

Summary
The inflammatory response to the colonic pathogen Clostridium difficile is characterized by the induction of inflammatory cytokines including Interleukin‐23 (IL‐23) and interferon‐γ (IFN‐γ) and the recruitment of myeloid cells including Ly6CH igh monocytes. IL‐23 knockout mice showed reduced expression of the monocyte chemokines Ccl4 and Ccl7, but not Ccl2, as well as reduced Ly6CH igh Ly6GM id monocyte recruitment to the colon in response to C. difficile colitis. Clostridium difficile‐infected CCR2−/− (CCR2 KO) mice showed a significant defect in Ly6CH igh Ly6GM id monocyte recruitment to the colon in response to C. difficile. Although there was no decrease in expression of the inflammatory cytokines Il1b, Il6 or Tnf or reduction in the severity of colonic histopathology associated with ablation of monocyte recruitment, Slpi and Inos expression was significantly reduced in the colons of these animals. Additionally, neutralization of IFN‐γ through the administration of anti‐IFN‐γ monoclonal antibody resulted in a significant reduction in the expression of the IFN‐γ‐inducible chemokines Cxcl9 and Cxcl10, but not a reduction in the neutrophil chemokines Cxcl1, Cxcl2 and Ccl3 or the monocyte chemokine Ccl2. Consistently, monocyte and neutrophil recruitment were unchanged following anti‐IFN‐γ treatment. Additionally, Inos and Slpi expression were unchanged following anti‐IFN‐γ treatment, suggesting that Inos and Slpi regulation is independent of IFN‐γ during C. difficile colitis. Taken together, these data strongly suggest that IL‐23 and CCR2 signalling are required for monocyte recruitment during C. difficile colitis. Additionally, these studies also suggest that monocytes, but not IFN‐γ, are necessary for full expression of Inos and Slpi in the colon.
Keywords: bacterial, cytokines, inflammation, mucosa
Introduction
Monocyte‐derived cells are key mediators of inflammatory responses in the gastrointestinal tract.1 Monocyte recruitment is often associated with the development of inflammation and epithelial damage at mucosal sites.2, 3, 4, 5, 6, 7, 8, 9 Monocytes and macrophages contribute to tumour necrosis factor‐α (TNF‐α) production during both pulmonary and colonic inflammation.6, 7 Monocyte recruitment is also required for full production of inflammatory cytokines including interleukin‐1β (IL‐1β) and IL‐6, during dextran sulphate sodium (DSS) colitis.9 Furthermore, the development of intestinal histopathology during DSS colitis is partially dependent on monocyte recruitment.8, 9
Clostridium difficile infection in mice results in innate large bowel inflammation, characterized by increased inflammatory cytokine expression, marked histopathology, and rapid, robust recruitment of innate immune cells including monocytes to the large bowel.2, 3, 4, 5, 10, 11, 12, 13, 14, 15, 16, 17, 18 MyD88 signalling is crucial for monocyte recruitment to the large intestine in response to C. difficile colitis, and CCR2‐deficient mice show a significant defect in monocyte recruitment when challenged with C. difficile.10 Studies suggest that monocyte recruitment to the large intestine is not required for host survival during C. difficile colitis,10 but the role of recruited monocytes in promoting inflammatory cytokine expression and epithelial damage, as well as the host signals driving monocyte recruitment during C. difficile colitis, remain poorly understood.
Interferon‐γ (IFN‐γ) is a potent mediator of innate inflammation at mucosal sites, including in the gastrointestinal tract.19, 20, 21 Interferon‐γ signalling is required for full recruitment of neutrophils and production of CXCL1 in response to Streptococcus pneumoniae infection in the lung.20 Interferon‐γ is also required for CCL2 production and neutrophil recruitment to the colon during chemically induced colitis.19 Furthermore, recruited neutrophils produce IFN‐γ in response to both C. difficile 15 and Salmonella typhimurium 22 infection. Specific to the host response to C. difficile, Ishida et al. reported reduced TNF‐α and CXCL1 expression in IFN‐γ knockout (KO) mice following administration of C. difficile Toxin‐A to ligated ileal loops.21 Consistently, numerous studies from our own laboratory have reported increased IFN‐γ expression in the colonic mucosa in response to C. difficile infection.5, 11, 23 Additionally, a recent study by Abt et al. has suggested a critical role for IFN‐γ‐producing type I innate lymphoid cells in mediating host survival during C. difficile colitis.24 However, the role of IFN‐γ in modulating innate inflammatory responses, especially myeloid cell recruitment and inflammatory cytokine and chemokine expression in response to infection with metabolically active C. difficile, is largely unknown.
Interleukin‐23 is a known driver of innate inflammation at mucosal sites.25, 26 It drives inflammatory myeloid cell recruitment to the lung in response to chemical27 and microbial28 challenge. Additionally, IL‐23 is required for the recruitment of inflammatory monocytes to the spleen in response to Listeria monocytogenes infection.29 Furthermore, CD11bHigh myeloid cell recruitment is markedly ablated during chemically induced colitis in the absence of IL‐23 signalling.25 Work by Buonomo et al. has suggested a clear role for IL‐23 in promoting severe outcomes during experimental C. difficile infection,18 and recent studies from our laboratory have demonstrated a role for IL‐23 in driving neutrophil recruitment and contributing to colonic histopathology during C. difficile infection.26 However, the role of IL‐23 in driving the recruitment of other myeloid populations during C. difficile colitis, including monocytes, is poorly understood.
In the current study, our initial goal was to determine the role of IL‐23 in driving Ly6CHigh Ly6GMid monocyte recruitment to the large intestine during C. difficile colitis. Having demonstrated reduced monocyte recruitment in IL‐23 KO mice, we next sought to determine if ablation of CCR2‐dependent monocyte recruitment alone was sufficient to ameliorate the severity of colonic inflammatory gene expression or colonic histopathology. Additionally, recent studies have suggested a role for IFN‐γ in mediating host protection during C. difficile infection,24 but the role of IFN‐γ in driving innate inflammatory responses to C. difficile colitis remains poorly understood. As such, we investigated the role of IFN‐γ signalling in driving neutrophil and monocyte recruitment, inflammatory cytokine expression, and colonic histopathology during acute C. difficile colitis in mice. Collectively, these studies reveal the role of IL‐23 in driving monocyte recruitment during C. difficile infection, as well as identify the roles of CCR2‐dependent monocyte recruitment and IFN‐γ in driving inflammatory cytokine expression, colonic histopathology, and inflammatory myeloid cell recruitment during acute C. difficile colitis.
Materials and methods
Animals and housing
C57BL/6 male mice aged 5–12 weeks from a colony maintained at the University of Michigan founded by Jackson breeders were used in the current study. Male and female CCR2−/− (CCR2KO) and p19−/− (IL‐23KO) mice on a C57BL/6 background aged 5–14 weeks were used in the current study. Both CCR2KO and IL‐23KO mice were obtained from in‐house breeding colonies at the University of Michigan. All mice were permitted autoclaved water and food ad libitum, and were maintained under specific pathogen‐free conditions with autoclaved bedding. All animal manipulations were performed in a laminar flow hood, and all experiments were performed in accordance with a protocol approved by the University Commission on the Use and Care of Animals at the University of Michigan.
Bacterial culture and growth conditions
Clostridium difficile spores were prepared for infection as previously described.11, 26 Briefly, an existing spore stock of C. difficile strain VPI 10463 was plated on Taurocholate Cefoxitin Cycloserine Fructose Agar and cultured overnight to generate vegetative cells. An individual colony was used to inoculate an overnight culture in Columbia broth. This overnight culture (2 ml) was then used to inoculate 40 ml of Clospore30 sporulation medium, and the culture was then allowed to grow for 7 days at 37° anaerobically. Spores were recovered by washing the resulting pellet at least four times to remove residual vegetative cells. Stocks were stored at 4° in water until use.
Quantification of C. difficile colonization
Mucosal C. difficile colonization was assessed as described previously4, 11, 31, 32 using a C. difficile‐specific PCR of DNA isolated from host colonic tissue. Reaction volumes, cycling conditions, and primer and probe sequences are identical to those used previously.4, 31, 32 Raw Ct values were normalized to a single copy per genome host gene used as an internal control to generate dCt values.32, 33 Normalized dCt values were then converted to ‘C. difficile genomes per gram of host tissue’ using a standard curve developed using known quantities of host tissue and vegetative C. difficile.
Antibiotic treatment and infection
For all experiments, animals were given cefoperazone (Sigma, St Louis, MO) at a concentration of 0·5 g/l in their drinking water for 5 days as described previously.11, 13, 23, 26 After the antibiotic treatment, mice were permitted a 2‐day recovery period on regular drinking water before infection with C. difficile. Untreated animals received neither C. difficile challenge nor antibiotic pretreatment.
For C. difficile infection studies, mice received approximately 106 colony‐forming units of VPI 10463 spores by oral gavage on Day 0. Infected animals were monitored for any sign of undue stress including lethargy, hunched posture and weight loss exceeding 20% of baseline body weight, and any moribund animals were humanely killed. All experimental samples were collected on Day 2.
Neutralizing antibody
In order to neutralize IFN‐γ in vivo, mice were given 500 μg of anti‐IFN‐γ monoclonal antibody (clone XMG.1.2) via intraperitoneal injection 1 day before and 1 day after infection (Day –1 and Day 1, respectively).
Colonic leucocyte isolation
Leucocytes were isolated from colonic tissue as described previously,4, 5, 23, 26, 34 with certain modifications. Briefly, colonic tissue was excised and physically disrupted using serrated scissors. Minced tissue was then incubated in 20 ml of Hanks’ balanced salt solution (HBSS) supplemented with 1 mm dithiothreitol, 5 mm EDTA, and 2·5% fetal calf serum for 20 min at 37°. Tissue was then washed, and subsequently incubated with 20 ml of HBSS supplemented with 0·5 mg/ml DNase (Roche, Basel, Switzerland), 400 U/ml collagenase type 3 (Worthington Biochemicals, Lakewood, NJ), and 2·5% fetal calf serum for 1 hr at 37°. After washing, samples were then resuspended in 20% Percol (Sigma) in PBS and spun at 900 g for 30 min without brake. The resulting single cell suspension was stained for surface marker expression by flow cytometry.
Flow cytometry staining and analysis
Flow cytometry staining was performed as described previously.4, 5, 23, 26 Briefly, cells were plated at a concentration of approximately 106 cells per well in a 96‐well plate, and were blocked with unlabelled FcRIII/II. After blocking, cells were stained with fluorescently labelled antibodies for 30 min at 4°. Cells were washed, and resuspended in stabilizing fixative (BD Biosciences, Franklin Lakes, NJ). All samples were acquired on a three‐laser FACSCanto II using FACS‐diva software (BD Biosciences, Franklin Lakes, NJ). All data analysis was performed in flowjo (Treestar, Ashland, OR). Cells were stained with the following antibody clones: CD45 (clone 30‐F11), CD11c (clone HL3), CD11b (clone M1/70), Ly6C (clone AL‐21), and Ly6G (clone 1A8). All antibodies were purchased from BD Biosciences or Biolegend (San Diego, CA).
For calculating number of CD11bHigh CD11cLow cells per 100 000 events, the frequency of CD45+ events was multiplied by the frequency of CD11bHigh CD11cLow events, and the resulting number was multiplied by 100 000.
For calculating the number of Ly6CHigh Ly6GMid monocytes or Ly6CMid Ly6GHigh neutrophils per 100 000 events, the frequency of CD45+ events was multiplied by the frequency of CD11bHigh CD11cLow events, and the frequency of either Ly6CHigh Ly6GMid monocytes or Ly6CMid Ly6GHigh neutrophils. The resulting number was then multiplied by 100 000..
The gating strategy for identification and enumeration of colonic leucocyte subsets is also highlighted in the Supplementary material (Fig. S1).
Preparation and examination of colonic histological sections
Excised colonic tissue was prepared for histological analysis as described previously.4, 5, 11, 23, 26 Colonic tissue was fixed in 10% formalin for a minimum of 24 hr and then transferred to 70% ethanol. Cassettes were processed, paraffin‐embedded, sectioned and used to prepare haematoxylin and eosin stained slides by McClinchey Histology Lab. Inc. (Stockbridge, MI).
Representative photomicrographs were acquired using an Olympus BX40 light microscope (Olympus corporation, Tokyo, Japan) using a QImaging MicroPublisher RTV 5.0 5 megapixel camera. All photomicrographs were acquired at a total magnification of 400×. qcapture suite PLUS version 3.1.3.10 (QImaging , Surrey, BC, Canada) was used for image acquisition. All panels were assembled in adobe photoshop CS5, version 12.0 (Adobe Systems Incorporated, San Jose, CA 95110‐2704). Processing of images was restricted to global adjustments of brightness, contrast and image size for each photomicrograph.
RNA isolation and expression analysis
RNA was purified from colonic tissue and gene expression was assessed as described previously.4, 5, 11, 23, 26 Briefly, samples of colonic tissue approximately 1 cm2 were excised from the midpoint of the colon and stored in RNAlater (Ambion, Austin, TX) for further analysis. To isolate RNA, samples were homogenized in TRIzol reagent (Life Technologies, Grand Island, NY), and RNA was purified using the RNeasy Mini Kit (Ambion) according to the manufacturers’ instructions. Purified RNA was assessed for concentration and purity using a nanodrop instrument (Thermo Fisher, Waltham, MA) and Agilent Bioanalyzer (Agilent, Santa Clara, CA), respectively. RNA was converted to cDNA using the RT2 first strand kit (Qiagen, Hilden, Germany) and colonic gene expression was assessed using RT2 Profiler PCR Assays (Qiagen). All PCR were run on a Roche LightCycler 480. For RT2 Profiler PCR Assays, cross card normalization was performed to control for card‐to‐card variability.35 ∆Ct (dCt) values were calculated by subtracting the mean Ct value of two internal control genes from the gene in question.33, 36
Statistical analysis
Statistically significant differences in gene expression were determined by performing a one‐way analysis of variance (anova) with Tukey's post‐hoc test on normalized dCt values.4, 11, 26 Statistically significant difference in colonic C. difficile colonization were likewise identified by performing a one‐way anova with Tukey's post‐hoc test on normalized dCt values.4 A one‐way anova with Tukey's post‐hoc test was also used to identify statistically significant differences in the number of particular cellular subsets per 100 000 cells. For all analyses, statistical significance was set at P ≤ 0·05.
Results
Effect of IL‐23 deficiency on colonic monocyte recruitment
To investigate the role of IL‐23 in promoting monocyte recruitment during C. difficile colitis, wild‐type (WT) and IL‐23KO mice were infected with C. difficile as described previously.26 Briefly, mice were given 0·5 g/l cefoperazone in their drinking water for 5 days and after a 2‐day recovery period on regular water were infected with spores of the C. difficile strain VPI 10463 by oral gavage. The mice were followed for an additional 2 days, at which point the infection was terminated and all samples were collected.
RT‐PCR analysis was used to examine the effect of IL‐23 deficiency on the expression of known monocyte‐recruiting chemokines in the colon following C. difficile infection. The absence of IL‐23 was associated with significantly reduced expression of Ccl4 and Ccl7, but not Ccl2, in the colonic mucosa in response to C. difficile infection (Fig. 1a). The absence of IL‐23 did not affect the C. difficile burden within the colon (see Supplementary material, Fig. S2). Flow cytometric analysis of colonic CD45+ leucocytes revealed a significant reduction in the frequency of CD11bHigh CD11cLow myeloid cells recruited to the colon in IL‐23KO mice compared with WT animals (Fig. 1b). Subsequent analysis of the CD11bHigh CD11cLow population demonstrated that IL‐23 deficiency was also associated with a significant defect in the recruitment of Ly6CHigh Ly6GMid monocytes (Fig. 1c). Taken together, these data suggest that IL‐23 promotes monocyte recruitment and the expression of monocyte‐recruiting chemokines in the colon during C. difficile colitis.
Figure 1.
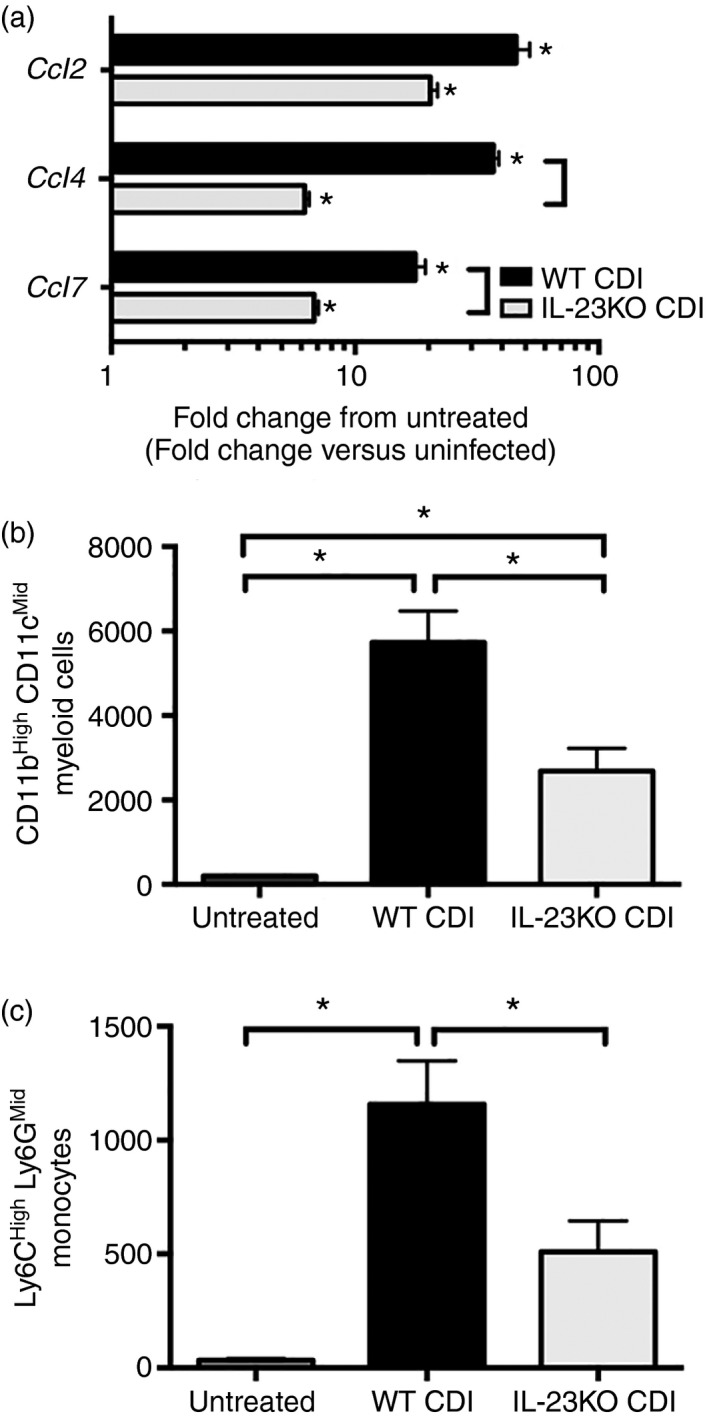
Colonic monocyte recruitment and colonic chemokine expression during Clostridium difficile infection in interleukin‐23 (IL‐23) ‐deficient mice. (a) Colonic chemokine expression as assessed by quantitative PCR. n ≥ 6 per group. Data are shown as fold change in gene expression in the indicated group compared with untreated wild‐type (WT) animals. Black bars: WT C. difficile‐infected (CDI), white bars: IL‐23 knockout (KO) CDI. *P < 0·05 for the difference between the indicated group and untreated WT animals. Square brackets indicate P < 0·05 for the differences in expression levels between the indicated groups. (b) Number of CD11bHigh CD11cLow and (c) number of Ly6CH igh Ly6GM id cells per 100 000 cells. Bars represent mean ± SEM number of the indicated cell type per 100 000 cells. n = 8 for all groups. *P < 0·05 for the differences between the indicated groups.
Colonic monocyte recruitment during C. difficile colitis in CCR2‐deficient mice
To further investigate the role of recruited monocytes during C. difficile colitis, CCR2−/− (CCR2KO) mice were infected as described in the Materials and methods. Clostridium difficile infection in WT animals was associated with significant recruitment of CD11bHigh CD11cLow myeloid cells (Fig. 2a centre panel and Fig. 3a). This CD11bHigh CD11cLow population contained both Ly6CHigh Ly6GMid monocytes and Ly6CMid Ly6GHigh neutrophils, and the frequency of both these populations was significantly increased above baseline following C. difficile infection (Fig. 2b centre panel and Fig. 3b,c). Whereas neither CD11bHigh CD11cLow myeloid cell nor Ly6CMid Ly6GHigh neutrophil recruitment were significantly altered in CCR2KO mice (Fig. 2a,b right column and Fig. 3a,c), Ly6CHigh Ly6GMid monocyte recruitment was significantly abrogated in CCR2KO mice infected with C. difficile (Fig. 2b right panel, Fig. 3b). There was no difference in the frequency of total CD45+ leucocytes between groups (data not shown). These data indicate that Ly6CHigh Ly6GMid monocyte recruitment, but not CD11bHigh CD11cLow myeloid cell or Ly6CMid Ly6GHigh neutrophil recruitment, is significantly reduced during C. difficile colitis in the absence of CCR2 signalling.
Figure 2.
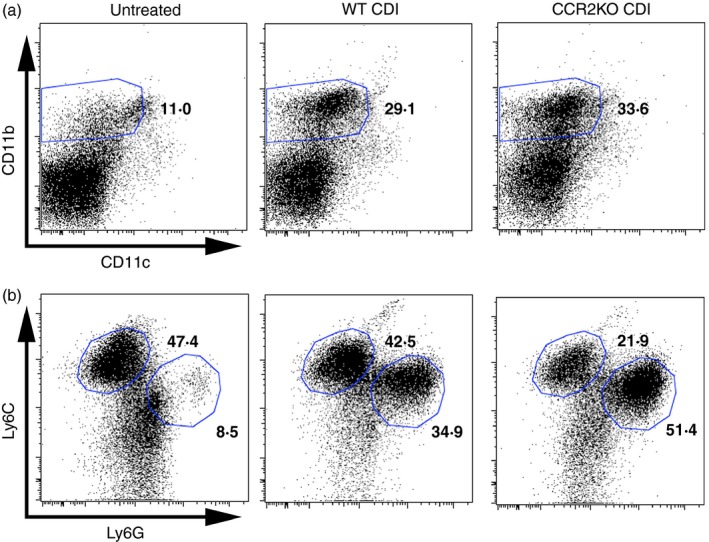
Flow cytometric analysis of colonic CD45+ leucocytes from untreated, wild‐type Clostridium difficile‐infected (WT CDI), and CCR2 knockout (KO) CDI mice. (a) Analysis of CD11b and CD11c expression profiles on isolated colonic CD45+ leucocytes. (b) Analysis of Ly6C and Ly6G expression profiles on the CD11bHigh CD11cLow population as defined in (a). The number in bold indicates the percentage of the parent population contained within the indicated gate. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 3.
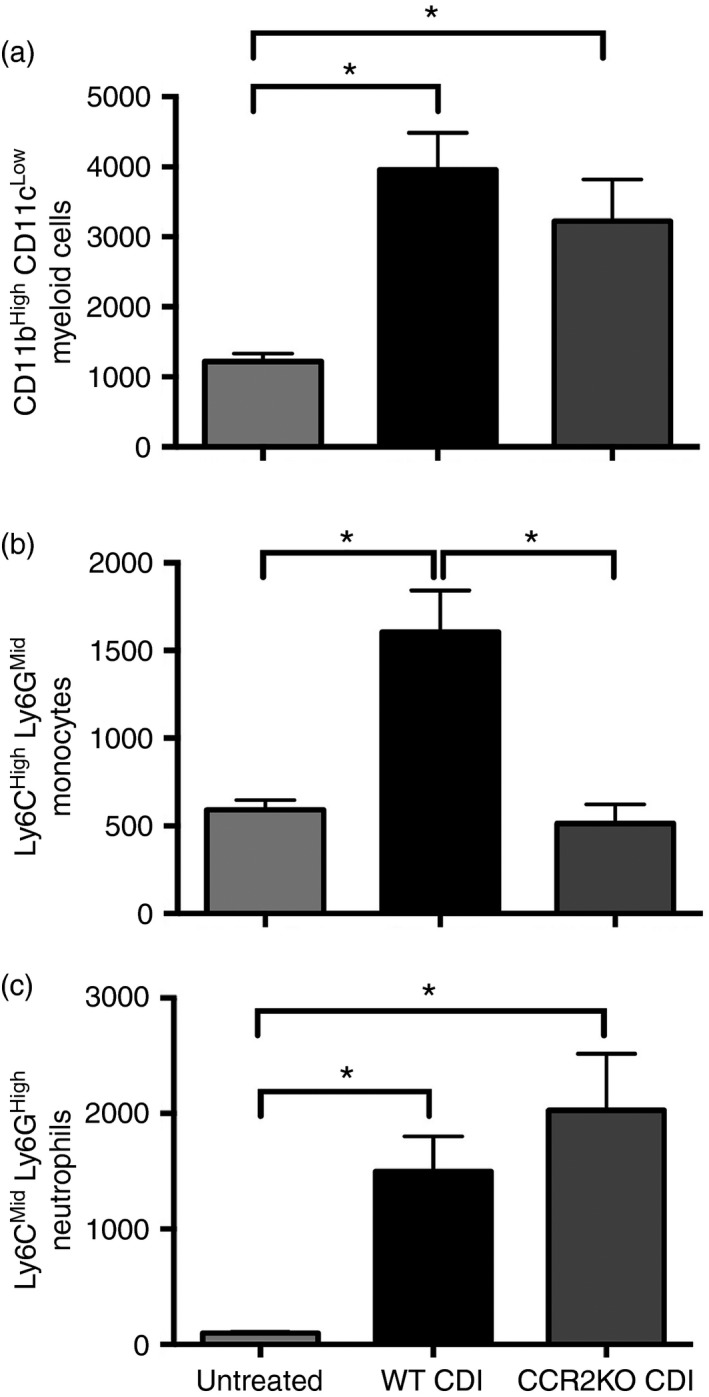
Quantification of colonic myeloid cell populations from Untreated, wild‐type Clostridium difficile‐infected (WT CDI), and CCR2 knockout (KO) CDI mice as defined in Fig. 2. (a) Number of CD11bHigh CD11cLow, (b) Ly6CH igh Ly6GM id and (c) Ly6CM id Ly6GH igh cells per 100 000 cells. All populations were defined as shown in Fig. 2. Bars represent mean ± SEM number of the indicated cell type per 100 000 cells. n ≥ 6 per group. CDI = C. difficile‐infected. *P < 0·05 for the differences between the indicated groups.
Effect of CCR2 deficiency on inflammatory cytokine and chemokine expression
RT‐PCR analysis was used to examine the effect of CCR2 deficiency on colonic inflammatory cytokine and chemokine expression in response to C. difficile colitis. Consistent with the significant influx of neutrophils observed in these animals (Figs 2b and 3c), C. difficile infection in WT mice was associated with increased expression of the neutrophil chemokines Ccl3, Cxcl1 and Cxcl2 (Fig. 4a). Expression of the inflammatory cytokines Il1b, Il6, Il17f and Tnf were all significantly increased in WT C. difficile infected animals (Fig. 4b). Arg1, Slpi and Inos were also significantly induced during C. difficile colitis (Fig. 4c).
Figure 4.
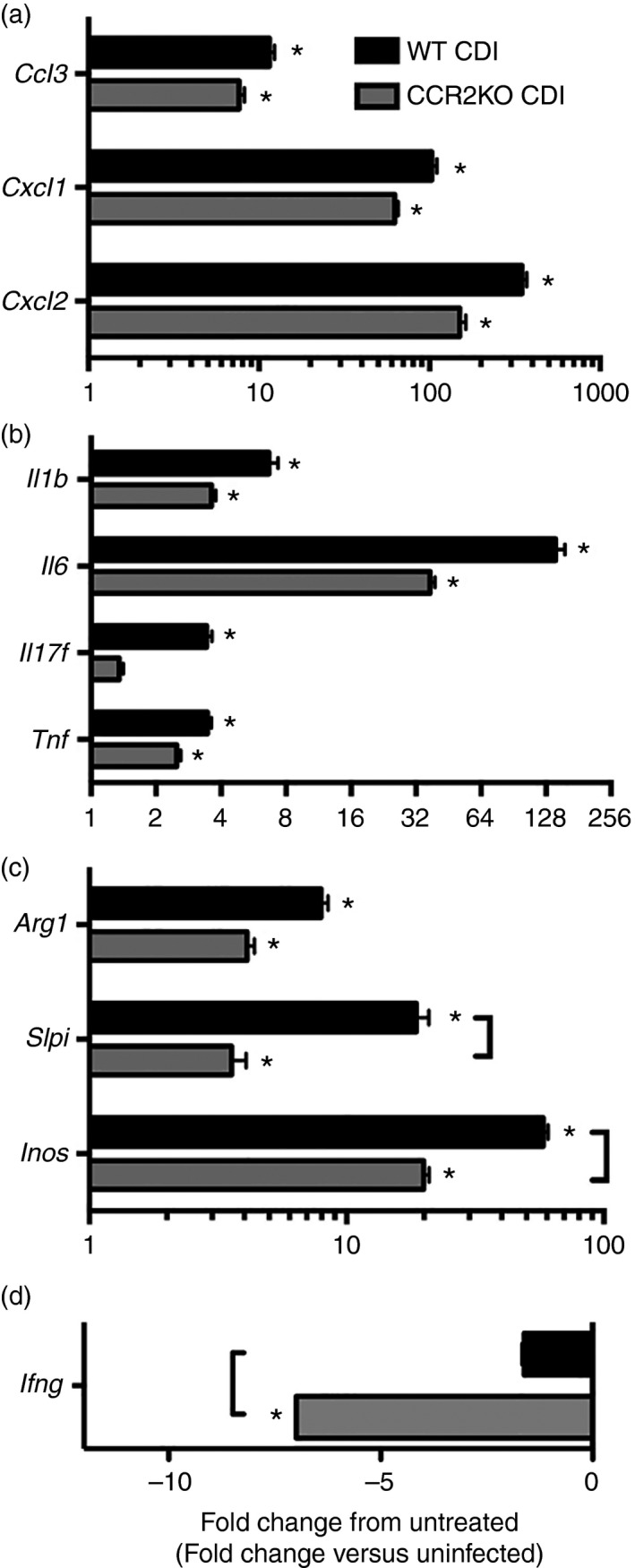
Inflammatory cytokine expression in wild‐type (WT) and CCR2 knockout (KO) mice infected with Clostridium difficile. Expression of inflammatory cytokine genes in the colon was assessed via quantitative PCR as outlined in the Materials and methods. Data are shown as mean ± SEM fold change gene expression in the indicated group compared with untreated WT animals. n ≥ 7 per group. Black bars: wild‐type C. difficile‐infected (WT CDI, grey bars: CCR2KO CDI. Brackets P < 0·05 for the differences in expression levels between the indicated groups. *P < 0·05 for the difference between the indicated group and untreated WT animals.
CCR2 deficiency did not result in any defects in the induction of the myeloid cell chemokines Ccl3, Cxcl1 or Cxcl2, (Fig. 4a). Furthermore, CCR2‐deficient animals displayed no reduction in Il1b, Il6, Tnf or Arg1 expression (Fig. 4b,c). The expression levels of Slpi and Inos, however, were significantly reduced in CCR2KO mice compared with WT animals infected with C. difficile (Fig. 4c). Consistent with the gene expression, we observed no reduction in the severity of colonic histopathology (see Supplementary material, Fig. S3, left and middle columns) or levels of C. difficile colonization in CCR2KO animals (see Supplementary material, Fig. S2). Additionally, the absence of CCR2 was associated with a significant decrease in ifng expression within the colonic mucosa in response to C. difficile infection (Fig. 4d). Collectively, these data suggest that while CCR2 signalling is not required for the induction of inflammatory mediators including Cxcl1, Cxcl2, Il1b and Il6, the full expression of Inos and Arg1 is dependent on CCR2 signalling during C. difficile colitis.
Effect of anti‐IFN‐γ treatment on myeloid cell recruitment and inflammatory cytokine expression within the colon
To investigate the role of IFN‐γ in promoting monocyte and neutrophil recruitment, as well as inflammatory cytokine and chemokine expression during C. difficile colitis, animals were treated with anti‐IFN‐γ monoclonal antibody 1 day before and 1 day after infection. As in previous experiments, all samples were collected 2 days after infection.
Analysis of CD45+ colonic leucocytes revealed no defect in CD11bHigh CD11cLow myeloid cell recruitment in anti‐IFN‐γ‐treated animals (Fig. 5a). Further analysis of Ly6C and Ly6G expression within the CD11bHigh CD11cLow myeloid cell population revealed equivalent levels of Ly6CHigh Ly6GMid monocyte and Ly6CMid Ly6GHigh neutrophil recruitment following anti‐IFN‐γ treatment (Fig. 5b). Taken together, these data suggest that the recruitment of inflammatory myeloid cells, including Ly6CHigh Ly6GMid monocytes and Ly6CMid Ly6GHigh neutrophils is independent of IFN‐γ signalling during acute C. difficile colitis.
Figure 5.
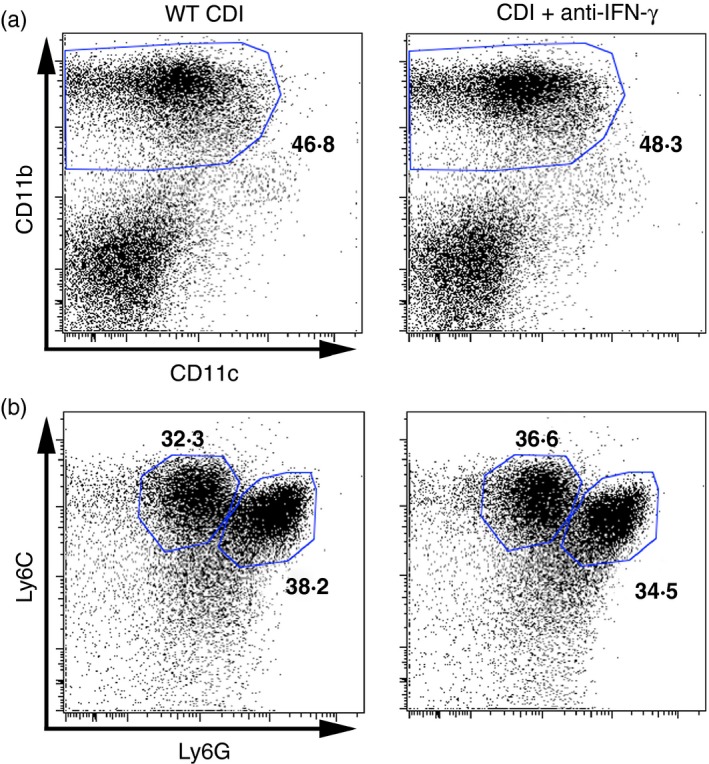
Flow cytometric analysis of colonic CD45+ leucocytes from wild‐type Clostridium difficile‐infected (WT CDI), and CDI+ anti‐interferon‐γ (IFN‐γ) ‐treated mice. (a) Analysis of CD11b and CD11c expression profiles on isolated colonic CD45+ leucocytes. (b) Analysis of Ly6C and Ly6G expression profiles on the CD11bHigh CD11cLow population as defined in (a). The number in bold indicates the percentage of the parent population contained within the indicated gate. [Colour figure can be viewed at wileyonlinelibrary.com]
Colonic inflammatory cytokine and chemokine expression following anti‐IFN‐γ treatment was also examined using RT‐PCR. Anti‐IFN‐γ treatment was associated with significantly reduced expression of the IFN‐γ‐inducible chemokines Cxcl9 and Cxcl10 in response to C. difficile infection (Fig. 6a). However, in agreement with the high level of monocyte and neutrophil recruitment seen in these animals, anti‐IFN‐γ treatment did not ablate expression of Ccl3, Cxcl1, Cxcl2, Ccl2 or Ccl4 (Fig. 6b,c). Consistently, the expression of inflammatory cytokines including Il6 was not reduced in anti‐IFN‐γ‐treated animals (Fig. 6d). Additionally, we observed no change in C. difficile burden or reduction in the severity of colonic histopathology following ablation of IFN‐γ (see Supplementary material, Figs S2 and S3).
Figure 6.
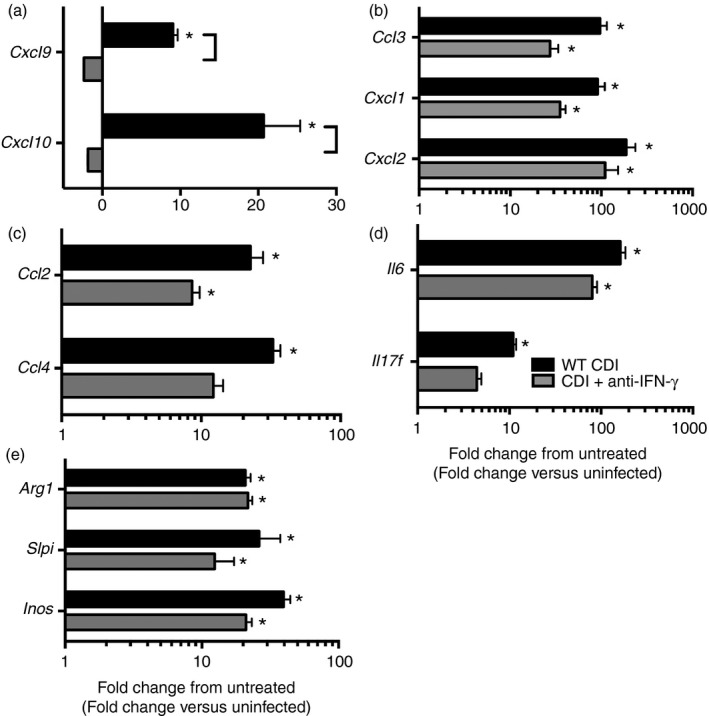
Effect of anti‐interferon‐γ (IFN‐γ) treatment on colonic inflammatory cytokine and chemokine expression. Data are shown as mean ± SEM fold change gene expression in the indicated group compared with untreated wild‐type (WT) animals. n ≥ 4 per group. CDI = Clostridium difficile‐infected. Black bars: WT CDI, light grey bars: CDI+ anti‐IFN‐γ. Brackets P < 0·05 for the differences in expression levels between the indicated groups. *P < 0·05 for the difference between the indicated group and untreated WT animals.
Furthermore, induction of Arg1 and Slpi was also unchanged following anti‐IFN‐γ treatment (Fig. 6e). Surprisingly, Inos expression was independent of anti‐IFN‐γ treatment as well (Fig. 6e). These data indicate that IFN‐γ is not a major driver of inflammatory cytokine and chemokine expression within the colon during acute C. difficile infection.
Discussion
In the current study, we reported a significant reduction in both CD11bHigh CD11cLow myeloid cell and Ly6CHigh Ly6GMid monocyte recruitment to the colon, as well as significant defects in the induction of the monocyte chemokines Ccl4 and Ccl7, within the colonic mucosa of IL‐23KO mice. Additionally, we observed significantly reduced monocyte recruitment in CCR2‐deficient mice infected with C. difficile. Despite the drastic reduction in monocyte recruitment, the induction of inflammatory cytokines and chemokines including Il1b, Il6, Cxcl1 and Cxcl2 were not significantly altered in CCR2‐deficient animals. Furthermore, CCR2KO mice were not protected from the development of severe intestinal histopathology during C. difficile colitis. Collectively, these data strongly suggest that IL‐23 signalling promotes Ly6CHigh Ly6GMid monocyte recruitment to the colon in response to C. difficile colitis, but that the monocytes themselves are not major drivers of inflammatory cytokine expression or intestinal histopathology during C. difficile colitis.
The reduced Ly6CHigh Ly6GMid monocyte influx seen in CCR2KO mice infected with C. difficile was associated with significantly reduced expression of Inos and Slpi in the colon. Inducible nitric oxide synthase production by CD11bHigh monocyte/macrophages has been previously reported in the lungs following lipopolysaccharide‐mediated pulmonary inflammation.37 Ly6CHigh monocytes recruited to the spleen following L. monocytogenes infection also produce inducible nitric oxide synthase.29 Furthermore, several in vitro studies have reported Slpi induction in macrophages following stimulation with microbial products including lipopolysaccharide38 and heat‐killed Mycobacterium tuberculosis.39 Consistent with these in vitro studies we observed decreased Inos and Slpi expression in CRR2‐deficient mice infected with C. difficile. However, Inos and Slpi expression were not affected by anti‐IFN‐γ treatment. These data suggest that Ly6CHigh Ly6GMid monocytes are a major source of IFN‐γ‐independent Inos and Slpi expression in the colon in response to C. difficile colitis.
We observed no reduction in inflammatory cytokine or chemokine expression in CCR2KO mice during C. difficile colitis, although Ly6CHigh Ly6GMid monocyte recruitment was significantly reduced in these animals. Reduction of macrophage/monocyte recruitment during bleomycin‐mediated pulmonary inflammation is associated with reduced TNF‐α production in the lungs.6 Likewise, TNF‐α + monocyte/macrophages are recruited to the colon in a CCR2‐dependent manner during DSS colitis.7 Furthermore, a recent study has suggested that Ly6CHigh monocytes are largely responsible for colonic IL‐6, IL‐1β and IFN‐γ production during DSS colitis.9 Specific to C. difficile, stimulation of monocytes with C. difficile toxins in vitro results in rapid production of inflammatory cytokines including IL‐8, IL‐1β and TNF‐α.40, 41 However, in the current study there was no reduction in inflammatory cytokine expression in CCR2‐deficient mice infected with C. difficile, suggesting that Ly6CHigh Ly6GMid monocytes are not a major source of inflammatory cytokine expression in the colon during acute C. difficile colitis.
Despite significantly reduced levels of Ly6CHigh Ly6GMid monocyte influx, CCR2KO mice were not protected against the development of severe colonic inflammation and histopathology in response to C. difficile colitis. Numerous studies have reported reduced intestinal histopathology during DSS colitis following interference with CCR2‐dependent monocyte influx.8, 9 Furthermore, robust monocyte/macrophage recruitment has been previously reported during C. difficile infection in association with marked intestinal histopathology.2, 3, 4, 5, 10 A previous study from our group also reported no decrease in the severity of intestinal histopathology following the depletion of both monocyte and neutrophil populations by anti‐Gr‐1 monoclonal antibody treatment.4 In the current study we report no protection from robust intestinal histopathology in CCR2KO mice. Hence, the data presented here suggest that Ly6CHigh Ly6GMid monocytes are not major drivers of intestinal histopathology during acute C. difficile colitis.
We report no reduction in monocyte and neutrophil recruitment or decreased inflammatory cytokine expression following anti‐IFN‐γ treatment. IFN‐γ drives neutrophil recruitment in response to insult at mucosal sites,19, 20, 21 and IFN‐γ + neutrophil influx has recently been reported in response to Salmonella typhimurium typhlocolitis22 as well as C. difficile colitis.15 Specific to the colon, neutrophil recruitment and CCL2 production during DSS colitis are both dependent upon IFN‐γ.19 Additionally, CXCL1 expression and neutrophil recruitment to the ileum were blunted in response to C. difficile toxin A in IFN‐γKO mice.21 Furthermore, a recent study has highlighted the crucial role of type 1 innate lymphoid cells for host survival during experimental C. difficile infection.24 However, in the current study, we observed no defect in myeloid cell recruitment or expression of Il6 and Il17f following anti‐IFN‐γ treatment, suggesting that innate IFN‐γ protects the host from mortality through other mechanisms.
One potential interpretation of the data presented in the current study is that IFN‐γ is largely redundant in the host response to C. difficile colitis. However, a recent study by Abt et al. has reported a marked defect in the induction of IFN‐γ in association with decreased survival in mice lacking Tbet+ type 1 innate lymphoid cells cells following challenge with C. difficile.24 Additionally, Rag−/− IFN‐γ −/− mice also demonstrated a large increase in mortality, strongly suggesting that innate IFN‐γ signalling is critical for host survival in response to C. difficile infection.24 Collectively, these data suggest that IFN‐γ is protective during C. difficile colitis via mechanisms independent of inflammatory cytokine expression and granulocyte recruitment.
We observed significantly reduced CD11bHigh CD11cLow myeloid cell and Ly6CHigh Ly6GMid monocyte recruitment to the colon during C. difficile colitis in IL‐23‐deficient mice. Interleukin‐23 has been previously demonstrated to drive TNFα + inflammatory monocyte recruitment to the spleen in response to Listeria monocytogenes infection.29 Specific to colonic inflammation, IL‐23 signalling promotes CD11bHigh myeloid cell recruitment during chemically induced colitis.25 We report significantly reduced Ly6CHigh Ly6GMid monocyte recruitment and expression of the monocyte chemotactic factors Ccl4 and Ccl7 in IL‐23‐deficient mice during C. difficile colitis. Hence, our data strongly suggest that IL‐23 signalling drives monocyte recruitment to the colon in response to C. difficile infection.
One model which explains our observations is that IL‐23 promotes the expression of the monocyte chemokines Ccl4 and Ccl7, but not Ccl2, contributing to the recruitment of Ly6CHigh Ly6GMid monocytes to the colon. These recruited monocytes are not required for the induction of inflammatory cytokines and chemokines including Cxcl1, Cxcl2 and Il1b or the development of severe intestinal histopathology, but are required for full expression of Slpi and Inos within the colon.
A previous study from our laboratory reported reduced neutrophil recruitment and inflammatory cytokine and chemokine expression in the absence of IL‐23 signalling,26 whereas an additional study noted no decrease in the severity of intestinal histopathology or inflammatory cytokine expression following the concomitant depletion of monocytes and neutrophils.4 Taken together, these studies strongly suggest that (i) IL‐23 is required for the recruitment of numerous myeloid populations including monocytes and neutrophils during C. difficile colitis, (ii) monocytes and neutrophils are not major drivers of intestinal histopathology or inflammatory cytokine expression during C. difficile infection, and (iii) the reduced intestinal histopathology and inflammatory cytokine production seen in IL‐23KO mice is independent of the reduced neutrophil and monocyte recruitment in these animals. Hence, IL‐23‐responsive host cells other than recruited myeloid populations are the main sources of inflammatory cytokine expression in the colon during C. difficile colitis.
Recent studies have reported a protective role of innate IFN‐γ 24 and a therapeutic benefit for enhanced eosinophil recruitment17 in response to C. difficile infection, strongly suggesting a protective role for Type 1 and Type 2 responses, respectively. The role for Type 17 responses, and specifically the role of the Type 17‐promoting cytokine IL‐23, remains more difficult to define. Interleukin‐23 has been implicated in driving severe intestinal histopathology and mortality during C. difficile infection,18, 26 suggesting that interference with IL‐23 signalling would be of therapeutic benefit to patients with clinical C. difficile infection. However, neutrophil recruitment, a host response repeatedly demonstrated to be critical for survival during C. difficile infection,3, 10 is also significantly reduced in the absence of IL‐23 signalling,26 As such, interference with IL‐23 signalling may ultimately worsen disease by impairing protective neutrophil recruitment, in addition to increasing the susceptibility of the patient to other infections. Therefore, any immunomodulatory treatment for C. difficile infection, either a biologically or microbiota‐based method, should seek to diminish the pathological aspects of IL‐23 signalling while still retaining robust neutrophil recruitment. Future studies will focus on better understanding the inflammatory cascades driven by IL‐23, with the ultimate goals of identifying IL‐23‐dependent host signals that promote histopathology and disease development but not neutrophil recruitment.
Author contributions
AJM and GBH conceived, designed and interpreted the experiments. VBY, NRF and RAM contributed to their design and interpretation. AJM, NRF, RAM, CRF and CRP performed the experiments. AJM, NRF, CRF and GBH analysed the data. AJM and GBH prepared the manuscript, and all other authors provided comments and advice on the manuscript.
Disclosures
The authors declare no financial conflicts of interest.
Supporting information
Figure S1. Gating strategy for flow cytometric analysis of colonic leucocytes.
Figure S2. Colonic mucosal Clostridium difficile colonization as determined by C. difficile‐specific quantitative PCR (2 days post‐infection).
Figure S3. Photomicrographs of representative haematoxylin and eosin‐stained colonic sections from Clostridium difficile‐infected wild‐type (WT) (left column), C. difficile infected CCR2 knockout (KO; centre column), and C. difficile‐infected mice treated with anti‐interferon‐γ.
Acknowledgements
We thank the University of Michigan Flow Cytometry Core for their invaluable assistance performing flow cytometry experiments. We thank Dr Merritt Gillilland III for critical review of this manuscript and Dr John Erb‐Downward for helpful discussions. This work was supported by National Institutes of Health grants U19 AI090871 (GBH and VBY) and P30 DK034933 (GBH and VBY). Support was also provided by the Host‐Microbiome Initiative (HMI) of the University of Michigan Medical School (GBH and VBY).
References
- 1. McDermott AJ, Huffnagle GB. The microbiome and regulation of mucosal immunity. Immunology 2014; 142:24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hasegawa M, Kamada N, Jiao Y, Liu MZ, Nunez G, Inohara N. Protective role of commensals against Clostridium difficile infection via an IL‐1β‐mediated positive‐feedback loop. J Immunol 2012; 189:3085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G et al Nucleotide‐binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 2011; 186:4872–80. [DOI] [PubMed] [Google Scholar]
- 4. McDermott AJ, Higdon KE, Muraglia R, Erb‐Downward JR, Falkowski NR, McDonald RA et al The role of Gr‐1+ cells and tumour necrosis factor‐α signalling during Clostridium difficile colitis in mice. Immunology 2015; 144:704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sadighi Akha AA, Theriot CM, Erb‐Downward JR, McDermott AJ, Falkowski NR, Tyra HM et al Acute infection of mice with Clostridium difficile leads to eIF2α phosphorylation and pro‐survival signalling as part of the mucosal inflammatory response. Immunology 2013; 140:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel WA, Kawasuji M et al C‐C chemokine receptor 2 (CCR2) deficiency improves bleomycin‐induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage‐derived matrix metalloproteinases. J Pathol 2004; 204:594–604. [DOI] [PubMed] [Google Scholar]
- 7. Platt AM, Bain CC, Bordon Y, Sester DP, Mowat AM. An independent subset of TLR expressing CCR2‐dependent macrophages promotes colonic inflammation. J Immunol 2010; 184:6843–54. [DOI] [PubMed] [Google Scholar]
- 8. Waddell A, Ahrens R, Steinbrecher K, Donovan B, Rothenberg ME, Munitz A et al Colonic eosinophilic inflammation in experimental colitis is mediated by Ly6Chigh CCR2+ inflammatory monocyte/macrophage‐derived CCL11. J Immunol 2011; 186:5993–6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zigmond E, Varol C, Farache J, Elmaliah E, Satpathy AT, Friedlander G et al Ly6Chi monocytes in the inflamed colon give rise to proinflammatory effector cells and migratory antigen‐presenting cells. Immunity 2012; 37:1076–90. [DOI] [PubMed] [Google Scholar]
- 10. Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88‐mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun 2012; 80:2989–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McDermott AJ, Frank CR, Falkowski NR, McDonald RA, Young VB, Huffnagle GB. Role of GM‐CSF in the inflammatory cytokine network that regulates neutrophil influx into the colonic mucosa during Clostridium difficile infection in mice. Gut Microbes 2014; 5:476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hasegawa M, Yada S, Liu MZ, Kamada N, Munoz‐Planillo R, Do N et al Interleukin‐22 regulates the complement system to promote resistance against pathobionts after pathogen‐induced intestinal damage. Immunity 2014; 41:620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. Cefoperazone‐treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2011; 2:326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN et al A mouse model of Clostridium difficile‐associated disease. Gastroenterology 2008; 135:1984–92. [DOI] [PubMed] [Google Scholar]
- 15. El‐Zaatari M, Chang YM, Zhang M, Franz M, Shreiner A, McDermott AJ et al Tryptophan catabolism restricts IFN‐γ‐expressing neutrophils and Clostridium difficile immunopathology. J Immunol 2014; 193:807–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A et al Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile‐induced colitis. Infect Immun 2012; 80:62–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Buonomo EL, Cowardin CA, Wilson MG, Saleh MM, Pramoonjago P, Petri WA Jr. Microbiota‐regulated IL‐25 increases eosinophil number to provide protection during Clostridium difficile infection. Cell Rep 2016; 16:432–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Buonomo EL, Madan R, Pramoonjago P, Li L, Okusa MD, Petri WA Jr. Role of interleukin 23 signaling in Clostridium difficile colitis. J Infect Dis 2013; 208:917–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ito R, Shin‐Ya M, Kishida T, Urano A, Takada R, Sakagami J et al Interferon‐γ is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol 2006; 146:330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rijneveld AW, Lauw FN, Schultz MJ, Florquin S, Te Velde AA, Speelman P et al The role of interferon‐γ in murine pneumococcal pneumonia. J Infect Dis 2002; 185:91–7. [DOI] [PubMed] [Google Scholar]
- 21. Ishida Y, Maegawa T, Kondo T, Kimura A, Iwakura Y, Nakamura S et al Essential involvement of IFN‐γ in Clostridium difficile toxin A‐induced enteritis. J Immunol 2004; 172:3018–25. [DOI] [PubMed] [Google Scholar]
- 22. Spees AM, Kingsbury DD, Wangdi T, Xavier MN, Tsolis RM, Baumler AJ. Neutrophils are a source of γ interferon during acute Salmonella enterica serovar Typhimurium colitis. Infect Immun 2014; 82:1692–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sadighi Akha AA, McDermott AJ, Theriot CM, Carlson PE Jr, Frank CR, McDonald RA et al Interleukin‐22 and CD160 play additive roles in the host mucosal response to Clostridium difficile infection in mice. Immunology 2015; 144:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Susac B et al Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microbe 2015; 18:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox JH, Kljavin NM, Ota N, Leonard J, Roose‐Girma M, Diehl L et al Opposing consequences of IL‐23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol 2012; 5:99–109. [DOI] [PubMed] [Google Scholar]
- 26. McDermott AJ, Falkowski NR, McDonald RA, Pandit CR, Young VB, Huffnagle GB. Interleukin‐23 (IL‐23), independent of IL‐17 and IL‐22, drives neutrophil recruitment and innate inflammation during Clostridium difficile colitis in mice. Immunology 2016; 147:114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, di Padova F et al IL‐1 and IL‐23 mediate early IL‐17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE 2011; 6:e23185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dubin PJ, Martz A, Eisenstatt JR, Fox MD, Logar A, Kolls JK. Interleukin‐23‐mediated inflammation in Pseudomonas aeruginosa pulmonary infection. Infect Immun 2012; 80:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Indramohan M, Sieve AN, Break TJ, Berg RE. Inflammatory monocyte recruitment is regulated by interleukin‐23 during systemic bacterial infection. Infect Immun 2012; 80:4099–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Perez J, Springthorpe VS, Sattar SA. Clospore: a liquid medium for producing high titers of semi‐purified spores of Clostridium difficile . J AOAC Int 2011; 94:618–26. [PubMed] [Google Scholar]
- 31. Luna RA, Boyanton BL Jr, Mehta S, Courtney EM, Webb CR, Revell PA et al Rapid stool‐based diagnosis of Clostridium difficile infection by real‐time PCR in a children's hospital. J Clin Microbiol 2011; 49:851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2011; 2:145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 2008; 3:1101–8. [DOI] [PubMed] [Google Scholar]
- 34. Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc 2007; 2:2307–11. [DOI] [PubMed] [Google Scholar]
- 35. Sadighi Akha AA, Harper JM, Salmon AB, Schroeder BA, Tyra HM, Rutkowski DT et al Heightened induction of proapoptotic signals in response to endoplasmic reticulum stress in primary fibroblasts from a mouse model of longevity. J Biol Chem 2011; 286:30344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A et al Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3: RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. D'Alessio FR, Tsushima K, Aggarwal NR, Mock JR, Eto Y, Garibaldi BT et al Resolution of experimental lung injury by monocyte‐derived inducible nitric oxide synthase. J Immunol 2012; 189:2234–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jin FY, Nathan C, Radzioch D, Ding A. Secretory leukocyte protease inhibitor: a macrophage product induced by and antagonistic to bacterial lipopolysaccharide. Cell 1997; 88:417–26. [DOI] [PubMed] [Google Scholar]
- 39. Ding A, Yu H, Yang J, Shi S, Ehrt S. Induction of macrophage‐derived SLPI by Mycobacterium tuberculosis depends on TLR2 but not MyD88. Immunology 2005; 116:381–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Linevsky JK, Pothoulakis C, Keates S, Warny M, Keates AC, Lamont JT et al IL‐8 release and neutrophil activation by Clostridium difficile toxin‐exposed human monocytes. Am J Physiol 1997; 273:G1333–40. [DOI] [PubMed] [Google Scholar]
- 41. Ng J, Hirota SA, Gross O, Li Y, Ulke‐Lemee A, Potentier MS et al Clostridium difficile toxin‐induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 2010; 139:542–52. e1‐3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Gating strategy for flow cytometric analysis of colonic leucocytes.
Figure S2. Colonic mucosal Clostridium difficile colonization as determined by C. difficile‐specific quantitative PCR (2 days post‐infection).
Figure S3. Photomicrographs of representative haematoxylin and eosin‐stained colonic sections from Clostridium difficile‐infected wild‐type (WT) (left column), C. difficile infected CCR2 knockout (KO; centre column), and C. difficile‐infected mice treated with anti‐interferon‐γ.