

Abstract
Next-generation sequencing technologies have dramatically increased the rate at which new genomes are sequenced. Accordingly, automated annotation programs have become adept at identifying and annotating protein coding regions, as well as common and conserved RNAs. Additionally, RNAseq techniques have advanced our ability to identify and annotate regulatory RNAs (sRNAs), which remain significantly understudied. Recently, our group catalogued and annotated all previously known and newly identified sRNAs in several Staphylococcus aureus strains. These complete annotation files now serve as tools to compare the sRNA content of S. aureus with other bacterial strains to investigate the conservation of their sRNomes. Accordingly, in this study we performed RNAseq on two staphylococcal species, Staphylococcus epidermidis and Staphylococcus carnosus, identifying 118 and 89 sRNAs in these organisms, respectively. The sRNA contents of all three species were then compared to elucidate their common and species-specific sRNA content, identifying a core set of between 53 and 36 sRNAs encoded in each organism. In addition, we determined that S. aureus has the largest set of unique sRNAs (137) while S. epidermidishas the fewest (25). Finally, we identify a highly conserved sequence and structural motif differentially represented within, yet common to, both S. aureus and S. epidermidis. Collectively, in this study, we uncover the sRNome common to three staphylococcal species, shedding light on sRNAs that are likely to be involved in basic physiological processes common to the genus. More significantly, we have identified species-specific sRNAs that are likely to influence the individual lifestyle and behaviour of these diverse staphylococcal strains.
Keywords: sRNA, small regulatory RNA; Staphylococcus aureus, Staphylococcus epidermidis, Staphylococcus carnosus; transcriptomics, RNA sequencing, RNAseq; genome annotation; comparative genomics; RNA structure
Data Summary
The RNAseq results have been deposited to the NCBI Gene Expression Omnibus; GEO submission GSE77567 (url – http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=olgjaisktxerfqz&acc=GSE77567).
The updated GenBank file, containing novel sRNA annotations (annotated as ‘misc. RNA') has been deposited to Figshare; DOI: 10.6084/m9.figshare.3385861 (url – https://figshare.com/s/ac122d912782908e6359).
Impact Statement
Staphylococcus aureus is a leading cause of nosocomial infections and exhibits profound levels of antimicrobial resistance. The importance of this pathogen has been well established, forming the subject of extensive research, but a comprehensive understanding of the regulatory processes governing its virulence has yet to be elucidated. Recently, our group has investigated the role of regulatory RNAs (sRNAs) by cataloguing and annotating them in S. aureus genomes. The study presented here continues this line of research by performing transcriptomic analyses with two closely related species, Staphylococcus epidermidis and Staphylococcus carnosus, to annotate, for the first time, their genomes for sRNAs. The sRNAs of all three organisms were then compared to determine the common and species-specific sRNA content of each genome. In addition, we identified a subset of sRNAs shared between S. aureus and S. epidermidis that demonstrate high sequence and structural conservation. This study provides a platform to guide studies on sRNAs that are important for the general physiology of staphylococci (shared sRNAs) as well as the unique lifestyles of each organism (species-specific sRNAs).
Introduction
The wide availability of sequenced genomes and the decreasing cost of producing such data have revolutionized the way molecular biology research is performed (Dark, 2013). With the increasing knowledge base of genomic information published each year there is an escalating demand for automated pipelines to identify and annotate genes within sequence data (Richardson & Watson, 2013). To highlight the vast amount of genetic information available, at the time of writing this manuscript, a total of 5443 completed prokaryotic genomes were available in the NCBI Genome database (http://www.ncbi.nlm.nih.gov/genome), with a further 65,259 partially completed genomes. Furthermore, the rate of publication continues to increase exponentially each year for studies on such topics (Tatusova et al., 2015). Traditionally, the pipelines used for de novo genome assembly involve prediction of protein-coding genes, rRNAs and tRNAs, followed by comparison with a reference genome to assign ORF function (Richardson & Watson, 2013). However, many drawbacks exist to such approaches, not the least of which is a lack of efficient detection for small, regulatory RNAs (sRNAs), some of which can also encode small peptides (<50 aa).
sRNAs as a class of molecule are increasingly recognized as playing important regulatory roles in bacteria (Beisel & Storz, 2010; Murphy et al., 2014; Caron et al., 2010; Geissmann et al., 2006; Harris et al., 2013; Hoe et al., 2013; Weiberg et al., 2015; Papenfort & Vanderpool, 2015; Oliva et al., 2015; Papenfort & Vogel, 2014; Weilbacher et al., 2003). For example, sRNAs regulate a wide variety of cellular processes, such as carbon metabolism and iron acquisition, and also have profound influence on virulence gene expression in many important pathogenic bacteria (Geissmann et al., 2006; Oliva et al., 2015; Caswell et al., 2012; Giangrossi et al., 2010; Broach et al., 2012; Hoe et al., 2013; Beisel & Storz, 2010; Weilbacher et al., 2003; Papenfort & Vogel, 2014). The wide-ranging effects of these molecules underlines the crucial need to fully annotate and study sRNAs in individual organisms, from both a functional and an evolutionary perspective.
The versatile genus Staphylococcus encompasses a diverse set of organisms that range from highly pathogenic to food-grade species. Staphylococci live on the mucous membranes of virtually all animals, as well as in aged meat products. Staphylococcus carnosus is an avirulent, coagulase-negative member of the staphylococci with the highest G+C content, and is commonly used as a starter culture for fermented sausages (Rosenstein et al., 2009; Schleifer & Fischer, 1982; Wagner et al., 1998). The genome of S. carnosus, an organism often regarded as an ancient and genetically simple species, generally has a lack of mobile genetic elements, especially in comparison with the other staphylococci. In contrast, Staphylococcus epidermidis is a coagulase-negative, opportunistic pathogen that is found as a part of the normal human flora of the skin and nares (Otto, 2009). S. epidermidis infections often occur through indwelling devices such as catheters, but are rarely life threatening or invasive (Otto, 2009). Staphylococcus aureus, also a normal part of the human flora, is a coagulase-positive member of the staphylococci, and is one of the leading causes of human infectious disease and death. S. aureus causes a wide variety of infections, ranging from minor cellulitis to life-threatening sepsis, and is capable of infecting all organ systems (Archer, 1998; Lowy, 1998). Compounding its extensive pathogenicity is the widespread prevalence of antibiotic-resistant isolates, which severely limits the number of viable treatment options (Lowy, 2003).
Collectively, the diversity of lifestyles and evolutionary relationships between the staphylococci (Fig. 1) make this a model genus to ask how regulatory molecules change and adapt across species; and how they develop specialized, and niche-specific functions within a given organism. As such, in this study we identified and annotated the sRNA content of both S. epidermidis and S. carnosus using next-generation sequencing technologies coupled with comparative genomics. These newly annotated sRNAs were analysed for homology to each other, and to those recently curated by our group for S. aureus (Carroll et al., 2016), to identify conserved and unique elements for each species. In total, we identified 118 total sRNAs in S. epidermidis and 89 in S. carnosus, compared with 303 in S. aureus (Carroll et al., 2016). A comparison of these datasets revealed that each genome contains between 36 and 53 sRNAs that are common to all three organisms. Finally, we uncovered the presence of several highly homologous sRNAs in S. epidermidis and S. aureus that share conserved sequences, and appear to retain common structural motifs. Collectively, our work shines a light on these complex and largely overlooked regulators, providing insight into staphylococcal speciation, and the evolution of pathogenesis within this genus.
Fig. 1.

Staphylococcal phylogeny and sRNA content. (a) Phylogenetic relationship was determined using the rpoB gene from a range of S. aureus isolates, alongside other species within the Staphylococcaceae. The three strains from this study (S. carnosus TM300, S. epidermidis RP62A and S. aureus USA300) are highlighted (blue, red and black, respectively) within the tree. The tree was created using the CLC Main Workbench software and default settings. The rpoB gene sequences were retrieved from NCBI. (b) Circos file representing the S. aureus USA300 genome with recently annotated sRNAs (Carroll et al., 2016). Depicted from the outermost semicircle inward are: the genome of S. aureus, sRNA annotations, expression level of each sRNA under standard conditions, genomic landmarks [SCCmec (purple), pathogenicity islands (red), prophages (orange) and other genomic islands (yellow)], sRNAs encoded on the forward strand and, innermost, the reverse strand.
Methods
Bacterial strains and growth conditions.
S. epidermidis RP62a (Gill et al., 2005) and S. carnosus TM300 (Schleifer & Fischer, 1982; Rosenstein et al., 2009) were cultivated in tryptic soy broth (TSB), with shaking (250 r.p.m.) at 37 °C overnight. Synchronous cultures were achieved as outlined by us previously (Kolar et al., 2011) before being grown for 3 h to the exponential phase.
RNAseq.
Transcriptomic experiments using an Ion Torrent Personal Genome Machine (PGM) system (Ion Torrent) were performed as described by us previously (Carroll et al., 2014). Briefly, total RNA was isolated from exponentially growing cultures using an RNeasy kit (Qiagen), with DNA removed using a TURBO DNA-free kit (Ambion). Next, RNA integrity was confirmed utilizing an Agilent 2100 Bioanalyzer system in combination with a RNA 6000 Nano Kit (Agilent). To remove rRNA from samples, a Ribo-Zero rRNA Removal Kit (Bacteria) (Epicentre) and MICROBExpress Bacterial mRNA Enrichment Kit (Ambion) were used in a sequential approach; complete removal of rRNA species was confirmed using an Agilent RNA 6000 Nano Kit. cDNA libraries were constructed from the enriched RNA with an Ion Total RNA-seq Kit v2 (Ion Torrent), before cDNA fragments were amplified onto Ion Sphere Particles (ISPs) using an Ion PGM Template OT2 200 Kit (Ion Torrent) and an Ion OneTouch 2 system (Ion Torrent). Template-positive ISPs were subsequently loaded onto Ion 318 v2 chips (Ion Torrent) and sequencing runs were performed utilizing an Ion PGM Sequencing 200 Kit v2 (Ion Torrent). After completion of each run, data were imported to the CLC Genomics Workbench software (CLC bio; Qiagen) and aligned to the publicly available S. epidermidis RP62a (NCBI accession number: NC_002976.3) and S. carnosus TM300 (NCBI accession number: AM295250.1) genomes. The addition of novel annotations to the S. epidermidis and S. carnosus genomes was performed according to guidelines outlined by us previously (Carroll et al., 2016; Weiss et al., 2015). Updated annotation files including novel sRNA transcripts for S. epidermidis RP62a and S. carnosus TM300 were deposited to Figshare (Data citation 1). The annotation files containing sRNA annotations were used to generate expression values calculated as RPKM (reads per kilobase material per million reads) in CLC Genomics Workbench. All downstream bioinformatic analyses (e.g. blast searches investigating sRNA similarities between different species) were also performed with CLC Genomics Workbench software. RNA structure predictions were performed using the mfold web server (Zuker, 2003).
Northern blots.
To confirm the presence of novel transcripts identified by RNAseq, we performed Northern blot analysis for selected sRNA candidates. Northern blots were performed as outlined previously (Caswell et al., 2012), as follows. RNA from exponentially growing cultures was isolated and DNA-depleted as described for RNAseq samples. RNA was electrophoretically separated in a 10 % polyacrylamide gel [1x× TBE (Tris/borate/EDTA) buffer, 7 m urea] and transferred to an Amersham Hybond N+ membrane (GE Healthcare) by electroblotting. Samples were crosslinked to membranes via UV radiation, followed by pre-hybridization in ULTRAhyb-Oligo buffer (Ambion) for 1 h at 43 °C in a rotating oven. Next, [γ-32P]-ATP end-labelled oligonucleotides specific for each target RNA sequence (Table S1, available in the online Supplementary Material) were added to membranes and hybridized overnight at 43 °C. The following day membranes were washed with 2×, 1× and 0.5× SSC (saline and sodium citrate) buffer for 30 min at 43 °C. Finally, membranes were exposed to X-ray film to detect radiolabelled and specifically bound probes.
Results
Annotation of sRNAs in the S. epidermidis RP62a and S. carnosus TM300 genomes
The goal of this study was to gain insight into the impact of sRNAs on staphylococcal species-specific adaptation. A set of organisms was chosen to represent the diverse lifestyles of staphylococcal species: S. aureus USA300-Houston, an epidemic community-associated methicillin-resistant strain isolated from the wrist abscess of a 36-year-old, HIV-positive, intravenous drug user; S. epidermidis RP62a, a methicillin-resistant strain isolated from a patient suffering from intravascular catheter-associated sepsis; and S. carnosus TM300, originally isolated from dry sausage in 1982 in Germany (Highlander et al., 2007; Gill et al., 2005; Schleifer & Fischer, 1982; Rosenstein et al., 2009). Importantly, these organisms are intermediately and distanty related species (Fig. 1a), representing the highly virulent (S. aureus), the mildly virulent (S. epidermidis) and the avirulent (S. carnosus). As such, they have the potential to provide significant insight into those sRNAs that are core to the staphylococci, as well as those that influence species-specific adaptation. Recently we re-annotated the genome of S. aureus (Carroll et al., 2016) to include all sRNAs from the literature, as well as several novel transcripts identified by our group using next-generation sequencing approaches (Fig. 1b). As such, we used a similar RNAseq-based approach to re-annotate the genomes of S. epidermidis RP62a and S. carnosus TM300.
To our knowledge, no sRNAs have been identified or studied in either S. epidermidis or S. carnosus to date, and neither published genome has any sRNAs currently annotated. Given the absence of any information regarding the sRNAs of these two species, a transcriptomic approach was used to identify sRNAs in these genomes. Initially, each RNAseq was performed on cultures grown to the mid-logarithmic phase, with all reads generated aligned to the published genomes of S. epidermidis RP62a and S. carnosus TM300 (Gill et al., 2005; Schleifer & Fischer, 1982; Rosenstein et al., 2009). Files were then reviewed for the presence of sRNA reads using criteria defined by us previously for S. aureus and Acinetobacter baumannii (Carroll et al., 2016; Weiss et al., 2015), as: antisense to previously annotated protein coding genes (Fig. S1a), in intergenic regions (Fig. S1b) or that showed differential expression from annotated genes with which they overlapped (Fig. S1c).
The first genome-wide identification of sRNAs in S. epidermidis and S. carnosus
In total, 118 and 89 sRNAs were identified in S. epidermidis RP62a and S. carnosus TM300, respectively (Tables 1 and 2). The sRNAs in each organism are distributed across their respective chromosomes, with the exception of a general lack of sRNAs in regions encoding prophages. The lack of sRNAs residing in these regions is perhaps to be expected, as these are relatively recent evolutionary events that have not yet been homogenized into the rest of the genome. To facilitate the addition of novel sRNA annotations in the future, an annotation system was created that does not relate to function, but instead acts only as an identifier (as described by us for S. aureus) (Carroll et al., 2016). As such, sRNAs from S. epidermidis were denoted as SERPs001–SERPs118, referring to their total number, for ease of sequential incorporation of new sRNA annotations in the genome. Similarly, in S. carnosus sRNAs were denoted as SCAs001–SCAs089. Newly annotated genes were given the gene names jointly annotated epidermidis loci (jaeL)1–118 and jointly annotated carnosus loci (jacL) 1–89 for S. epidermidis and S. carnosus, respectively. To confirm the size and expression of sRNAs discovered in S. epidermidis and S. carnosus, several representative transcripts were chosen for Northern blot validation (Fig. 2). Each of the sRNAs analysed produced a single, probe-specific band at the size suggested by RNAseq, and as annotated herein. These findings suggest that the methods used by our group to identify and annotate novel sRNAs are both robust and reproducible.
Table 1. Newly annotated non-coding RNAs of S epidermidis RP62a .
| Locus ID | Gene name | Position | Upstream* | Orientation | Downstream* | RPKM† |
|---|---|---|---|---|---|---|
| SERPs001 | jaeL-1 | 34999..35245 | SERPs117 | < | SERP0038 | 247.23 |
| SERPs002 | jaeL-2 | 42907..43025 | SERP0050 | < | SERP0051 | 125.96 |
| SERPs003 | jaeL-3 | 953853..954050 | SERPs113 | > | trpE | 11.22 |
| SERPs004 | jaeL-4 | 60337..60509 | guaA | < | SERP0071 | 327.32 |
| SERPs005 | jaeL-5 | 586811..586861 | SERPs022 | < | SERP0593 | 21.77 |
| SERPs006 | jaeL-6 | 183818..183969 | rplA | > | rplJ | 2081.83 |
| SERPs007 | jaeL-7 | 212119..212293 | SERP0201 | < | SERP0202 | 367.99 |
| SERPs008 | jaeL-8 | 233449..233600 | SERP0223 | < | SERP0224 | 1281.97 |
| SERPs009 | jaeL-9 | 233917..234080 | SERP0224 | > | SERP0225 | 67.7 |
| SERPs010 | jaeL-10 | 241408..241559 | SERP0234 | > | SERP0235 | 927.69 |
| SERPs011 | jaeL-11 | 307532..307673 | SERP0304 | < | SERP0305 | 328.4 |
| SERPs012 | jaeL-12 | 370776..370941 | SERP0373 | < | pabA | 153.84 |
| SERPs013 | jaeL-13 | 377942..378056 | SERP0379 | < | SERP0380 | 77.24 |
| SERPs014 | jaeL-14 | 393987..394130 | SERP0391 | > | SERP0392 | 34.7 |
| SERPs015 | jaeL-15 | 443311..443466 | SERP0438 | > | SERP0439 | 1633.44 |
| SERPs016 | jaeL-16 | 469574..469690 | SERP0466 | < | SERP0467 | 469.75 |
| SERPs017 | jaeL-17 | 475770..475890 | SERP0477 | < | SERP0478 | 50.47 |
| SERPs018 | jaeL-18 | 480754..480892 | SERP0488 | > | SERP0489 | 7045.28 |
| SERPs019 | jaeL-19 | 526358..526540 | SERP0542 | < | SERP0543 | 54.61 |
| SERPs020 | jaeL-20 | 570220..570273 | trpS | > | SERP0576 | 452.35 |
| SERPs021 | jaeL-21 | 576526..576580 | SERPs082 | > | SERP0581 | 2967.56 |
| SERPs022 | jaeL-22 | 586669..586828 | SERP0592 | > | SERPs121 | 190.83 |
| SERPs023 | jaeL-23 | 592434..592529 | SERP0599 | < | SERP0600 | 763.34 |
| SERPs024 | jaeL-24 | 658364..658539 | SERP0662 | < | SERP0663 | 466.83 |
| SERPs025 | jaeL-25 | 695170..695337 | SERP0697 | < | SERP0698 | 168.53 |
| SERPs026 | jaeL-26 | 717301..717513 | SERP0720 | > | pheS | 297.13 |
| SERPs027 | jaeL-27 | 817973..818158 | sucD | < | SERP0815 | 984.95 |
| SERPs028 | jaeL-28 | 964074..964190 | femB | < | SERP0948 | 332.14 |
| SERPs029 | jaeL-29 | 981290..981497 | SERP0962 | > | lysC | 10366.45 |
| SERPs030 | jaeL-30 | 1007973..1008125 | SERP0990 | > | SERP0991 | 486.21 |
| SERPs031 | jaeL-31 | 1099406..1099581 | SERP1053 | < | srrB | 1526.68 |
| SERPs032 | jaeL-32 | 1150188..1150367 | SERP1109 | > | SERP1110 | 77.1 |
| SERPs033 | jaeL-33 | 1227591..1227843 | SERP1191 | < | aspS | 4785.74 |
| SERPs034 | jaeL-34 | 1231240..1231434 | hisS | < | SERP1194 | 1773.65 |
| SERPs035 | jaeL-35 | 1279465..1279597 | infC | < | SERP1245 | 346.45 |
| SERPs036 | jaeL-36 | 1281335..1281543 | SERP1245 | < | thrS | 1500.78 |
| SERPs037 | jaeL-37 | 1308770..1308884 | dnaE | < | SERP1267 | 1042.73 |
| SERPs038 | jaeL-38 | 1318633..1318805 | SERP1273 | < | SERP1274 | 67.39 |
| SERPs039 | jaeL-39 | 1337905..1338103 | SERP1292 | > | tyrS | 544 |
| SERPs040 | jaeL-40 | 1379890..1380079 | leuS | < | SERP1319 | 978.83 |
| SERPs041 | jaeL-41 | 1389878..1390104 | ribD | < | SERP1329 | 2298.88 |
| SERPs042 | jaeL-42 | 1409579..1409745 | metK | < | pckA | 1618.92 |
| SERPs043 | jaeL-43 | 1518292..1518416 | pheA | < | SERP1453 | 31.09 |
| SERPs044 | jaeL-44 | 1542695..1542748 | SERP1479 | > | SERP1480 | 318.7 |
| SERPs045 | jaeL-45‡ | 1557626..1557897 | SERP1488 | < | hld | 289.82 |
| SERPs046 | jaeL-46 | 1647845..1649295 | SERP1623 | > | SERP1624 | 3008.01 |
| SERPs047 | jaeL-47 | 292616..292825 | SERPs119 | < | sitC | 795.72 |
| SERPs048 | jaeL-48 | 1744046..1744193 | SERP1701 | < | sceD | 213.81 |
| SERPs049 | jaeL-49 | 1769544..1769767 | murAB | < | fbaA | 2614.68 |
| SERPs050 | jaeL-50 | 1774668..1774750 | pyrG | < | rpoE | 3083.45 |
| SERPs051 | jaeL-51 | 1790875..1791019 | SERP1753 | < | SERP1754 | 298.64 |
| SERPs052 | jaeL-52 | 292457..292605 | SERP0289 | < | SERPs120 | 1538.79 |
| SERPs053 | jaeL-53 | 1905371..1905605 | SERP1880 | < | nhaC | 1946.59 |
| SERPs054 | jaeL-54 | 1917116..1917271 | SERP1894 | < | SERP1895 | 327.4 |
| SERPs055 | jaeL-55 | 1935344..1935518 | SERP1914 | < | SERP1915 | 396.54 |
| SERPs056 | jaeL-56 | 1997129..1997489 | sarZ | > | SERP1980 | 522.86 |
| SERPs057 | jaeL-57 | 2014397..2014589 | SERP1994 | < | SERP1995 | 175.46 |
| SERPs058 | jaeL-58 | 2096196..2096389 | SERP2069 | > | SERP2070 | 354.84 |
| SERPs059 | jaeL-59 | 2110878..2111312 | SERP2083 | < | aldA-2 | 98.27 |
| SERPs060 | jaeL-60 | 2118226..2118357 | SERP2091 | < | SERP2092 | 786.47 |
| SERPs061 | jaeL-61 | 2184407..2184483 | ldh | < | SERP2157 | 439.8 |
| SERPs062 | jaeL-62 | 2206812..2206918 | SERP2175 | < | betA | 15.57 |
| SERPs063 | jaeL-63 | 2251118..2251304 | SERP2212 | > | SERPs064 | 724.37 |
| SERPs064 | jaeL-64 | 2251300..2251353 | SERPs063 | < | SERP2213 | 586 |
| SERPs065 | jaeL-65 | 2258818..2259071 | cadC | > | SERPs066 | 657.88 |
| SERPs066 | jaeL-66 | 2259072..2259128 | SERPs065 | < | SERP2223 | 2084.27 |
| SERPs067 | jaeL-67 | 2266286..2266340 | SERP2235 | < | SERP2236 | 403.75 |
| SERPs068 | jaeL-68 | 2301741..2301875 | SERP2268 | > | SERPs069 | 16.45 |
| SERPs069 | jaeL-69 | 2301905..2301964 | SERPs068 | < | SERP2269 | 120.28 |
| SERPs070 | jaeL-70 | 2352134..2352256 | mqo-3 | < | SERP2313 | 216.65 |
| SERPs071 | jaeL-71 | 2400262..2400406 | SERP2353 | < | SERP2354 | 72.74 |
| SERPs072 | jaeL-72 | 2508708..2508954 | SERP2454 | < | SERP2455 | 384.34 |
| SERPs073 | jaeL-73 | 2519862..2520195 | SERP2465 | < | SERP2466 | 28.26 |
| SERPs074 | jaeL-74 | 2550198..2550302 | kdeP | < | SERP2491 | 158.62 |
| SERPs075 | jaeL-75 | 2574243..2574391 | SERP2518 | > | mecI | 21766.54 |
| SERPs076 | jaeL-76 | 2600571..2600779 | SERP2541 | < | SERP2542 | 159.37 |
| SERPs077 | jaeL-77 | 2605191..2605352 | SERP2546 | < | SERP2547 | 376.96 |
| SERPs078 | jaeL-78 | 297416..298093 | tagA | < | SERP0296 | 133.47 |
| SERPs079 | jaeL-79 | 484345..485243 | SERP0494 | > | SERP0495 | 397.69 |
| SERPs080 | jaeL-80 | 1799010..1799177 | SERP1761 | < | glmM | 218.1 |
| SERPs081 | jaeL-81 | 1848280..1848453 | SERP1803 | < | rplQ | 618.97 |
| SERPs082 | jaeL-82 | 576366..576526 | pepF | > | SERPs021 | 303.44 |
| SERPs083 | jaeL-83 | 1702694..1702924 | SERP1664 | > | ilvD | 4.81 |
| SERPs084 | jaeL-84 | 1862469..1862606 | rpsJ | < | SERP1833 | 1890.75 |
| SERPs085 | jaeL-85 | 1844698..1844868 | rplM | < | truA | 879.81 |
| SERPs086 | jaeL-86 | 755335..755547 | SERP0757 | > | ileS | 364.89 |
| SERPs087 | jaeL-87 | 1283598..1283838 | thrS | < | dnaI | 377.78 |
| SERPs088 | jaeL-88 | 2475827..2476027 | SERP2413 | < | SERP2414 | 116 |
| SERPs089 | jaeL-89 | 1146822..1146987 | gcvT | < | aroK | 662.17 |
| SERPs090 | jaeL-90 | 185734..185859 | SERP0182 | > | rpoB | 74.9 |
| SERPs091 | jaeL-91 | 603425..603663 | prfC | > | SERP0610 | 308.94 |
| SERPs092 | jaeL-92 | 1406055..1406237 | SERP1349 | > | SERP1350 | 21.24 |
| SERPs093 | jaeL-93 | 1238590..1238761 | recJ | < | SERP1200 | 22.59 |
| SERPs094 | jaeL-94 | 1449425..1449591 | SERP1393 | > | SERP1394 | 13.3 |
| SERPs095 | jaeL-95 | 2020634..2020835 | fmhA | < | SERP2002 | 118.18 |
| SERPs096 | jaeL-96 | 632659..632761 | SERP0638 | > | SERP0639 | 21.56 |
| SERPs097 | jaeL-97 | 1932904..1933097 | SERP1912 | > | SERP1913 | 57.23 |
| SERPs098 | jaeL-98 | 2173475..2173629 | SERP2146 | > | SERP2147 | 25.07 |
| SERPs099 | jaeL-99 | 550036..550252 | SERP0558 | < | SERP0559 | 28.14 |
| SERPs100 | jaeL-100 | 367959..368097 | SERP0369 | > | SERP0370 | 27.96 |
| SERPs101 | jaeL-101 | 950460..950575 | SERP0933 | > | dmpI | 23.93 |
| SERPs102 | jaeL-102 | 1142437..1142535 | SERP1099 | > | SERPs104 | 33.65 |
| SERPs103 | jaeL-103 | 1444172..1444269 | SERP1386 | < | fumC | 385.21 |
| SERPs104 | jaeL-104 | 1142550..1142729 | SERPs102 | < | SERP1100 | 259.07 |
| SERPs105 | jaeL-105 | 2192309..2192465 | SERP2163 | > | SERP2164 | 17.68 |
| SERPs106 | jaeL-106 | 2360082..2360248 | SERP2323 | < | SERP2324 | 9.97 |
| SERPs107 | jaeL-107 | 54838..54966 | SERP0066 | > | xpt | 210.87 |
| SERPs108 | jaeL-108 | 617908..618225 | SERP0626 | > | menA | 80.31 |
| SERPs109 | jaeL-109 | 485246..485535 | SERP0495 | > | sufC | 45.94 |
| SERPs110 | jaeL-110 | 1263896..1264162 | valS | < | SERP1229 | 380.5 |
| SERPs111 | jaeL-111 | 1339993..1340256 | tyrS | < | SERP1294 | 172.43 |
| SERPs112 | jaeL-112 | 328604..328788 | SERP0323 | < | scdA | 96.03 |
| SERPs113 | jaeL-113 | 953678..953822 | tyrA | > | SERPs122 | 7.66 |
| SERPs114 | jaeL-114 | 176298..176556 | gltX | > | cysE | 182.19 |
| SERPs115 | jaeL-115 | 1217236..1217468 | alaS | < | SERP1183 | 457.47 |
| SERPs116 | jaeL-116 | 1170711..1170867 | SERP1131 | > | glyS | 495.04 |
| SERPs117 | jaeL-117 | 34833..34995 | SERP0037 | < | SERPs001 | 136.23 |
| SERPs118 | jaeL-118 | 847682..847797 | ribF | > | rpsO | 641.3 |
*Gene.
†RPKM, reads per kilobase material per million reads.
‡Region corresponds to portion of RNAIII.
Table 2. Newly annotated non-coding RNAs of S. carnosus TM300.
| Locus ID | Gene name | Position | Upstream* | Orientation | Downstream* | RPKM† |
|---|---|---|---|---|---|---|
| SCAs001 | jacL-1 | 9628..9890 | SCA_0010 | < | SCA_0011 | 329.72 |
| SCAs002 | jacL-2 | 16355..16590 | SCAs079 | < | SCA_0015 | 489.92 |
| SCAs003 | jacL-3 | 68516..68644 | fcbC | > | SCA_0067 | 1331.56 |
| SCAs004 | jacL-4 | 114686..114974 | SCA_0114 | > | SCA_0115 | 1318.64 |
| SCAs005 | jacL-5 | 144461..144729 | SCA_0141 | > | SCAs006 | 212.44 |
| SCAs006 | jacL-6 | 144466..144711 | SCAs005 | < | SCA_0142 | 4.05 |
| SCAs007 | jacL-7 | 195809..196025 | gltX | > | cysE | 736.45 |
| SCAs008 | jacL-8 | 239896..240240 | proP | < | thiD | 1253.87 |
| SCAs009 | jacL-9 | 248359..248731 | SCA_0240 | > | SCA_0241 | 26355.27 |
| SCAs010 | jacL-10 | 286557..286798 | tagA | > | thrS | 6.86 |
| SCAs011 | jacL-11 | 302807..302992 | SCA_0293 | < | SCA_0294 | 7754.21 |
| SCAs012 | jacL-12 | 349042..349212 | norA | > | SCA_0342 | 443 |
| SCAs013 | jacL-13 | 370812..371006 | SCA_0365 | > | SCA_0366 | 386.77 |
| SCAs014 | jacL-14 | 386729..386902 | SCA_0377 | > | SCAs090 | 324.61 |
| SCAs015 | jacL-15 | 409625..409751 | SCA_0399 | > | SCA_0400 | 7319.89 |
| SCAs016 | jacL-16 | 435453..435577 | SCA_0420 | > | SCA_0421 | 459.83 |
| SCAs017 | jacL-17 | 439576..439686 | gapA | > | pgk | 12059.66 |
| SCAs018 | jacL-18 | 449553..449930 | smpB | > | NEW_REGION_507 | 40323.14 |
| SCAs019 | jacL-19 | 465254..465398 | SCA_0454 | > | SCA_0455 | 6846.57 |
| SCAs020 | jacL-20 | 475448..475663 | int | > | SCA_0464 | 189.2 |
| SCAs021 | jacL-21 | 478847..479060 | SCA_0469 | < | SCA_0470 | 172.33 |
| SCAs022 | jacL-22 | 519236..519323 | glpQ | > | SCA_0522 | 211.43 |
| SCAs023 | jacL-23 | 704781..704974 | SCA_0700 | < | SCA_0701 | 438.43 |
| SCAs024 | jacL-24 | 736699..736810 | SCA_0733 | < | SCA_0734 | 3740.74 |
| SCAs025 | jacL-25 | 754215..755140 | SCA_0752 | > | SCA_0753 | 236.45 |
| SCAs026 | jacL-26 | 809208..809442 | rluD | > | pyrR | 10723.79 |
| SCAs027 | jacL-27 | 954424..954574 | mutS | > | mutL | 28.6 |
| SCAs028 | jacL-28 | 1026107..1026242 | SCA_1009 | < | tyrA | 2460.09 |
| SCAs029 | jacL-29 | 1028624..1028907 | SCA_1011 | > | trpE | 926.55 |
| SCAs030 | jacL-30 | 1055393..1055592 | SCA_1036 | > | lysC | 3098.2 |
| SCAs031 | jacL-31 | 1080768..1080936 | SCA_1060 | > | SCA_1061 | 139.58 |
| SCAs032 | jacL-32 | 1098995..1099432 | SCA_1079 | < | SCA_1080 | 1725.71 |
| SCAs033 | jacL-33 | 1103951..1104094 | pbp2 | < | SCA_1085 | 230.73 |
| SCAs034 | jacL-34 | 1136220..1136427 | SCA_1116 | < | srrB | 1380.1 |
| SCAs035 | jacL-35 | 1239320..1239485 | alaS | < | SCA_1231 | 28.02 |
| SCAs036 | jacL-36 | 1249565..1249804 | SCA_1240 | < | aspS | 5994.28 |
| SCAs037 | jacL-37 | 1252944..1253215 | hisS | < | SCA_1243 | 2385.58 |
| SCAs038 | jacL-38 | 1294117..1294234 | infC | < | lysP | 23251.64 |
| SCAs039 | jacL-39 | 1339350..1339494 | SCA_1325 | > | rpsD | 1271.7 |
| SCAs040 | jacL-40 | 1354036..1354126 | fhs | < | acsA | 200.81 |
| SCAs041 | jacL-41 | 1373449..1373678 | SCA_1356 | < | dat | 1246.65 |
| SCAs042 | jacL-42 | 1383627..1383700 | leuS' | > | SCA_1366 | 9195.15 |
| SCAs043 | jacL-43 | 1393914..1394102 | ribD | < | SCA_1376 | 1063.54 |
| SCAs044 | jacL-44 | 1406587..1406766 | metK | < | pckA | 2198.37 |
| SCAs045 | jacL-45 | 273470..273659 | sarA | < | SCA_0267 | 34.97 |
| SCAs046 | jacL-46 | 1546771..1546977 | SCA_1526 | < | SCA_1527 | 500.78 |
| SCAs047 | jacL-47 | 1562210..1562443 | SCA_1544 | > | agrB | 985.38 |
| SCAs048 | jacL-48 | 1585607..1585749 | ilvA | < | 23S rRNA D | 2012.07 |
| SCAs049 | jacL-49 | 1664220..1664368 | SCA_1648 | > | SCA_1649 | 196.23 |
| SCAs050 | jacL-50 | 1824275..1824411 | SCA_1822 | < | SCA_1823 | 637.82 |
| SCAs051 | jacL-51 | 1917867..1918006 | SCA_1912 | < | SCA_1913 | 832.99 |
| SCAs052 | jacL-52 | 1929303..1929527 | SCA_1921 | < | SCA_1922 | 84.17 |
| SCAs053 | jacL-53 | 1941508..1941755 | opuCA | < | SCA_1935 | 515.79 |
| SCAs054 | jacL-54 | 2031863..2032724 | SCA_2018 | > | SCA_2019 | 7319.06 |
| SCAs055 | jacL-55 | 2088262..2088525 | SCA_2075 | > | SCA_2076 | 761.4 |
| SCAs056 | jacL-56 | 2199217..2199338 | SCA_2172 | < | SCA_2173 | 1628.55 |
| SCAs057 | jacL-57 | 2224729..2224907 | SCA_2191 | < | SCA_2192 | 848.25 |
| SCAs058 | jacL-58 | 2266552..2266687 | SCA_2211 | > | SCAs059 | 4277.68 |
| SCAs059 | jacL-59 | 2266688..2266868 | SCAs058 | < | SCAs060 | 2180.71 |
| SCAs060 | jacL-60 | 2267000..2267107 | SCAs059 | > | SCAs061 | 1239.77 |
| SCAs061 | jacL-61 | 2267108..2267271 | SCAs060 | < | tatA | 1847.62 |
| SCAs062 | jacL-62 | 2294415..2294594 | SCA_2236 | < | SCA_2237 | 265.8 |
| SCAs063 | jacL-63 | 2303997..2304203 | SCA_2247 | > | SCAs064 | 1295.28 |
| SCAs064 | jacL-64 | 2304288..2304494 | SCAs063 | > | SCA_2248 | 166.93 |
| SCAs065 | jacL-65 | 2312205..2312796 | SCA_2253 | < | SCAs066 | 2442.46 |
| SCAs066 | jacL-66 | 2313086..2313451 | SCAs065 | > | SCA_2254 | 2055.21 |
| SCAs067 | jacL-67 | 2322081..2322191 | acsA | > | putP | 1655.25 |
| SCAs068 | jacL-68 | 2375256..2375403 | SCA_2309 | > | SCAs069 | 60.61 |
| SCAs069 | jacL-69 | 2375475..2375602 | SCAs068 | > | SCAs070 | 3569.06 |
| SCAs070 | jacL-70 | 2375660..2375782 | SCAs069 | < | opuD | 27.01 |
| SCAs071 | jacL-71 | 387163..387583 | SCAs014 | > | nrdI | 153.1 |
| SCAs072 | jacL-72 | 2550964..2551426 | SCA_2464 | > | serS | 45.21 |
| SCAs073 | jacL-73 | 1295825..1296074 | SCA_1288 | < | thrS | 1306.39 |
| SCAs074 | jacL-74 | 1732302..1732452 | SCA_1707 | < | rplQ | 4134.38 |
| SCAs075 | jacL-75 | 756990..757182 | SCA_0755 | > | pheS | 165.26 |
| SCAs076 | jacL-76 | 1639798..1639959 | rpmE | < | rho | 514.78 |
| SCAs077 | jacL-77 | 2552910..2553191 | serS | < | hutH | 153.16 |
| SCAs078 | jacL-78 | 1574476..1574699 | SCA_1558 | > | ilvD | 467.22 |
| SCAs079 | jacL-79 | 16200..16350 | metC | < | SCAs002 | 699.7 |
| SCAs080 | jacL-80 | 203095..203214 | rplA | > | rplJ | 2129.15 |
| SCAs081 | jacL-81 | 1298095..1298341 | thrS | < | dnaI | 793.63 |
| SCAs082 | jacL-82 | 1728725..1728877 | rplM | < | truA | 297.5 |
| SCAs083 | jacL-83 | 1200679..1200816 | SCA_1186 | > | glyS | 334.65 |
| SCAs084 | jacL-84 | 1674226..1674513 | glmS | < | mtlA | 57.68 |
| SCAs085 | jacL-85 | 365862..366036 | SCA_0360 | < | SCA_0361 | 32.28 |
| SCAs086 | jacL-86 | 1648746..1648822 | pyrG | < | SCA_1631 | 207.11 |
| SCAs087 | jacL-87 | 1278047..1278317 | valS | < | tag | 202.29 |
| SCAs088 | jacL-88 | 605369..605416 | pepF | > | SCA_0600 | 699.1 |
| SCAs089 | jacL-89 | 1811579..1811734 | SCA_1806 | < | SCA_1807 | 10.65 |
*Gene.
†RPKM, reads per kilobase material per million reads.
Fig. 2.
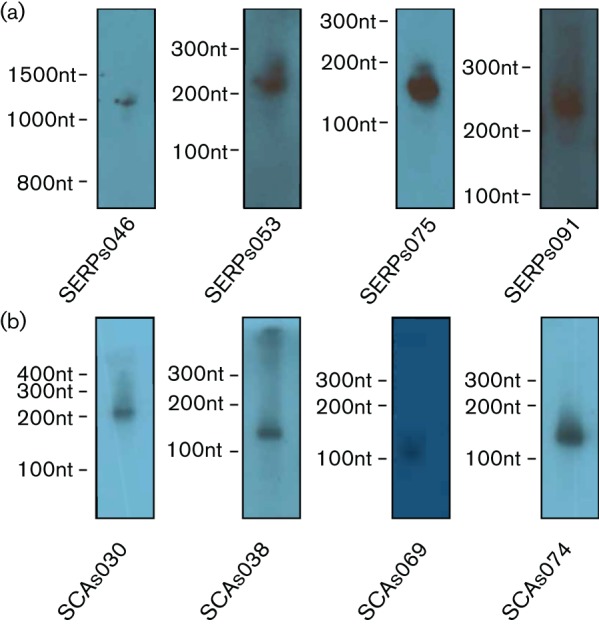
Northern blot analysis of sRNAs in S. epidermidis and S. carnosus. Total RNA was isolated from S. epidermidis RP62a (a) and S. carnosus TM300 (b) cultures grown to the mid-logarithmic phase. Samples were analysed using DNA probes specific to each transcript. Size markers, and the RNA probed for, are denoted on each gel.
Defining the core staphylococcal sRNA content
Given that a primary goal of this study was to better understand the sRNAs that are specific to each species, and that may contribute to their individual lifestyles, we first set out to elucidate the shared sRNA content of the staphylococci (Fig. 3 and Table S2). An sRNA in one genome was considered homologous to another gene if blast searches returned an E-value ≤10−10 in a region that had been annotated. As such, we queried all sRNAs from each organism in a nucleotide blast search against the genomes of the other two staphylococcal species to gain a comprehensive overview of the shared and unique sRNAs encoded by each genome.
Fig. 3.

Shared and unique sRNA content amongst the staphylococci. (a) Depicted from the outermost semicircle inward are: the genome of S. aureus, sRNA annotations, expression level of each sRNA under standard conditions, genomic landmarks [SCCmec (purple), pathogenicity islands (red), prophages (orange) and other genomic islands (yellow)], sRNAs encoded on the forward strand and, innermost, the reverse strand. The inner links connect sRNAs that have sequence conservation. Red and blue links show homologous sRNAs between S. aureus and either S. carnosus or S. epidermidis, respectively; and the black link indicates a single homologous sRNA shared between S. epidermidis and S. carnosus but with no relation to any in S. aureus. (b) Pie charts representing the portion of sRNA content that is shared with each of the species in this study. The total sRNA content of each genome is indicated. (c) Numbers used to generate images in (b). Shown is the number of sRNAs shared between a given species pairing (upper section of each cell) as well as the number of sRNAs unique to a given species pairing (lower section of cells). For example, S. aureus has 105 sRNAs in common with S. epidermidis, but only 52 of these 105 are unique to S. aureus and S. epidermidis (i.e. not found in S. carnosus). When viewing these data, an organism-specific point-of-view must be employed to understand the differences in numbers from similar comparisons. Specifically, the numbers are different for S. aureus vs. S. epidermidis (105 and 52 sRNAs shared and specific, respectively) compared with S. epidermidis vs. S. aureus (92 and 56 sRNAs shared and specific, respectively) because S. aureus has 52 sRNAs that are homologous to 56 sRNAs in S. epidermidis.
A confounding issue to this approach, however, is that there does not appear to be a 1 : 1 ratio of sRNAs from one organism to another. For example, a number of sRNAs from S. aureus have significant sequence homology to several sRNAs from S. epidermidis (described in more detailed below ). Indeed, this is not a lone occurrence as each organism comparison results in several such relationships. Accordingly, the unique and shared sRNA content of the staphylococci can only be specifically calculated from one genome to another, rather than across the genus as a whole. Such analyses are visually represented in Fig. 3(a) where links represent a homologous relationship between sRNAs of S. aureus and S. epidermidis (blue) or S. carnosus (red). A single sRNA exists in S. epidermidis and S. carnosus that shares homology to each other but has no relationship to any in S. aureus (black link). The relative (Fig. 3b) and absolute (Fig. 3c) number of shared sRNAs by genome vary significantly. At first glance it is readily apparent that nearly two-thirds of the sRNAs (187 of 303) previously identified in S. aureus are unique to this organism (Fig. 3b, c). S. carnosus has the next highest number of unique sRNAs, 41 of 89 (Fig. 3b, c). The high percentage of unique sRNAs in S. carnosus (~46 %) is perhaps to be expected, as it is the most distantly related of the three organisms in this study. In contrast, S. epidermidis has the least number of unique sRNAs, at 25 of 118 (~21 %) (Fig. 3c), meaning that nearly 79 % of its sRNA content is shared with S. aureus and/or S. carnosus (Fig. 3b, c). Collectively, we identified 53 core and 187 unique sRNAs in S. aureus, 36 core and 25 unique sRNAs in S. epidermidis and 39 core and 41 unique sRNAs in S. carnosus (Fig. 3b, c). The conservation of sequence and expression suggests that these sRNAs may be involved in more central and conserved processes, such as metabolism. As such, unique sRNAs may represent elements that are probably involved in individual, species-specific adaptation, which, in the case of S. aureus, suggests virulence processes.
A consideration with these data is that ours is the first study to evaluate S. epidermidis and S. carnosus sRNAs, which are derived from a single transcriptomic experiment. Conversely, studies by many groups, using a wealth of different approaches, have contributed to the 303 S. aureus sRNAs identified thus far. This is placed in context when one considers that the S. aureus sRNA content is greater than that from S. epidermidis (118 in total) and S. carnosus (89 in total) combined. As such, the possibility remains that several other sRNAs exist in these latter two species, but are not expressed under the conditions tested in our study. Accordingly, all sRNAs from S. aureus that showed significant sequence homology (E-value ≤10−10) to regions in the S. epidermidis or S. carnosus chromosomes were identified and denoted (Fig. 4a, Table S2). These regions were not annotated as sRNAs in the newly generated genome annotations, but their locations have been recorded (Table S2). While these loci did not show any transcriptional activity in S. epidermidis or S. carnosus in our study, they do share high sequence homology to known sRNAs of S. aureus, and thus may be expressed under different conditions not examined within this study. These transcriptionally inactive regions are linked to their homologous sRNA in S. aureus using blue and red links (S. epidermidis and S. carnosus, respectively) as before (Fig. 4a). When one factors these homologous, transcriptionally inactive regions into the shared and unique calculations, a very different picture appears (Fig. 4b, c). Specifically, the number of shared sRNAs increases greatly, elevating the putative S. aureus core-sRNA content from 53 to 87, whilst at the same time decreasing the number of unique sRNAs from 187 to 137.
Fig. 4.

The identification of transcriptionally silent S. aureus sRNAs in S. epidermidis and S. carnosus. (a) Data are arranged in the same manner as Fig. 3(a), with the following differences: links connect annotated sRNAs of S. aureus to homologous regions within the chromosomes of S. epidermidis (blue) and S. carnosus (red) that do not show transcriptional activity in these latter species. Regions were considered homologous if blast search returned an E-value of <10–10. (b) Pie chart showing the total shared and unique sRNA content of S. aureus including the expressed sRNAs from Fig. 3 and the homologous unexpressed from (a). (c) Numbers used to generate images in (b). Shown are the number of small RNAs shared between a given species pairing (upper section of cells) as well as the number of sRNAs unique to a given species pairing (lower section of cells).
ORF prediction and conservation
The genomes of S. aureus USA300, S. epidermidis RP62a and S. carnosus TM300 have previously been annotated for standard genomic features, including origin of replication, tRNAs, rRNAs and protein-coding genes. During the automated annotation process, ORFs smaller than 50 codons in length are generally dismissed, but the importance of small peptides (those smaller than 50 aa) encoded by small ORFs is becoming increasingly recognized (Hobbs et al., 2011; Storz et al., 2014). As such, we examined the predicted ORF content of the newly annotated transcripts, as our annotation process does not exclude potential protein-coding genes (Tables S3 and S4). In S. epidermidis, only a single newly annotated transcript had a predicted ORF of 50 codons or longer, whilst 111 had predicted ORFs between five and 50 codons, and six had no predicted ORFs of five or more codons. Similarly, in S. carnosus six newly annotated transcripts had predicted ORFs greater than 50 codons, 73 had ORFs between five and 50 codons long and 11 sRNAs had no identifiable ORFs of five or more codons. Importantly, none of the predicted ORFs within each of the organisms examined had any significant homology to any protein with known function aside from the S. aureus Δ-hemolysin. Furthermore, the predicted ORFs from all three organisms also have very little similarity to each other, suggesting that these may not be translated (Tables S5 and S6). S. epidermidis and S. carnosus have a similar number of predicted ORFs per sRNA (3.3 ORFs and 3.9 ORFs per sequence, respectively) whereas the S. aureus sRNAs contain a much higher number of predicted ORFs (11.2 ORFs per sequence). This discrepancy is probably due to a difference in the average size of annotated sRNAs, as S. aureus has an average sRNA size of 506 nt compared with S. epidermidis and S. carnosus with 190 and 217 nt, respectively. As a note, the algorithm used to predict potential ORFs can predict more than one ORF per sRNA but does not evaluate the presence or absence of a ribosomal binding site. As such, the presence of an ORF does not provide any information on the likelihood of translation.
An interspecies conserved and recurring sRNA structural motif
Initial investigations into the overall conservation of sRNA content in the staphylococci revealed the presence of a number of S. epidermidis elements with homology to sRNAs from S. aureus (Fig. 3a). Twenty-one sRNAs from S. epidermidis and three from S. aureus demonstrate a higher than random level of homology as first identified by blast analysis (Fig. 3a), and confirmed by sequence alignments (Figs 5a, 6a and S2). The sRNAs identified in S. epidermidis have a significantly higher level of nucleotide identity to each other (as determined by pairwise comparisons) than to the sRNAs of S. aureus, or that the S. aureus sRNAs do with each other (Figs S2–S4). Furthermore, while sequence conservation does exist between SAUSA300s206 and the other 23 sRNAs identified, it is the most divergent sequence (Figs 6a, S2 and S3).
Fig. 5.

Sequence and structural conservation of several highly related and newly identified S. epidermidis sRNAs. (a) A sequence alignment of 21 newly identified sRNA genes from S. epidermidis, with a particular focus on the most conserved region within each. Within the zoomed in region, conservation at the nucleotide level is shown, with a consensus sequence generated from the alignment presented (SERPsCon). The level of conservation of each nucleotide is indicated by colour, and the number of sequences containing the conserved residue from all 21 sRNAs. Purple, conservation in all 21 sequences; dark green, 20/21; yellow, 19/21; orange, ≤ 18/21; and white, not conserved, and not included in the consensus sequence. Alignment, conservation analysis and consensus sequence generation were performed using the CLC Genomics Workbench software. (b) RNA secondary structure prediction for the consensus sequence generated in (a), with each residue colour coded to its level of conservation, as detailed in (a). RNA secondary structure predictions were generated using the mfold software. (c) RNA secondary structure predictions for each of the 21 sRNAs from the alignment. The most highly conserved region of each [from the zoomed in area in (a)] is highlighted in red. RNA secondary predictions were again generated using the mfold software.
Fig. 6.

The S. epidermidis sequence and structural motif is conserved in homologous sRNAs in S. aureus. (a) Sequence alignment of three sRNA genes (SAUSA300s205, SAUSA300s206 and SAUSA300s288) from S. aureus and the consensus sequence generated (SERPsCon) in Fig. 4(a). Sequence annotations are shown on the left, and on the right total sequence length. Zoomed in areas show nucleotide conservation amongst the four sequences, with the conservation for each residue indicated by colour. Purple is 100 % conservation, green is 75 % and yellow is 50 % or below. (b) As in (a), but containing the reverse complement region of SAUSA300s206 (RC-SAUSA300s206) instead of its native orientation. (c) Secondary structure predictions for each S. aureus sRNA as well as RC-SAUSA300s206. Regions sharing a high level of homology to SERPsCon as determined in (a) (SAUSA300s206) or (b) (the rest) were highlighted in each structure prediction. RNA secondary predictions were generated using the mfold software.
The 21 highly related sRNA genes in S. epidermidis have a higher relative G+C content, ranging from 32.6 % for SERPs014 to 44.2 % for SERPs106, than the relative G+C content of the S. epidermidis genome (32.2 %). They also span a range of sizes from 98 bp (SERPs103) to 217 bp (SERPs099), with this variation seemingly attributable to differences in their 5' regions (Fig. 5a). Conversely, each sequence shares a key region of high conservation that extends approximately from the middle of the sequence to its 3′ end. This common region demonstrates nucleotide-level conservation of 71.4–100 %, not including the 3 bp insertion found in SERPs013. Using this information, we generated a consensus sequence (SERPsCon) that reflects the nucleotide identity of >71 % of the sequences in the conserved region.
The SERPsCon sequence and each S. epidermidis sRNA were subjected to secondary structure prediction using the mfold software (Fig. 5b, c, respectively) (Zuker, 2003). The predicted SERPsCon structure includes a stem with two single-stranded regions, and a terminal loop (bracketed) near the 5′ end of the molecule that includes 28 of the 38 residues conserved in all 21 sequences (Fig. 5a, b). The terminal single-stranded region of the SERPsCon structure within the bracket has a 10 nt sequence that is variable only at the ninth residue, and defined by the sequence motif 5′-GAAGACUAYA (Fig. 5b). Furthermore, mfold secondary structure predictions performed on the 21 S. epidermidis sRNAs suggest the sequence homology extends to structural conservation. The secondary structure predictions suggest that in all 21 of these elements, the region corresponding to SERPsCon (Fig. 5c, red regions) includes an extended stem-loop structure that is identical (for 17 of the 21) to the motif defined in SERPsCon (5′-GAAGACUAYA). The remaining four sRNAs have the same sequence motif at the terminus of a stem, although the optimal structure, as predicted by mfold, suggests less single-strandedness. The conserved region and terminal loop do not appear to be related to any known RNA families or motifs as determined by an Rfam analysis (http://rfam.xfam.org/) (Nawrocki et al., 2015), and thus may constitute a new regulatory RNA family.
Initial sequence analysis of SAUSA300s205, SAUSA300s206 and SAUSA300s288 determined that these sRNAs share less sequence similarity than their S. epidermidis counterparts (Figs S3 and S4). SAUSA300s205 and SAUSA300s288 are more similar to each other than to SAUSA300s206 (Figs 6a, S2 and S3), although upon further examination it was noted that SAUSA300s205 and SAUSA300s288 share more similarity with the reverse complement of SAUSA300s206 (RC-SAUSA300s206) (Figs 6b, S2 and S3). Secondary structure predictions suggest at least partial structural conservation between SAUSA300s205 and SAUSA300s288 in relation to SERPsCon and the other S. epidermidis sRNAs (Fig. 6c). The predicted secondary structure of SAUSA300s288 has the highest level of structure and sequence conservation, with nine residues in the terminal loop structure, seven of which are perfectly conserved in relation to SERPsCon (Fig. 5b). SAUSA300s205 contains a 59 nt insert within the region corresponding to SERPsCon that necessarily shifts the structure, resulting in a slightly lower level of sequence conservation in the terminal loop (six of nine residues) (Fig. 6c). Folding predictions of SAUSA300s206 suggest very little, if any, structural conservation, mirroring the lack of sequence similarity, with the other sRNAs (Fig. 6a, c). However, as perhaps is expected, mfold analysis of RC-SAUSA300s206 suggests structural conservation including the terminal loop (six of nine residues) (Fig. 6c).
The antisense nature of SAUSA300s206 in comparison with SAUSA300s205 and SAUSA300s288 hints at the possibility of an interaction between SAUSA300s206 and the other two sRNAs. To evaluate this potential, we queried the SAUSA300s206 sequence against the target sequences SAUSA300s205 and SAUSA300s288 using RNA–RNA interaction prediction software, IntaRNA (http://rna.informatik.uni-freiburg.de/) (Busch et al., 2008; Wright et al., 2014). Perhaps unsurprisingly, the predicted areas of interaction between SAUSA300s206 and both of the other sRNAs are extensive, and have a very low free energy (−181.7 and −95.3 kcal mol−1 for SAUSA300s205 and SAUSA300s288, respectively) thus making these interactions energetically favorable.
Functional prediction of S. aureus-specific sRNAs
A major goal of this study was to differentiate sRNA content between the staphylococci (Table S2), and to garner a better understanding of the potential physiological role for unique elements, particularly in the context of S. aureus pathogenesis. As such, the complete set of S. aureus-specific sRNAs (Table S7) was subjected to target prediction using TargetRNA2 (http://cs.wellesley.edu/~btjaden/TargetRNA2/) (Kery et al., 2014). The resulting list of putative targets (Table S8) was subjected to ontological classification, to identify those that are known virulence factors. Of note, 85 of the 137 (62 %) sRNAs unique to S. aureus were found to have the capacity to interact with at least one virulence-related transcript. Interestingly, the gene with the highest number of predicted sRNA regulators (10 different sRNAs) was splA, which encodes a well-characterized serine protease (Stec-Niemczyk et al., 2009). This is particularly compelling as S. aureus proteases have a major role in pathogenesis via the global modulation of virulence determinant stability (Kolar et al., 2013). As such, this clearly suggests potential for sRNA-based regulation of the infectious process in S. aureus. Ultimately, each of the predictions generated require further experimental verification to assess specific functional roles. However, we suggest that the data presented herein represent an important first step in exploring the influence of sRNAs in the staphylococci, and their impact on species-specific adaptation.
Discussion
The advent of next-generation sequencing technologies has resulted in a vast amount of genomic and transcriptomic data available for all domains of life. This flood of data has resulted in the need for automated annotation software (Dark, 2013; Richardson & Watson, 2013). While automated annotation has become fairly robust for protein-coding regions, tRNAs and rRNAs, the ability to accurately predict the presence of other non-coding RNAs lags behind, which necessitates the manual curation of such genes (Sridhar & Gunasekaran, 2013). Collectively, sRNAs are of growing interest, as the diverse roles they play in regulating carbon metabolism, virulence gene expression, iron acquisition and many other cellular processes becomes increasingly apparent (Hoe et al., 2013; Beisel & Storz, 2010; Murphy et al., 2014; Caron et al., 2010; Geissmann et al., 2006; Harris et al., 2013; Papenfort & Vanderpool, 2015; Oliva et al., 2015). The inability to efficiently identify and annotate these elements hinders research on sRNAs and creates a need for transcriptomic-based approaches to supplement automated annotation software pipelines. To this end, our group has begun manually cataloguing and curating these molecules into their respective genomes within the staphylococci.
In the present work, we have identified and annotated sRNAs in the genomes of both S. epidermidis RP62a and S. carnosus TM300 using RNAseq methodologies. The total sRNA contents of S. epidermidis and S. carnosus were compared with our previous work in S. aureus, generating a fully comprehensive comparison of the shared and unique sRNA content of these common staphylococci. In so doing, we identified and annotated 118 and 89 novel sRNAs in S. epidermidis and S. carnosus, respectively. The sRNA content of these two genomes initially appears strikingly small compared with S. aureus (303 annotated sRNAs). The difference in the number of sRNAs between these organisms is probably not due to differences in genome size (2 872 769, 2 616 530 and 2 566 424 bp, respectively), but rather an artifact of the overall number of conditions tested for sRNA expression within each species. For S. epidermidis and S. carnosus, our study is the first assessment of their sRNA content, based on a single growth condition (mid-logarithmic phase, TSB at 37 °C), whereas those sRNAs for S. aureus are derived from a wealth of different studies and experimental conditions (Pichon & Felden, 2005; Marchais et al., 2009; Geissmann et al., 2009; Abu-Qatouseh et al., 2010; Bohn et al., 2010; Beaume et al., 2010; Nielsen et al., 2011; Xue et al., 2014; Anderson et al., 2006, 2010; Olson et al., 2011; Howden et al., 2013; Carroll et al., 2016). The discrepancy in the number of studies that have examined the sRNA content of these three organisms also underlies the very different proportion of sRNAs common to the staphylococci in each genome. For example, considering only the transcriptionally active sRNA comparisons, S. aureus has a common sRNA set of 53 (~17.5 %) while S. epidermidis and S. carnosus have 36 (~30.5 %) and 39 (~43.8 %), respectively. The sRNAs represented in all three genomes probably have similar roles within the cell, speculatively involved in evolutionary conserved processes such as basic metabolism and maintenance of cellular homeostasis. While the number of sRNAs shared by S. aureus increases to 87 (~28.7 %) if the homologous, but transcriptionally inactive, regions of S. epidermidis and S. carnosus are included, this is still a smaller proportion of the sRNAs compared with S. epidermidis, and considerably smaller than that of S. carnosus (~30.5 % and ~43.8 %, respectively). One could hypothesize that these sRNAs may be involved in conserved processes that are perhaps unnecessary under the conditions tested. Conversely, and of some interest, several regions within the S. aureus genome show high sequence similarity to newly annotated sRNAs from S. epidermidis and S. carnosus, despite themselves being transcriptionally silent (data not shown). Either the presence of such regions suggests an evolutionary event that has silenced expression from these loci, or, perhaps a more likely scenario, we have yet to elucidate the permissive conditions for their expression in S. aureus. As such, a need exists for further research into lifestyle-specific and patho-physiologically relevant transcriptomic conditions and effects within the staphylococci.
The presence of a set of highly conserved sRNAs from S. epidermidis and S. aureus is seemingly quite unusual. The high level of sequence similarity within these sRNAs also results in a conserved structural motif that takes the form of a stem and multi-loop region, ending in a terminal hairpin with an unpaired, conserved 9 – 10 nt motif. Conservation of the multi-loop stem and terminal loop would suggest a common function for these sRNAs as a group and/or for the region of homology. Several possibilities for general function present themselves with such sequence and structure conservation. For example, it is possible that these structures act to bind and sequester proteins, as is the case for the CsrB/C sRNAs. CsrB/C sRNAs were originally identified in Escherichia coli as binding to and sequestering the CsrA protein through a conserved, repeated RNA motif, ultimately affecting carbon utilization and virulence gene expression (Liu et al., 1997; Jonas & Melefors, 2009). A second scenario, that has been demonstrated for several of the Rsa sRNAs in S. aureus (first characterized for their UCCC motif), is that the terminal hairpin serves to bind conserved regions within a target RNA, and the surrounding, less conserved regions confer target specificity (Geissmann et al., 2009). More work is necessary to elucidate the function of each of these individual sRNAs as well as the conserved domain that characterizes them. Curiously, the homology searches also identified SAUSA300s206 within this group, although further in silico analysis demonstrated SAUSA300s206 has high sequence complementarity to SAUSA300s205 and SAUSA300s288 as well as SERPsCon. The presence of high levels of sequence complementarity begs the question: is SAUSA300s206 a regulator of SAUSA300s205 and SAUSA300s288? Regulation of one sRNA by another (so-called anti-sRNAs) is not unprecedented in the literature. In E. coli the molecular mechanism for interaction of two such anti-sRNAs, AsxR and AgvB, with their targets has recently been elucidated (Tree et al., 2014). AsxR binds the sRNA FnrS, which normally represses the expression of a heme oxygenase, ChuS; thus, AsxR acts to enhance expression of ChuS (Tree et al., 2014). In the context of AgvB, it binds the sRNA GcvB, repressing the GcvB-dependent repression of DppA expression (Tree et al., 2014). In direct parallel to this, our group recently identified a set of highly transcribed, highly homologous sRNAs in A. baumannii, termed Group 1 sRNAs, for which there appears to be an anti-sRNA, ABUWs043 (Weiss et al., 2015). ABUWs043 is encoded in an antisense fashion to ABUWs042, and thus may regulate ABUWs042 through several means, including promoter interference and/or complementary binding (Weiss et al., 2015). Importantly, ABUWs043 has a high level of sequence complementarity to the rest of the Group 1 sRNAs (21 such elements exist in the A. baumannii genome), albeit lower than that found for ABUWs042, suggesting ABUWs043 may regulate the rest of the Group 1 sRNAs in an anti-sRNA fashion (Weiss et al., 2015). SAUSA300s206 shares many characteristics with these confirmed and putative anti-sRNAs, but ultimately more work must be done to characterize its function within S. aureus. Finally and perhaps the most intriguing observation about these sRNAs is the absence of an identified anti-sRNA encoded in S. epidermidis that shares homology with SAUSA300s206. The possibility that such an sRNA exists cannot be excluded, although the potential that this is an S. aureus specific adaptation is a potentially fascinating point of evolution. Regardless, a better understanding of the function of these 24 sRNAs may underlie basic physiological and regulatory differences between S. aureus and S. epidermidis, and further our understanding of the staphylococci in general.
Acknowledgements
This study was supported in part by grant AI080626 (to L.N.S.) from the National Institute of Allergy and Infectious Diseases.
Supplementary Data
Supplementary File 1
Supplementary Data
Supplementary File 2
Abbreviation:
- sRNA
small regulatory RNA
References
- Abu-Qatouseh L. F., Chinni S. V., Seggewiss J., Proctor R. A., Brosius J., Rozhdestvensky T. S., Peters G., von Eiff C., Becker K.(2010). Identification of differentially expressed small non-protein-coding RNAs in Staphylococcus aureus displaying both the normal and the small-colony variant phenotype. J Mol Med (Berl) 88565–575. 10.1007/s00109-010-0597-2 [DOI] [PubMed] [Google Scholar]
- Anderson K. L., Roberts C., Disz T., Vonstein V., Hwang K., Overbeek R., Olson P. D., Projan S. J., Dunman P. M.(2006). Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol 1886739–6756. 10.1128/JB.00609-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K. L., Roux C. M., Olson M. W., Luong T. T., Lee C. Y., Olson R., Dunman P. M.(2010). Characterizing the effects of Inorganic Acid and Alkaline shock on the Staphylococcus aureus Transcriptome and Messenger RNA turnover. FEMS Immun Med Mic 60208–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer G. L.(1998). Staphylococcus aureus: a well-armed pathogen. Clin Infect Dis 261179–1181. [DOI] [PubMed] [Google Scholar]
- Beaume M., Hernandez D., Farinelli L., Deluen C., Linder P., Gaspin C., Romby P., Schrenzel J., Francois P.(2010). Cartography of methicillin-resistant S. aureus transcripts: detection, orientation and temporal expression during growth phase and stress conditions. PLoS One 5e10725. 10.1371/journal.pone.0010725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beisel C. L., Storz G.(2010). Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol Rev 34866–882. 10.1111/j.1574-6976.2010.00241.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn C., Rigoulay C., Chabelskaya S., Sharma C. M., Marchais A., Skorski P., Borezée-Durant E., Barbet R., Jacquet E., other authors(2010). Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nuc Acid Res 386620–6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broach W. H., Egan N., Wing H. J., Payne S. M., Murphy E. R.(2012). VirF-independent regulation of Shigella virB transcription is mediated by the small RNA RyhB. PLoS One 7e38592. 10.1371/journal.pone.0038592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch A., Richter A. S., Backofen R.(2008). IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. B Informatics 242849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron M. P., Lafontaine D. A., Massé E.(2010). Small RNA-mediated regulation at the level of transcript stability. RNA Biol 7140–144. [DOI] [PubMed] [Google Scholar]
- Carroll R. K., Weiss A., Shaw L. N.(2014). RNA-Sequencing of Staphylococcus aureus Messenger RNA [Online]. The Genetic Manipulation of Staphylococci. Vol. 1373 131–141. Edited by Bose J. L.New York, NY: Springer New York; Available from: [Accessed 25 November 2015]. [DOI] [PubMed] [Google Scholar]
- Carroll R. K., Weiss A., Broach W. H., Wiemels R. E., Mogen A. B., Rice K. C., Shaw L. N.(2016). Genome-wide Annotation, Identification, and Global Transcriptomic Analysis of Regulatory or Small RNA Gene Expression in Staphylococcus aureus. MBio 7e01990–15. 10.1128/mBio.01990-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caswell C. C., Gaines J. M., Ciborowski P., Smith D., Borchers C. H., Roux C. M., Sayood K., Dunman P. M., Roop Ii R. M.(2012). Identification of two small regulatory RNAs linked to virulence in Brucella abortus 2308. Mol Microbiol 85345–360. . 10.1111/j.1365-2958.2012.08117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dark M. J.(2013). Whole-Genome sequencing in bacteriology: state of the art. Infect Drug Res 6115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann T., Possedko M., Huntzinger E., Fechter P., Ehresmann C., Romby P.(2006). Regulatory RNAs as mediators of virulence Gene expression in Bacteria. Exp Pharmacol 1739–43. [DOI] [PubMed] [Google Scholar]
- Geissmann T., Chevalier C., Cros M.-J., Boisset S., Fechter P., Noirot C., Schrenzel J., Francois P., Vandenesch F., et al. (2009). A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nuc Acids Res 377239–7257. 10.1093/nar/gkp668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangrossi M., Prosseda G., Tran C. N., Brandi A., Colonna B., Falconi M.(2010). A novel antisense RNA regulates at transcriptional level the virulence gene icsA of Shigella flexneri. Nucleic Acids Res 383362–3375. 10.1093/nar/gkq025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S. R., Fouts D. E., Archer G. L., Mongodin E. F., Deboy R. T., Ravel J., Paulsen I. T., Kolonay J. F., Brinkac L., other authors(2005). Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol 1872426–2438. 10.1128/JB.187.7.2426-2438.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. F., Micheva-Viteva S., Li N., Hong-Geller E.(2013). Small RNA-Mediated regulation of host-pathogen interactions. Virul 4785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highlander S. K., Hultén K. G., Qin X., Jiang H., Yerrapragada S., Mason E. O., Shang Y., Williams T. M., Fortunov R. M., other authors(2007). Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol 799. 10.1186/1471-2180-7-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs E. C., Fontaine F., Yin X., Storz G.(2011). An expanding universe of small proteins. Cur Opinion Microbiol 14167–173. 10.1016/j.mib.2011.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe C. H., Raabe C. A., Rozhdestvensky T. S., Tang T. H.(2013). Bacterial sRNAs: regulation in stress. Int J Med Microbiol 303217–229. 10.1016/j.ijmm.2013.04.002 [DOI] [PubMed] [Google Scholar]
- Howden B. P., Beaume M., Harrison P. F., Hernandez D., Schrenzel J., Seemann T., Francois P., Stinear T. P.(2013). Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Anti Agents Chemother 573864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas K., Melefors O.(2009). The Escherichia coli CsrB and CsrC Small RNAs Are Strongly Induced during Growth in Nutrient-Poor Medium. FEMS Microbiol Letters 29780–86. [DOI] [PubMed] [Google Scholar]
- Kery M. B., Feldman M., Livny J., Tjaden B.(2014). TargetRNA2: identifying targets of small regulatory RNAs in bacteria. Nucleic Acids Res, 42 W124–W129. 10.1093/nar/gku317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolar S. L., Nagarajan V., Oszmiana A., Rivera F. E., Miller H. K., Davenport J. E., Riordan J. T., Potempa J., Barber D. S., et al. (2011). NsaRS is a cell-envelope-stress-sensing two-component system of Staphylococcus aureus. Microbiology 1572206–2219. 10.1099/mic.0.049692-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolar S. L., Ibarra J. A., Rivera F. E., Mootz J. M., Davenport J. E., Stevens S. M., Horswill A. R., Shaw L. N.(2013). Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen 218–34. 10.1002/mbo3.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M. Y., Gui G., Wei B., Preston J. F., Oakford L., Yüksel U., Giedroc D. P., Romeo T.(1997). The RNA molecule CsrB binds to the global regulatory protein CsrA and antagonizes its activity in Escherichia coli. J Biol Chem 27217502–17510. [DOI] [PubMed] [Google Scholar]
- Lowy F. D.(1998). Staphylococcus aureus infections. N Engl J Med 339520–532. 10.1056/NEJM199808203390806 [DOI] [PubMed] [Google Scholar]
- Lowy F. D.(2003). Antimicrobial resistance: the example of Staphylococcus aureus. J Clin Invest 1111265–1273. 10.1172/JCI18535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchais A., Naville M., Bohn C., Bouloc P., Gautheret D.(2009). Single-pass classification of all noncoding sequences in a bacterial genome using phylogenetic profiles. Genome Res 191084–1092. 10.1101/gr.089714.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E., Broach W., Kouse A. B.(2014). Prokaryotic Gene Regulation by Small RNAs [Online]. Molecular Life Sciences. 1–6. Edited by Bell E.New York, NY: Springer New York. [Google Scholar]
- Nawrocki E. P., Burge S. W., Bateman A., Daub J., Eberhardt R. Y., Eddy S. R., Floden E. W., Gardner P. P., Jones T. A.(2015). Rfam 12.0: updates to the RNA families database. Nucleic Acids Res 43D130–D137. 10.1093/nar/gku1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J. S., Christiansen M. H., Bonde M., Gottschalk S., Frees D., Thomsen L. E., Kallipolitis B. H.(2011). Searching for small σB-regulated genes in Staphylococcus aureus. Arch Microbiol 19323–34. 10.1007/s00203-010-0641-1 [DOI] [PubMed] [Google Scholar]
- Oliva G., Sahr T., Buchrieser C.(2015). Small RNAs, 5' UTR elements and RNA-binding proteins in intracellular bacteria: impact on metabolism and virulence. FEMS Microbiol Rev 39331–349. 10.1093/femsre/fuv022 [DOI] [PubMed] [Google Scholar]
- Olson P. D., Kuechenmeister L. J., Anderson K. L., Daily S., Beenken K. E., Roux C. M., Reniere M. L., Lewis T. L., Weiss W. J.(2011). Small molecule inhibitors of Staphylococcus aureus RnpA alter cellular mRNA turnover, exhibit antimicrobial activity, and attenuate pathogenesis. PLoS Pathog 7e1001287. 10.1371/journal.ppat.1001287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M.(2009). Staphylococcus epidermidis--the 'accidental' pathogen. Nat Rev Microbiol 7555–567. 10.1038/nrmicro2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K., Vogel J.(2014). Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front Cell Infect Microbiol 4Available from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4098024/. Accessed 23 August 2015. 10.3389/fcimb.2014.00091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K., Vanderpool C. K.(2015). Target activation by regulatory RNAs in bacteria. FEMS Microbiol Rev 39362–378. 10.1093/femsre/fuv016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichon C., Felden B.(2005). Small RNA genesexpressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci U S A 10214249–14254. 10.1073/pnas.0503838102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson E. J., Watson M.(2013). The automatic annotation of bacterial genomes. Brief Bioinform 141–12. 10.1093/bib/bbs007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstein R., Nerz C., Biswas L., Resch A., Raddatz G., Schuster S. C., Gotz F.(2009). Genome Analysis of the Meat Starter Culture Bacterium Staphylococcus carnosus TM300. Applied and Environmental Microbiology. 75811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifer K. H., Fischer U.(1982). Description of a New Species of the Genus Staphylococcus: Staphylococcus carnosus. Int J Syst Bacteriol 32153–156. [Google Scholar]
- Sridhar J., Gunasekaran P.(2013). Computational small RNA prediction in bacteria. Bioinform Biol Insights 783–95. pp. . 10.4137/BBI.S11213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stec-Niemczyk J., Pustelny K., Kisielewska M., Bista M., Boulware K. T., Stennicke H. R., Thogersen I. B., Daugherty P. S., Enghild J. J.(2009). Structural and functional characterization of SplA, an exclusively specific protease of Staphylococcus aureus. Biochem J 419555–564. 10.1042/BJ20081351 [DOI] [PubMed] [Google Scholar]
- Storz G., Wolf Y. I., Ramamurthi K. S.(2014). Small proteins can no longer be ignored. Annu Rev Biochem 83753–777. 10.1146/annurev-biochem-070611-102400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova T., Ciufo S., Federhen S., Fedorov B., McVeigh R., O'Neill K., Tolstoy I., Zaslavsky L.(2015). Update on RefSeq Microbial Genomes Resources. Nucleic Acids Res, 43D599–D605. 10.1093/nar/gku1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree J. J., Granneman S., McAteer S. P., Tollervey D., Gally D. L.(2014). Identification of bacteriophage-encoded anti-sRNAs in pathogenic Escherichia coli. Mol Cell 55199–213. 10.1016/j.molcel.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner E., Doskar J., Götz F.(1998). Physical and genetic map of the genome of Staphylococcus carnosus TM300. Microbiology 144509–517. 10.1099/00221287-144-2-509 [DOI] [PubMed] [Google Scholar]
- Weiberg A., Bellinger M., Jin H.(2015). Conversations between kingdoms: small RNAs. Curr Opin Biotechnol 32207–215. 10.1016/j.copbio.2014.12.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weilbacher T., Suzuki K., Dubey A. K., Wang X., Gudapaty S., Morozov I., Baker C. S., Georgellis D., Babitzke P., Romeo T.(2003). A novel sRNA component of the carbon storage regulatory system of Escherichia coli. Mol Microbiol 48657–670. [DOI] [PubMed] [Google Scholar]
- Weiss A., Broach W. H., Lee M. C., Shaw L. N.(2015). Towards the Complete Small RNome of Acinetobacter baumannii. Microbial Genomics [Online]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. R., Georg J., Mann M., Sorescu D. A., Richter A. S., Lott S., Kleinkauf R., Hess W. R., Backofen R.(2014). CopraRNA and IntaRNA: predicting small RNA targets, networks and Interaction domains. Nucleic Acids Res, 42W119–W123. 10.1093/nar/gku359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue T., Zhang X., Sun H., Sun B.(2014). ArtR, a novel sRNA of Staphylococcus aureus, regulates α-toxin expression by targeting the 5' UTR of sarT mRNA. Med Microbiol Immunol 2031–12. 10.1007/s00430-013-0307-0 [DOI] [PubMed] [Google Scholar]
- Zuker M.(2003). Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 313406–3415. 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Data Bibliography
- Broach, W. H., Weiss, A. & Shaw, L. N. NCBI Gene Expression Omnibus, GSE77567 (2016).
- Broach, W. H., Weiss, A. & Shaw, L. N. Figshare 10.6084/m9.figshare.3385861 (2016).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1
Supplementary File 2