

Abstract
Although tremendous efforts have been made on targeted drug delivery systems, current therapy outcomes still suffer from low circulating time and limited targeting efficiency. The integration of cell-mediated drug delivery and theranostic nanomedicine can potentially improve cancer management in both therapeutic and diagnostic applications. By taking advantage of innate immune cell’s ability to target tumor cells, we developed a novel drug delivery system by using macrophages as both nanoparticle-carriers and navigators to achieve cancer-specific drug delivery. Theranostic nanoparticles were fabricated from a unique polymer, biodegradable photoluminescent poly (lactic acid) (BPLP-PLA), which possesses strong fluorescence, biodegradability, and cytocompatibility. In order to minimize the toxicity of cancer drugs to immune cells and other healthy cells, an anti-BRAF V600E mutant melanoma specific drug (PLX4032) was loaded into BPLP-PLA nanoparticles. Muramyl tripeptide (MTP) was also conjugated onto the nanoparticles to improve the nanoparticle loading efficiency. The resulting nanoparticles were internalized within macrophages, which were tracked via the intrinsic fluorescence of BPLP-PLA. Macrophages carrying nanoparticles delivered drugs to melanoma cells via cell-cell binding. Pharmacological studies also indicated that the PLX4032 loaded nanoparticles effectively killed melanoma cells. Our “self-powered” immune cell-mediated drug delivery system demonstrates a potentially significant advancement in targeted theranostic cancer nanotechnologies.
Keywords: Nanoparticles, Immune cells, Theranostics, Melanoma, Drug Delivery
Graphical abstract
1. Introduction
Nanotechnology plays a crucial role in modern medicine, especially in targeted cancer drug delivery and imaging[1]. Nanocarriers provide protection for chemotherapeutics, genes, and imaging agents against the harsh environment encountered in systemic circulation[2]. Compared to conventional systematic delivery and passive targeting, active targeted delivery of nanoparticles improves the therapeutic index and reduces side effects as well as minimizes immunogenicity[3, 4]. Most existing active targeting strategies utilize surface molecules such as antibodies, proteins, aptamers, peptides, or small molecules to recognize receptors that are expressed or overexpressed in cancer cells or cancer microenvironments[5, 6]. However, despite tremendous efforts towards discovering surface markers and targeting molecules, existing nanomedicine still fails to meet the expectations of efficient delivery of therapeutics to specific tumors via intravenous injection[5, 7]. The major limitations are low circulation time and poor target selectivity for a specific disease or cancer. The complexity of living systems makes specific recognition chance-dependent, which compromises the effectiveness of drug delivery systems. Thus there is an urgent need to develop “true” specific targeting strategies for cancer treatment.
In recent years, circulating cells such as immune cells have gained interest as novel “living” delivery vehicles[8, 9]. Research has shown that leukocytes are capable of homing into tumor sites and regulating metastasis by adhesion molecule-mediated interactions with circulating tumor cells (CTCs)[10–12]. Such evidence points to the rational design of using immune cells as “smart” vehicles for cancer targeting and therapeutics delivery. For example, Choi et al. demonstrated that macrophages could be manipulated as “Trojan Horses” to deliver gold nanoparticles to breast tumors and brain metastases[13, 14]. A similar concept was used to guide gold nanoshells to brain gliomas for photothermal therapy via macrophages[15]. These studies, however, only focused on using macrophages as delivery carriers to reach tumor sites. As such, the potential of leukocytes targeting circulating cancer cells under dynamic blood stream conditions remained relatively unknown. Recently, leukocytes were functionalized with tumor necrosis factor-related apoptosis-inducing ligands (TRAIL)/E-selectin adhesion receptors to kill cancer cells in the circulation[16–18], providing insight into immune cell-meditated targeting. These studies suggest that utilizing immune cells as carriers to mediate therapeutic nanoparticles is an attractive approach for targeted drug delivery.
Today, significant leaps are being made in nanomedicine by integrating controlled drug delivery with imaging-based diagnosis, resulting in a new, emerging field of theranostic nanomedicine. Advances in fluorescence imaging techniques have enabled early detection of cancer and real-time monitoring of the drug delivery processes. However, living cell-mediated theranostic drug delivery has not been widely investigated, partly because fluorescence imaging agents such as organic dyes, green fluorescence proteins, and quantum dots generally suffer from poor stability, solubility, and potential cellular toxicity[19]. Due to these major drawbacks, traditional fluorescence imaging materials cannot be used as drug delivery carriers alone, but must undergo conjugation or encapsulation steps, contributing to unfavorable drug release profiles that are difficult to control.
To overcome these unmet challenges, we herein report the development of theranostic immune cell-mediated biodegradable polymeric nanoparticles with intrinsic fluorescence in order to enable carrier cell imaging with improved photostability and reduced cytotoxicity, as well as better control over cancer-specific drug release. This novel polymeric nanoparticle is synthesized from biodegradable photoluminescent poly (lactic acid) (BPLP-PLA), which is a fully degradable polymer with tunable intrinsic fluorescence as we reported previously[20]. BPLP-PLA possesses strong and stable fluorescence for optical imaging, along with good cytocompatibility on par with widely used PLA in FDA approved devices [20]. The degradation rates of BPLP-PLA can also be easily tuned to achieve controlled drug delivery, making BPLP-PLA an ideal biomaterial for theranostic drug delivery. To the best of our knowledge, no study has reported controlled delivery of biodegradable polymeric nanoparticles by using immune cells for cancer treatment.
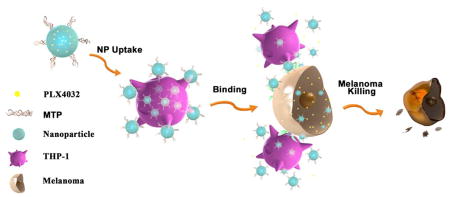
For this study, THP-1 cells were selected as the nanoparticle carrier in order to model monocyte/macrophage systems, while the theranostic nanoparticles were equipped with melanoma-specific drugs to target melanoma cells. THP-1 is a human leukemia cell line that is widely used as a monocytic model for mnonocyte-macrophage differentiation and has affinities to cancer cells and other inflammatory cells[21]. To minimize the adverse effects on leukocytes and maximize the antitumor effects on cancer cells, PLX4032 (also known as Vemurafenib; a drug specifically designed for treating BRAF V600E mutated melanomas) was selected as the therapeutic agent and encapsulated within BPLP-PLA nanoparticles (BPLP-PLA-PLX4032)[22]. To further improve the THP-1 cellular uptake efficiency of nanoparticles, muramyl tripeptide (MTP)[23] was conjugated onto drug-loaded nanoparticles to form a complex named MTP-BPLP-PLA-PLX4032. The design of our nanoparticles and immune cell-mediated targeting strategy for melanoma cells is illustrated in Figure 1.
Figure 1.

Schematic illustration of the immune cell-mediated nanoparticle (NP) delivery system targeting to cancer cells. Specifically, PLX4032, an anti-BRAF V600E mutant melanoma drug, are loaded within BPLP-PLA nanoparticles. MTP-conjugated BPLP-PLA-PLX4032 nanoparticles bind to THP-1 macrophages first and they are then delivered to melanoma cells via interactions between THP-1 macrophages and melanoma cells. The delivered nanoparticles eventually release the PLX4032 drug to kill cancer cells.
2. Results and Discussions
2.1. Fabrication and Characterization of BPLP-PLA Nanoparticles
Biodegradable BPLP-PLA was used in this study for immune cell-mediated delivery to bridge the limitations of existing immune cell-mediated nanoparticle delivery strategies that utilize inorganic nanoparticles, as well as of liposomes that lack theranostic capabilities and a controlled drug release mechanism[13, 14, 16]. BPLP-PLA copolymer with intrinsic fluorescence was synthesized as we reported previously[20, 24]. As mentioned above, varying the BPLP to L-lactide molar ratio allows control over the degradation rate[20]. A BPLP to L-lactide ratio of 1:50 was used in this study to ensure minimal degradation and drug release in the initial 24 hours, which is the time window for immune cells to uptake nanoparticles and further bind to melanoma cells[25]. BPLP-PLA nanoparticles were fabricated by a single emulsion method[20, 24]. In order to increase THP-1 cell targeting efficiency, we further modified drug-laden nanoparticles with MTP, which has macrophage immuno-potentiating effects without significant cytotoxicity[26–28]. MTP was successfully conjugated with BPLP-PLA and BPLP-PLA-PLX4032 nanoparticles by carbodiimide chemistry, as confirmed by FTIR (Supporting Information Figure S1). The increments of –NH stretching and –(C=O)N- stretching indicate the presence of peptides on BPLP-PLA nanoparticles.
PLX4032 was selected as a drug for encapsulation as it is specific for inhibiting BRAF (V600E) mutation melanoma[22, 29, 30]. PLX4032-encapsulated BPLP-PLA nanoparticles were fabricated by the same single emulsion method by mixing PLX4032 into BPLP-PLA solution with a ratio of 1:5 w/w. The drug loading efficiency was 54% as determined by HPLC (Supporting Information Figure S2). Scanning electron microscopy (SEM) images of PLX4032-loaded nanoparticles (BPLP-PLA-PLX4032) and MTP-modified BPLP-PLA-PLX4032 nanoparticles (MTP-BPLP-PLA-PLX4032) are shown in Figure 2A. The average diameters of BPLP-PLA-PLX4032 and MTP-BPLP-PLA-PLX4032 nanoparticles as determined by dynamic light scattering (DLS) were 217.2 nm (PDI: 0.147) and 209.1 nm (PDI: 0.121) (Figure. 2B and 2C), respectively. The results were consistent with those from SEM images. Zeta potentials of our nanoparticles were -30.3 mV and -36.2 mV, suggesting that they can be stable in physiological solutions[31].
Figure 2.

Characterization of BPLP-PLA-PLX4032 and MTP-BPLP-PLA-PLX4032 nanoparticles. SEM images of BPLP-PLA-PLX4032 (A1) and MTP-BPLP-PLA-PLX4032 (A2) nanoparticles, scale bar = 500 nm. (B) Size distributions of BPLP-PLA-PLX4032 (Red) and MTP-BPLP-PLA-PLX4032 (Blue) nanoparticles by DLS. (C) The average sizes, polydisperse indexes (PDI), and zeta potentials of BPLP-PLA-PLX4032 and MTP-BPLP-PLA-PLX4032 nanoparticles measured by DLS. (D) Fluorescence emission spectra of MTP-BPLP-PLA-PLX4032 nanoparticle suspension (20 μg/ml) at different excitation wavelengths.
Finally, we examined the effects of nanoparticle fabrication on the fluorescence properties of BPLP-PLA. In our previous work, we found that BPLP-PLA exhibits intrinsic fluorescence and band shifting emission with different excitation wavelengths[20, 24]. Here, our nanoparticles maintained strong fluorescence emission, tunable up to 700 nm by varying the excitation wavelength (Figure 2D). The intrinsic fluorescence of nanoparticles enables in vitro visualization without secondary labeling with traditional imaging agents such as organic dyes and quantum dots that often demonstrate significant toxicity. BPLP-PLA also possesses excellent photostability, which is desirable for cell tracking applications [20, 32].
2.2. THP-1 Cellular Uptake of Drug-loaded Nanoparticles
As a well-established native monocyte-derived macrophage model[33], THP-1 cell was chosen for this study to demonstrate macrophage uptake of drug-laden nanoparticles. Hence BPLP-PLA-PLX4032 or MTP-conjugated nanoparticles were incubated with THP-1 cells for two hours on a rocker, followed by washing steps to remove unbound nanoparticles. Preliminary confocal microscopy studies suggested that our nanoparticles were surface-bound and internalized by THP-1 cells, and that cellular fluorescence could be detected in both FITC and PE-Texas Red channels due to the intrinsic variable fluorescence of BPLP-PLA nanoparticles (Figure 3A). Indeed, flow cytometry confirmed that both FITC and PE-Texas Red signals from nanoparticle-laden THP-1 cells increased after the extracellular fluorescence was quenched by trypan blue, further suggesting that the nanoparticles were internalized by THP-1 cells (Figure 3B). These results demonstrate the versatility and effectiveness of BPLP-PLA nanoparticles in cellular imaging and tracking, since the band shifting behavior resulted from the use of different excitation wavelengths (Figure 2D) enables a wide range of detection channels, even to red fluorescence.
Figure 3.

THP-1 cellular uptake of nanoparticles. (A) Confocal images of MTP-BPLP-PLA-PLX4032 nanoparticles taken up and internalized by THP-1 cells. Nuclei were stained by DAPI; nanoparticles were shown in the FITC and PE-Texas Red channels, CD11b was immunostained by Alexa647 (pseudo color in pink). (B) FACS analysis of THP-1 cells before and after treated with BPLP-PLA-PLX4032 and MTP-BPLP-PLA-PLX4032 nanoparticles (C) FITC and PE-Texas Red average fluorescence intensity of THP-1 cells, THP-1 cells with BPLP-PLA-PLX4032 and MTP-BPLP-PLA-PLX4032 nanoparticles. *, p<0.01 compare to the THP-1 control. (D) THP-1 cell binding efficiency as the percentage of THP-1 cells that were labeled by BPLP-PLA-PLX4032 and MTP-BPLP-PLA-PLX4032 nanoparticles. *, p<0.01.
Since MTP is a macrophage activator known to enhance nanoparticle uptake[23], MTP was conjugated to BPLP-PLA-PLX4032 to correlate enhancement of FITC and PE-Texas Red signals to nanoparticle internalization. It is clear that both FITC and PE-Texas Red fluorescence increased with MTP-conjugated nanoparticles from a greater extent of nanoparticle internalization by THP-1 cells (Figure 3C–D). Quantitatively, the average FITC and PE-Texas Red intensities of THP-1 cells treated with MTP-BPLP-PLA-PLX4032 nanoparticles were stronger than those with just BPLP-PLA-PLX4032 nanoparticles (Figure 3C), suggesting that MTP conjugation increased the nanoparticle loading efficiency of THP-1 cells. The nanoparticle loading efficiency (how many cell have nanoparticles attached or internalized) was determined by the percentage of cells in Q1, Q2, and Q3 that had increased fluorescence. About 96% of the THP-1 cells were labeled by MTP-conjugated BPLP-PLA-PLX4032 nanoparticles, whereas 61% of the THP-1 cells were marked by pristine BPLP-PLA-PLX4032 nanoparticles. Thus MTP conjugation was shown to improve nanoparticle binding to THP-1 cells. Additionally, the protein corona effect is considered to play a negligible role in affecting the overall internalization efficiency of THP-1 cells even in physiological relevant conditions (i.e. protein-containing medium) based on a recent study, which suggests that a protein-containing medium or complete cell medium does not result in significant change on the overall internalization efficiency of differentiated, macrophage-like THP-1 cells[34].
We also studied CD11b (an alpha chain of the β2 integrin MAC-1) expression to determine whether nanoparticle internalization affects THP-1’s macrophage-like functionality. Studies have indicated that polymorphonuclear neutrophils (PMNs) bind to melanoma cells in the blood circulation through β2 integrins of PMNs and intercellular adhesion molecule-1 (ICAM-1) of melanoma cells[35]. We hypothesized that macrophages such as THP-1, upon PMA (Phorbol 12-myristate 13-acetate)-induced CD11b expression, have this same melanoma binding ability through ICAM-1[36–38]. Thus, the maintenance of CD11b would be critical in binding melanoma. First, we showed that macrophage-like phenotype from differentiated THP-1 cells (induced by PMA) could be identified by CD11b expression (from the fluorescence of Alexa647) (Supporting Information Figure S3). Next, confocal microscopy and 3D flow cytometry plots showed strong immunofluorescence from Alexa647 (in addition to the FITC and PE-Texas Red fluorescence signals from the nanoparticles), indicating that THP-1 cells still expressed CD11b after taking up nanoparticles (Figure 3A and Supporting Information Figure S4), thereby suggesting that the encapsulation of nanoparticles did not alter THP-1’s functionality as a macrophage-like cell. These results showed that BPLP-PLA-PLX4032 nanoparticles were picked up and internalized by THP-1 cells while MTP conjugation increased loading efficiency without affecting THP-A macrophage-like functionality. Based on these results, MTP-BPLP-PLA-PLX4032 nanoparticles were deemed suitable for follow-up experiments on melanoma binding and therapeutic studies.
2.3. THP-1 Cell-mediated Nanoparticle delivery to Melanoma Cells
Tumor progression is a complex and dynamic process involving immune cells and tumor cells, as tumor cell subpopulations are susceptible to recognition and destruction by immune cells[39]. In particular, macrophages are major players in the tumor microenvironment and are involved in chronic inflammation, matrix remodeling, tumor cell invasion, intravasation, angiogenesis, and metastasis[40]. This central role of leukocytes on tumor cell immunosurveillance highlights the value of utilizing macrophages as nanoparticle “carriers” to target cancer cells and deliver drugs.
To demonstrate such value in immune cell-mediated theranostic drug delivery, the next step was to assess the effectiveness of THP-1 cell binding and MTP-BPLP-PLA-PLX4032 nanoparticle delivery to melanoma cells. 1205Lu cells and WM35 cells were chosen as models for high and low metastatic melanoma cells respectively, and co-culture studies were performed under both static and dynamic conditions to model laminar and shear flow conditions[41]. First, bare THP-1 cells without nanoparticles were co-cultured with either type of melanoma cells for two hours under static conditions (on a rocker) to demonstrate that THP-1 cells are able to attach to melanoma cells (Supporting Information Figure S5). Next, THP-1 cells loaded with MTP-BPLP-PLA-PLX4032 nanoparticles (with a two hour incubation and washing steps) were co-cultured with either type of melanoma cells for two hours under static or dynamic conditions.
Nanoparticle-laden THP-1 cells were able to successfully bind to GFP-1205Lu cells as determined by confocal microscopy (Figure 4A). To better show THP-1/melanoma cell binding for confocal microscopy and quantitative flow cytometry analysis, 1205Lu cells were tagged with Green Fluorescent Protein (GFP-1205Lu) and WM35 cells were stained with CellTrace™ CFSE. Since GFP intensity is much greater than BPLP-PLA nanoparticle intensity at the FITC channel, nanoparticle-labeled THP-1 cells emit minimal fluorescence at the FITC channel at an exposure time suitable for GFP-labeled melanoma cells (Figure 4A). Likewise, GFP-labeled melanoma cells emit minimal red fluorescence compared to nanoparticle-labeled THP-1. Thus, intensity along the FITC channel indicated melanoma cells, whereas intensity along the PE-Texas Red channel indicated BPLP-PLA nanoparticles, thereby enabling qualitative and quantitative discriminatory analysis. Next, flow cytometer analysis was performed to quantitatively evaluate this binding. Compared to untreated GFP-1205Lu cells that show no fluorescence in the PE-Texas Red channel, co-culturing with nanoparticle-bearing THP-1 cells induced the population of GFP-1205Lu cells to shift into the Q2 region with a greater degree of red fluorescence (Figure 4B). This enhanced red fluorescence indicates binding of GFP-1205Lu cells with nanoparticle-bearing THP-1 cells to form leukocyte/nanoparticle/melanoma complexes, or transference of nanoparticles from THP-1 to GFP-1205Lu upon binding. In fact, confocal microscopy images showed trace red fluorescence within GFP-1205Lu cells, supporting nanoparticles released from THP-1 cells to GFP-1205Lu cells (Figure 4A). It is likely that nanoparticles were released via exocytosis, owing to the equilibrium of engulfing foreign substances and liberating engulfed particles, particularly since BPLP-PLA nanoparticles are constructed by a polyester copolymer with minimal charges[42]. Previous studies suggested that neutrally charged nanoparticles were able to transport out of macrophages significantly faster than cationic nanoparticles[43].
Figure 4.

THP-1 mediated melanoma binding and nanoparticles delivery. THP-1 pre-treated with MTP-BPLP-PLA nanoparticles for two hours and then co-cultured with GFP-tagged 1205Lu for one hour under static conditions and dynamic conditions with shear rates varied from 50 s−1 to 200 s−1. (A) Confocal images of THP-1/GFP-1205Lu binding and nanoparticles (PE-Texas Red) delivery, scale bar: 20 μm. (B) FACS analysis of THP-1 control, GFP-1205Lu ITcontrol, THP-1/nanoparticle/1205Lu complexes after static incubation and dynamic binding. (C) Average fluorescence intensity of PE-Texas Red and FITC within the Q1 and Q2 areas of Figure 4B. *, p<0.01.
In order to mimic the dynamic shear-flow environments of the bloodstream, GFP-1205Lu cells were exposed to nanoparticle-carrying THP-1 cells on a cone-plate viscometer. As shown in Figure 4, confocal microscopy and flow cytometry results showed that THP-1 cells were bound to GFP-1205Lu cells even under shear flow. Lastly, FACS analysis was performed to quantify the binding efficiency of THP-1 and melanoma cells, which is defined as the ratio of the cells in Q2 to the cells in both Q1 and Q2. Over 90% of 1205Lu cells were bound to nanoparticle-laden THP-1 cells or received nanoparticles via exocytosis when shear rates ranged from 50 s−1 to 200 s−1, which is the relatively lower range of the blood wall shear rate[44]. These numbers suggest that nanoparticle-carrying THP-1 cells adhere to melanoma cells. This macrophage-mediated delivery strategy is also verified by a low metastatic melanoma cell line, WM35. Similar results were observed, indicating THP-1/WM35 binding and nanoparticle transportation to WM35, as shown in Supporting Information Figure S6 Thus, in vitro binding was demonstrated between THP-1 cells and melanoma cells, as well as THP-1 cell-mediated nanoparticle delivery.
2.4. Pharmacological Studies
Since we have verified the binding and nanoparticle delivery capabilities of THP-1 cells to melanoma cells, the final step was to examine the safety of our immune cell-mediated nanoparticle delivery system and its pharmacological effects on cancer cells. In order to minimize the potential damage to immune cells and normal tissues, PLX4032 was used as an anti-cancer drug that specifically inhibits the BRAF oncogene of V600E-mutated positive melanomas[45, 46] which prevents melanoma cell extravasation and subsequent metastasis[29, 30]. We investigated two melanoma cell lines, 1205Lu (high metastatic) and WM35 (low metastatic), which are both BRAF mutants with V600E expression[22, 46]. First, we found that free PLX4032 itself selectively killed 1205Lu and WM35 at concentrations of 50 ng/mL (Figure 5A). With PLX4032 concentration above 5 μg/mL, almost 100% death of melanomas was achieved. However, no significant reduction in viability of THP-1 cells was observed even with concentrations as high as 100 μg/mL. Thus, PLX4032 was determined to be an ideal drug for immune cell-mediated drug delivery to melanoma cells, presenting minimal toxicity to the carrier immune cells. Second, in vitro drug release studies showed sustained release of PLX4032 from our nanoparticles (Supporting Information Figure S7). No clear burst release was observed in the release curve, which is important in minimizing side effects and achieving high therapeutic outcomes. Third, we tested the effects of drug-free MTP-BPLP-PLA nanoparticles and PLX4032-loaded MTP-BPLP-PLA nanoparticles on THP-1 and melanoma cells. As shown in Figure 5B, pristine BPLP-PLA nanoparticles lacking the drug did not significantly reduce the THP-1 cell viability at nanoparticle concentrations as high as 1000 μg/mL. For 1205Lu and WM35 melanomas, however, pristine nanoparticles exhibited toxicity at 500 μg/mL. After 7 days of incubation, PLX4032 loaded nanoparticles killed far more melanomas than pristine nanoparticles, especially for WM35, which indicated significant difference at 50 μg/mL of nanoparticles. The cell viability of both 1205Lu and WM35 melanoma cells were significantly reduced to around 30% at 1000 μg/mL MTP-BPLP-PLA-PLX4032 nanoparticle concentrations after seven days of incubation, suggesting that the drug released from nanoparticles were effective in killing melanoma cells. Finally, we assessed the effectiveness of the complete THP-1-mediated MTP-BPLP-PLA-PLX4032 nanoparticle delivery system. THP-1 cells were treated with PLX4032 loaded nanoparticles, and after removal of free nanoparticles, THP-1 cells were co-cultured with 1205Lu or WM35 cells (at 2000 cells per well each) with different THP-1 cell to melanoma cell ratios, including 5000, 10000, 25000, 50000, and 100000 THP-1 per well, for seven days. Bare THP-1 cells lacking nanoparticles served as control. Compared with pristine THP-1 cells, nanoparticle-bearing THP-1 significantly decreased the viability of both 1205Lu and WM35 cells; even at the lowest THP-1 number (5,000 cells per well), as shown in Figure 5C. With increasing number of THP-1 cells added, more melanoma cells were killed, likely due to activated macrophages releasing tumor necrosis factor that kill cancer cells[47]. Again, THP-1 cells that engulfed PLX4032-loaded nanoparticles further reduced both 1205Lu and WM35 cells viabilities than that of pristine THP-1 cells even at higher ratios. Since we have previously noted that nanoparticles can be transported from THP-1 cells into melanoma cells (Figure 4A), it is likely that PLX4032 was released to effectively treat melanomas. Noticeably, THP-1 cell-mediated drug delivery was more effective in killing WM35 cells than 1205Lu cells, probably because PLX4032 is more effective on WM35 cells[22]. In conclusion, our immune cell-mediated nanoparticle delivery strategy is an effective approach to transport therapeutics to melanoma cells.
Figure 5.

Pharmacological studies of effects of free drugs, nanoparticles and nanoparticle-bearing THP-1 cells on melanoma cells (1205Lu and WM35). Cell viability tests were conducted by CCK-8 assay (n=6). (A) Toxicity of PLX4032 to THP-1, WM35 and 1205Lu cells after 24 hours of incubation. (B) Toxicity of MTP-BPLP-PLA and MTP-BPLP-PLA-PLX4032 nanoparticles to THP-1, WM35 and 1205Lu cells after seven days of incubation. (C) THP-1 mediated nanoparticles delivery and drug release effects on melanoma cells with different THP-1 to melanoma cell (2,000 cells per well) ratios after seven days of incubation. #, p<0.01 compared to controls; *, p<0.01 between two groups.
3. Conclusions
In this paper, we developed a novel targeted nanomedicine strategy based on a “living” nanoparticle delivery, mediated by immune cells such as macrophages. Our novel biodegradable polymeric theranostic nanoparticles encapsulated melanoma specific therapeutics, PLX4032, to provide cell tracking capabilities, safe protection towards macrophage model cells, THP-1, and controlled release of drugs to cancer cells. High THP-1 uptake of nanoparticles was achieved by modifying particles with MTP peptides. The active binding of THP-1 cells to melanoma cells was confirmed with and without the presence of nanoparticles. Guided by THP-1 cells, nanoparticles were delivered to melanoma cells and consequently released drug contents to kill cancer cells. In summary, our immune cell-mediated nanoparticle targeting strategy enabled a self-powered effective targeted and traceable cancer drug delivery by taking advantage of the nature of interactions between immune cells and tumors. This work may potentially innovate immune cell-mediated systems not only for cancer drug delivery and imaging, but also for other diseases that involve innate immune responses.
4. Experimental Section
Materials
Chemicals for BPLP-PLA synthesis were purchased from Sigma-Aldrich. THP-1 cells were purchased from ATCC. Human melanoma cells, WM35, 1205Lu, and GFP-tagged 1205Lu melanoma cells were purchased from the Wistar Institute. RPMI-1640 medium, Leibovitz’s L-15 medium, 2-mercaptoethanol, Alexa Fluor® 647 Goat Anti-Rat IgG (H+L), and cell dissociation solutions were all obtained from Life Technologies. Fetal Bovine Serum (FBS) was obtained from Atlanta Biologicals. Phorbol 12-myristate 13-acetate (PMA), MCDB 153 medium, lipopolysaccharide (LPS) from Escherichia coli and Dulbecco’s phosphate-buffered saline (DPBS), and other chemicals were all purchased from Sigma-Aldrich. CCK-8 assay kit was obtained from Dojindo. PLX4032 drug was purchased from Chemie-Tech. Muramyl tripeptide (MTP) was obtained from InvivoGen. Integrin αM antibody (M1/70) (CD11b) was purchased from Santa Cruz Biotechnology.
Cell Culture
THP-1 and WM35 cells were maintained with RPMI-1640 medium supplemented with 10% FBS and 0.05 mM 2-mercaptoethanol at 37°C under 5% CO2. Before THP-1 uptake, cell binding, and pharmacological studies, THP-1 cells were first differentiated by 200 nM PMA in RPMI-1640 medium for three days followed by one-day culture in PMA-free medium. Then, 1 μg/mL LPS was applied to stimulate differentiated THP-1 for 24 hours. 1205Lu and GFP-tagged 1205Lu cells were cultured in a tumor medium containing a 4:1 mixture of MCDB 153 medium with 1.18 g/L sodium bicarbonate and Leibovitz’s L-15 medium with 2 mM L-glutamine. The mixed medium were supplemented with 0.005 mg/mL bovine insulin, 1.68 mM CaCl2, and 2% FBS. Both 1205Lu and GFP-tagged 1205Lu cells were cultured at 37°C under 5% CO2.
Polymer Synthesis and Nanoparticle Fabrication
BPLP was synthesized according to our previous protocol with a simple polycondensation of reacting citric acid, 1,8-octanediol, and L-serine at 140°C[24]. Next, BPLP-PLA was synthesized via a ring-opening polymerization according to our previously reported method[20]. The feeding molar ratio of BPLP to L-lactide monomers was 1:50. The characterization of BPLP-PLA can be found in our previous reports[20]. BPLP-PLA nanoparticles were prepared by a single emulsion method. Briefly, 50 mg BPLP-PLA polymer was dissolved in 2 mL chloroform solution, which was added drop-wise into 20 mL 5wt% poly(vinyl alcohol) (87% hydrolyzed, Mw of 87 kDa) solution during sonication. The solution was stirred vigorously overnight for solvent evaporation. Resulting nanoparticles were centrifuged at 10, 000 rpm and washed with DI water for three times before lyophilized. PLX4032 drug loaded nanoparticles (BPLP-PLA-PLX4032) was prepared by dissolving 20 wt% (to BPLP-PLA) of PLX4032 in 200 μL DMSO and mixing with the polymer solution, followed by the same single emulsion and washing procedure to obtain drug loaded nanoparticles. MTP was conjugated to nanoparticles by carbodiimide chemistry, according to an established protocol[48]. Specifically, 40 mg nanoparticles were dispersed in 20 mL MES buffer (pH 4.5) by sonication. 20 mg 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) and 20 mg N-hydroxysuccinimide (NHS) were added sequentially to activate the carboxyl groups of BPLP-PLA nanoparticles under stirring for one hour each at room temperature. Then, 100 μg MTP was then added into the mixture and stirred for four hours. MTP-conjugated nanoparticles were washed by DI water for three times before lyophilized as well.
Nanoparticle Characterization
The particle size, size distribution, and zeta potential of various nanoparticles were measured by dynamic light scattering (DLS, Malvern Zetasizer ZS). The chemical structures and morphology of nanoparticles were characterized by Fourier transform infrared spectroscopy (FTIR, Bruker Vertex V70) and scanning electron microscopy (SEM, FEI Nova NanoSEM 630). Fluorescence spectra were measured by a fluorescence spectroscope (Horiba FMax-4) with a slit size of 2 nm by 2 nm. The concentration of nanoparticles was 20 μg/mL in DPBS. The drug loading efficiency was measured by using a high-performance liquid chromatography (Shimadzu) equipped with a photodiode array detector (Shimadzu) and a Phenomenex Kinetex C18 column. The mobile phase was a mixture of 40% acetonitrile and 60% DI water, and the flow rate was 1 ml/min. PLX4032 concentration was determined by reading the absorbance at 270 nm, and a calibration curve was built on same conditions (Supporting Information Figure S2). For drug release tests, 50 mg PLX4032 loaded nanoparticles were dispersed in 5 ml of 50 mM PBS in a tube and shaken at 37°C. At each time point, the nanoparticles suspension were centrifuged and then 0.5 ml of release solution was removed for HPLC measurement. Afterwards, 0.5 ml of fresh PBS was added into the tube and followed by re-dispersion.
THP-1 Uptake
Differentiated THP-1 cells were lifted by cell dissociation buffer and used for further studies. THP-1 uptake studies were carried out by incubating 1×106 differentiated THP-1 cells and 200 μg/ml BPLP-PLA and MTP-BPLP-PLA nanoparticles in 1 ml DPBS at 37°C for 2 hours on a rocker, respectively. Afterwards, THP-1 cells were washed gently by DPBS for three times, and then subjected for characterization and further studies.
Immunofluorescence Staining
To prevent non-specific binding, THP-1 cells were blocked by 1% BSA for 1 hr at room temperature and incubated with 2 μg/ml CD11b rat anti-mouse mAb overnight at 4 °C. Cells were then stained with Alexa Fluor® 647 Goat anti-rat IgG (H+L) (2 μg/ml) for 1 hr at room temperature. THP-1 cells incubated with secondary antibodies served as controls. The cells were fixed by 4% paraformaldehyde at room temperature for 30 minutes and subjected to flow cytometry and confocal microscopy. For confocal microscopy, DAPI was used to stain the nuclei of THP-1 cells.
Cell Binding Studies under Static Conditions
Two melanoma cell lines, 1205Lu (high metastatic) and WM35 (low metastatic) cells, were selected as BRAF positive mutant melanomas. One million nanoparticle-loaded THP-1 cells were incubated with one million GFP-tagged 1205Lu cells in 1 ml DPBS at 37°C for two hours on a rocker. The resulting cells were gently washed with DPBS for three times and fixed with 4% paraformaldehyde at room temperature for 30 minutes. Cell binding was analyzed by using a BD Fortessa LSRII flow cytometry and FACS analysis was performed by using FlowJo 10. Confocal microscopy was performed in inverted mode on an Olympus Fluorview 100 confocal microscope. For both flow cytometry and microscopy, the FITC channel was set to detect the fluorescence from GPF-tagged 1205Lu cells and the Texas Red channel was used to detect the fluorescence from BPLP-PLA nanoparticles. For binding studies between THP1 and WM35 cells, the WM35 cells were stained by CellTrace™ CFSE (Life Technologies) to obtain green fluorescence.
Cell Binding Studies under Shear-Flow Conditions
To simulate the shear-flow conditions of the blood flow, cell binding studies were performed in a uniform shear flow by using a cone-plate viscometer (Thermo Scientific). One million nanoparticle-loaded THP-1 cells were mixed with GFP-tagged 1205Lu cells at 1:1 ratio in 1 ml DPBS. The cell mixtures were immediately added into the cone-plate viscometer and exposed to shear flows at a range of shear rates that varied from 50 s−1 to 200 s−1 at room temperature for one hour. Then, the cells were removed from the cone-plate viscometer and washed twice by DPBS. The cells were fixed by 4% paraformaldehyde at room temperature for 30 minutes and subjected for confocal and flow cytometry studies as described in the section above.
Pharmacological Studies
First, toxicity and selectivity of PLX4032 were confirmed by adding PLX4032 solutions at different concentrations into 96 well plates with THP-1, 1205Lu and WM35 cells separately (cell seeding density = 5,000 cells per well). After 24 hours of incubation, the cells were washed with PBS twice and supplemented with 10 μl CCK-8 in 100 μl RPMI-1640 medium in each well. After two hours of incubation, the absorbance at 450 nm of each well was measured by a micro-plate reader (TECAN, infinite M200 PRO) and converted to cell viability by normalized to the control (tissue culture plates). Next, MTP-BPLP-PLA nanoparticles with and without PLX4032 were dispersed in RPMI-1640 medium at various concentrations, followed by incubation with THP-1, 1205Lu, and WM35 cells (2,000 cells per well) separately in same conditions for seven days. At last, MTP-BPLP-PLA-PLX4032 loaded THP-1 cells and pristine THP-1 cells were seeded with 1205Lu or WM35 cells (2,000 cells per well) together with different ratios for seven days, respectively. Cell viability was tested by CCK-8 assays as well.
Statistical Analysis
All data was recorded as mean ± standard error, unless otherwise stated. All statistical analyses were performed via one-way ANOVA on GraphPad Prism 6.0. For all studies, n equals to 6, unless specifically stated.
Supplementary Material
Acknowledgments
The authors are grateful for the funding support from the National Institutes of Health Award (NIBIB EB012575, NCI CA182670, NHLBI HL118498), National Science Foundation (NSF) Awards DMR 1313553, CMMI 1266116, and CBET-BME 1330663. The authors also want to acknowledge the instrumental help from the Penn State Microscopy and Cytometry Facility at University Park, PA.
References
- 1.Chow EKH, Ho D. Sci Transl Med. 2013;5:216rv4. doi: 10.1126/scitranslmed.3005872. [DOI] [PubMed] [Google Scholar]
- 2.Barreto JA, O’malley W, Kubeil M, Graham B, Stephan H, Spiccia L. Adv Mater. 2011;23:H18. doi: 10.1002/adma.201100140. [DOI] [PubMed] [Google Scholar]
- 3.Petros RA, Desimone JM. Nat Rev Drug Discov. 2010;9:615. doi: 10.1038/nrd2591. [DOI] [PubMed] [Google Scholar]
- 4.Cole AJ, Yang VC, David AE. Trends Biotechnol. 2011;29:323. doi: 10.1016/j.tibtech.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu MK, Park J, Jon S. Theranostics. 2012;2:3. doi: 10.7150/thno.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kona S, Dong JF, Liu Y, Tan J, Nguyen KT. Int J Pharm. 2012;423:516. doi: 10.1016/j.ijpharm.2011.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kobayashi H, Longmire MR, Ogawa M, Choyke PL. Chem Soc Rev. 2011;40:4626. doi: 10.1039/c1cs15077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porada CD, Almeida-Porada G. Adv Drug Deliv Rev. 2010;62:1156. doi: 10.1016/j.addr.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su Y, Xie Z, Kim GB, Dong C, Yang J. ACS Biomater Sci Eng. 2015;1:201. doi: 10.1021/ab500179h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spicer JD, Mcdonald B, Cools-Lartigue JJ, Chow SC, Giannias B, Kubes P, Ferri LE. Cancer Res. 2012;72:3919. doi: 10.1158/0008-5472.CAN-11-2393. [DOI] [PubMed] [Google Scholar]
- 11.Huh SJ, Liang S, Sharma A, Dong C, Robertson GP. Cancer Res. 2010;70:6071. doi: 10.1158/0008-5472.CAN-09-4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong C, Robertson GP. Biorheology. 2009;46:265. doi: 10.3233/BIR-2009-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi MR, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, Halas NJ, Clare SE. Nano Letters. 2007;7:3759. doi: 10.1021/nl072209h. [DOI] [PubMed] [Google Scholar]
- 14.Choi MR, Bardhan R, Stanton-Maxey KJ, Badve S, Nakshatri H, Stantz KM, Cao N, Halas NJ, Clare SE. Cancer Nanotechnol. 2012;3:47. doi: 10.1007/s12645-012-0029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madsen SJ, Baek SK, Makkouk AR, Krasieva T, Hirschberg H. Ann Biomed Eng. 2012;40:507. doi: 10.1007/s10439-011-0415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell MJ, Wayne E, Rana K, Schaffer CB, King MR. Proc Natl Acad Sci USA. 2014;111:930. doi: 10.1073/pnas.1316312111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wayne EC, Chandrasekaran S, Mitchell MJ, Chan MF, Lee RE, Schaffer CB, King MR. J Control Release. 2016;223:215. doi: 10.1016/j.jconrel.2015.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Ai Y, Wang L, Bu P, Sharkey CC, Wu Q, Wun B, Roy S, Shen X, King MR. Biomaterials. 2016;76:52. doi: 10.1016/j.biomaterials.2015.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Yang J. J Mater Chem B Mater Biol Med. 2013;1:132. doi: 10.1039/C2TB00071G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xie Z, Zhang Y, Liu L, Weng H, Mason RP, Tang L, Nguyen KT, Hsieh JT, Yang J. Adv Mater. 2014;26:4491. doi: 10.1002/adma.201306070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auwerx J. Experientia. 1991;47:22. doi: 10.1007/BF02041244. [DOI] [PubMed] [Google Scholar]
- 22.Lee JT, Li L, Brafford PA, Van Den Eijnden M, Halloran MB, Sproesser K, Haass NK, Smalley KS, Tsai J, Bollag G, Herlyn M. Pigment Cell Melanoma Res. 2010;23:820. doi: 10.1111/j.1755-148X.2010.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nardin A, Lefebvre ML, Labroquere K, Faure O, Abastado JP. Curr Cancer Drug Targets. 2006;6:123. doi: 10.2174/156800906776056473. [DOI] [PubMed] [Google Scholar]
- 24.Yang J, Zhang Y, Gautam S, Liu L, Dey J, Chen W, Mason RP, Serrano CA, Schug KA, Tang L. Proc Natl Acad Sci USA. 2009;106:10086. doi: 10.1073/pnas.0900004106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagayama S, Ogawara KI, Fukuoka Y, Higaki K, Kimura T. Int J Pharm. 2007;342:215. doi: 10.1016/j.ijpharm.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 26.Nagao S, Nakanishi M, Kutsukake H, Yagawa K, Kusumoto S, Shiba T, Tanaka A, Kotani S. Infect Immun. 1990;58:536. doi: 10.1128/iai.58.2.536-542.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wahl S, Wahl L, Mccarthy J, Chedid L, Mergenhagen S. J Immunol. 1979;122:2226. [PubMed] [Google Scholar]
- 28.Fevrier M, Birrien J, Leclerc C, Chedid LT, Liacopoulos P. Eur J Immunol. 1978;8:558. doi: 10.1002/eji.1830080804. [DOI] [PubMed] [Google Scholar]
- 29.Liang S, Sharma A, Peng HH, Robertson G, Dong C. Cancer Res. 2007;67:5814. doi: 10.1158/0008-5472.CAN-06-4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma A, Tran MA, Liang S, Sharma AK, Amin S, Smith CD, Dong C, Robertson GP. Cancer Res. 2006;66:8200. doi: 10.1158/0008-5472.CAN-06-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doane TL, Chuang CH, Hill RJ, Burda C. Acc Chem Res. 2011;45:317. doi: 10.1021/ar200113c. [DOI] [PubMed] [Google Scholar]
- 32.Leung CWT, Hong Y, Chen S, Zhao E, Lam JWY, Tang BZ. J Am Chem Soc. 2012;135:62. doi: 10.1021/ja310324q. [DOI] [PubMed] [Google Scholar]
- 33.Daigneault M, Preston JA, Marriott HM, Whyte MK, Dockrell DH. PloS one. 2010;5:e8668. doi: 10.1371/journal.pone.0008668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan Y, Gause KT, Kamphuis MM, Ang CS, O’brien-Simpson NM, Lenzo JC, Reynolds EC, Nice EC, Caruso F. ACS Nano. 2013;7:10960. doi: 10.1021/nn404481f. [DOI] [PubMed] [Google Scholar]
- 35.Liang S, Slattery MJ, Wagner D, Simon SI, Dong C. Ann Biomed Eng. 2008;36:661. doi: 10.1007/s10439-008-9445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mittar D, Paramban R, Mcintyre C. Flow Cytometry and High-Content Imaging to Identify Markers of Monocyte-Macrophage Differentiation. https://http://www.bdbiosciences.com/documents/BD_Multicolor_MonocyteMacrophageDiff_AppNote.pdf.
- 37.Schwende H, Fitzke E, Ambs P, Dieter P. J Leukoc Biol. 1996;59:555. [PubMed] [Google Scholar]
- 38.Zhang P, Ozdemir T, Chung CY, Robertson GP, Dong C. J Immunol. 2011;186:242. doi: 10.4049/jimmunol.1000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Nature Immunol. 2002;3:991. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 40.Condeelis J, Pollard JW. Cell. 2006;124:263. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Peng HH, Liang S, Henderson AJ, Dong C. Exp Cell Res. 2007;313:551. doi: 10.1016/j.yexcr.2006.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chithrani BD, Chan WCW. Nano Letters. 2007;7:1542. doi: 10.1021/nl070363y. [DOI] [PubMed] [Google Scholar]
- 43.Oh N, Park JH. ACS Nano. 2014;8:6232. doi: 10.1021/nn501668a. [DOI] [PubMed] [Google Scholar]
- 44.Dong C, Cao J, Struble EJ, Lipowsky HH. Ann Biomed Eng. 1999;27:298. doi: 10.1114/1.143. [DOI] [PubMed] [Google Scholar]
- 45.Garber K. Science. 2009;326:1619. doi: 10.1126/science.326.5960.1619. [DOI] [PubMed] [Google Scholar]
- 46.Comin-Anduix B, Chodon T, Sazegar H, Matsunaga D, Mock S, Jalil J, Escuin-Ordinas H, Chmielowski B, Koya RC, Ribas A. Clin Cancer Res. 2010;16:6040. doi: 10.1158/1078-0432.CCR-10-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Urban JL, Shepard HM, Rothstein JL, Sugarman BJ, Schreiber H. Proc Natl Acad Sci USA. 1986;83:5233. doi: 10.1073/pnas.83.14.5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukherjee K, Parashuraman S, Krishnamurthy G, Majumdar J, Yadav A, Kumar R, Basu SK, Mukhopadhyay A. J Cell Sci. 2002;115:3693. doi: 10.1242/jcs.00034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.