

Abstract
Allele-specific expression (ASE) of the Adenomatous Polyposis Coli (APC) gene occurs in up to one-third of families with adenomatous polyposis (FAP) that have screened mutation-negative by conventional techniques. To advance our understanding of the genomic basis of this phenomenon, 54 APC mutation-negative families (21 with classical FAP and 33 with attenuated FAP, AFAP) were investigated. We focused on four families with validated ASE and scrutinized these families by sequencing of the blood transcriptomes (RNA-seq) and genomes (WGS). Three families, two with classical FAP and one with AFAP, revealed deep intronic mutations associated with pseudoexons. In all three families, intronic mutations (c.646-1806T>G in intron 6, c.1408+729A>G in intron 11, and c.1408+731C>T in intron 11) created new splice donor sites resulting in the insertion of intronic sequences (of 127 bp, 83 bp, and 83 bp, respectively) in the APC transcript. The respective intronic mutations were absent in the remaining polyposis families and the general population. Premature stop of translation as the predicted consequence as well as co-segregation with polyposis supported the pathogenicity of the pseudoexons. We conclude that next generation sequencing on RNA and genomic DNA is an effective strategy to reveal and validate pseudoexons that are regularly missed by traditional screening methods and is worth considering in apparent mutation-negative polyposis families.
Keywords: familial adenomatous polyposis, APC, pseudoexon, RNA-seq, allele-specific expression
INTRODUCTION
Familial adenomatous polyposis (FAP; OMIM #175100) is characterized by a dominant predisposition to multiple adenomatous polyps throughout the colon and rectum as a consequence of germline mutations in the Adenomatous Polyposis Coli (APC) gene [1]. While FAP mostly represents an inherited disease, up to 25% may result from de novo mutations of APC without any family history of the disease [2]. The number of adenomatous polyps in the bowel is used to stratify APC-associated polyposis into a classical form (FAP; 100 adenomas or more) and attenuated form (AFAP; below 100 adenomas). These two phenotypes additionally differ relative to the onset of polyposis (in the second or third decades of life in FAP vs. later in AFAP), colonic location (left-sided disease in FAP vs. frequently right-sided disease in AFAP), and life-time risk of colorectal cancer (100% in FAP vs. up to 70% in AFAP) [1, 3].
The APC gene has 16 exons and translation starts from exon 2 (http://insight-database.org/genes/APC). More than 1,500 unique germline mutations in APC are known [4]. The frequency of detectable APC mutations in polyposis patients varies a lot depending on the method of ascertainment of the patients and families, and the strategies used for mutation screening. In a large cohort of individuals who had undergone clinical genetic testing because of a personal or family history of polyposis, 58% (851/1457) of those with classic polyposis and 9% (376/4223) of those with AFAP had APC mutations by exon-specific sequencing and large rearrangement analysis of the APC gene [3]. Moreover, Grover et al. [3] found that the APC mutation rate progressively increased with the cumulative adenoma count (being 80% in individuals with at least one thousand adenomas), while the mutation rate of MUTYH, which is another polyposis-associated gene, remained constant (below 10 percent) across all polyp number categories. Sanger sequencing of genomic DNA to examine the coding exons and intron-exon boundaries of APC, combined with multiplex ligation-dependent probe amplification (MLPA) for large rearrangements is the standard mutation screening strategy adopted by most laboratories [4]. The protein truncating test (PTT) was commonly used in previous years and may be beneficial in certain situations [5]. In a typical PTT design, APC exons are examined in RNA, except for the last exon that is investigated in genomic DNA. Nevertheless, over 20% of classical FAP and up to 80% of AFAP patients remain APC mutation-negative, which may be attributable to methodological shortcomings in association with particular types of mutations [5–8], nontruncating alterations with uncertain pathogenic significance [2], and susceptibility associated with other genes than APC, such as MUTYH [3], POLE and POLD [9], and AXIN2 [10].
Unbalanced expression of the two parental alleles, due to loss-of-function mutations or various cis- or trans-acting factors, may facilitate the identification of susceptibility genes for human diseases [11]. APC mutations occurring prior to the last exon of the gene are associated with allele-specific expression (ASE) [12]. ASE imbalance of APC has been found in blood samples from 9 – 31% of adenomatous polyposis families without any detectable APC mutations by conventional techniques, suggesting the existence of hidden mutations [12–14]. Moreover, ASE of APC may contribute to common forms of colorectal cancer, as colorectal cancer risk has been shown to increase along with increasing ASE imbalance [15].
This study was undertaken to address the underlying basis of predisposition in 54 APC mutation-negative adenomatous polyposis families from Finland, with a particular focus on families with constitutionally unbalanced mRNA expression of APC alleles by Single Nucleotide Primer Extension (SNuPE) [13]. Interrogation of the latter four families by whole transcriptome (RNA-seq) and whole-genome (WGS) sequencing revealed deep intronic mutations associated with pseudoexons in three of four families.
RESULTS
Identification of pseudoexons by RNA-seq and deep intronic mutations as their underlying causes
We focused on three FAP families (42, 85, and 103) from the research-based cohort (Figure 1 and Table 1). The families were associated with ASE imbalance of APC by SNuPE but no identifiable causative change in APC had been detected by PTT, Sanger sequencing of all exons and intron/exon borders, MLPA, and promoter mutation and methylation analyses (ref. [8] and this study). Only family 85 included several affected members. Of these, 85-1 [13] and 85-2 (Supplementary Figure S1) showed ASE imbalance, whereas 85-3 was uninformative in ASE analysis due to homozygosity for polymorphisms. No RNA was available from 85-4.
Figure 1. Pedigrees of ASE families.
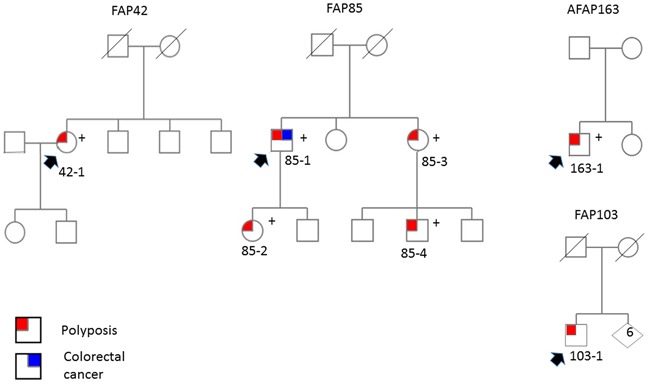
Pedigrees of adenomatous polyposis families with ASE. Individuals with polyposis and/or colorectal cancer are indicated (see Table 1 for additional clinical details). Plus sign denotes carriers of deep intronic mutations associated with pseudoexons of APC. Index persons are marked with arrows.
Table 1. Clinical and molecular characteristics of polyposis cases investigated.
| Case IDa | ASE statusb | Inheritance pattern | Number of polyps | Age at diagnosisc | Extracolonic manifestationsd | Classification of familye | Large rearrangement by MLPAf | APC methylation by MS-MLPAg | |
|---|---|---|---|---|---|---|---|---|---|
| RESEARCH BASED | 42 | ASE | Sporadic | 100-1000 | 40 | No | FAP | No | No |
| 78 | N | Sporadic | 50 | 55 | No | AFAP | No | No | |
| 85-1 | ASE | dominant | 2000 | 38 | Yes | FAP | No | No | |
| 85-2 | ASE | dominant | 100-200 | 16 | No | FAP | No | No | |
| 85-3 | NI | dominant | 2000 | 44 | No | FAP | NA | No | |
| 85-4 | dominant | 100-200 | 12 | No | FAP | NA | No | ||
| 88 | N | sporadic | 100-1000 | 58 | No | FAP | No | No | |
| 92 | N | sporadic | 200 | 51 | No | FAP | No | No | |
| 96 | NI | sporadic | 561 | 48 | No | FAP | No | No | |
| 97 | N | sporadic | > 1000 | 58 | No | FAP | No | No | |
| 98 | dominant | 100-1000 | 30 | No | FAP | No | No | ||
| 100 | NI | sporadic | 30 | 62 | No | AFAP | No | No | |
| 103 | (ASE) | sporadic | > 100 | 51 | No | FAP | No | No | |
| 104 | N | dominant? | 210 | 54 | Yes | FAP | No | No | |
| 111 | NI | sporadic | 30-40 | 36 | No | AFAP | No | No | |
| 123 | NI | sporadic | 2100 | 37 | No | FAP | No | No | |
| 125 | N | sporadic | 300 | 31 | No | FAP | No | No | |
| CLINIC BASED | 134 | sporadic | 200-300 | 55 | No | FAP | No | No | |
| 136 | sporadic | >100 | 67 | Yes | FAP | No | No | ||
| 139 | sporadic | 100 | 71 | No | FAP | No | No | ||
| 145 | recessive | 20-50 | 61 | No | AFAP | No | No | ||
| 148 | sporadic | 150-200 | 50 | No | FAP | No | No | ||
| 158 | sporadic | 50 | 49 | Yes | AFAP | No | No | ||
| 159 | sporadic | 200 | 50 | No | FAP | No | No | ||
| 162 | sporadic | >50 | 52 | No | AFAP | No | No | ||
| 163 | (ASE) | sporadic | 10-20 | 16 | Yes | AFAP | No | No | |
| 165-1 | dominant? | Colon cancer x 2 | 50 | NA | AFAP | No | No | ||
| 165-2 | dominant? | 20-30 | 33 | Yes | AFAP | No | No | ||
| 168 | sporadic | 100 | 56 | Yes | FAP | No | No | ||
| 177 | sporadic | 100-200 | 52 | No | FAP | No | No | ||
| 179 | dominant? | >10 | 23 | No | AFAP | No | No | ||
| 180 | NA | >100 | 38 | NA | FAP | No | No | ||
| 1001 | dominant | 10 | 48 | NA | AFAP | No | No | ||
| 1003 | sporadic | 20-30 | 70 | NA | AFAP | No | No | ||
| 1005 | dominant | 10-20 | 68 | Yes | AFAP | No | No | ||
| 1006 | sporadic | 20 | 60 | No | AFAP | No | No | ||
| 1007 | sporadic | 20 | 30 | No | AFAP | No | No | ||
| 1010 | dominant | 5-10 | 68 | NA | AFAP | No | No | ||
| 1011 | NA | 60-100 | 31 | NA | FAP | No | No | ||
| 1013 | sporadic | >100 | 48 | NA | FAP | No | No | ||
| 1015 | sporadic | 10 | 47 | Yes | AFAP | No | No | ||
| 1017 | sporadic? | 10-20 | 57 | NA | AFAP | No | No | ||
| 1018 | sporadic | 20-30 | 74 | NA | AFAP | No | No | ||
| 1019 | sporadic | 2-3 | 30 | Yes | AFAP | No | No | ||
| 1020 | sporadic | 3 | 35 | Yes | AFAP | No | No | ||
| 1021 | sporadic | 30 | 72 | NA | AFAP | No | No | ||
| 1022 | dominant | 3 | 65 | Yes | AFAP | No | No | ||
| 1023 | sporadic | 40 | 33 | NA | AFAP | No | No | ||
| 1024 | sporadic | 20 | 72 | NA | AFAP | No | No | ||
| 1025 | sporadic | 20-30 | 67 | NA | AFAP | No | No | ||
| 1026 | sporadic | 10-20 | 51 | NA | AFAP | No | No | ||
| 1029 | sporadic | 20-30 | 56 | No | AFAP | No | No | ||
| 1030 | sporadic | >10 | 59 | No | AFAP | No | No | ||
| 1032 | NA | 8 | 63 | Yes | AFAP | No | No | ||
| 1034 | NA | >10 | 62 | No | AFAP | No | No | ||
| 1035 | sporadic | 20-30 | 71 | No | AFAP | No | No | ||
| 1036 | dominant | >10 | 61 | No | AFAP | No | No | ||
| 1037 | sporadic | ~10 | 52 | No | AFAP | No | No |
Identification number of family, followed by identification number of individual if several family members were studied.
ASE, shows allele-specific expression of APC; (ASE), putative ASE (see Materials and Methods); N, no ASE; NI, not informative (homozygous); blank, no RNA available
Polyposis or colorectal carcinoma, whichever comes first
Desmoids and duodenal adenomas in particular
Based on the number of intestinal adenomas with 100 as the cut-off
P043-C1 assay from MRC-Holland
ME001-C1 assay from MRC-Holland
NA, information not available
Blood RNA specimens from the three ASE families were subjected to RNA-seq. Data analysis revealed aberrant splice junctions which raised a suspicion of pseudoexons, i.e., inclusion of intronic sequence in the mature mRNA, in families 42 and 85 (Figure 2). To verify pseudoexons, APC cDNA was amplified in five overlapping fragments with primers described in Spier et al. [6] in addition to which primers from exons 11 (forward) and 13 (reverse) were used to evaluate the suspected pseudoexon in family 85 (Supplementary Table S1 and Figure 3). Sequencing of reverse transcription (RT)-PCR products (fragment 2 in family 42 and fragment 4 as well as the exon 11-13-specific fragment in family 85) revealed a 127-bp insertion from intron 6 in family 42 and an 83-bp insertion from intron 11 in family 85.
Figure 2. RNA-seq (42, 85-2, and 163).
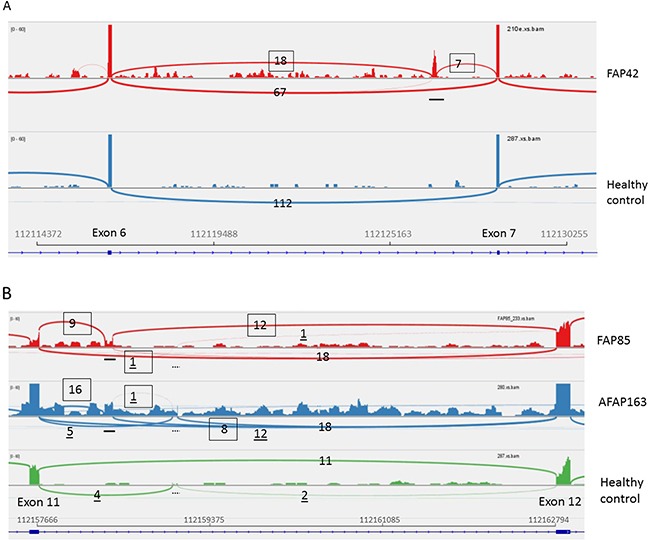
Sashimi plots to visualize splice junctions. IGV display of RNA-seq data is provided for an affected representative of each family and a healthy control individual for reference for each region. Sequence alignments are based on TopHat. The region between APC exons 6 and 7 (GRCh37/Hg19) is shown for FAP42 (Figure 2A) and that between exons 11 and 12 for FAP85 (individual 85-2) and AFAP163 (Figure 2B). The locations of pseudoexons are indicated by horizontal bars. A 54-bp in-frame insertion present in the normal reference sample, too, and not associated with any genomic change is denoted by a dashed bar (Figure 2B). The same insertion was discovered in an earlier investigation [12]. Numbers on the plots indicate APC exon coverages expressed as junction depth. Splice events corresponding to pseudoexons are boxed and those associated with the 54-bp insertion are underlined; the remaining ones represent canonical splicing.
Figure 3. RT-PCR (42, 85-1, 163, 103).
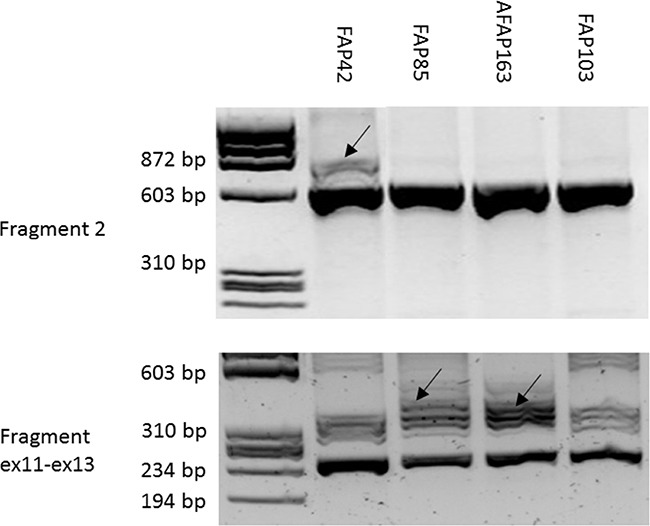
RT-PCR analysis of samples from ASE families. RT-PCR products separated by gel electrophoresis are shown. Arrows denote fragments with intronic insertions (pseudoexons). Fragment 2 (upper panel) encompasses a 615-bp cDNA segment from exon 4 to exon 9 [6] and shows a heterozygous 127-bp insertion in family 42. The wild-type size of the exon 11 - exon 13 fragment (lower panel) is 246 bp (Supplementary Table S1). An identical 83-bp insertion in families 85 (case 85-1) and 163 is evident. The RT-PCR products from the index persons and healthy controls were cloned and sequenced to verify their DNA sequences. In the exon 11 - exon 13 fragment, a 54-bp in-frame insertion (see legend for Figure 2) accompanied the pseudoexon and wild-type sequences in a proportion (up to one-third) of all clones and likely contributed to the multiplicity of fragments seen after gel electrophoresis.
As the predisposing mutations of the families were unknown, WGS on blood DNA was applied. At the outset, mutations in the APC coding region and exon/intron borders had been screened for (see Materials and Methods). Particularly, WGS offered the opportunity to investigate the entire introns of APC as well as regions outside APC. Families 42 and 85 revealed deep intronic mutations, both creating new splice donor sites (/gt): c.646-1806T>G in intron 6 and c.1408+731C>T in intron 11 of APC, respectively (Figure 4). The changes were validated by Sanger sequencing.
Figure 4. WGS (42 and 85-2) + Sanger seq.
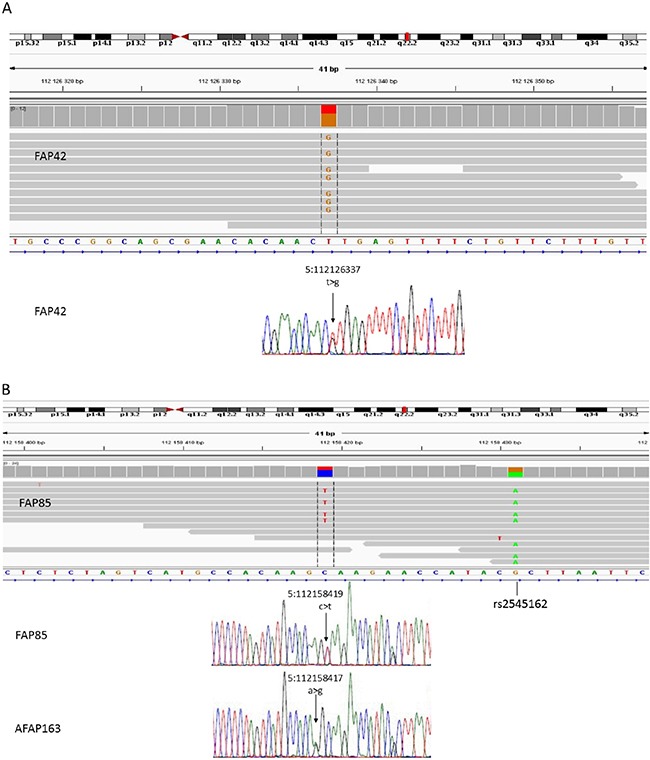
Deep intronic mutations in APC. Upper panels provide IGV display of WGS data for intron 6 in FAP42 (Figure 4A) and intron 11 in FAP85, individual 85-2 (Figure 4B). Lower panels show Sanger sequence tracings of the mutations. In Figure 4B, the Sanger sequencing result of AFAP163 is also given (AFAP163 was not included in WGS analysis).
The remaining 51 families (Table 1) were subsequently screened by Sanger sequencing with primers from introns 6 and 11 of APC (Supplementary Table S1) to examine the presence of the deep intronic mutations identified in families 42 and 85. These particular mutations were absent in the remaining families. Incidentally, however, family 163 revealed another nucleotide substitution (c.1408+729A>G) two nucleotides upstream of the mutation present in family 85 (Figure 4). The nucleotide change in family 163 was predicted to activate the same cryptic splice donor site (AG/gt) as the mutation in family 85 (the nucleotide substitutions created an apparently viable AG/ and /gt, respectively) (Figure 5). Family 163 represented a clinic-based cohort for which only DNA was routinely available. However, we were able to obtain RNA from the single affected family member in a separate effort. RNA-seq (Figure 2) and RT-PCR (Figure 3) identified an 83-bp insertion from intron 11, identical to that in family 85. Furthermore, the c.1408+729A>G mutation was part of the resulting transcript unlike the deep intronic mutations of families 42 and 85 (Figure 5). Analysis of the individual RNA reads with pseudoexons validated the presence of variant nucleotide (G) at the position of the mutation, indicating that the variant nucleotide was specifically associated with pseudoexon formation (Supplementary Figure S2). Finally, SNuPE analysis of cDNA from the index individual from family 163 showed putative ASE with the value of 1.7 for the ratio of allelic peak areas in cDNA relative to genomic DNA at rs2229992 (Supplementary Figure S1).
Figure 5. Schematic diagrams of pseudoexons.
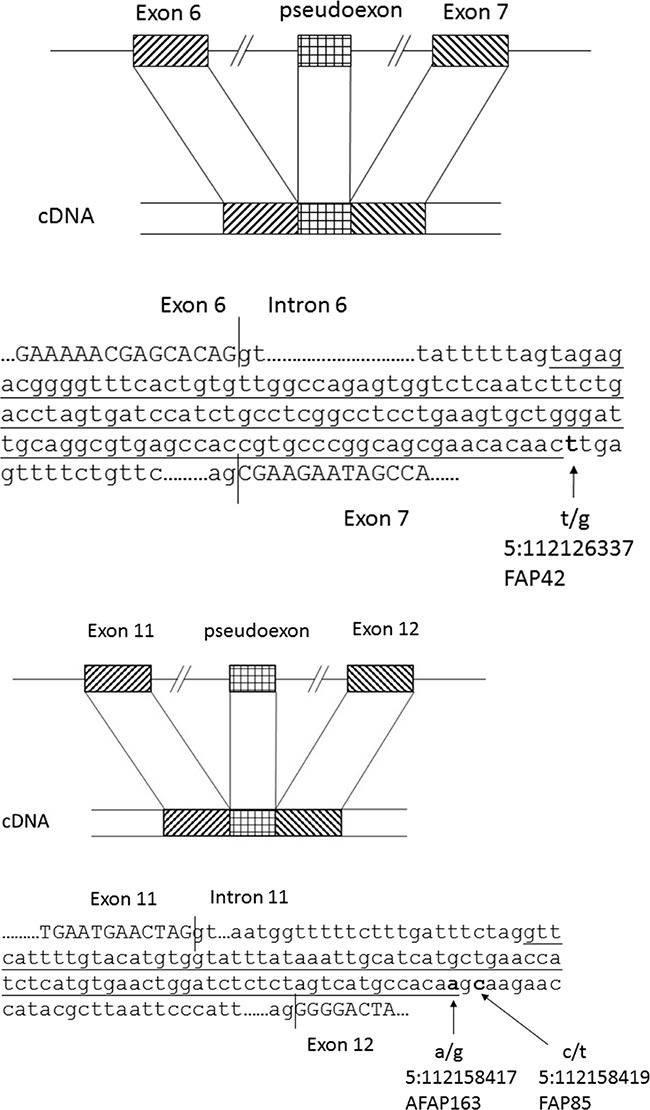
Schematic diagrams of APC pseudoexons identified. The canonical splice sites at the exon/intron borders, pseudoexons (underlined), and the responsible deep intronic mutations (in bold) are highlighted.
Pathogenicity of pseudoexons
The pseudoexon findings are summarized in Table 2. The pseudoexons in families 42, 85, and 163 were all predicted to cause premature stop of translation; the very first three nucleotides of the pseudoexon in family 42 coded for a stop of translation, whereas in families 85 and 163 the pseudoexon caused a frameshift and a premature stop 55 codons later. The following evidence supports the idea that the pseudoexons underlay polyposis predisposition in all three families. First, the splice prediction program BDGP (Materials and Methods) indicated a splice efficiency of 99% for the new splice donor sites introduced in intron 6 in family 42 and intron 11 in family 85. The new splice donor site in intron 11 in family 163 did not match with the canonical splice site model and was therefore not recognized by the splice prediction programs. However, RNA-seq and our cloning experiment showed that all pseudoexon-containing transcripts had the variant nucleotide G in the 3' end of the pseudoexon (Supplementary Figure S2). Moreover, our cloning experiment (on the ex 11 – 13 fragment, see legend to Figure 3) combined with haplotype analysis (with SNuPE markers) suggested that all transcripts representing the mutant allele, as inferred from haplotypes, had the pseudoexon inserted (data not shown). Second, the intronic variant showed a complete co-segregation with polyposis in family 85 (Figure 1). The variants were also absent in the general population (ExAC Browser Beta, SISu and Ensembl databases and our investigation of 300 anonymous blood donors from Finland). Finally, WGS data available for families 42 and 85 revealed no other apparently pathogenic mutations in established cancer genes as possible alternative explanations for polyposis predisposition.
Table 2. Summary of the variants.
| Family | Location in APC (GRCh37/GRCh38) | Insertion length (bp) | Genomic variant | RNA alteration | Predicted protein alteration |
|---|---|---|---|---|---|
| FAP42 | intron 6 (5:112126337/5:112790640) | 127 | c.646-1806T>G | r.645_646ins646-1933_646-1807 | p.Arg216* |
| FAP85 | intron 11 (5:112158419/5:112822720) | 83 | c.1408+731C>T | r.1408_1409ins1408+647_1408+729 | p.Gly471Serfs*55 |
| AFAP163 | intron 11 (5:112158417/5:112822722) | 83 | c.1408+729A>G | r.1408_1409ins1408+647_1408+729 | p.Gly471Serfs*55 |
Note: An updated APC nomenclature based on 16 exons (NM_012583R138.4, ENST012583R10257430) was used for exon annotation.
SNuPE vs. RNA-seq in the detection of ASE
The ASE diagnoses of the four families (42, 85, 103, and 163) with unbalanced expression of APC alleles in our series (Table 1) were initially based on SNuPE. To evaluate if ASE imbalance was also recoverable in RNA-seq data, a genome-wide ASE imbalance analysis was performed as described in Materials and Methods. The results are given in Supplementary Table S2. Applying stringent criteria for ASE, FAP42 and FAP85 (individual 85-2) revealed unequivocal ASE for APC (q-value < 0.05). Three APC-mutation-positive cases not belonging to the study series specified in Table 1 were also included, and ASE was detected in one (the remaining two were uninformative). FAP85 (individual 85-1) and FAP103, as well as healthy control sample 3, showed borderline ASE which was, however, not statistically significant after multiple hypothesis correction (q value > 0.05 and ≤ 0.15). The ASE value for APC in AFAP163 did not reach statistical significance. As shown in Supplementary Table S2, the overall concordance between ASE results by SNuPE and RNA-seq was high.
DISCUSSION
Canonical splice-site sequences at the intron/exon borders define exons. The canonical 5' (splice donor) site has a consensus sequence AG/gtragtand the 3' (splice acceptor) site poly(y)nyag/G (where capital letters indicate exonic and lowercase letters intronic sequence, r denotes purine, y pyrimidine, and n any nucleotide, and the nearly invariant nucleotides are underlined) [16]. Pseudoexons are intronic sequences of 50 – 300 bp in length that have apparent 5' and 3' splice sites, but are normally ignored by the splicing machinery [17, 18]. Pseudoexons can be activated by mutations that create viable splice donor or acceptor sites by different mechanisms, resulting in the insertion of intronic sequences in the mature mRNA [19]. Such mutations can be inherited and may cause predisposition to cancer syndromes, including ataxia-telangiectasia (ATM) [20], breast and ovarian cancer (BRCA2) [21], Lynch syndrome (MSH2) [22] and familial adenomatous polyposis (APC) (ref. [6] and this study). From the therapeutic point of view, location far outside the coding sequence makes deep intronic mutations excellent candidates for correction by antisense oligonucleotides to restore the production of normal protein [20, 21].
Using RNA-seq and WGS, we discovered two different pseudoexons (127-bp insertion from intron 6 and 83-bp insertion from intron 11) caused by three different heterozygous germline mutations in APC. To our knowledge, our effort is the first one successfully identifying pseudoexons in APC using next-generation sequencing and the second ever to reveal APC-related pseudoexons in FAP. The study by Spier et al. [6] was the first report and described two different APC pseudoexons (167-bp insertion from intron 5 and 83-bp insertion from intron 11). These pseudoexons were caused by three different heterozygous germline mutations. By RT-PCR screen of APC cDNA from 125 APC- and MUTYH mutation-negative adenomatous polyposis cases from Germany, a frequency of 6.4% (8/125 individuals) was obtained for cases with an identifiable genomic change underlying pseudoexon formation. Interestingly, the pseudoexon in intron 11 occurring in our family 85 in association with c.1408+731C>T nucleotide substitution was on genomic DNA and RNA level precisely the same as that present in two unrelated German patients [6]. The region around position +731 in intron 11 may be prone to pseudoexon formation in general, given the existence of two additional pseudoexon-associated nucleotide substitutions in this region, one located two nucleotides upstream (our study) and another one six nucleotides downstream of position +731 [6]. The overall frequency of APC pseudoexons in our series from Finland (3/54 index patients, 5.5%) may be an underestimate since our full pseudoexon screen focused on four index patients with unbalanced expression of APC alleles, whereas the remaining index patients (with mainly DNA available only) underwent a targeted screen for the same mutations identified in the former patients.
Diagnostic strategies mostly target coding regions in DNA [4]. Detection of disease-associated pseudoexons in turn requires simultaneous RNA- and DNA-based evidence to demonstrate the insertion of extraneous sequence in mRNA and distinguish transcriptional post-modification errors from deep intronic mutation in genomic DNA as the mechanistic basis of insertion. Hence, validated disease-associated pseudoexons have remained scarce [6, 20–22], despite the fact that potential pseudoexons are frequent in introns of human genes [18]. The pseudoexons in families 42 and 85 were missed by our original PTT screen [23]. Family 42 did reveal a visible truncation, but the subsequent search of a causative change by Sanger sequencing of genomic DNA did not extend deep into the introns [23]. On the other hand, no convincing extra fragment was visible for FAP85. This is likely attributable to some commonly observed disadvantages of PTT, such as decreased RNA stability and assay artifacts [24]. Instead of PTT, family 163 originally underwent an exon-by-exon screen in genomic DNA [8] that, obviously, was not able to capture deep intronic mutations.
Family 103 showed putative ASE imbalance (Table 1, Supplementary Table S2), but neither RNA-seq nor WGS revealed variants that might underlie the suggestive ASE phenotype. This apparently sporadic case with classical FAP (Figure 1) might be explained by a mosaicism for APC mutation; such mutations are challenging to detect and verify [5]. Eventual in-cis or in-trans regulatory changes or complex rearrangements escaping detection by sequencing would be examples of other theoretical possibilities to consider in future investigations. The ASE phenotype in FAP103 affected many other genes beyond APC (Supplementary Table S2), offering possible candidate genes to be tested for germline alterations. It is important to note that up to ~20% of all informative genes expressed in lymphoblastoid cells/blood may show ASE even in healthy control individuals [25, 26], and the underlying cause remains elusive for most genes.
The site of germline mutation in the APC gene is known to correlate with the disease phenotype [1]. Our family 42 with pseudoexon 6/7 was associated with classical FAP in agreement with genotype-phenotype expectations. Among the two pseudoexon 11/12 families, family 85 complied with established genotype-phenotype correlations by showing classical FAP like the two German families with the same mutation. Family 163 was classified as AFAP based on polyp count (10 – 20), but also showed features more typical (although not exclusive) of a profuse form of FAP such as low age at onset (16 years) and presence of extracolonic manifestations (mandibular osteomas) (Table 1). In FAP85, we demonstrated co-segregation of the respective genomic change with polyposis (Figure 1). Unfortunately, segregation studies were not possible in the remaining two families because of the lack of additional affected members.
Next-generation sequencing techniques are changing the screening for predisposing mutations. Targeted gene panels capturing the entire introns in addition to exons and combined with deep sequencing are likely to replace current screening protocols that rely on exon-specific Sanger sequencing and MLPA [4, 27]. We show that deep intronic mutations of the APC gene explained three out of four FAP and AFAP families displaying ASE imbalance and remaining mutation-negative by traditional methods. This indicates that our strategy to use ASE for pre-selection of cases for pseudoexon testing was effective and could even serve as a proxy for the initial screening of out-of-frame pseudoexon insertion events in FAP and AFAP. Unavailability of RNA made ASE and pseudoexon screening impossible in a significant fraction of our polyposis families (Table 1); hence, investigation of larger series is necessary for a reliable determination of the frequency and clinical significance of ASE and pseudoexon events in this disease. In the clinical context, pathogenicity of pseudoexons requires special attention. Considerations we point out (see Results above) as well as recommendations valid to any splicing aberrations [28] would apply. In our experience, next generation sequencing on RNA and genomic DNA facilitate pseudoexon identification and provide valuable tools to explore the genetic basis of mutation-negative families.
MATERIALS AND METHODS
Patients and samples
The series consisted of 54 unrelated families/cases from Finland, including 21 with classical FAP and 33 with AFAP (Table 1). Fourteen families represented a research-based cohort from the nation-wide Hereditary Colorectal Cancer Registry of Finland [13] lacking APC point mutations by PTT and exon-specific screening methods (heteroduplex analysis and Sanger sequencing) and large rearrangements by MLPA [8] (P043-C1). The remaining 40 families represented a clinic-based cohort of consecutive index cases with newly diagnosed FAP or AFAP and overlapped with the series described in ref. [8]. These cases were recruited via clinical genetic units of Finnish university hospitals, and cases remaining APC mutation-negative after exon-specific sequencing and MLPA were eligible (additionally, APC epimutations were excluded by methylation-specific multiplex ligation-dependent probe amplification [8]). MUTYH-positive cases and occasional cases with mutations in other polyposis-related genes were excluded. Cases with allele-specific expression (ASE) of APC were 42, 85-1, 85-2, 103, and 163 (ref. [13] and this study). No ASE was detected in cases 78, 88, 92, 97, 104, and 125 [13]. The remaining families/cases were uninformative or not tested for ASE because of the lack of RNA (as a rule, no RNA was available for clinic-based cases).
DNA and RNA were extracted from lymphocytes or EBV-transformed lymphoblasts as described [13]. This study was approved by the institutional review board of the Helsinki University Central Hospital (Helsinki, Finland).
Single nucleotide primer extension (SNuPE)
SNuPE uses a single dideoxynucleotide (ddNTP) and a combination of three dNTPs for an extension reaction where the incorporation of a ddNTP yields differential extension of primers attached close to the polymorphic site [13]. Four coding single nucleotide polymorphisms (SNPs) in APC were used to study APC allele-specific expression (cDNA compared with gDNA) as described in Pavicic et al. [8]. ASE ratios (R) were validated against SNuPE results from individuals not carrying any APC mutation [8]. Ratios R≤0.6 or R≥1.67 were considered to indicate unequivocal ASE (40% reduction of one allele relative to the other allele) and 0.6<R<0.8 or 1.25<R<1.67 putative ASE (21 – 39% reduction of one allele relative to the other allele). The ASE statuses in Table 1 were assigned according to the highest ASE ratio yielded by any of the four coding polymorphisms.
Transcriptome sequencing (RNA-seq) and transcriptome data-analysis
RNA-seq libraries were prepared using the ribo-depletion protocol from 12 DNAse treated total RNA samples, including three from FAP family 85 (individuals 85-1, 85-2, and 85-3), three from the index persons from families 42, 103 and 163, and three from proven mutation carriers from APC-mutation positive families 3, 93, and 63. RNA-seq data for three healthy individuals were generated for comparison. Sequencing of samples was done using Illumina HiSeq 2000 at the Institute for Molecular Medicine Finland (FIMM) (Helsinki, Finland). The bioinformatics workflow included correction of the sequence data for adapter sequences, bases with low quality, and reads less than 36-bp in length using Trimmomatics [29]. Paired-end reads passing the pre-processing were aligned to human reference genome build 38 (EnsEMBL v82) using STAR [30] with the default 2-pass multi-sample mapping settings, except that alignSJstitchMismatchNmax was set to 0 -1 -1 -1, outSJfilterCountUniqueMin to 6 2 2 2, outSJfilterCountTotalMin to 6 2 2 2, and outSJfilterDistToOtherSJmin to 10 0 0 0 in order to allow a more sensitive recovery of mutations at splice sites. Duplicate reads were marked with the Picard tools (http://picard.sourceforge.net) and strandedness information added with Bamutils [31]. Transcripts were assembled using StringTie [32] using the EnsEMBL v82 reference annotation file. Transcript predictions across all 12 samples were combined to a non-redundant set of transcripts using default parameters, except that minimum input transcript TPM and FPKM were set to 0.5.
RNA-sequencing data variant calling and ASE analysis
Allele-specific expression of genes was quantified using Genome Analysis Toolkit (GATK) package [33] and ASE deceptions algorithm MBASED [34]. Briefly, pre-processed and mapped reads were split into exon segments using GATK SplitNCigarReads, local indel realignment was performed around indels using GATK IndelRealigner, and base qualities were recalibrated using GATK BaseQualityScoreRecalibration. Variants were called using GATK HaplotypeCaller and filtered using GATK VariantFiltration according to the best practice recommendations regarding the RNA-seq variant analysis workflow. Multi allelic sites were removed with GATK SelectVariants and non-heterozygous variants and variants falling outside of StringTie-called exon regions extended by 3 bp discareded with GATK VariantFiltration. The ASE deceptions algorithm MBASED [34] was then applied for each variant set to infer the probability of ASE in genes listed in EnsEMBL v82 and having ≥ 2 variants. Default non-phased ASE calling settings were used, except that dispersion estimate was set to 0.004 and the probability to detected haplotype 1 supporting reads was set to the average fraction of aligned reads supporting haplotype 1 variants with coverage ≥ 30 in the given sample. Sequence data was visualized using Integrative Genomics Viewer (IGV) browser [35]. Supplementary Table S3 outlines the performance of our RNA-seq and ASE experiments.
Whole genome sequencing (WGS)
WGS was applied to DNAs from individuals 85-1, 85-2, and 85-3 from FAP family 85 as well as index patients from FAP families 42 and 103. Briefly, DNA was extracted from blood samples and KAPA and ThruPLEX sequencing libraries prepared according to the manufacturer's instructions. Sequencing was then conducted using Illumina HiSeq 2000 platform with KAPA and ThruPLEX libraries at the Institute for Molecular Medicine Finland (FIMM) (Helsinki, Finland). Sequencing data was analyzed by the FIMM variant calling pipeline version (VCP) 3.1 [36], including quality control of raw reads before and after alignment, pre-processing of reads for sequencing artifacts, alignment of reads to the human reference genome (build 19) using the Burrows-Wheeler Alignment (BWA) software [37], and calling of variants with the samtools package [38]. The minimum acceptable read depth for a variant was 7. Variant data were then analyzed by the VarSeq® software version 1.3.2 (Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com). Genotype quality (difference between the Phred-scale likelihoods of the two most likely genotypes) was assessed on a scale between 0 and 99 and variants with genotype quality less than 70 excluded. All common variants with minor allele frequency (MAF) ≥0.001 were removed. Only heterozygote variants were considered in agreement with dominant inheritance in FAP family 85 and any inheritance pattern was accepted in the index patients from FAP families 42 and 103 (sporadic cases, Table 1). The identified variants were checked against ExAC (http://exac.broadinstitute.org) and SISu databases (www.sisuproject.fi) as well as Ensembl database (http://www.ensembl.org) to assess population frequencies. Sequence data was visualized using Integrative Genomics Viewer (IGV) browser [35]. Supplementary Table S4 lists some essential performance characteristics for the WGS experiments.
Verfication of pseudoexons by sanger sequencing
To verify the pseudoexons identified by RNA-seq, relevant fragments of APC cDNA were amplified with primers from Spier et al. [6] and Sanger sequenced. The 11/12 pseudoexon was additionally verified from a cDNA fragment from exon 11 to exon 13 (amplified with primers given in Supplementary Table S1). Moreover, cDNA fragments 2 and exon 11 – 13 (Figure 3, Supplementary Table S1) were cloned into a pCR2.1 TOPO vector using the TOPO TA Cloning system (Invitrogen, Carlsbad, CA, USA) and DNAs extracted from the resulting white colonies were sequenced. The genomic variants discovered by WGS were confirmed by Sanger sequencing using primers around the respective nucleotide substitutions in introns 6 and 11 of APC (Supplementary Table S1).
URL addresses for web resources used
Berkeley Drosophila Gene Project (BDGP), http://www.fruitfly.org/seq_tools/splice.html
Ensembl, http://www.ensembl.org
Exome Aggregation Consortium (ExAC), http://exac.broadinstitute.org
InSiGHT, http://insight-group.org
Sequencing Initiative Suomi (SISu), www.sisuproject.fi
PICARD, http://picard.sourceforge.net
GATK, https://software.broadinstitute.org/gatk/
VarSeqTM, http://www.goldenhelix.com
SUPPLEMENTARY FIGURES AND TABLES
Acknowledgments
We thank the patients and responsible clinical experts for participation. Tuula Lehtinen and Beatriz Alcala-Repo are thanked for collecting clinical data and Saila Saarinen for expert technical assistance.
Footnotes
CONFLICTS OF INTEREST
No conflicts of interests.
GRANT SUPPORT
This work was supported by the European Research Council (FP7-ERC-232635), the Academy of Finland (grants no. 257795 and 294643), the Finnish Cancer Organizations, the Sigrid Juselius Foundation, the Nordic Cancer Union, Jane and Aatos Erkko Foundation, Maud Kuistila Memorial Foundation, and Biocentrum Helsinki.
REFERENCES
- 1.Leoz ML, Carballal S, Moreira L, Ocana T, Balaguer F. The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl Clin Genet. 2015;8:95–107. doi: 10.2147/TACG.S51484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aretz S, Stienen D, Friedrichs N, Stemmler S, Uhlhaas S, Rahner N, Propping P, Friedl W. Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP) Hum Mutat. 2007;28:985–992. doi: 10.1002/humu.20549. [DOI] [PubMed] [Google Scholar]
- 3.Grover S, Kastrinos F, Steyerberg EW, Cook EF, Dewanwala A, Burbidge LA, Wenstrup RJ, Syngal S. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308:485–492. doi: 10.1001/jama.2012.8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A, Working Group of the American College of Medical Genetics. Genomics (ACMG) Laboratory Quality Assurance Committee ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis) Genet Med. 2014;16:101–116. doi: 10.1038/gim.2013.166. [DOI] [PubMed] [Google Scholar]
- 5.Necker J, Kovac M, Attenhofer M, Reichlin B, Heinimann K. Detection of APC germ line mosaicism in patients with de novo familial adenomatous polyposis: a plea for the protein truncation test. J Med Genet. 2011;48:526–529. doi: 10.1136/jmg.2011.089474. [DOI] [PubMed] [Google Scholar]
- 6.Spier I, Horpaopan S, Vogt S, Uhlhaas S, Morak M, Stienen D, Draaken M, Ludwig M, Holinski-Feder E, Nothen MM, Hoffmann P, Aretz S. Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Hum Mutat. 2012;33:1045–1050. doi: 10.1002/humu.22082. [DOI] [PubMed] [Google Scholar]
- 7.Shirts BH, Salipante SJ, Casadei S, Ryan S, Martin J, Jacobson A, Vlaskin T, Koehler K, Livingston RJ, King MC, Walsh T, Pritchard CC. Deep sequencing with intronic capture enables identification of an APC exon 10 inversion in a patient with polyposis. Genet Med. 2014;16:783–786. doi: 10.1038/gim.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pavicic W, Nieminen TT, Gylling A, Pursiheimo JP, Laiho A, Gyenesei A, Jarvinen HJ, Peltomaki P. Promoter-specific alterations of APC are a rare cause for mutation-negative familial adenomatous polyposis. Genes chromosomes cancer. 2014;53:857–864. doi: 10.1002/gcc.22197. [DOI] [PubMed] [Google Scholar]
- 9.Bellido F, Pineda M, Aiza G, Valdes-Mas R, Navarro M, Puente DA, Pons T, Gonzalez S, Iglesias S, Darder E, Pinol V, Soto JL, Valencia A, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med. 2016:325–332. doi: 10.1038/gim.2015.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–1050. doi: 10.1086/386293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Chapelle A. Genetic predisposition to human disease: allele-specific expression and low-penetrance regulatory loci. Oncogene. 2009;28:3345–3348. doi: 10.1038/onc.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castellsague E, Gonzalez S, Guino E, Stevens KN, Borras E, Raymond VM, Lazaro C, Blanco I, Gruber SB, Capella G. Allele-specific expression of APC in adenomatous polyposis families. Gastroenterology. 2010;139:439–47. doi: 10.1053/j.gastro.2010.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renkonen ET, Nieminen P, Abdel-Rahman WM, Moisio AL, Jarvela I, Arte S, Jarvinen HJ, Peltomaki P. Adenomatous polyposis families that screen APC mutation-negative by conventional methods are genetically heterogeneous. J Clin Oncol. 2005;23:5651–5659. doi: 10.1200/JCO.2005.14.712. [DOI] [PubMed] [Google Scholar]
- 14.Aceto GM, Fantini F, De Iure S, Di Nicola M, Palka G, Valanzano R, Di Gregorio P, Stigliano V, Genuardi M, Battista P, Cama A, Curia MC. Correlation between mutations and mRNA expression of APC and MUTYH genes: new insight into hereditary colorectal polyposis predisposition. J Exp Clin Cancer Res. 2015;34:131. doi: 10.1186/s13046-015-0244-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curia MC, De Iure S, De Lellis L, Veschi S, Mammarella S, White MJ, Bartlett J, Di Iorio A, Amatetti C, Lombardo M, Di Gregorio P, Battista P, Mariani-Costantini R, et al. Increased variance in germline allele-specific expression of APC associates with colorectal cancer. Gastroenterology. 2012;142:71–77. doi: 10.1053/j.gastro.2011.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maquat LE. Defects in RNA splicing and the consequence of shortened translational reading frames. Am J Hum Genet. 1996;59:279–286. [PMC free article] [PubMed] [Google Scholar]
- 17.Faustino NA, Cooper TA. Pre-mRNA splicing and human disease. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 18.Dhir A, Buratti E. Alternative splicing: role of pseudoexons in human disease and potential therapeutic strategies. FEBS J. 2010;277:841–855. doi: 10.1111/j.1742-4658.2009.07520.x. [DOI] [PubMed] [Google Scholar]
- 19.Romano M, Buratti E, Baralle D. Role of pseudoexons and pseudointrons in human cancer. Int J Cell Biol. 2013:810572. doi: 10.1155/2013/810572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavalieri S, Pozzi E, Gatti RA, Brusco A. Deep-intronic ATM mutation detected by genomic resequencing and corrected in vitro by antisense morpholino oligonucleotide (AMO) Eur J Hum Genet. 2013;21:774–778. doi: 10.1038/ejhg.2012.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anczukow O, Buisson M, Leone M, Coutanson C, Lasset C, Calender A, Sinilnikova OM, Mazoyer S. BRCA2 deep intronic mutation causing activation of a cryptic exon: opening toward a new preventive therapeutic strategy. Clin Cancer Res. 2012;18:4903–4909. doi: 10.1158/1078-0432.CCR-12-1100. [DOI] [PubMed] [Google Scholar]
- 22.Clendenning M, Buchanan DD, Walsh MD, Nagler B, Rosty C, Thompson B, Spurdle AB, Hopper JL, Jenkins MA, Young JP. Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam Cancer. 2011;10:297–301. doi: 10.1007/s10689-011-9427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moisio AL, Jarvinen H, Peltomaki P. Genetic and clinical characterisation of familial adenomatous polyposis: a population based study. Gut. 2002;50:845–850. doi: 10.1136/gut.50.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Half E, Bercovich D, Rozen P. Familial adenomatous polyposis. Orphanet J Rare Dis. 2009;4:22. doi: 10.1186/1750-1172-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lappalainen T, Sammeth M, Friedlander MR, 't Hoen PA, Monlong J, Rivas MA, Gonzalez-Porta M, Kurbatova N, Griebel T, Ferreira PG, Barann M, Wieland T, Greger L, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, Bhardwaj N, Rubin M, Snyder M, et al. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol Syst Biol. 2011;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pritchard CC, Smith C, Salipante SJ, Lee MK, Thornton AM, Nord AS, Gulden C, Kupfer SS, Swisher EM, Bennett RL, Novetsky AP, Jarvik GP, Olopade OI, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn. 2012;14:357–366. doi: 10.1016/j.jmoldx.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spurdle AB, Couch FJ, Hogervorst FB, Radice P, Sinilnikova OM, IARC Unclassified Genetic Variants Working Group Prediction and assessment of splicing alterations: implications for clinical testing. Hum Mutat. 2008;29:1304–1313. doi: 10.1002/humu.20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breese MR, Liu Y. NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets. Bioinformatics. 2013;29:494–496. doi: 10.1093/bioinformatics/bts731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayba O, Gilbert HN, Liu J, Haverty PM, Jhunjhunwala S, Jiang Z, Watanabe C, Zhang Z. MBASED: allele-specific expression detection in cancer tissues and cell lines. Genome Biol. 2014;15:405. doi: 10.1186/s13059-014-0405-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sulonen AM, Ellonen P, Almusa H, Lepisto M, Eldfors S, Hannula S, Miettinen T, Tyynismaa H, Salo P, Heckman C, Joensuu H, Raivio T, Suomalainen A, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011;12:R94. doi: 10.1186/gb-2011-12-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.