

Summary
We report a new mouse strain with a single point mutation in the type 2 transporter associated with antigen processing (TAP2). This strain randomly arose in one of our C57BL/6J mouse colonies and was initially discovered because of the lack of CD8+ T cells in the periphery. Following our observation, we subsequently revealed a lack of cell surface MHC‐I expression, derived from TAP2 protein deficiency. Our strain, named eightless, has a C to T substitution in exon 5 resulting in a glutamine to stop codon substitution at position 285 in the TAP2 protein. Interestingly, in addition to the expected lack of CD8+ T cell phenotype, eightless mice have a diminished number of macrophages in their peritoneum. Moreover, following peritoneal inflammation, elicited eightless macrophages showed impaired survival both in vivo and ex vivo. Our study describes the first ever TAP2 complete knockout mouse strain and provides a possible explanation for why patients with TAP2 deficiency syndrome present clinical manifestations that would suggest a phagocyte defect rather than a lack of CD8+ T cells.
Keywords: immunodeficiency diseases, major histocompatibility complex, T cells
Introduction
Bare lymphocyte syndrome (BLS) is characterized by a severe down‐regulation of MHC class I (MHC‐I) and/or class II molecules.1 In type 1 BLS, the defect is confined to MHC‐I, a complex consisting of a highly polymorphic membrane‐spanning heavy chain associated with a light chain, the β2‐microglobulin (β2m). The MHC‐I complex binds peptides that are 8–11 amino acids long and are generated from the cytosolic proteasome‐dependent degradation of cellular proteins.2 After proteolysis, these peptides are transferred to the lumen of the endoplasmic reticulum (ER) by the transporter associated with antigen presentation (TAP) and finally are loaded onto the MHC‐I complex and relocated to the cell surface.3 TAP, a member of the ATP‐binding cassette (ABC) superfamily transporters, is a heterodimer consisting of two protein subunits, TAP1 and TAP2.4 Deletion of or mutation in TAP1 and/or TAP2 severely affects the translocation of peptides into the ER as MHC‐I molecules are retained within the ER, a phenomenon that can be reverted in vitro by cellular incubation at 26°.5
MHC‐I molecules are expressed on the cell surface of all nucleated cells and in the thymus are crucial for the presentation of self‐peptides to developing CD8+ thymocytes undergoing positive selection.6 In the periphery, they support naive CD8+ T cell survival,7 cytotoxic T‐cell generation and activation8 and are able to modulate γδ T and natural killer (NK) cells by interacting with MHC‐I binding receptors.9 The generation of mutants carrying gene deletions within the MHC‐I complex processing and antigen presentation pathway, such as β2m−/− and TAP1−/− mice,10, 11, 12 has been useful to characterize the mechanisms behind these pathways and to provide information about CD8+ T and NK cell development.13, 14 Surprisingly, despite being low in number, CD8+ T cells were clearly detectable in these mice.15, 16, 17
Patients with TAP1 or TAP2 deficiency have been included within a specific BLS‐I subgroup in which reduced HLA‐I surface expression is associated with recurrent bacterial infections of the upper and lower airways eventually evolving to bronchiectasis, often associated with necrotizing granulomatous skin lesions.18, 19 Inexplicably, severe viral infections do not occur. Clinicians prefer to refer to this condition as TAP deficiency syndrome rather than BLS‐I.20 Indeed percentages of CD8+ T cells in these patients vary from more or less severe reductions to normal or even elevated values,21, 22, 23 while there is an expansion of T cell receptor‐γδ + (TCR‐γδ +) T cells18, 23 and NK cells,18 which were found to be autoreactive in four patients studied.18, 24 However, functional tests revealed that NK cells from TAP‐deficient patients have no cytotoxic activity towards HLA‐I‐deficient targets.24
These clinical manifestations and immunological features are difficult to interpret if we consider the biological role of TAP, so the physiopathology of BLS‐I still remains mysterious.25, 26
The mutation that we identified in our mouse strain, which we named eightless, very closely resembles the genetic lesions reported in humans, as BLS‐I patients harbour a TAP2 premature stop codon, resulting in a truncated and non‐functional protein.23 We therefore decided to use this model in order to elucidate the physiopathology of human TAP deficiency syndrome.
In our study we found that these mice contain fewer peritoneal resident macrophages and when we induced peritoneal inflammation we noted that the newly generated macrophages died prematurely.
Materials and methods
Development of mouse strain
The eightless strain randomly arose in our colony from C57BL/6J mice that were purchased from Jackson Laboratories (Bar Harbor, ME) and were routinely screened by flow cytometry for CD4 and CD8 ratios in the blood. Outlying mice or their littermates were bred to confirm heritability of the trait, and to establish the strain. Both males and females (8–12 weeks) were used to define the described phenotypes. The entire work was approved by the Animal Ethics Committee, and carried out in accordance with institutional guidelines.
Identification of the eightless mutation
Spleen RNA was extracted by Trizol reagent (Thermo Fisher Scientific, Waltham, MNA) and cDNA generated by ProtoScript first‐strand cDNA synthesis kit (New England BioLabs, Ipswich, MA). β2m, Tap1 and Tap2 cDNAs were then amplified by PCR and sequenced using 16–20 bp complementary primers (sequences available on request).
Flow cytometry
Blood, lymph node, thymus and spleen samples, as well as peritoneum lavage cells, were depleted of erythrocytes by incubation with red blood cell lysis buffer (10 mm KHCO3, 150 mm NH4Cl, 0·1 mm EDTA, pH 8·0), re‐suspended in FACS buffer (PBS, 2% fetal bovine serum, 3 mm EDTA) and stained with fluorochrome‐conjugated antibodies (BioLegend, San Diego, CA). For survival assay after staining and wash, the samples were re‐suspended in 10 μg/ml propidium iodide (Sigma‐Aldrich, St Louis, MO) FACS buffer. Samples were collected on a Gallios cytometer (Beckman Coulter, Brea, CA) and the data were analysed using kaluza flow analysis software v1.2 (Beckman Coulter).
Real‐time PCR
Total RNA from cultured macrophages was extracted by Trizol reagent (Thermo Fisher Scientific) and cDNA was synthesized using ProtoScript First Strand cDNA Synthesis Kit (New England BioLabs, Ipswich, MA). Quantitative real‐time PCR was then performed using QuantStudio 12K Flex Real‐Time PCR system with a Power SYBR green PCR master mix kit (Applied Biosystems, Foster City, CA). The primers sequences used in this study have been described elsewhere.27
Exogenous addition of peptide and splenocytes culture
Spleens from euthanized mice were cut in small fragments with a blade and then smashed with a syringe plunger on a 40‐μm cell strain, filtered on a 50‐ml Falcon tube, pelleted and red blood cells were lysed. Erythrocyte‐depleted splenocytes were then incubated for 3–9 hr at 37° in medium (RPMI‐1640, 10% fetal bovine serum, 1% peptomycin‐streptomycin, 50 nm 2‐mercaptoethanol) either untreated or with 1–2·5 × 10−6 m synthetic SIINFEKL peptide. Alternatively, the splenocytes were incubated overnight at 26° without peptide. After this time, cells were stained with anti‐H‐2Kb‐phycoerythrin (clone AF6 88.5) and the corresponding isotype control in FACS buffer and analysed by flow cytometry.
Peritoneal macrophage culture
Residential peritoneal macrophages were directly collected from anaesthetized mice by lavage with 5 ml PBS using a 5‐ml syringe with 19G needle. When elicited macrophages were needed, the mice were injected intraperitoneally with 1 ml 4% thioglycollate at least 36 hr in advance. Then, 5 × 105 erythrocyte‐depleted lavage‐derived cells were plated on a tissue culture treated polystyrene 24‐well plate (Corning, NY) in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 1% peptomycin–streptomycin, 50 nm 2‐mercaptoethanol. After 2 hr, medium was removed, plate wells were washed twice in PBS and 1 ml media was added to remove unattached cells and select for macrophages. Cells were cultured for 24–48 hr in a humified incubator with 5% CO2. For survival assay macrophages were detached using Enzyme‐Free Cell Dissociation Buffer (Gibco, Grand Island, NY), counted by microscopy and immediately processed for flow cytometry analysis as previously described.
Statistical analysis
All data were analysed using unpaired t‐tests and were represented as mean ± SEM. All statistical analyses were carried out using prism software (GraphPad Software, San Diego, CA) and the α level was set at 0·05 for all tests.
Results
Diminished CD8+ T cells in eightless mutant mice
The recessive eightless phenotype was initially randomly detected in our C57BL/6J mouse colony among mice with very low CD8+ T cell proportions (about 0·5%) in the blood. However, these mice had normal total leucocyte numbers in the spleen, thymus, lymph nodes and blood.
To further characterize the immunological defect caused by the eightless mutation, we immunophenotyped cells from spleen, lymph node and thymus by flow cytometry. Low percentages and absolute numbers of CD8+ T cells were evident in the lymph nodes (Fig. 1a,b), spleen (Fig. 1c,d) and thymus (Fig. 1f,g) relative to those of wild‐type mice. In addition, relative to wild‐type mice, eightless mice were found to have a significantly increased percentage and absolute number of CD4+ T cells but equal quantities of B cells in both spleen and lymph nodes (Fig. 1a–d). Finally, a slight increment in the number of NK but not NKT cells was found in eightless spleen (Fig. 1d) and, compared with the wild‐type, eightless NK cells expressed substantially higher Ly49C/I levels (Fig. 1e).
Figure 1.
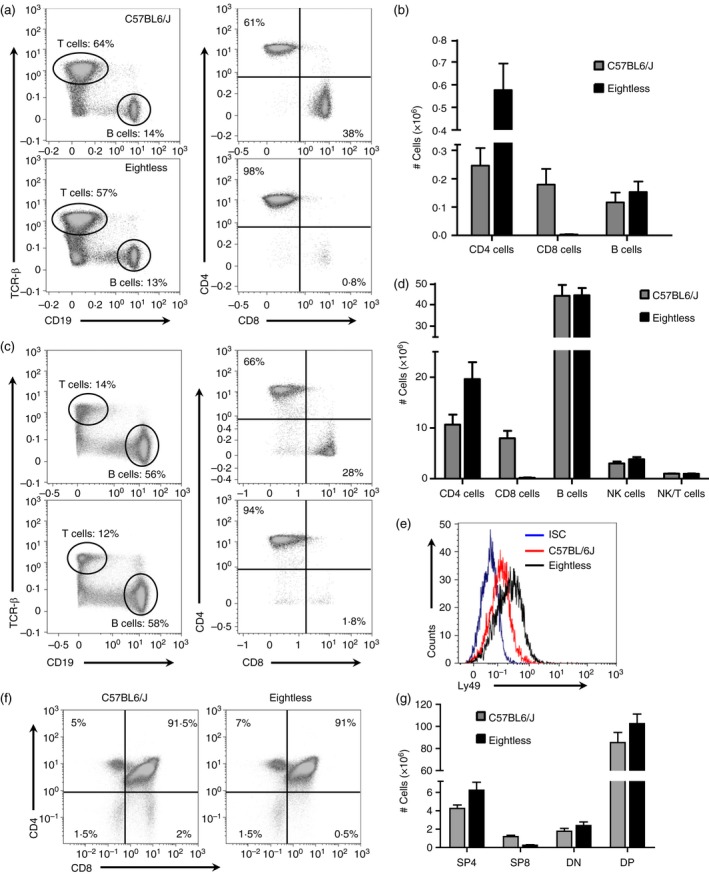
Diminished CD8+ T cells in eightless mutant mice. (a). Flow cytometry analysis of the expression of CD19 versus T cell receptor‐β (TCR‐β) by cells from two inguinal lymph nodes (left panels), and CD8 versus CD4 (right panels, gated on TCR‐β + cells), from wild‐type or eightless mice. Numbers in quadrants indicate per cent cells in each. (b) Graph presenting absolute numbers of CD4 T, CD8 T and B cells in the spleen of wild‐type and eightless mice. (c) Same as in (a) using cells from the spleen. (d) Absolute numbers of CD4 T, CD8 T, natural killer (NK), NKT and B cells in the spleens of wild‐type and eightless mice. (e) Histogram plot of Ly49 expression level on NK cells (gated on NK1.1+ TCR‐β − cells) from wild‐type (red line) or eightless (black line) mice. ISC‐isotype control (blue line). (f) Flow cytometry analysis of the expression of CD8 versus CD4 by thymocytes from wild‐type or eightless mice. (g) Absolute numbers of CD4 and CD8 T double‐negative, double‐positive and either CD4 or CD8 single‐positive thymocytes in the thymus of wild‐type and eightless mice. (n = 6 mice, a two tailed unpaired t‐test was used for statistical analysis). Scale bars represent ± SEM. [Colour figure can be viewed at wileyonlinelibrary.com]
Eightless mice lack MHC‐I cell surface expression
Reduced numbers of the CD8+ T cell population can be caused by either lack of CD8 expression or reduced MHC‐I expression on the cell surface. As CD8 expression was found to be normal on CD4 CD8 double‐positive thymocytes (Fig. 1e) we excluded CD8 protein expression as the cause for the eightless phenotype.
To test the involvement of MHC‐I expression in the phenotype, MHC‐I cell‐surface expression was detected in splenocytes by flow cytometry using an anti‐H‐2Kb antibody. Eightless mice were found to express substantially lower levels of MHC‐I than wild‐type mice, with levels comparable to the isotype‐matched control antibody (Fig. 2a,b).
Figure 2.
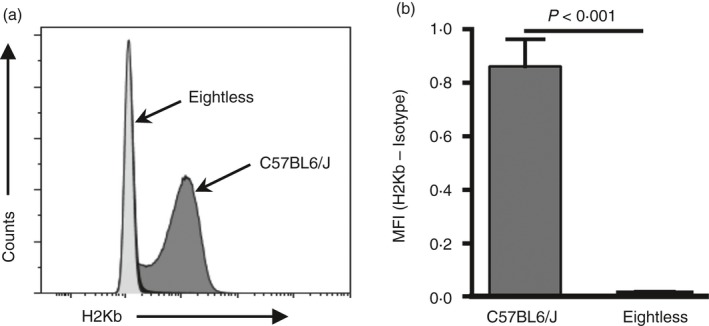
Eightless mice lack MHC‐I cell surface expression. (a) Representative histogram of MHC‐I expression (H2Kb) on the cell surface of total splenocytes from eightless (light grey) and wild‐type (dark grey) mice. (b) Graph summarizing the results presented in (a). Normalized values (H2Kb‐Isotype) of wild‐type and eightless splenocyte MHC‐I mean fluorescence intensity (MFI) (n = 6 mice, a two‐tailed unpaired t‐test was used for statistical analysis). Scale bars represent ± SEM.
In addition, as expected for C57BL/6J mice, H‐2Db molecules were not expressed (data not shown), though other non‐classical molecules that might depend on TAP transport, such as Qa or M3, were not tested.
Identification of the eightless mutation
Several genes have been reported to have a role in the cell surface expression of the MHC‐I complex. Among these genes are β2m, TAP1, TAP2 and the MCH‐I encoding genes themselves.
It is unlikely that the phenotype is caused by mutations in MHC‐I genes, as three separate mutations would be required to inactivate all three mouse MHC‐I genes (H‐2K, H‐2D and H‐2L). Since β2m, TAP1 and TAP2 proteins are all crucial for efficient MHC‐I complex formation, peptide loading and subsequent valid cell surface expression, we decided to sequence these genes. A single point mutation was detected in TAP2 consisting of a C to T transition of base 314 in exon 5 (Fig. 3a). This shift generated a glutamine to stop codon substitution at position 285 in the TAP2 protein (Fig. 3b).
Figure 3.
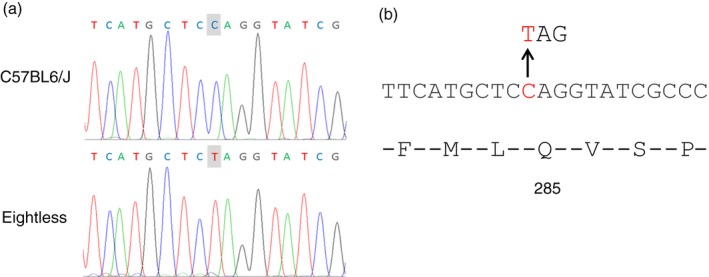
Identification of the eightless mutation. (a) Genomic sequence from part of the TAP2 gene reveals a C > T point mutation in eightless mice. (b) Diagram of the TAP2 amino acids and nucleotide sequences demonstrating the nonsense mutation in eightless leading to the conversion of glutamine in position 285 to a stop codon. [Colour figure can be viewed at wileyonlinelibrary.com]
Exogenous peptide or cell incubation at 26° rescues MHC‐I surface expression
As mutation in TAP2 severely affects the translocation of peptides into the ER, eightless mice clearly have a defect in the transport of antigenic peptides into the ER. Therefore, it could be expected that the rest of the MHC‐I antigen presentation pathway would be intact. TAP‐deficient cell lines exogenously incubated with MHC‐I‐binding peptides or incubated at 26° partially restored their surface MHC‐I expression.5, 28
Therefore, we next tested the ability of splenocytes from eightless mice cultured with an H‐2Kb‐binding ovalbumin‐derived peptide (SIINFEKL) to increase levels of surface H‐2Kb measured by flow cytometry. As predicted for cells with a sole defect in TAP activity, cell culture with the peptide partially restored MHC‐I expression in splenocytes from eightless mice (Fig. 4a), whereas MHC‐I expression was only minimally affected in wild‐type cells. Moreover, we found that the increase in expression was strongly dependent on the duration of incubation with the peptide, and less so on the dosage (Fig. 4a). The relevance of incubation time is emphasized by the evidence that during the time of our culture (3–9 hr), normal splenocytes tended to lose surface MHC‐I (Fig. 4b) whereas MHC‐I expression in eightless cells kept increasing (Fig. 4a). These data are compatible with the idea that several empty MHC‐I complexes are normally present on the surface of TAP2‐deficient cells but these constantly undergo rapid turn‐over unless stabilized by the addition of exogenous peptide. In fact, kinetic studies have shown that empty MHC‐I molecules are quickly internalized from the cell surface.29
Figure 4.
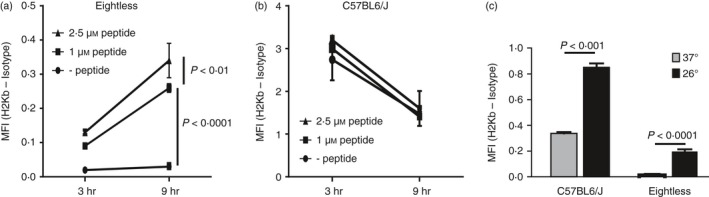
Exogenous peptide or incubation at 26° rescues eightless MHC‐I surface expression. (a, b) MHC‐I cell surface expression on splenocytes from eightless (a) or wild‐type (b) mice cultured with or without SIINFEKL peptide for 3 and 9 hr. (c) MHC‐I cell surface expression on splenocytes from eightless or wild‐type mice incubated overnight in vitro at 26° (black bars) or 37° (grey bars). (n = 3 mice, a two‐tailed unpaired t‐test was used for statistical analysis). Scale bars represent ± SEM.
The presence of empty MHC‐I complexes on the cellular surface was strongly enhanced by overnight incubation at 26° in cells derived from both wild‐type and eightless mice (Fig. 4c). The MHC‐I expression rescue we detected in the mutated samples implies that eightless cells are genuinely TAP defective. In fact it is known that some peptide‐receptive MHC‐I molecules commonly escape ER retention to reach the cell surface and this flux can be enhanced by lowering temperature.5 Incubation at 26° also enhanced surface expression of MHC‐I molecules in wild‐type splenocytes, but the proportional enhancement of Kb expression was less dramatic, most probably because these cells had higher baseline levels of pre‐existing cell surface Kb molecules.
Reduced residential peritoneal macrophages in eightless mice
Given that patients with TAP syndrome clinically present with symptoms typical of an innate immune defect and that the peritoneal cavity contains various immune cell populations crucial for the innate immune response, we decided to inspect the cellular composition of the peritoneum of eightless mice. Whereas the total number of CD45+ cells recovered from lavage was similar in wild‐type and eightless mice (data not shown), the proportion of the macrophage population out of total CD45+ cells in eightless mice was reduced (Fig. 5a–c) and this difference resulted in a much lower absolute number of macrophages in eightless compared with wild‐type mice (Fig. 5d). Also, residential peritoneal macrophage viability in eightless mice was slightly reduced (Fig. 5e,f).
Figure 5.
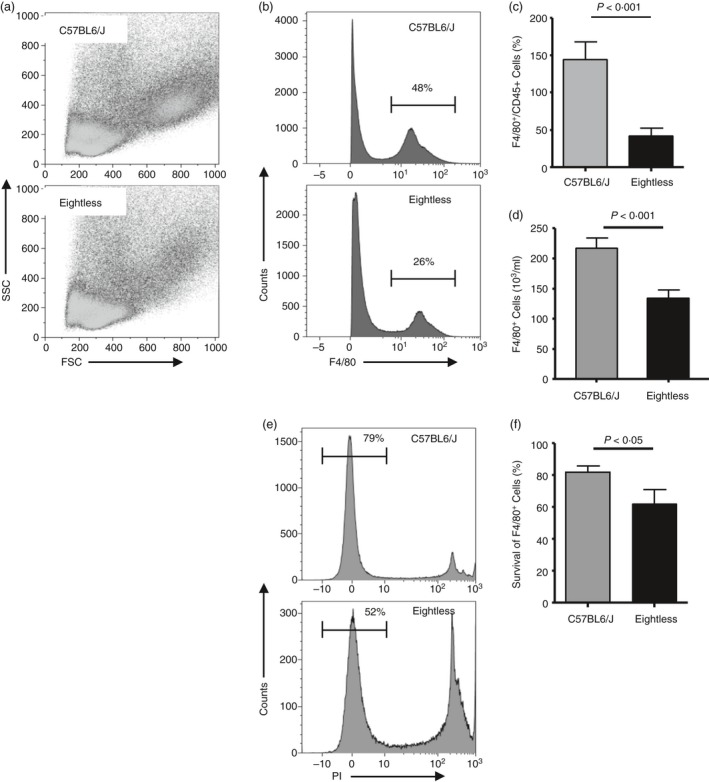
Reduced peritoneal resident macrophages in eightless mice. (a) Flow cytometry density plots of forward scatter versus side scatter of total residential immune cells (gated on CD45+ population) from the peritoneal cavity of wild‐type (upper panel) and eightless (lower panel) mice. (b) Representative histogram plots of F4/80 staining of peritoneal CD45+ cells from wild‐type (upper panel) or eightless (lower panel) mice. Numbers above bracketed lines indicate per cent macrophages. (c) Percentage of peritoneal resident macrophages in wild‐type (grey bar) or eightless (black bar) mice. (d) The absolute numbers of peritoneal resident macrophages in wild‐type (grey bar) or eightless (black bar) mice. (e) Representative histogram plots of propidium iodide staining of peritoneal‐resident macrophages from wild‐type (upper panel) or eightless (lower panel) mice. Numbers above bracketed lines indicate per cent of live macrophages. (f) Bar graph summarizing the data presented in (e). (c, d and f n = 10 mice, a two‐tailed unpaired t‐test was used for statistical analysis). Scale bars represent ± SEM.
Thioglycollate‐elicited eightless macrophages are recruited normally into the peritoneum but are short‐lived
As eightless mice have a reduced number of residential peritoneal macrophages, we decided to investigate whether this might be a consequence of reduced precursor recruitment or due to a defect in the mature macrophage itself. To distinguish between these two possibilities, we challenged mice with 1 ml intraperitoneal 4% thioglycollate and analysed their blood, bone marrow, spleen and peritoneal lavages at serial intervals from 36 hr up to 12 days (Fig. 6a–d). No significant differences in the number of recruited inflammatory monocytes and neutrophils were observed in the blood and spleen of eightless compared with wild‐type mice, at all time‐points tested (Fig. 6a,b). In addition, we observed no accumulation of monocyte and neutrophil precursors in the bone marrow of eightless mice (Fig. 6c). However, starting from around 4 days from thioglycollate injection, a significant reduction in both number and viability of macrophages was evident in the peritoneum cavity of eightless mice compared with that of wild‐type mice (Fig. 6d,e). This tendency was conserved throughout the successive time–course of analysis and by the 12th day, most eightless macrophages had already disappeared (Fig. 6d).
Figure 6.
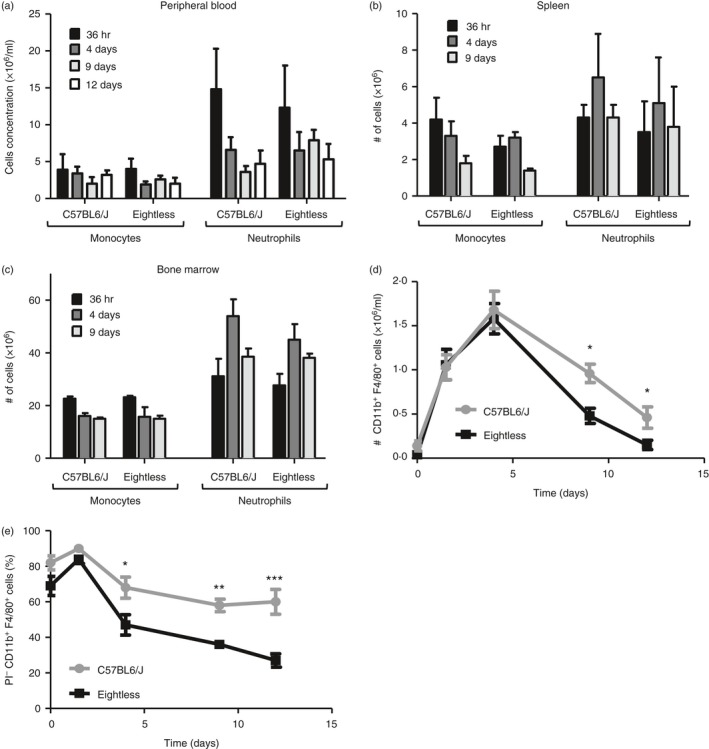
Thioglycollate‐elicited eightless macrophages are recruited normally into the peritoneum but are short‐lived. (a–c) Absolute numbers of neutrophils (CD11b+ Ly6Clow Ly6G+) and monocytes (CD11b+ Ly6Chi Ly6G−) in the peripheral blood (a), spleen (b) and bone marrow (c) of eightless or wild‐type mice, at different time‐points after thioglycollate injection (n = 3 mice, a two‐tailed unpaired t‐test was used for statistical analysis; none of the differences are statistically significant). Scale bars represent ± SEM. (d) Absolute number of macrophages (CD11b+ F4/80+) from mouse peritoneum at different time‐points after thioglycollate injection. Some differences between eightless and wild‐type are significant. (e) Percentages of live macrophages (propidium iodide‐negative, CD11b+ F4/80+) from mouse peritoneum cavity at different time‐points after thioglycollate injection. (e,f, n = 3 mice for each time‐point, a two‐tailed unpaired t‐test was used for statistical analysis). Scale bars represent ± SEM, *P < 0·05, **P < 0·01 and ***P < 0·001
These results suggest that following thioglycollate challenge, monocytes are recruited into the eightless peritoneum normally, and there they differentiate into macrophages. However, these cells seem to have a reduced survival capability.
Thioglycollate‐elicited eightless macrophages show reduced survival in vitro
To demonstrate that in eightless mice inflammatory macrophages that develop in the peritoneum following thioglycollate challenge are more susceptible to cell death, we cultured these cells and checked their survival over time in vitro. For this purpose we used macrophages collected 5 days after thioglycollate injection, as we noted that their number peaked on the fifth day and because up to the fourth day there was no evident difference in the number or viability of these cells (Fig. 6d,e). After 24 hr in culture, eightless macrophages (F4/80+ CD45+, Fig. 7a) had a lower forward‐scatter–side‐scatter profile compared with those from wild‐type mice (Fig. 7b). In addition, the percentage of eightless propidium iodide‐negative macrophages decreased after 24 hr in culture (Fig. 7d). We collected cells after either 24 or 48 hr of culture and we found no difference between these two time‐points (Fig. 7d). Therefore in vitro, some peritoneal macrophages die during the first 24 hr of culture whereas the remainder survive.
Figure 7.
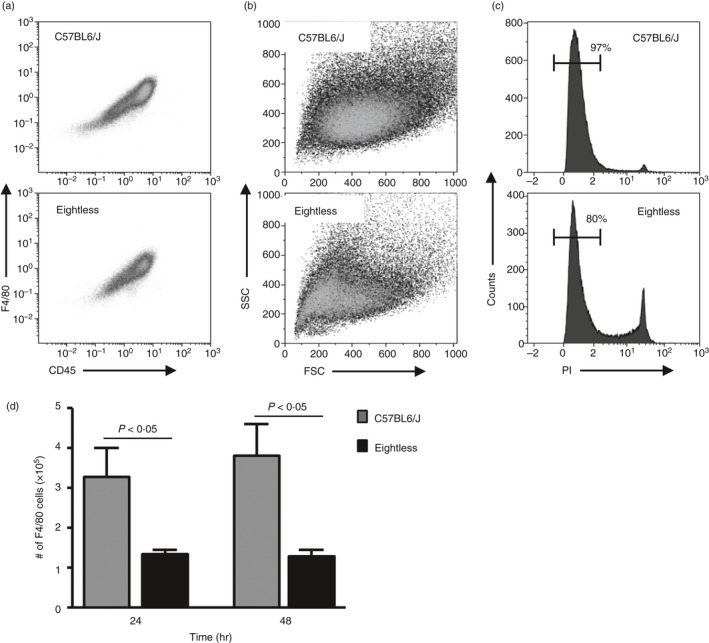
Thioglycollate‐elicited eightless macrophages show reduced survival in vitro. (a–c) Flow cytometric analysis of wild‐type or eightless peritoneal macrophages 5 days after thioglycollate that were incubated in vitro for 24 hr. (a) CD45 versus F4/80 staining. (b) Forward/side scatter profile of cells presented in (a). (c) propidium iodide staining of the cells presented in (a). Numbers above bracketed lines indicate per cent of live macrophages. (d) Graph bar summarizing the numbers of F4/80+ cells from peritoneal macrophages 5 days after thioglycollate that were incubated in vitro (0·5 × 105 per well) for 24 or 48 hr (n = 8 mice, a two‐tailed unpaired t‐test was used for statistical analysis). Scale bars represent ± SEM.
These data support our notion that during inflammation, most of the newly generated TAP‐deficient macrophages are defective and therefore die prematurely.
Thioglycollate‐elicited eightless macrophages show intact ER stress response
TAP deletion or mutation, severely affects the translocation of peptides into the ER and as a result MHC‐I molecules are retained within the ER. The accumulation of MHC‐I proteins in the ER raises the possibility that the death of macrophages observed in eightless mice is the result of an ER stress response. Specifically, the development of chronic, unresolved ER stress can lead to induction of cellular death, primarily through the activation of the transcription regulators, CHOP or XBP1s.30 Therefore to examine whether the ER stress response is activated in eightless macrophages, the expressions of ER stress genes and splicing of XBP1 mRNA were measured in peritoneal macrophages. For this, the peritoneal macrophages from wild‐type and eightless mice were tested under steady‐state conditions and upon Toll‐like receptor 4 activation by lipopolysaccharide treatment. To positively control the experiment, the expressions and splicing of XBP1 mRNA were also measured in tunicamycin‐treated macrophages from wild‐type mice. Interestingly, ER stress response genes were not found to be elevated in eightless macrophages in comparison with wild‐type cells (Fig. 8a–d). Similarly, the ratio between the spliced and unspliced transcripts of XBP1 was elevated in both wild‐type and eightless lipopolysaccharide‐treated macrophages to the same level (Fig. 2e). These results indicate that the ER stress response is not elevated in eightless macrophages and therefore suggest that chronic ER stress can be excluded as the mechanism governing eightless macrophage cellular death.
Figure 8.
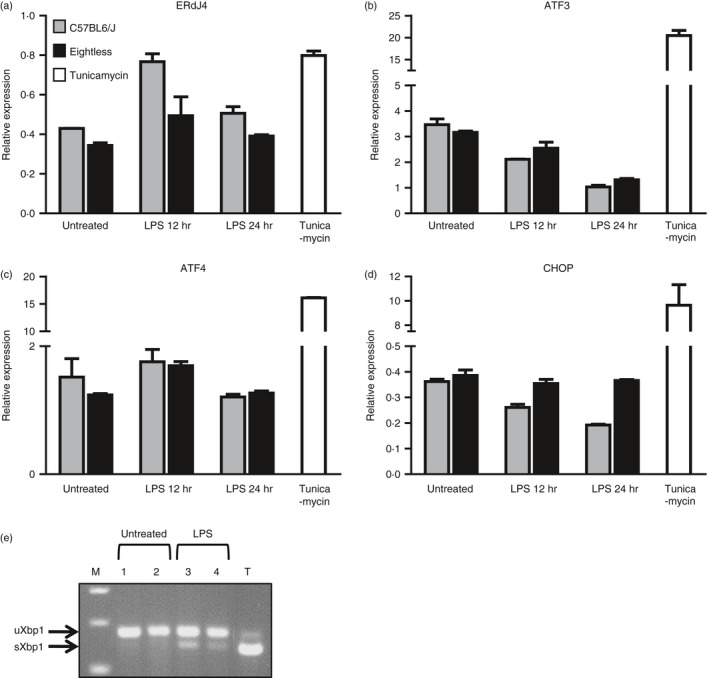
Thioglycollate‐elicited eightless macrophages show intact endoplasmic reticulum (ER) stress response. (a–d) RT‐PCR analysis for ERDJ4 (a), ATF3 (b), ATF4 (c) and CHOP (D) mRNA relative expression normalized to the UBC housekeeping gene from treated (12–24 hr) and untreated wild‐type (grey bar) or eightless (black bar) peritoneal macrophages (n = 3). Tunicamycin‐treated wild‐type peritoneal macrophages served as a positive control for ER stress (white bar). (e) Representative results of unspliced and spliced XbpI in wild‐type (lanes 1 and 3) or eightless (lanes 2 and 4) macrophages untreated or treated with lipopolysaccharide (12 hr). T‐Tunicamycin (4 hr) treated wild‐type peritoneal macrophages. M‐DNA marker.
Discussion
The lack of CD8+ T cells in eightless mice closely recapitulates the phenotype reported in other MHC‐I‐deficient mice.10, 11, 12, 31 Recently a chemically induced point mutation in TAP2 gene, jasmine, was generated. Mice homozygous for this mutation showed a severe reduction of the TAP2 protein expression.31 To our knowledge, jasmine is the only existing mouse model besides eightless harbouring a TAP2 defect. However, in jasmine TAP2 expression is only reduced, whereas in eightless this gene is fully disrupted. Therefore, an animal model harbouring a genuinely disrupted version of the TAP2 gene, such as eightless, should be extremely useful to address a series of unresolved questions regarding TAP complex functionality.
Our results confirm that, similarly to TAP1, TAP2 has a non‐redundant role in peptide loading of MHC‐I molecules indicating that the TAP complex truly works as a heterodimer. Although this confirms previous in vitro data, we cannot rule out that TAP1 and TAP2 might also have distinct functions as suggested by other in vitro studies.32, 33, 34, 35
In line with previous data, eightless mice were shown to have residual CD8+ T cells.10, 11, 31 In both TAP1−/− and β2m−/− mice, this limited population has the ability to recognize allo‐antigens in vitro, though the level of reactivity among the two strains is different.36 Several possible explanations have been proposed to clarify this phenomenon. The explanation most supported by both researchers and clinicians is that very limited amounts of MHC‐I molecules are expressed on the surface of TAP‐deficient cells, an idea supported by the detection of a polyclonal CD8+ T cell repertoire displaying both diversity and peptide specificity in TAP1−/− mice and in TAP2‐deficient patients.37, 38 However, the occurrence of a minor population of functional CD8+ T cells even in H‐2Kb−/−/Db−/− mice, animal models which by definition are completely deficient of classical MHC‐I molecules,39 would imply that there could be an additional developmental pathway entirely independent of the H‐2K and D locus. The different amount of CD8+ T cells in humans and mice can be easily explained by a more extensive exposure to pathogens in humans compared with laboratory animals. Nevertheless, the presence of a residual number of CD8+ T cells in mouse strains defective for MHC‐I expression is still not fully understood. Eightless cells were able to increase their MHC‐I expression on their surface following incubation with exogenous peptide in a timely dependent manner. In addition, a strong increase in Kb expression was detected after incubation of eightless splenocytes at 26°. These results support the notion that a limited amount of empty MHC‐I molecules is constantly present and continuously recruited on the eightless cell surface. Interestingly, TAP1−/− macrophages have been reported to express lower levels of pre‐existing, cell‐surface, peptide‐receptive MHC‐I molecules compared with wild‐type and these molecules also have a reduced half‐life, because they do not include complexes bound to easily dissociable and replaceable low‐affinity peptides.40 This scenario is also explained by the fact that empty and fully conformed MHC‐I complexes use distinct recycling routes resulting in a higher rate of open MHC‐I conformer internalization.41 Therefore TAP‐deficient cells would essentially harbour particularly unstable truly empty MHC‐I molecules that can be readily stabilized by encountering exogenous peptide, which might be sufficient to support CD8+ T cell survival and activation.
Patients affected by TAP syndrome usually suffer from recurrent bacterial infections of the upper respiratory tract such as chronic purulent rhinitis, sinusitis and otitis media during the first 6 years of life.20, 21, 22 Interestingly, necrotizing granulomatous lesions, which strictly resemble Wegener's granulomatosis, are often detectable in paranasal sinuses of these patients.18 Involvement of the lower respiratory tract typically manifests in the second decade of life with recurrent spastic bronchitis, bronchiolitis, bacterial pneumonia and eventually bronchiectasis leading to a clinical scenario that resembles chronic granulomatous disease. Severe viral infections are noticeably absent and normal titres of antibodies against viruses are present in the blood.19, 20, 21, 22 These clinical manifestations would imply a B cell or phagocyte rather than CD8+ T‐cell defect. As in TAP syndrome titres of antibodies towards common viruses and total immunoglobulin levels are typically normal, or even increased,20, 22 a humoral defect can be reasonably excluded. Here we showed that the number of in situ macrophages in eightless mice is reduced, and that during inflammation monocyte precursors are recruited normally into their peritoneum, where the newly generated macrophages die prematurely. These results indicate that the clinical symptoms of TAP2 deficiency can result from a macrophage survival defect. To our knowledge any specific alteration in the phagocyte cellular system in patients or in animal models of TAP deficiency has never been previously reported.
Nevertheless, the mechanisms behind the limited survival of these cells are unknown. Here we tested the possibility that the death of macrophages observed in eightless mice is mediated by unresolved ER stress response due to the accumulation of improperly conformed MHC‐I molecules in the ER. Our results indicate comparable ER stress responses in eightless and wild‐type macrophages, both under steady state and upon activation. Therefore, chronic ER stress can be excluded as the mechanism governing TAP‐deficient macrophages cellular death. Other possible explanations would be that the TAP complex directly supports macrophage survival, or that the cytosolic deposition and accumulation of peptides that cannot be transferred to the ER might be toxic to these cells.
To conclude, we report here for the first time the existence of a specific survival defect in TAP2‐deficient macrophages. In addition, we present a novel unique animal model for human TAP syndrome consisting of a TAP2 gene stop codon that results in disruption of this gene. Finally, this study implies that a better comprehension of TAP syndrome should include consideration of the innate branch of the immune system.
Disclosures
The authors declare no conflict of interest.
Acknowledgements
This work was supported by grants from the Israel Science Foundation grant No. 1275/12, Israel Cancer Research Fund grant No. 13/726/RCDA, Marie Curie People grant No. 322006 and Concern Foundation.
References
- 1. Touraine JL, Betuel H, Souillet G, Jeune M. Combined immunodeficiency disease associated with absence of cell‐surface HLA‐A and ‐B antigens. J Pediatr 1978; 93:47–51. [DOI] [PubMed] [Google Scholar]
- 2. Hulpke S, Tampe R. The MHC I loading complex: a multitasking machinery in adaptive immunity. Trends Biochem Sci 2013; 38:412–20. [DOI] [PubMed] [Google Scholar]
- 3. Abele R, Tampe R. Peptide trafficking and translocation across membranes in cellular signaling and self‐defense strategies. Curr Opin Cell Biol 2009; 21:508–15. [DOI] [PubMed] [Google Scholar]
- 4. Procko E, Gaudet R. Antigen processing and presentation: TAPping into ABC transporters. Curr Opin Immunol 2009; 21:84–91. [DOI] [PubMed] [Google Scholar]
- 5. Ljunggren HG, Stam NJ, Ohlen C, Neefjes JJ, Hoglund P et al Empty MHC class I molecules come out in the cold. Nature 1990; 346:476–80. [DOI] [PubMed] [Google Scholar]
- 6. Kisielow P, Teh HS, Bluthmann H, von Boehmer H. Positive selection of antigen‐specific T cells in thymus by restricting MHC molecules. Nature 1988; 335:730–3. [DOI] [PubMed] [Google Scholar]
- 7. Nesic D, Vukmanovic S. MHC class I is required for peripheral accumulation of CD8+ thymic emigrants. J Immunol 1998; 160:3705–12. [PubMed] [Google Scholar]
- 8. Williams MA, Bevan MJ. Effector and memory CTL differentiation. Annu Rev Immunol 2007; 25:171–92. [DOI] [PubMed] [Google Scholar]
- 9. Lanier LL, Phillips JH. Inhibitory MHC class I receptors on NK cells and T cells. Immunol Today 1996; 17:86–91. [DOI] [PubMed] [Google Scholar]
- 10. Koller BH, Marrack P, Kappler JW, Smithies O. Normal development of mice deficient in β2M, MHC class I proteins, and CD8+ T cells. Science 1990; 248:1227–30. [DOI] [PubMed] [Google Scholar]
- 11. Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH et al β2‐microglobulin deficient mice lack CD4‐8+ cytolytic T cells. Nature 1990; 344:742–6. [DOI] [PubMed] [Google Scholar]
- 12. Van Kaer L, Ashton‐Rickardt PG, Ploegh HL, Tonegawa S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4‐8+ T cells. Cell 1992; 71:1205–14. [DOI] [PubMed] [Google Scholar]
- 13. Joncker NT, Shifrin N, Delebecque F, Raulet DH. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med 2010; 207:2065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elliott JM, Wahle JA, Yokoyama WM. MHC class I‐deficient natural killer cells acquire a licensed phenotype after transfer into an MHC class I‐sufficient environment. J Exp Med 2010; 207:2073–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lamouse‐Smith E, Clements VK, Ostrand‐Rosenberg S. β2M−/− knockout mice contain low levels of CD8+ cytotoxic T lymphocyte that mediate specific tumor rejection. J Immunol 1993; 151:6283–90. [PubMed] [Google Scholar]
- 16. Glas R, Ohlen C, Hoglund P, Karre K. The CD8+ T cell repertoire in beta 2‐microglobulin‐deficient mice is biased towards reactivity against self‐major histocompatibility class I. J Exp Med 1994; 179:661–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Aldrich CJ, Ljunggren HG, Van Kaer L, Ashton‐Rickardt PG, Tonegawa S et al Positive selection of self‐ and alloreactive CD8+ T cells in Tap‐1 mutant mice. Proc Natl Acad Sci USA 1994; 91:6525–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moins‐Teisserenc HT, Gadola SD, Cella M, Dunbar PR, Exley A et al Association of a syndrome resembling Wegener's granulomatosis with low surface expression of HLA class‐I molecules. Lancet 1999; 354:1598–603. [DOI] [PubMed] [Google Scholar]
- 19. Donato L, de la Salle H, Hanau D, Tongio MM, Oswald M et al Association of HLA class I antigen deficiency related to a TAP2 gene mutation with familial bronchiectasis. J Pediatr 1995; 127:895–900. [DOI] [PubMed] [Google Scholar]
- 20. Gadola SD, Moins‐Teisserenc HT, Trowsdale J, Gross WL, Cerundolo V. TAP deficiency syndrome. Clin Exp Immunol 2000; 121:173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. de la Salle H, Hanau D, Fricker D, Urlacher A, Kelly A et al Homozygous human TAP peptide transporter mutation in HLA class I deficiency. Science 1994; 265:237–41. [DOI] [PubMed] [Google Scholar]
- 22. Zimmer J, Andres E, Donato L, Hanau D, Hentges F et al Clinical and immunological aspects of HLA class I deficiency. QJM 2005; 98:719–27. [DOI] [PubMed] [Google Scholar]
- 23. de la Salle H, Saulquin X, Mansour I, Klayme S, Fricker D et al Asymptomatic deficiency in the peptide transporter associated to antigen processing (TAP). Clin Exp Immunol 2002; 128:525–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zimmer J, Donato L, Hanau D, Cazenave JP, Tongio MM et al Activity and phenotype of natural killer cells in peptide transporter (TAP)‐deficient patients (type I bare lymphocyte syndrome). J Exp Med 1998; 187:117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shrestha D, Szollosi J, Jenei A. Bare lymphocyte syndrome: an opportunity to discover our immune system. Immunol Lett 2012; 141:147–57. [DOI] [PubMed] [Google Scholar]
- 26. Zimmer J, Andres E. Comments on type I bare lymphocyte syndrome. Immunol Lett 2012; 143:218–9. [DOI] [PubMed] [Google Scholar]
- 27. Uzi D, Barda L, Scaiewicz V, Mills M, Mueller T et al CHOP is a critical regulator of acetaminophen‐induced hepatotoxicity. J Hepatol 2013; 59:495–503. [DOI] [PubMed] [Google Scholar]
- 28. Schumacher TN, Heemels MT, Neefjes JJ, Kast WM, Melief CJ et al Direct binding of peptide to empty MHC class I molecules on intact cells and in vitro. Cell 1990; 62:563–7. [DOI] [PubMed] [Google Scholar]
- 29. Zagorac GB, Mahmutefendic H, Tomas MI, Kucic N, Le Bouteiller P et al Early endosomal rerouting of major histocompatibility class I conformers. J Cell Physiol 2012; 227:2953–64. [DOI] [PubMed] [Google Scholar]
- 30. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep 2006; 7:880–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Theodoratos A, Whittle B, Enders A, Tscharke DC, Roots CM et al Mouse strains with point mutations in TAP1 and TAP2. Immunol Cell Biol 2010; 88:72–8. [DOI] [PubMed] [Google Scholar]
- 32. Leonhardt RM, Keusekotten K, Bekpen C, Knittler MR. Critical role for the tapasin‐docking site of TAP2 in the functional integrity of the MHC class I‐peptide‐loading complex. J Immunol 2005; 175:5104–14. [DOI] [PubMed] [Google Scholar]
- 33. Gabathuler R, Reid G, Kolaitis G, Driscoll J, Jefferies WA. Comparison of cell lines deficient in antigen presentation reveals a functional role for TAP‐1 alone in antigen processing. J Exp Med 1994; 180:1415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Powis SJ. Major histocompatibility complex class I molecules interact with both subunits of the transporter associated with antigen processing, TAP1 and TAP2. Eur J Immunol 1997; 27:2744–7. [DOI] [PubMed] [Google Scholar]
- 35. Antoniou AN, Ford S, Pilley ES, Blake N, Powis SJ. Interactions formed by individually expressed TAP1 and TAP2 polypeptide subunits. Immunology 2002; 106:182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ljunggren HG, Van Kaer L, Ashton‐Rickardt PG, Tonegawa S, Ploegh HL. Differential reactivity of residual CD8+ T lymphocytes in TAP1 and β2‐microglobulin mutant mice. Eur J Immunol 1995; 25:174–8. [DOI] [PubMed] [Google Scholar]
- 37. Sandberg JK, Chambers BJ, Van Kaer L, Karre K, Ljunggren HG. TAP1‐deficient mice select a CD8+ T cell repertoire that displays both diversity and peptide specificity. Eur J Immunol 1996; 26:288–93. [DOI] [PubMed] [Google Scholar]
- 38. Bachmann MF, Oxenius A, Pircher H, Hengartner H, Ashton‐Richardt PA et al TAP1‐independent loading of class I molecules by exogenous viral proteins. Eur J Immunol 1995; 25:1739–43. [DOI] [PubMed] [Google Scholar]
- 39. Vugmeyster Y, Glas R, Perarnau B, Lemonnier FA, Eisen H et al Major histocompatibility complex (MHC) class I KbDb−/− deficient mice possess functional CD8+ T cells and natural killer cells. Proc Natl Acad Sci USA 1998; 95:12492–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Song R, Porgador A, Harding CV. Peptide‐receptive class I major histocompatibility complex molecules on TAP‐deficient and wild‐type cells and their roles in the processing of exogenous antigens. Immunology 1999; 97:316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mahmutefendic H, Zagorac GB, Tomas MI, Groettrup M, Momburg F et al Endosomal trafficking of open Major Histocompatibility Class I conformers – implications for presentation of endocytosed antigens. Mol Immunol 2013; 55:149–52. [DOI] [PubMed] [Google Scholar]