

Summary
Acetyl-coenzyme A (acetyl-CoA) is a critical metabolic signaling molecule that regulates gluconeogenesis, pyruvate oxidation, protein acetylation, steroid and fatty acid biosynthesis (Fig. 1); however, measurements of this metabolite using standard biochemical approaches are technically demanding and there is currently no method to non-invasively assess hepatic acetyl-CoA content in vivo. To this end, we developed and validated a method to non-invasively detect differences in hepatic acetyl-CoA content in vivo across a five-fold range of physiological acetyl-CoA concentrations by assessing the turnover of [13C4]β-hydroxybutyrate (β–OHB). Here we show a strong correlation (R2=0.86, P<0.0001) between hepatic acetyl-CoA content and β–OHB turnover in rats with varying degrees of fasting hyperglycemia and insulin resistance. These studies demonstrate that β–OHB turnover can be used as a surrogate to non-invasively assess hepatic acetyl-CoA content, thereby allowing researchers to further elucidate the role of this metabolite in the regulation of hepatic gluconeogenesis and other metabolic processes in vivo.
Introduction
Acetyl-CoA is a key metabolic signaling molecule, which influences a variety of important hepatic cellular processes such as gluconeogenesis, glucose oxidation, protein acetylation, steroid and fatty acid biosynthesis (Pietrocola et al., 2015). In this regard we have recently demonstrated that reduction in hepatic acetyl-CoA content is a key mediator of insulin’s ability to acutely suppress hepatic gluconeogenesis in vivo (Perry et al., 2015a) and that increases in hepatic acetyl-CoA are both necessary and sufficient to promote increased rates of hepatic gluconeogenesis in rodent models of poorly controlled type 1 diabetes (T1D) and type 2 diabetes (T2D) (Perry et al., 2015a; Perry et al., 2015b; Perry et al., 2014). However, each of these studies required rapid (<10 sec) in situ freeze clamping of liver tissue in aluminum tongs pre-chilled in liquid nitrogen prior to the LC-MS/MS analyses because of acetyl-CoA’s rapid degradation ex vivo. While it is possible to obtain liver tissue from human subjects undergoing intra-abdominal surgical procedures (e.g. bariatric surgery, cholecystectomy, etc.) the relatively long delay (>30 seconds) in removing the liver tissue prior to freeze-clamping makes this biochemical approach infeasible in humans.
Given the great interest in understanding the role of hepatic acetyl-CoA in the regulation of hepatic intermediary metabolism, we examined whether β–OHB turnover, assessed by an infusion of [13C4]β–OHB, would serve as a reliable surrogate of hepatic acetyl-CoA content in vivo. Using this approach we found a strong correlation (R2=0.86, P<0.0001) between hepatic acetyl-CoA content and β–OHB turnover in five rat models (overnight fasted, chow- and high fat diet [HFD]-fed; HFD-fed rats treated with a low dose of streptozotocin [STZ] to induce whole-body insulin resistance associated with modest relative insulinopenia mimicking type 2 diabetes; STZ-induced severely insulinopenic rat model of poorly-controlled type 1 diabetes [T1D]; and rats undergoing a hyperinsulinemic-euglycemic clamp) with varying degrees of fasting hyperglycemia and insulin resistance, which represent the entire spectrum of hepatic acetyl-CoA content (40–250 nmol/g) reported in our prior studies (Perry et al., 2015a; Perry et al., 2015b; Perry et al., 2014). Taken together these data demonstrate that β–OHB turnover can be used as a non-invasive surrogate of hepatic acetyl-CoA content in vivo.
Results
Hepatic acetyl-CoA concentrations are increased in hyperglycemic rats
Hepatic acetyl-CoA degrades rapidly (<30 sec) after excision of the liver tissue (Fig. 2), demonstrating the need for a method to measure hepatic acetyl-CoA content in vivo that does not rely on collecting tissue samples during surgical interventions. In order to assess the potential of β-OHB turnover to predict hepatic acetyl-CoA content, we studied five rat models with widely varying fasting plasma glucose and insulin concentrations and rates of hepatic glucose production, including chow and high fat fed rats, poorly-controlled rat models of T1D and T2D (Reed et al., 2000), as well as rats undergoing a hyperinsulinemic-euglycemic clamp (Fig. S1A–D). These animals exhibited a 4.5-fold difference in hepatic glucose production, ranging from 6±1 μmol/min in clamped animals to 33±1 μmol/min in T1D rats (Fig. 3A). These differences in hepatic glucose production were reflected in a 3.5-fold difference in hepatic acetyl-CoA content between the groups (Fig. 3B). Both plasma β-OHB concentrations and whole-body β-OHB turnover tracked closely with hepatic acetyl-CoA content and rates of HGP, with all four parameters lowest in clamped rats, then chow fed controls, then high fat fed rats (Fig. 3C–D). Rates of HGP and β-OHB turnover as well as plasma β-OHB concentrations and hepatic acetyl-CoA content were increased in HFD-STZ rats and highest in poorly controlled T1D animals (Fig. 3A–D). Identical plasma β-OHB enrichment between 100 and 120 minutes of the clamp study validated that our 120 min [13C4]β-OHB infusion protocol achieved steady-state plasma β-OHB enrichment suitable for measuring β-OHB turnover rates in our rats, without altering plasma β-OHB concentrations (Fig. S1E–F).
Figure 2.
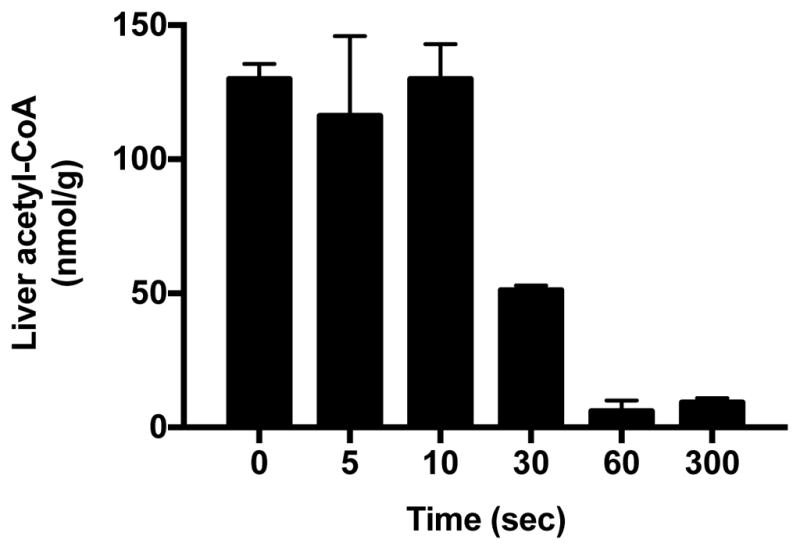
Figure 3.

Hepatic acetyl-CoA content correlates with β-OHB turnover, plasma β-OHB concentrations and hepatic glucose production in 16h fasted rats
Our data demonstrate a linear correlation between whole-body β-OHB turnover and hepatic acetyl-CoA content in vivo (Fig. 4A; R2=0.86, P<0.0001). In addition, there was a linear correlation between the means of β-OHB turnover rates and hepatic acetyl-CoA concentrations in the five groups studied, with an R2 of 0.95 (Fig. 4B; P=0.005). In addition, we found that β-OHB turnover correlated with hepatic acetyl-CoA concentrations within three of the groups: clamped (R2=0.63, P=0.01), HFD±STZ (R2=0.53, P=0.0006), and T1D rats (R2=0.81, P<0.0001). Plasma β-OHB concentrations also correlated linearly with hepatic acetyl-CoA content (Fig. 4C; R2=0.71, P<0.0001), and manifested a non-linear correlation (R2=0.70) with β-OHB turnover (Fig. S2A), reflecting reduced β-OHB clearance at higher plasma β-OHB concentrations. Next we examined the relationship between β-OHB turnover, hepatic acetyl-CoA content, and hepatic glucose production. Our data showed a strong correlation (R2=0.79, P<0.0001) between rates of HGP and hepatic acetyl-CoA content (Fig. 5A), consistent with a critical role for acetyl-CoA in allosteric activation of pyruvate carboxylase (Perry et al., 2015a; Utter and Keech, 1963; Utter et al., 1964), thereby regulating rates of hepatic gluconeogenesis. It should be noted that all of these studies were performed following a 16-hour overnight fast, which almost totally depletes hepatic glycogen content. Therefore rates of hepatic glucose production in these studies almost entirely reflect rates of hepatic gluconeogenesis. Finally, we observed linear correlations between rates of whole-body β-OHB turnover and both HGP (Fig. 5B; R2=0.75, P<0.0001) and fasting plasma glucose concentrations (Fig. S2B; R2=0.47, P<0.0001), although the latter correlation is weaker than that between rates of HGP and β-OHB turnover, likely due to other factors that are known to influence fasting plasma glucose concentrations in addition to HGP.
Figure 4.

Figure 5.

Hepatic acetyl-CoA is dissociated from hepatic glucose production under certain conditions
Next we studied rats infused with glycerol [60 μmol/(kg-min)], which led to a three-fold increase in plasma glycerol concentrations (Fig. 6A). This intervention resulted in increased hepatic glucose production, which in turn resulted in an increase in plasma glucose concentrations and a compensatory increase in plasma insulin concentrations (Fig. 6B–D). Hyperinsulinemia was associated with suppression of plasma non-esterified fatty acid concentrations, plasma β-OHB concentrations, β-OHB turnover and liver acetyl-CoA content (Fig. 6E–H). In order to examine the predictive value of β-OHB turnover to assess hepatic acetyl-CoA content under normal physiologic conditions of feeding and fasting, we next compared 6 hr food withdrawn (hepatic glycogen replete) rats to 48 hr fasted (hepatic glycogen depleted) animals and found that 48 hr fasted rats exhibited lower plasma glucose and insulin concentrations as well as lower rates of hepatic glucose production, despite increases in β-OHB turnover, β-OHB concentrations, and hepatic acetyl-CoA content as compared to recently fed rats (Fig. 7A–F). β-OHB turnover also correlated strongly with hepatic acetyl-CoA (R2=0.59, P=0.002) in hyperinsulinemic-euglycemic clamped and 48 hr fasted rats.
Figure 6.

Figure 7.

Discussion
Hepatic acetyl-CoA plays a key role in the regulation of several important hepatocellular functions such as gluconeogenesis, glucose oxidation, protein acetylation, steroid and fatty acid biosynthesis. However understanding its role in the regulation of these processes has been hampered by the inability to measure this metabolite due to the practical difficulties of measuring such a labile and relatively low-abundance molecule in vivo.
To address this unmet need we examined whether β–OHB turnover would be a useful non-invasive surrogate for hepatic acetyl-CoA content in vivo. We measured rates of whole-body β–OHB turnover, assessed by an infusion of [13C4]β–OHB, and hepatic acetyl-CoA content, assessed by LC-MS/MS (Perry et al., 2014), in five rat models with varying degrees of fasting hyperglycemia and insulin resistance. These five rodent models represent the entire spectrum of hepatic acetyl-CoA content (40–250 nmol/g) reported in our prior studies (Perry et al., 2015a; Perry et al., 2015b; Perry et al., 2014). Using this approach we observed a strong linear correlation between rates of whole-body β–OHB turnover and hepatic acetyl-CoA content (R2=0.86, P<0.0001). Furthermore plasma β-OHB concentrations also showed a strong, but less robust, linear correlation (R2=0.71, P<0.0001) with hepatic acetyl CoA content, which in part could be attributed to a non-linear relationship between rates of β-OHB turnover and β-OHB concentrations at high plasma β-OHB concentrations (Fig. S2A). It may be possible to improve these correlations by measuring plasma acetoacetate concentrations in addition to β-OHB concentrations; further studies will be needed to examine this possibility. Taken together these data demonstrate that β–OHB turnover is a useful non-invasive surrogate for hepatic acetyl CoA content in vivo. Furthermore plasma β–OHB concentrations may also serve as a simple but less robust alternative surrogate for hepatic acetyl-CoA content that may be useful in the clinic or in large population-based studies where β–OHB turnover measurements are not feasible.
These results stand in contrast to a previous study by Hans Krebs and coworkers nearly five decades ago that failed to observe a relationship between blood ketone concentrations and hepatic acetyl CoA content. These workers found that intramuscular injection of glycerol suppressed plasma acetoacetate and β-OHB concentrations without significantly altering hepatic acetyl-CoA content in rats, and consequently concluded that the main reason for the antiketogenic effect of glycerol was a consequence of its rapid metabolism and the provision of an increased supply of three-carbon intermediates for conversion to oxaloacetate (Williamson et al., 1969). The reason for the discrepancy between their findings and our findings is unclear; however, in contrast to our studies, which obtained freeze clamped liver tissue within 10 seconds following intravenously administered anesthesia in free-ranging, chronically catheterized rats and measured acetyl-CoA directly by LC-MS/MS, Krebs and his coworkers obtained liver tissue after an unspecified amount of time following decapitation and blood collection from the trunk and employed a coupled enzymatic method to measure hepatic acetyl-CoA content. Furthermore in contrast to the study by Krebs and coworkers we also assessed rates of in vivo hepatic β-OHB turnover, which we found was a more robust predictor of hepatic acetyl-CoA content than plasma ketone concentrations. Interestingly Krebs and coworkers did observe a strong tendency for hepatic acetyl-CoA content to be suppressed following intramuscular glycerol administration, which was associated with an increase in plasma glucose concentrations. We hypothesized that glycerol administration would result in hyperglycemia due to increased hepatic gluconeogenesis (Previs et al., 1999), resulting in compensatory hyperinsulinemia, which in turn would suppress lipolysis thus reducing hepatic acetyl-CoA content. To test this hypothesis, we infused glycerol into awake chronically catheterized rats and observed an increase in rates of hepatic glucose production, which was associated with hyperglycemia, hyperinsulinemia, reductions in plasma fatty acid concentrations and suppression of both hepatic acetyl-CoA content and β-OHB turnover. These data mirror the findings of Rawat and Menahan who showed that intramuscular injection of gluconeogenic substrates (fructose, glyceraldehyde, and sorbitol) raised blood glucose concentrations and suppressed both hepatic acetyl-CoA content and blood ketone concentrations (Rawat and Menahan, 1975).
We also examined the relationship between β-OHB turnover and hepatic acetyl-CoA content in 6 hr food withdrawn (hepatic glycogen repleted) and 48 hr fasted (hepatic glycogen depleted) rats. As compared to 6 hr food withdrawn rats, 48 hr fasted animals exhibited increased β-OHB turnover and increased hepatic acetyl-CoA concentrations despite suppression of whole-body glucose turnover, demonstrating that, in the physiologically relevant comparison between starved and recently fed animals, rates of whole-body glucose production do not track with hepatic acetyl-CoA content. In contrast when comparing the fed and fasted states, assessment of β–OHB turnover was a far more reliable assessment of hepatic acetyl-CoA content.
Taken together with the prior results (Rawat and Menahan, 1975; Williamson et al., 1969), these data demonstrate that in contrast to the robust correlation between rates of β-OHB turnover and hepatic acetyl-CoA content in all of our models, rates of whole-body glucose turnover correlate with hepatic acetyl CoA content only when glucose turnover is mostly derived from pyruvate carboxylase flux. However whole-body glucose turnover and hepatic acetyl-CoA content can be dissociated under conditions when net hepatic glycogenolysis, carbohydrate absorption from the gut, and/or glycerol conversion to glucose contributes significantly to rates of whole-body glucose turnover. Therefore, in contrast to β-OHB turnover, whole body glucose production cannot be used as a reliable surrogate for hepatic acetyl-CoA content.
The strong relationship that we observed between rates of hepatic β-OHB turnover and hepatic acetyl-CoA content in fed/fasted and diabetic rats is consistent with some but not all prior studies. Foster observed that during the first 12 hours of fasting hepatic acetyl CoA concentrations approximately doubled and continued to increase slightly after 24 hours of fasting. However, after 48 hours of fasting, he found that hepatic acetyl CoA concentrations surprisingly showed a marked decrease, whereas blood acetoacetate continued to increase to about twice the concentrations found at 24 hours. Furthermore, 15 minutes after intravenous administration of glucose and insulin hepatic acetyl-CoA concentrations remained completely unchanged despite a marked reduction in blood acetoacetate concentrations. From these data Foster concluded that acetoacetate synthesis in the liver cannot be related primarily to hepatic acetyl CoA concentrations (Foster, 1967). Wieland and Weiss demonstrated a modest correlation between blood acetoacetate concentrations and hepatic acetyl-CoA content in high fat-fed and cortisol-treated, alloxan-induced diabetic rats (Wieland and Weiss, 1963); however they failed to observe a relationship between these parameters in a subsequent study (Menahan et al., 1968). The reason for these discrepancies remains unclear but similar to the studies performed by Krebs and colleagues described above, may be attributed to difficulty in obtaining freeze-clamped liver tissue within 30 seconds in free-ranging unstressed animals as well as the limitation in the analytical methods available for assessing hepatic acetyl CoA content at that time. Furthermore in contrast to the current study, which assessed in vivo rates of β-OHB turnover, previous studies only assessed blood or liver ketone concentrations, which we show do not correlate as well with hepatic acetyl-CoA content as β-OHB turnover rates.
In summary, we report here that rates of β–OHB turnover correlate very strongly with hepatic acetyl-CoA content in vivo (R2=0.86, P<0.0001) across a five-fold range of acetyl-CoA concentrations in five rat models with varying degrees of fasting hyperglycemia and insulin resistance and that this parameter can be used as a reliable non-invasive surrogate for hepatic acetyl-CoA content. Given the key role of hepatic acetyl-CoA in regulating hepatic gluconeogenesis, pyruvate oxidation, protein acetylation, steroid and fatty acid biosynthesis and the difficulties in measuring this metabolite we anticipate that this simple, non-invasive stable isotope tracer method should be very useful in further elucidating the role of hepatic acetyl-CoA content in the regulation and dysregulation of these processes in humans and rodent models of diabetes and other metabolic diseases in vivo. Furthermore, plasma β–OHB concentrations may serve as a simple biomarker for hepatic acetyl-CoA content, which can be easily and inexpensively measured in population-based studies when β–OHB turnover measurements are untenable.
Experimental Procedures
Animals
All protocols were approved by the Yale University Institutional Animal Care and Use Committee. Male rats (~250 g) were purchased from Charles River and, following 1 week of acclimation, underwent surgery under general isoflurane anesthesia to place catheters in the common carotid artery and jugular vein (PE50 and PE90 tubing, Instech Laboratories, Plymouth Meeting, PA). Unless otherwise specified, rats were given ad lib access to regular chow (Harlan Teklad #2018). In high fat fed rats, insulin resistance was induced by giving ad lib access to a safflower oil-based high fat diet (59% calories from fat/26% carbohydrate/15% protein, Dyets #112245, Bethlehem, PA) for 4 weeks. Type 1 diabetes was induced by an intraperitoneal (IP) injection of 65 mg/kg streptozotocin (Sigma, St. Louis, MO) in overnight fasted rats 24 hr before the start of the tracer study. A rat model of type 2 diabetes was induced in overnight fasted high fat fed rats by IP injection of 40 mg/kg streptozotocin 15 min after an IP injection of 80 mg/kg nicotinamide (Sigma, St. Louis, MO), 24 hr prior to a study. In both STZ-treated models, food was replaced immediately after injection, and the rats were given ad lib access to food for the next 10 hours. Unless otherwise specified, rats were fasted overnight prior to studies. Separate groups of chow fed rats were fasted for 6 and 48 hrs prior to tracer infusions as described below.
In the hyperinsulinemic-euglycemic clamps, rats received a bolus of insulin (40 mU/kg) through the arterial catheter, and a continuous infusion of insulin (4 mU/[kg-min]) was immediately begun. A variable infusion of [1,2,3,4,5,6,6-2H7]glucose (2.5% APE) was administered, with rates adjusted to maintain euglycemia (100–110 mg/dL). Blood was sampled for measurement of plasma glucose and insulin concentrations and β–OHB enrichment. Throughout the clamp, sodium [13C4]β–OHB was also infused at the rates listed in the following section.
β–OHB turnover studies
Rats underwent a 120 min infusion of sodium [13C4]β–OHB [0.1 mg/(kg-min)] (Cambridge Isotopes, Tewksbury, MA) through the arterial catheter. 200 μL whole blood was drawn from the venous catheter at 100, 110, and 120 min and used to measure steady-state [13C4]β–OHB enrichment, as described below. The animals were then euthanized with intravenous pentobarbital and their livers immediately freeze clamped in situ and stored at −80°C pending further analysis.
Glycerol infusions
In the glycerol infusion studies, rats were infused with glycerol (60 μmol/[kg-min]) through an arterial catheter for the duration of a 120 min infusion of [13C4]β–OHB and [1,2,3,4,5,6,6-2H7]glucose at the rates listed above. Blood samples were taken at 0 and 120 min, and rats were sacrificed as described above.
Acetyl-CoA degradation
To analyze acetyl-CoA degradation ex vivo, we euthanized rats and removed their livers, separating them into six pieces upon excision and left them on the bench top at room temperature for varying periods of time. Liver samples were freeze clamped at the times indicated after excision and stored at −80°C to await measurement of acetyl-CoA concentrations as described below.
Tissue and plasma analysis
Plasma glucose concentrations were measured using the YSI Glucose Analyzer (Yellow Springs, OH), plasma insulin concentrations were measured by ELISA (Mercodia, Winston Salem, NC), and plasma β–OHB concentrations were measured by COBAS (Roche Diagnostics, Indianapolis, IN). Plasma NEFA concentrations were measured using the Wako Diagnostics NEFA-HR(2) reagent (Richmond, VA).
Hepatic acetyl-CoA concentrations were measured by LC-MS/MS and plasma glycerol concentration and enrichment was measured by GC/MS as we have described (Perry et al., 2015a). To measure plasma β–OHB enrichment, samples (25 μL plasma) were dried under N2 gas and derivatized with 75 μL n-butanol 4N HCl (Sigma), then heated for 60 min at 65°C and dried under N 2 gas. The samples were then reacted with 100 μL of trifluoroacetic acid (Thermo Fisher Scientific)-methylene chloride (Sigma) (1:7). [13C4]β–OHB enrichment was measured by GC/MS (CI mode, m/z 257 [m+0] and 261 [m+4]).
To measure hepatic glucose production, steady-state plasma samples were deproteinized in methanol (5x volume), dried, and derivatized by adding 75 μL of 1:1 acetic anhydride:pyridine and heating to 65°C for 2 0 min. After cooling the samples, 25 μL of methanol were added, and glucose enrichment was measured on the GC/MS (CI mode, m/z 331 [m+0], 332 [m+1], 333 [m+2], 334 [m+3], 335 [m+4], 336 [m+5], 337 [m+6], 338 [m+7]). We calculated whole-body β–OHB and glucose turnover using the plasma and infusate enrichments as follows:
Data analysis
GraphPad Prism version 7.0 (La Jolla, CA) was used for all data analysis. Data are presented as the mean ± S.E.M. of the numbers indicated in the figure legends.
Supplementary Material
Figure 1.

Acknowledgments
The authors thank Xian-Man Zhang, Jianying Dong, Wanling Zhu, and Mario Kahn for their invaluable technical contributions. This study was funded by grants from the National Institutes of Health (R01 DK-40936, R01 AG-23686, P30 DK-45735, T32 DK-101019) and an investigator-initiated grant from Gilead Sciences, Inc.
Footnotes
Author Contributions
All authors contributed to the design of the study. Experiments were performed and data analyzed by L.P., R.J.P., and G.W.C. The manuscript was written by R.J.P. and G.I.S. with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Foster DW. Studies in the ketosis of fasting. The Journal of clinical investigation. 1967;46:1283–1296. doi: 10.1172/JCI105621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menahan LA, Ross BD, Wieland O. Acetyl CoA level in perfused rat liver during gluconeogenesis and ketogenesis. Biochemical and biophysical research communications. 1968;30:38–44. doi: 10.1016/0006-291x(68)90709-2. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, Abudukadier A, Han MS, Zhang XM, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015a;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Lee S, Ma L, Zhang D, Schlessinger J, Shulman GI. FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nature communications. 2015b;6:6980. doi: 10.1038/ncomms7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RJ, Zhang XM, Zhang D, Kumashiro N, Camporez JP, Cline GW, Rothman DL, Shulman GI. Leptin reverses diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nature medicine. 2014;20:759–763. doi: 10.1038/nm.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell metabolism. 2015;21:805–821. doi: 10.1016/j.cmet.2015.05.014. [DOI] [PubMed] [Google Scholar]
- Previs SF, Cline GW, Shulman GI. A critical evaluation of mass isotopomer distribution analysis of gluconeogenesis in vivo. The American journal of physiology. 1999;277:E154–160. doi: 10.1152/ajpendo.1999.277.1.E154. [DOI] [PubMed] [Google Scholar]
- Rawat AK, Menahan LA. Antiketogenic action of fructose, glyceraldehyde, and sorbitol in the rat in vivo. Diabetes. 1975;24:926–932. doi: 10.2337/diab.24.10.926. [DOI] [PubMed] [Google Scholar]
- Reed MJ, Meszaros K, Entes LJ, Claypool MD, Pinkett JG, Gadbois TM, Reaven GM. A new rat model of type 2 diabetes: the fat-fed, streptozotocin-treated rat. Metabolism: clinical and experimental. 2000;49:1390–1394. doi: 10.1053/meta.2000.17721. [DOI] [PubMed] [Google Scholar]
- Utter MF, Keech DB. Pyruvate Carboxylase. I. Nature of the Reaction. The Journal of biological chemistry. 1963;238:2603–2608. [PubMed] [Google Scholar]
- Utter MF, Keech DB, Scrutton MC. A possible role for acetyl CoA in the control of gluconeogenesis. Advances in enzyme regulation. 1964;2:49–68. doi: 10.1016/s0065-2571(64)80005-4. [DOI] [PubMed] [Google Scholar]
- Wieland O, Weiss L. Increase in liver acetyl-coenzyme A during ketosis. Biochemical and biophysical research communications. 1963;10:333–339. doi: 10.1016/0006-291x(63)90534-5. [DOI] [PubMed] [Google Scholar]
- Williamson DH, Veloso D, Ellington EV, Krebs HA. Changes in the concentrations of hepatic metabolites on administration of dihydroxyacetone or glycerol to starved rats and their relationship to the control of ketogenesis. The Biochemical journal. 1969;114:575–584. doi: 10.1042/bj1140575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.