

Abstract
Oral squamous cell carcinoma (OSCC) is a highly invasive and metastatic malignancy. The nerve growth factor receptor (NGFR) has been observed to be expressed on a subset of cells in OSCC, and NGFR+ cells have greater tumor-initiating capacity in vivo. Further, inhibition of NGFR reduces tumor growth, indicating a functional role of this receptor; however, the mechanisms by which NGFR confers enhanced tumor formation are not known. Here, we used an established murine model of OSCC and gene expression array analysis to identify ESM1 as a downstream target gene of NGFR, critical for tumor invasion and metastasis. ESM1 encodes a protein called endocan, which has the property of regulating proliferation, differentiation, migration, and adhesion of different cell types. Incubation of NGFR+ murine OSCC cells with nerve growth factor resulted in increased expression of ESM1. Importantly, ESM1 overexpression conferred an enhanced migratory, invasive, and metastatic phenotype, similar to what has been correlated with NGFR expression. Conversely, shRNA knockdown of ESM1 in NGFR overexpressing OSCC cells abrogated the tumor growth kinetics and the invasive and metastatic properties associated with NGFR. Together, our data indicate that NGFR plays an important role in the pathogenesis and progression of OSCC via regulation of ESM1.
Keywords: nerve growth factor receptor, CD271, HNSCC, metastasis, endocan
INTRODUCTION
Oral squamous cell carcinoma (OSCC) accounts for more than 90% of all malignant lesions of the mouth [1]. Despite advances in therapy, survival rates have not dramatically improved [2]. A series of molecular drivers contribute not only to the initiation of this malignancy but also to its invasive and metastatic properties [3]. In normal oral mucosa epithelium, a sub population of basal cells with stem cell-like properties has been shown to express the nerve growth factor receptor (NGFR), and recent reports indicate that NGFR contributes to the tumor-initiating capacity of a number of malignancies [4, 5]. We have previously shown that NGFR+ cells in human OSCC possess the greatest tumor-initiating capacity in this malignancy and that inhibition of NGFR has profound negative effects on the ability of these tumor-initiating cells (TIC) to form tumors in vivo [4].
NGFR, also known as p75 neurotrophin receptor (p75NTR) and CD271, is a cell surface receptor that belongs to the tumor necrosis factor receptor superfamily. There are two general classes of neurotrophin receptors: the high-affinity nerve growth factor tyrosine kinase receptors Trk A, B and C (encoded by NTRK1, NTRK2, and NTRK3, respectively) and the low-affinity nerve growth factor receptor NGFR. TrkA binds NGF, while TrkB binds BDNF and NT4, and TrkC binds NT3. Activation of TrkA can inhibit angiogenesis, induce differentiation and growth arrest and mediate apoptosis [6, 7]. In contrast, high intratumoral expression of TrkB and its specific ligand, BDNF, enhances proliferation, metastatic behavior and chemoresistance in neuroblastoma cells [8]. Activation of NGFR results in activation of the NF-κB (nuclear factor-κB) [9, 10], Jun kinase [11, 12] and other signaling pathways. Dependent on the cell type, cell differentiation status, neurotrophin binding, availability of intracellular adaptor molecules, and interacting transmembrane co-receptors and post-translational modification expression, NGFR signal transduction pathways are extremely variable [13]. These variable pathways lead to different cellular responses, such as cell survival [14], apoptosis [14, 15], neurite outgrowth and retraction [16], myelination [17], cell cycle regulation [18], cell migration and invasion [19, 20], and progenitor differentiation [5]. As mentioned, NGFR is expressed not only in nervous tissue, but also in non-neuronal normal and cancer cells, such as head and neck squamous cell carcinoma [4] and breast cancer [21], where it enhances proliferation and promotes cancer metastasis. The precise function of NGFR in OSCC remains unclear. Here, we investigated the mechanism by which NGFR affects invasion and tumor progression in a murine oral squamous cell carcinoma (MOC) model and identified an important downstream target of NGFR to be a gene called ESM1.
RESULTS
NGFR expression correlates with tumor growth kinetics and invasion in a murine model of oral squamous cell carcinoma
Expression of NGFR in human OSCC is heterogeneous [4]. Examination of three murine OSCC cell lines (MOC2, MOC2-7 and MOC2-10), derived from oral squamous cell carcinoma tumors arising from topical administration of 7,12-dimethylbenz(a)anthracene (DMBA) in the oral cavities of mice [22], also revealed heterogeneous expression of NGFR (Figure 1A). Consistent with previous observations that NGFR contributes to tumor-initiating capacity [4], the degree to which NGFR was expressed in these cell lines appeared to correlate with both tumor cell invasion and migration capacity in transwell invasion assay and tumor growth kinetics in vivo (Figure 1B and 1C).
Figure 1. NGFR expression correlates with tumor growth kinetics and invasion in a murine model of oral squamous cell carcinoma.
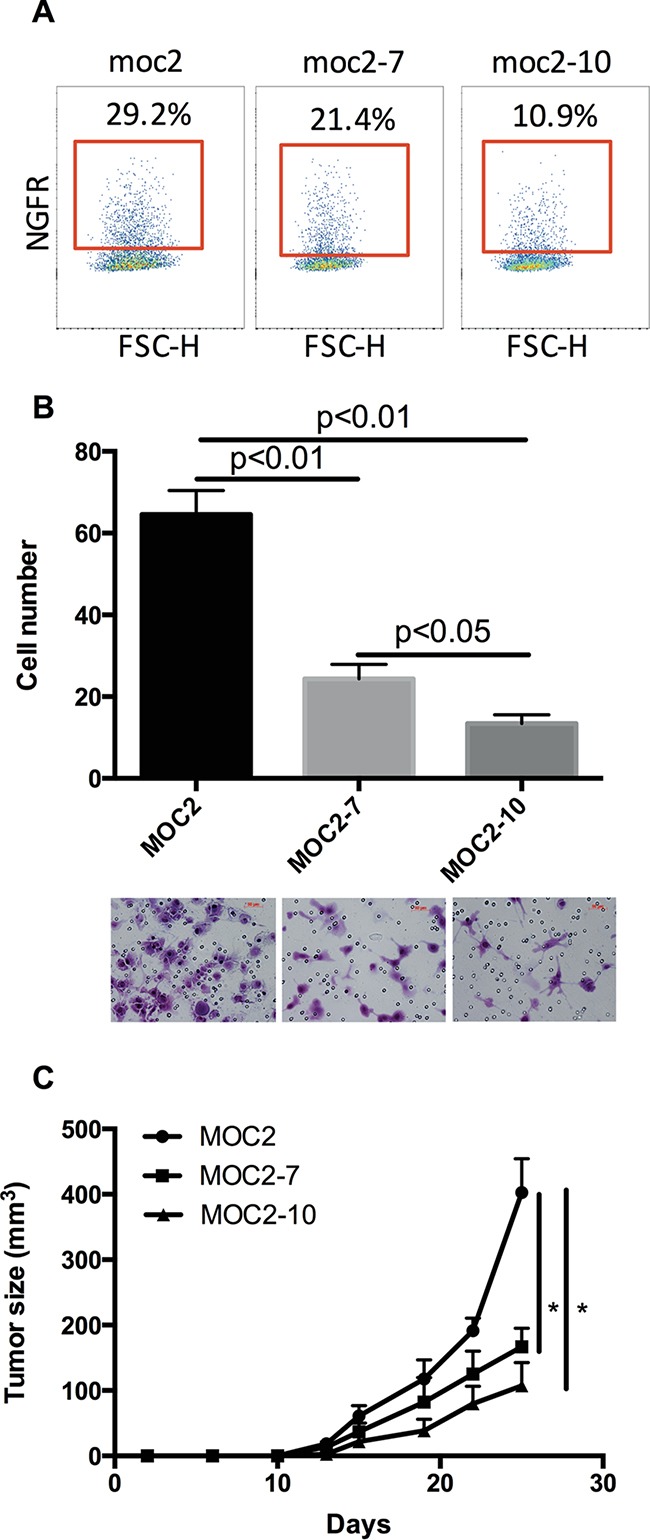
A. NGFR surface protein expression on MOC2, MOC2-7 and MOC2-10 cells, assessed by flow cytometry, gated on DAPI− cells. B. The invasive phenotype of MOC2, MOC2-7 and MOC2-10 cell lines was evaluated by transwell assay in vitro. Representative images of crystal violet-stained invasive cells after incubation. Data represent the mean±SEM. C. Tumor growth kinetics were assessed after subcutaneous injection with 1×104 cells/mouse of MOC2, MOC2-7 and MOC2-10 in B10; B6-Rag2−/−II2rg−/− mice. Each cohort consisted of 5 mice. Data represent the mean±SEM, *p<0.05.
To understand the role of NGFR in tumorigenicity, we assessed NGFR expression and related neurotrophin receptors, as well as neurotrophin ligands that bind these receptors, in the murine OSCC cell lines and found that NGFR is expressed in all three cell lines (Figure 2). The expression of other neurotrophin receptors (NTRK1, NTRK2 and NTRK3) was very low relative to NGFR. Since cells often co-express both neurotrophin receptors and cognate neurotrophin ligands (NGF, BDNF, NT-3 and NT-4) to create an autocrine loop, we examined the expression of these neurotrophins in the MOC cell lines. Analysis of transcripts showed that NGF and BDNF are expressed in all three cell lines (Figure 2). These data suggest a role for NGFR signaling in the pathogenic properties of MOC cells.
Figure 2. Neurotrophin receptor and neurotrophin expression in murine oral squamous cell carcinoma cell lines.
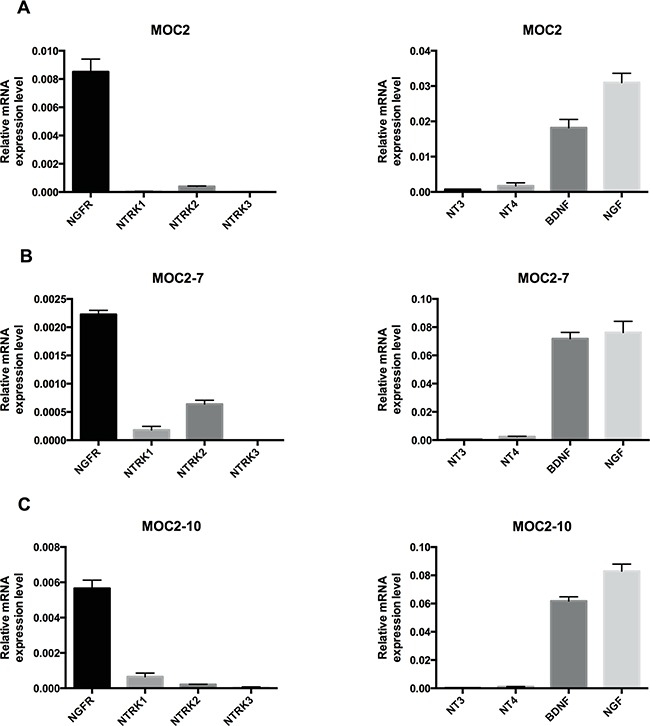
Quantitative RT-PCR analysis of NGFR, NTRK1, NTRK2, NTRK3, NT3, NT4, BDNF and NGF in murine OSCC cell lines: MOC2 A. MOC2-7 B. and MOC2-10 C. Results are presented as units defined as the n-fold difference relative to the control gene HPRT1. Data represent the mean±SEM.
NGFR activation upregulates ESM1 expression in MOC cells
To explore the potential function of NGFR in MOC cells, we overexpressed NGFR in MOC2 cells and compared the gene expression profiles between MOC2 and an NGFR overexpressing clone of MOC2 (MOC2T) by microarray analysis. Among 45,281 probes analyzed, 38 genes were differentially expressed by at least a two-fold difference (Figure 3A). Of these, 7 were up-regulated, and the rest were down-regulated in MOC2T cells. ESM1, one of the most up-regulated genes in the NGFR overexpressing MOC2T cells, was of particular interest given its known role in cancer progression [23]. The ESM1 differential expression, which was observed with the gene microarray, was confirmed in these cells by qRT-PCR (Figure 3B) and ELISA (Figure 3C).
Figure 3. NGFR regulates expression of ESM1.
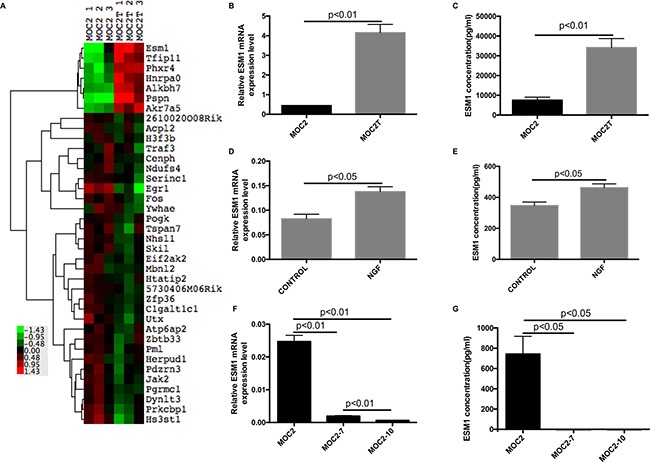
A. Heat map of hierarchical clustering analysis of differentially expressed genes in MOC2 cells and MOC2 cells that overexpress NGFR (MOC2T), selected at p≤0.05 followed by 2-fold cutoff change. Each column represents data from a single cohort with shades of red and green indicating up- or down- regulated genes according to the color scheme shown beside. B, C. ESM1 mRNA expression, assessed by qRT-PCR, and ESM1 soluble protein expression, assessed by ELISA, in MOC2 and MOC2T cells. Data represent the mean±SEM. D, E. ESM1 mRNA expression, assessed by qRT-PCR, and ESM1 soluble protein expression, assessed by ELISA, in MOC2 cells that were incubated in vitro with or without 100 ng/ml recombinant human NGF for 24 hours. Data represent the mean±SEM. F, G. Transcriptional expression of ESM1 mRNA, assessed by qRT-PCR, and ESM1 soluble protein expression, assessed by ELISA, in mouse oral squamous cell lines-MOC2, MOC2-7 and MOC2-10. Data represent the mean±SEM. The qRT-PCR results are presented as units defined as the n-fold difference relative to the control gene HPRT1.
To assess the effect of NGFR on ESM1 expression, MOC2 cells were cultured with recombinant human NGF for 24 hours. A significant increase in the expression of ESM1 was observed with NGF treatment, indicating that NGFR signaling was contributing to the expression of ESM1 in MOC2 (Figure 3D-3E). Further, comparison of ESM1 expression in MOC2, MOC2-7, and MOC2-10 cells revealed a correlation with the extent of NGFR expression and the tumor growth kinetics and invasive phenotype observed in the MOC cell lines (Figure 3F-3G and Figure 1). Among the three cell lines, ESM1 was most highly expressed in MOC2 and least in MOC2-10. Correspondingly, MOC2 was also the most invasive cell line, as measured by in vitro transwell invasion assay, and MOC2-10 the least invasive (Figure 1). Since ESM1 has been shown to contribute to tumor progression in multiple tumor types [24–26], these data suggested that ESM1 expression may also have a functional role in oral squamous cell carcinoma.
ESM1 modulates the invasive phenotype of MOC cells
To examine the functional role of ESM1 in MOC cells, shRNA targeting ESM1 was stably transduced into MOC2 cells (ESM1-SH) to knockdown expression of ESM1, and an ESM1 expression construct was also transduced into MOC2 cell line (ESM1-OVER) to overexpress ESM1. ESM1 knockdown or overexpression was confirmed by qRT-PCR (Figure 4A and 4C). ESM1 knockdown was also confirmed at the protein level by ELISA (Figure 4B). The effect of ESM1 expression on cell proliferation/viability was only modest (Figure 4D and 4E); however, there was a profound effect of ESM1 expression on the invasive phenotype of MOC2. Using transwell chamber assays, we assessed the ability of ESM1-SH and ESM1-OVER for their ability to invade and migrate through a Matrigel matrix. The ESM1 knockdown MOC2 cells showed a reduction in invasion, compared to the control cells (Figure 4F). Conversely, with the ESM1 overexpressing MOC2 cells, there was a significant increase in invasion that was observed (Figure 4F). These data indicate that ESM1 contributes to the invasive phenotype of MOC cells.
Figure 4. ESM1 modulates the invasive phenotype in MOC cells.

A, B. ESM1 mRNA expression, measured by qRT-PCR, and ESM1 soluble protein expression, measured by ELISA, in MOC2 cells after knockdown by ESM1 shRNA lentiviral transduction. ESM1 mRNA expression is normalized to HPRT1 expression. C. ESM1 mRNA expression in MOC2 cells after overexpression by ESM1 cDNA lentiviral transduction. ESM1 expression is normalized to HPRT1 expression. D. Cell proliferation/viability of control MOC2 cells and MOC2 cells expressing shRNA targeting ESM1 (ESM1-SH), measured by MTT assay. E. Cell proliferation/viability of control MOC2 cells and MOC2 cells overexpressing ESM1 (ESM1-OVER), measured by MTT assay. F. Cell invasive and migratory capacity, assessed by transwell assay, of MOC2 cells, compared to MOC2 cells either expressing shRNA targeting ESM1 (ESM1-SH) or overexpressing ESM1 (ESM1-OVER). Representative images of crystal violet-stained invasive cells after incubation. Data represent the mean±SEM.
ESM1 knockdown inhibits MOC tumor growth and metastasis in vivo
To assess the effect of ESM1 on tumor growth in vivo, MOC2 cells, in which ESM1 was knocked down (ESM1-SH), and control MOC2 cells were implanted subcutaneously into the flanks of mice, and the growth of the resultant primary tumors was monitored. A significant reduction in tumor growth kinetics and tumor volume was observed in the ESM1 knockdown cells (Figure 5A). At day 31, the average tumor volume of the control MOC2 cohort was 707.3±78.0 mm3, whereas the average tumor volume of the ESM1 knockdown cohort was 151.6±35.7 mm3. These data demonstrated that ESM1 knockdown effectively suppressed MOC2 tumor growth, despite only modest changes in cell proliferation/viability measured in vitro (Figure 4D). Further, there was a profound effect of ESM1 knockdown on metastasis (Figure 5B). The number of pulmonary metastases resulting from MOC2 primary tumors was significantly higher than that induced by ESM1 knockdown MOC2 cells (p<0.05). Histologic examination of lung sections confirmed that the pulmonary nodules were invasive squamous cell carcinoma. To assess whether ESM1 may participate in angiogenesis, VEGF expression was assessed in the tumor sections (Figure 5C), and VEGF expression was observed to be reduced in the ESM1 knockdown MOC2 tumors. Collectively, these data indicate that ESM1 contributes to the tumorigenicity, angiogenesis, invasiveness, and metastatic capacity of MOC cells.
Figure 5. ESM1 knockdown inhibits MOC tumor growth and metastasis in vivo.
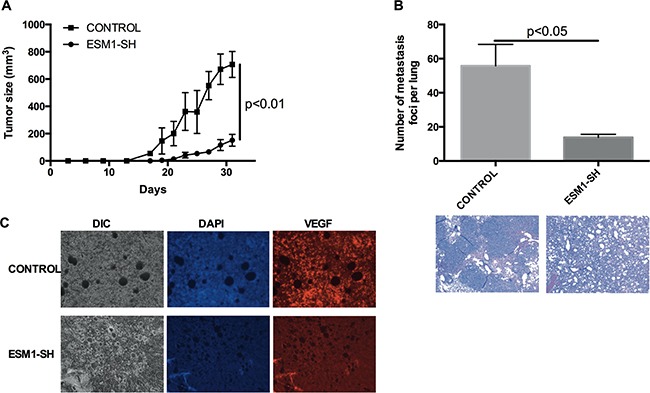
A. MOC2 cells and MOC2 cells transduced with ESM1 shRNA (ESM1-SH) were injected into the subcutaneous compartment over the flanks of mice (3×103 cells/mouse), and subsequent tumor growth was assessed. Each cohort consisted of 5 mice. Data represented the mean±SEM. B. The lungs of tumor-bearing mice were assessed for metastatic foci. Data represented the mean±SEM. Representative H&E stains of lungs of the MOC2 control and ESM1 shRNA knockdown cohorts are shown. C. Immunofluorescence imaging of VEGF of representative tumors from the control cohort and the ESM1 shRNA knockdown cohort are shown.
ESM1 knockdown abrogates the invasive and metastatic phenotype induced by NGFR in MOC cells
To investigate the functional relationship between ESM1 and NGFR, ESM1 shRNA was transduced into MOC2T cells (which overexpress NGFR), and the ESM1 knockdown clones (termed MOC2T-ESM1-SH) were selected by both puromycin resistance and GFP positive sorting. ESM1 and NGFR mRNA expression levels in MOC2T-ESM1-SH were examined and confirmed by qRT-PCR (Figure 6A). Knockdown of ESM1 resulted in a modest decrease in cell proliferation/viability compared to parental MOC2T cells. However, ESM1 knockdown had a significant negative effect on the invasive/migratory capacity of MOC2T cells (Figure 6C). These data indicated that the NGFR-enhanced migratory and invasive capacity of MOC2 cells is dependent on ESM1 expression.
Figure 6. ESM1 knockdown abrogates the invasive and metastatic phenotype induced by NGFR in MOC cells.
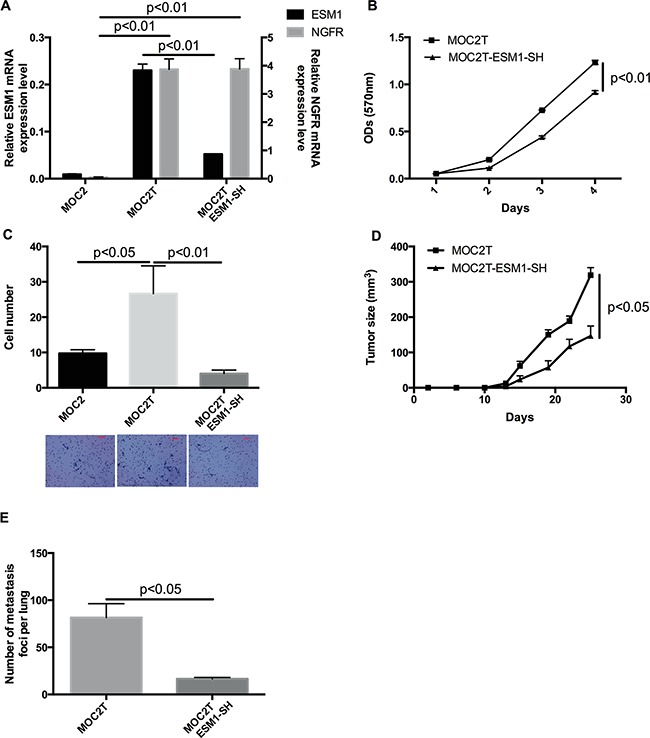
A. Expression of ESM1 and NGFR in MOC2, MOC2T (which overexpress NGFR) and MOC2T-ESM1-SH cells (which express an shRNA targeting ESM1 in the MOC2T cells) measured by qRT-PCR. Data represent the mean±SEM. Results are presented as units defined as the n-fold difference relative to the control gene HPRT1. B. Cell proliferation/viability of MOC2T and MOC2T-ESM1-SH cells as measured by MTT assay. Data represent the mean±SEM. C. Cell invasive and migratory capacity of MOC2, MOC2T, and MOC2T-ESM1-SH cells, measured by transwell invasion/migration assays. Representative images of crystal violet-stained cells after incubation. Data represent the mean±SEM. D. Tumor growth after MOC2T and MOC2T-ESM1-SH cells were injected (1×104 cells/mouse) subcutaneously into the flanks of mice. The MOC2T cohort consists of 7 mice and MOC2T-ESM1-SH cohort consists of 8 mice. (p<0.05). Data represented the mean±SEM. E. The number of metastatic foci counted on the surface of the lungs harvested from tumor-bearing mice. Data represented the mean±SEM.
To determine whether ESM1 plays an important role in the tumorigenicity and metastasis of MOC2T cells in vivo, we injected MOC2T and MOC2T-ESM1-SH cells subcutaneously into mice. Consistent with the effect of altered ESM1 expression on migration and invasion of MOC2T cells in vitro, knockdown of ESM1 significantly inhibited tumor growth (Figure 6D) and reduced the number of lung metastases (Figure 6E) associated with MOC2T cells in vivo. These data confirmed that the NGFR-enhanced migratory and invasive capacity of MOC2 cells is dependent on ESM1 expression.
DISCUSSION
Cells with tumor-initiating capacity have the ability to propagate tumor formation in vivo, and it has been proposed that these resilient cells contribute to tumor recurrence and poor clinical outcome, given their associated intrinsic metastatic capacity and resistance to chemotherapy and radiation [27–31]. NGFR has been identified as a marker of cells with these properties in multiple malignancies, including head and neck squamous cell carcinoma [4, 32]. Here, we report the identification of a novel downstream target gene of NGFR, ESM1. In cell culture, ESM1 overexpression in murine oral squamous cell carcinoma cells enhanced invasion and migration, whereas ESM1 knockdown resulted in reduced invasive and migratory capacity. To a limited extent, proliferation of the cells was also affected by altering ESM1 expression; however, the effects on proliferation were much more modest. In vivo transplantation of ESM1-knockdown cells led to a significant reduction in NGFR-induced tumor growth and pulmonary metastases. Collectively, our data demonstrate that the NGFR-enhanced invasive and migratory capacity of MOC2 cells is dependent on ESM1 expression.
ESM1, which encodes for a protein called endocan, has been previously characterized as an endothelium derived soluble dermatan sulfate proteoglycan (DSPG). It was cloned from a human umbilical vein endothelial cell (HUVEC) cDNA library. ESM1 is mainly expressed in the vascular endothelium and constitutively circulates in the bloodstream of healthy subjects. It has been found to be dramatically increased in the context of inflammation and cancer, including non-small cell lung cancer, renal clear cell carcinoma, and colorectal cancer etc. [24–26, 33–42]. As a biomarker of neovascularization, ESM1 overexpression is also often used as an indicator of tumor progression and metastasis in certain malignances, such as glioblastoma, renal cell carcinoma, and non-small cell lung cancer [24–26, 36]. Further, ESM1 has been found to be strongly associated with tumor invasion in pituitary adenomas [34, 43].
ESM1 is also known to be overexpressed in tumor cells themselves and can promote tumor growth [23, 44, 45]. ESM1 interacts with several growth factors, such as hepatocyte growth factor/scatter factor (HGF/SF), by presence of a glycan chain, thereby eliciting epithelial cell migration and growth in vitro [46]. In addition, ESM1 regulates the lymphocyte function-associated antigen-1 (LFA-1)/intercellular adhesion molecule-1 (ICAM-1) pathway; thus, it may play an important role in the migration of leukocytes into tumor tissues [47]. ESM1 overexpression in non-tumorigenic epithelial cells induces tumor formation in SCID mice [44]. In accordance with these studies, our in vivo and in vitro data both indicate that altered ESM1 expression plays an important role in tumor formation and metastasis of murine oral squamous carcinoma. Inhibited expression of ESM1 in MOC2 was associated with much reduced tumor growth, compared to control cells. These data indicate that ESM1 may a viable target for antitumor therapy.
Cancer metastasis is a complex process, and its cellular and molecular mechanisms remain unclear. VEGF, a secreted dimeric glycoprotein, is an important regulator of tumor angiogenesis. Previous reports have demonstrate a feedback loop where VEGF-A positively regulates ESM1 expression, which in turn enhances VEGF-A mediated signaling [38]. Consistent with this, we found that ESM1 knockdown resulted in significantly lower VEGF expression.
Taken together, our data demonstrate ESM1 to be a gene target downstream of NGFR and that ESM1 has an important functional role in the invasive phenotype of murine OSCC. We have demonstrated that ESM1 knockdown reduces the metastatic capacity of MOC2 cells in vivo, providing rationale to investigate the therapeutic potential of targeting NGFR and/or ESM1 in oral SCC. Current efforts to understand the signaling intermediates between NGFR activation and ESM1 expression are underway. Finally, ESM1 expression, alone or in combination with NGFR expression, might serve as novel prognostic biomarker for oral SCC.
MATERIALS AND METHODS
Cell lines
The MOC2, MOC2-7, and MOC2-10 murine oral squamous cell carcinoma cell lines were provided by Dr. Ravindra Uppaluri at Washington University in St. Louis, who developed the cell lines from murine oral squamous cell carcinomas induced by topical 7,12-dimethylbenz(a)anthracene (DMBA) administration. Cells were cultured in complete DMEM/F12 medium containing 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
RNA purification and quantitative RT-PCR analysis
Isolated cells were seeded at a density of 1×105 cells/well in a 6-well plate for one day prior to RNA preparation. For NGF-induced gene expression, cells were starved one day in a serum-free medium before treatments with NGF. Then, the cells were treated with or without 100 ng/ml recombinant human NGF (Sigma) in serum-free medium for 24 hours prior to harvest. Total RNA was extracted from the samples using a RNeasy Mini Kit (Qiagen). The cDNA synthesis was performed using a Maxima First Strand cDNA Synthesis Kit (Thermo) following the manufacturer's protocol. The relative amount of gene mRNA was analyzed by quantitative real-time reverse transcription-Polymerase Chain Reaction (qRT-PCR). The qRT-PCR was performed using Luminaris Color Probe High ROX qPCR Master Mix (Thermo). Mouse HPRT1 was amplified as control. Gene expression was expressed as arbitrary units defined as the n-fold difference relative to the control gene HPRT1.
Enzyme-linked immunosorbent assay
ESM1 protein expression level was measured by enzyme-linked immunosorbent assay (ELISA), using a Mouse ESM1 PicoKine ELISA kit (Boster Biological Technology, Pleasanton, CA). Plates came pre-coated with monoclonal rat anti-ESM1 antibody. Standards and cell culture supernatants were diluted in sample diluent buffer and incubated for 90 minutes at 37°C. Biotinylated polyclonal goat anti-ESM1antibody was diluted in antibody diluent buffer and incubated for 1 hour at 37°C. After washing steps in PBS, Avidin-Biotin-Peroxidase Complex was diluted in ABC diluent buffer and incubated for 45 minutes at 37°C. After washing steps in PBS, TMB color developing agent was added to each well and incubated in the dark for 20 minutes at 37°C. TMB stop solution was added to each well, and absorbance was measured at a wavelength of 450 nm using a microplate (ELISA) reader (SpectraMax M3, Molecular Devices). A six-point standard curve was used to calculate the concentration (pg/mL) of ESM1 in the samples.
Flow cytometry analysis
Cells were harvested from culture flasks and the single-cell suspensions were incubated with anti-mouse NGFR antibody (mu p75, ATS Bio). DAPI was used to allow exclusion of non-viable cells. NGFR expression was assessed by fluorescence activated cell sorting analysis on a BD LSRFortessa or BD FACSAria II. Events collected were analyzed using FlowJo Version 9.6.4 software (Tree Star).
Microarray anaylsis
Illumina MouseWG-6 v2 Expression BeadChip was used following the manufacturer's instructions by Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine. We conducted an unpaired student's t-test to compare gene differences between MOC2 and NGFR overexpression MOC2 (MOC2T) cells. Gene expression that showed at least 2-fold increase or decrease and had a significance level of p<0.05 was considered significantly altered after NGFR overexpression.
Cell proliferation and growth assays
MTT assays were carried out following the manufacturer's instruction (Cell Proliferation Kit I (MTT), Roche). Briefly, cells were seeded into 96-well plates at a concentration of 1,000 cells per well in 100 μl of culture medium. Wells without cells were used as blank controls. Cells were incubated for 1, 2, 3, 4 and 5 days at 37 deg C and 5% CO2, and medium was changed every two days. After the incubation period, 10 μl of the MTT labeling reagent was added to each well (final concentration 0.5 mg/ml). After a 4-hour incubation, 100 μl of the solubilization solution was added to each well. Cells were incubated overnight and then tested for complete solubilization of the purple formazan crystals through measuring the absorbance (OD) of the samples at 570nm using a microplate (ELISA) reader (SpectraMax M3, Molecular Devices). All final data were normalized to the OD of the blank controls.
Lentiviral plasmids
RNAi Consortium (TRC) LentiviralTM shRNA was purchased from GE Healthcare Dharmacon Inc. The shRNA against ESM1 (5′-TCTTTGCATTCCATCCCGAAG-3′) is in pLKO.1-based lentiviral vector. A cDNA encoding ESM1 from ATG codon to the stop codon of ESM1 was PCR cloned (Forward primer: 5′-GAATTCATGAAGAGCCTCTTGCTGCT-3′; reverse primer: 5′-GGATCCTCAGCGTGGATTTAACCATTTCA-3′) and subcloned BamHI/EcoRI fragments into the pHIV-Zsgreen expression construct, provided by Dr. Michael Clarke (Stanford). This was used for overexpression of ESM1 in the MOC cells.
Lentiviral production and transduction
For the production of the lentiviral particles, the HEK 293 cell line was transfected with the packaging plasmid pCMVR8.74, the envelope plasmid pCMV-VSVG and the lentiviral construct containing the shRNA or the transgene, using Lipofectamine® 2000 according to the manufacturer's instructions. Medium was changed 16 hours after the transfection. Virus-containing culture supernatant was collected after 24 hours and centrifuged. Virus was used immediately to infect cells, which were seeded at 3×105 cells per well in a 6-well plate 24 hours prior. Polybrene (8 μg/ml) was also added to enhance the lentiviral transduction efficiency. Medium was changed after 24 hours. In the case of the cells transduced with the pLKO.1 puro vectors, the cell cultures were treated with 1 μg/ml puromycin for one week after media change.
Invasion and migration assay
To assess the invasive and migratory capacity of the tumor cells, 1×104 cells in 500 μl of serum-free DMEM/F12 was added into the upper chamber and 500 μl of complete medium was placed into the lower chamber (Corning® BioCoatTM Matrigel® Invasion Chamber, Corning). Cells were incubated at 37 deg C for 48 hours, before the non-invading cells were removed from the upper surface of the membrane. After fixation in 95% ethanol for 5 min, the cells still on the opposite surface of the filter membrane were stained with 1% crystal violet for 10 min. The migratory cells were counted in five microscope fields and averaged.
Animal studies
The B10; B6-Rag2−/−II2rg−/− mice (6-11 weeks old, Taconic) were housed in laminar flow cabinets under specific pathogen-free conditions and fed ad libitum. All procedures were performed in accordance with protocols approved by the Administrative Panel on Laboratory Animal Care at Stanford University. Tumor cells were injected subcutaneous in mice. Tumor volume (in mm3) was determined by caliper measurements performed every two to three days and calculated by using the following formula: volume=length×width2×0.5. The tumors that arose in those mice were harvested when they reached about 1.0 cm in diameter. Lungs were harvested, fixed, and stained with Bouin's solution. Metastatic colonies on the surface of lungs were counted. Tissues were fixed with 10% formalin, and then the paraffin-embedded. H&E stained slides were made by the Department of Comparative Medicine Histology Service Center at Stanford.
Immunofluorescence
After the formalin-fixed paraffin-embedded specimens were deparaffinized and rehydrated, antigen retrieval was performed. Samples were blocked at 4 deg C overnight in 5% FBS/PBST, and then incubated for 4 hours at room temperature in Anti-VEGF antibody (Novus biologicals) (1:50) diluted with 5% FBS/PBST. Negative control of 5% FBS/PBST was also used.
Statistical analysis
Data were expressed as mean ±standard error of mean (SEM) and statistically analyzed by t-test. Statistical analysis was performed by IBM SPSS 22 software package. Values of p<0.05 were considered to be statistically significant.
Footnotes
CONFLICTS OF INTEREST
The authors have no relevant conflicts of interest to disclose.
GRANT SUPPORT
J.B.S. is supported by funding from the National Institutes of Health, the Stanford Cancer Institute, and by gifts from Peder and Kathy Knudsen, Jill and John Friedenrich, and the Harold Simmons Foundation.
REFERENCES
- 1.Pérez S. Genetic and molecular alterations associated with oral squamous cell cancer (Review) Oncology reports. 2009:22. doi: 10.3892/or_00000565. [DOI] [PubMed] [Google Scholar]
- 2.Rapidis AD, Gullane P, Langdon JD, Lefebvre JL, Scully C, Shah JP. Major advances in the knowledge and understanding of the epidemiology, aetiopathogenesis, diagnosis, management and prognosis of oral cancer. Oral oncology. 2009;45:299–300. doi: 10.1016/j.oraloncology.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371:1695–1709. doi: 10.1016/S0140-6736(08)60728-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murillo-Sauca O, Chung MK, Shin JH, Karamboulas C, Kwok S, Jung YH, Oakley R, Tysome JR, Farnebo LO, Kaplan MJ, Sirjani D, Divi V, Holsinger FC, et al. CD271 is a functional and targetable marker of tumor-initiating cells in head and neck squamous cell carcinoma. Oncotarget. 2014;5:6854–6866. doi: 10.18632/oncotarget.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura T, Endo K, Kinoshita S. Identification of human oral keratinocyte stem/progenitor cells by neurotrophin receptor p75 and the role of neurotrophin/p75 signaling. Stem cells (Dayton, Ohio) 2007;25:628–638. doi: 10.1634/stemcells.2006-0494. [DOI] [PubMed] [Google Scholar]
- 6.Eggert A, Ikegaki N, Liu X, Chou TT, Lee VM, Trojanowski JQ, Brodeur GM. Molecular dissection of TrkA signal transduction pathways mediating differentiation in human neuroblastoma cells. Oncogene. 2000;19:2043–2051. doi: 10.1038/sj.onc.1203518. [DOI] [PubMed] [Google Scholar]
- 7.Eggert A, Grotzer MA, Zuzak TJ, Wiewrodt BR, Ho R, Ikegaki N, Brodeur GM. Resistance to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in neuroblastoma cells correlates with a loss of caspase-8 expression. Cancer research. 2001;61:1314–1319. [PubMed] [Google Scholar]
- 8.Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucchesi JC. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanfgaster. Genetics. 1968;59:37–44. doi: 10.1093/genetics/59.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schallner N, Ulbrich F, Engelstaedter H, Biermann J, Auwaerter V, Loop T, Goebel U. Isoflurane but not sevoflurane or desflurane aggravates injury to neurons in vitro and in vivo via p75NTR-NF-kB activation. Anesthesia and analgesia. 2014;119:1429–1441. doi: 10.1213/ANE.0000000000000488. [DOI] [PubMed] [Google Scholar]
- 11.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 12.Linggi MS, Burke TL, Williams BB, Harrington A, Kraemer R, Hempstead BL, Yoon SO, Carter BD. Neurotrophin receptor interacting factor (NRIF) is an essential mediator of apoptotic signaling by the p75 neurotrophin receptor. The Journal of biological chemistry. 2005;280:13801–13808. doi: 10.1074/jbc.M410435200. [DOI] [PubMed] [Google Scholar]
- 13.Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nature reviews Neuroscience. 2005;6:603–614. doi: 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- 14.Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low-affinity NGF receptor. Science. 1993;261:345–348. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- 15.Coulson EJ, Reid K, Shipham KM, Morley S, Kilpatrick TJ, Bartlett PF. The role of neurotransmission and the Chopper domain in p75 neurotrophin receptor death signaling. Progress in brain research. 2004;146:41–62. doi: 10.1016/S0079-6123(03)46003-2. [DOI] [PubMed] [Google Scholar]
- 16.Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999;24:585–593. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- 17.Notterpek L. Neurotrophins in myelination: a new role for a puzzling receptor. Trends in neurosciences. 2003;26:232–234. doi: 10.1016/S0166-2236(03)00099-7. [DOI] [PubMed] [Google Scholar]
- 18.Chittka A, Arevalo JC, Rodriguez-Guzman M, Perez P, Chao MV, Sendtner M. The p75NTR-interacting protein SC1 inhibits cell cycle progression by transcriptional repression of cyclin E. The Journal of cell biology. 2004;164:985–996. doi: 10.1083/jcb.200301106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrmann JL, Menter DG, Hamada J, Marchetti D, Nakajima M, Nicolson GL. Mediation of NGF-stimulated extracellular matrix invasion by the human melanoma low-affinity p75 neurotrophin receptor: melanoma p75 functions independently of trkA. Molecular biology of the cell. 1993;4:1205–1216. doi: 10.1091/mbc.4.11.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston AL, Lun X, Rahn JJ, Liacini A, Wang L, Hamilton MG, Parney IF, Hempstead BL, Robbins SM, Forsyth PA, Senger DL. The p75 neurotrophin receptor is a central regulator of glioma invasion. PLoS biology. 2007;5:e212. doi: 10.1371/journal.pbio.0050212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forsberg LA, Rasi C, Pekar G, Davies H, Piotrowski A, Absher D, Razzaghian HR, Ambicka A, Halaszka K, Przewoznik M, Kruczak A, Mandava G, Pasupulati S, et al. Signatures of post-zygotic structural genetic aberrations in the cells of histologically normal breast tissue that can predispose to sporadic breast cancer. Genome Res. 2015;25:1521–1535. doi: 10.1101/gr.187823.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Judd NP, Winkler AE, Murillo-Sauca O, Brotman JJ, Law JH, Lewis JS, Jr, Dunn GP, Bui JD, Sunwoo JB, Uppaluri R. ERK1/2 regulation of CD44 modulates oral cancer aggressiveness. Cancer research. 2012;72:365–374. doi: 10.1158/0008-5472.CAN-11-1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sarrazin S, Adam E, Lyon M, Depontieu F, Motte V, Landolfi C, Lortat-Jacob H, Bechard D, Lassalle P, Delehedde M. Endocan or endothelial cell specific molecule-1 (ESM-1): a potential novel endothelial cell marker and a new target for cancer therapy. Biochimica et biophysica acta. 2006;1765:25–37. doi: 10.1016/j.bbcan.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 24.Maurage CA, Adam E, Mineo JF, Sarrazin S, Debunne M, Siminski RM, Baroncini M, Lassalle P, Blond S, Delehedde M. Endocan expression and localization in human glioblastomas. Journal of neuropathology and experimental neurology. 2009;68:633–641. doi: 10.1097/NEN.0b013e3181a52a7f. [DOI] [PubMed] [Google Scholar]
- 25.Leroy X, Aubert S, Zini L, Franquet H, Kervoaze G, Villers A, Delehedde M, Copin MC, Lassalle P. Vascular endocan (ESM-1) is markedly overexpressed in clear cell renal cell carcinoma. Histopathology. 2010;56:180–187. doi: 10.1111/j.1365-2559.2009.03458.x. [DOI] [PubMed] [Google Scholar]
- 26.Grigoriu BD, Depontieu F, Scherpereel A, Gourcerol D, Devos P, Ouatas T, Lafitte JJ, Copin MC, Tonnel AB, Lassalle P. Endocan expression and relationship with survival in human non-small cell lung cancer. Clinical cancer research. 2006;12:4575–4582. doi: 10.1158/1078-0432.CCR-06-0185. [DOI] [PubMed] [Google Scholar]
- 27.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–1115. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annual review of cell and developmental biology. 2007;23:675–699. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 30.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adorno-Cruz V, Kibria G, Liu X, Doherty M, Junk DJ, Guan D, Hubert C, Venere M, Mulkearns-Hubert E, Sinyuk M, Alvarado A, Caplan AI, Rich J, Gerson SL, Lathia J, Liu H. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer research. 2015;75:924–929. doi: 10.1158/0008-5472.CAN-14-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imai T, Tamai K, Oizumi S, Oyama K, Yamaguchi K, Sato I, Satoh K, Matsuura K, Saijo S, Sugamura K, Tanaka N. CD271 defines a stem cell-like population in hypopharyngeal cancer. PloS one. 2013;8:e62002. doi: 10.1371/journal.pone.0062002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voiosu T, Balanescu P, Bengus A, Voiosu A, Baicus CR, Barbu M, Ladaru A, Nitipir C, Mateescu B, Diculescu M, Voiosu R. Serum endocan levels are increased in patients with inflammatory bowel disease. Clinical laboratory. 2014;60:505–510. doi: 10.7754/clin.lab.2013.130333. [DOI] [PubMed] [Google Scholar]
- 34.Matano F, Yoshida D, Ishii Y, Tahara S, Teramoto A, Morita A. Endocan, a new invasion and angiogenesis marker of pituitary adenomas. J Neurooncol. 2014;117:485–491. doi: 10.1007/s11060-014-1377-6. [DOI] [PubMed] [Google Scholar]
- 35.Lv Z, Fan Y, Chen H, Zhao D. Endothelial cell-specific molecule-1: a potential serum marker for gastric cancer. Tumour biology. 2014;35:10497–10502. doi: 10.1007/s13277-014-2319-9. [DOI] [PubMed] [Google Scholar]
- 36.Ziol M, Sutton A, Calderaro J, Barget N, Aout M, Leroy V, Blanc JF, Sturm N, Bioulac-Sage P, Nahon P, Nault JC, Charnaux N, N'Kontchou G, Trinchet JC, Delehedde M, Seror O, et al. ESM-1 expression in stromal cells is predictive of recurrence after radiofrequency ablation in early hepatocellular carcinoma. Journal of hepatology. 2013;59:1264–1270. doi: 10.1016/j.jhep.2013.07.030. [DOI] [PubMed] [Google Scholar]
- 37.Yu PH, Chou SF, Chen CL, Hung H, Lai CY, Yang PM, Jeng YM, Liaw SF, Kuo HH, Hsu HC, Chen JY, Wang WB. Upregulation of endocan by Epstein-Barr virus latent membrane protein 1 and its clinical significance in nasopharyngeal carcinoma. PloS one. 2013;8:e82254. doi: 10.1371/journal.pone.0082254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roudnicky F, Poyet C, Wild P, Krampitz S, Negrini F, Huggenberger R, Rogler A, Stohr R, Hartmann A, Provenzano M, Otto VI, Detmar M. Endocan is upregulated on tumor vessels in invasive bladder cancer where it mediates VEGF-A-induced angiogenesis. Cancer research. 2013;73:1097–1106. doi: 10.1158/0008-5472.CAN-12-1855. [DOI] [PubMed] [Google Scholar]
- 39.El Behery MM, Seksaka MA, Ibrahiem MA, Saleh HS, El Alfy Y. Clinicopathological correlation of endocan expression and survival in epithelial ovarian cancer. Archives of gynecology and obstetrics. 2013;288:1371–1376. doi: 10.1007/s00404-013-2863-3. [DOI] [PubMed] [Google Scholar]
- 40.Kim JH, Park MY, Kim CN, Kim KH, Kang HB, Kim KD, Kim JW. Expression of endothelial cell-specific molecule-1 regulated by hypoxia inducible factor-1alpha in human colon carcinoma: impact of ESM-1 on prognosis and its correlation with clinicopathological features. Oncology reports. 2012;28:1701–1708. doi: 10.3892/or.2012.2012. [DOI] [PubMed] [Google Scholar]
- 41.Port M, Boltze C, Wang Y, Roper B, Meineke V, Abend M. A radiation-induced gene signature distinguishes post-Chernobyl from sporadic papillary thyroid cancers. Radiation research. 2007;168:639–649. doi: 10.1667/RR0968.1. [DOI] [PubMed] [Google Scholar]
- 42.Hatfield KJ, Lassalle P, Leiva RA, Lindas R, Wendelboe O, Bruserud O. Serum levels of endothelium-derived endocan are increased in patients with untreated acute myeloid leukemia. Hematology. 2011;16:351–356. doi: 10.1179/102453311X13127324303434. [DOI] [PubMed] [Google Scholar]
- 43.Miao Y, Zong M, Jiang T, Yuan X, Guan S, Wang Y, Zhou D. A comparative analysis of ESM-1 and vascular endothelial cell marker (CD34/CD105) expression on pituitary adenoma invasion. Pituitary. 2016 doi: 10.1007/s11102-015-0698-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scherpereel A, Gentina T, Grigoriu B, Senechal S, Janin A, Tsicopoulos A, Plenat F, Bechard D, Tonnel AB, Lassalle P. Overexpression of endocan induces tumor formation. Cancer research. 2003;63:6084–6089. [PubMed] [Google Scholar]
- 45.Yang J, Yang Q, Yu S, Zhang X. Endocan: A new marker for cancer and a target for cancer therapy. Biomedical reports. 2015;3:279–283. doi: 10.3892/br.2015.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bechard D, Gentina T, Delehedde M, Scherpereel A, Lyon M, Aumercier M, Vazeux R, Richet C, Degand P, Jude B, Janin A, Fernig DG, Tonnel AB, Lassalle P. Endocan is a novel chondroitin sulfate/dermatan sulfate proteoglycan that promotes hepatocyte growth factor/scatter factor mitogenic activity. The Journal of biological chemistry. 2001;276:48341–48349. doi: 10.1074/jbc.M108395200. [DOI] [PubMed] [Google Scholar]
- 47.Béchard D, Scherpereel A, Hammad H, Gentina T, Tsicopoulos A, Aumercier M, Pestel J, Dessaint J-P, Tonnel A-B, Lassalle P. Human endothelial-cell specific molecule-1 binds directly to the integrin CD11a/CD18 (LFA-1) and blocks binding to intercellular adhesion molecule-1. The Journal of Immunology. 2001;167:3099–3106. doi: 10.4049/jimmunol.167.6.3099. [DOI] [PubMed] [Google Scholar]