2001) Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 128: 5099–5108 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File