

Abstract
KRas is mutated in a significant number of human cancers and so there is an urgent therapeutic need to target KRas signaling. To target KRas in lung cancers we used a systems approach of integrating a genome-wide miRNA screen with patient-derived phospho-proteomic signatures of the KRas downstream pathway, and identified miR-193a-3p which directly targets KRas. Unique aspects of miR-193a-3p biology include two functionally independent target sites in the KRas 3′UTR and clinically significant correlation between miR-193a-3p and KRas expression in patients. Rescue experiments with mutated KRas 3′UTR showed very significantly that the anti-tumor effect of miR-193a-3p is via specific direct targeting of KRas and not due to other targets. Ex vivo and in vivo studies utilizing nanoliposome packaged miR-193a-3p demonstrated significant inhibition of tumor growth, circulating tumor cell viability, and decreased metastasis. These studies show the broader applicability of using miR-193a-3p as a therapeutic agent to target KRas-mutant cancer.
Keywords: miRNA, proteomics, lung cancer, KRas, metastasis
Introduction
The KRAS gene encodes for the small GTPase KRas, which functions to transmit signals through intracellular pathways, thereby regulating key cellular processes such as the cell cycle and proliferation (reviewed in(1)). Although it was demonstrated in 1983 that mutations in the KRAS gene can lead to oncogenic transformation of cells(2), and that approximately 30% of human malignancies contain mutations in a Ras family member (reviewed in(3)), targeted therapies to treat KRas-mutant cancers remain elusive. KRas-mutant tumors have been found to be resistant to many currently available therapies, including tyrosine kinase inhibitors(4–6), EGFR-directed monoclonal antibodies(7), paclitaxel(8) and radiation(9). It is, therefore, of critical importance to find novel methods of clinical intervention in KRas-mutant cancers. RNA interference represents one such potential technology(10). Recent clinical trials have included miRNA replacement therapy using a derivative of miR-34 in hepatocellular carcinoma (http://clinicaltrials.gov/ct2/show/NCT01829971), suggesting that miRNA-based therapy provides a promising strategy. While miRNA targeting KRas have been previously identified(11), here we show for the first time a miRNA (miR-193a-3p) that has two target sites on KRas which function independently of each other and neither of which have been shown to be mutated in cancers, unlike the Let-7a target sites(12). The effect of miR-193a-3p is directly mediated via targeting of KRas as demonstrated by rescue experiments with site directed mutants of the KRas 3′UTR. Additionally, we show using novel ex vivo and in vivo studies the high translational potential of nanoliposome packaged miR-193a-3p to target KRas tumors and inhibit both circulating tumor cells and metastasis, making these finding highly relevant to a broader biological and clinical use.
Recent advances in proteomic profiling have demonstrated the importance of changes in protein and phospho-protein levels in determining aberrant signaling events, and thereby novel therapeutic targets, in tumor cells(13). We therefore elected to isolate a phospho-protein signature associated with oncogenic Ras signaling, and sought to identify miRNA which would be able to act as therapeutic agents by inhibiting the signature. Using a screen of 879 miRNA by Reverse Phase Protein Array (RPPA), we identified miR-193a-3p as being capable of reversing this signature as well as being lost in the majority of lung cancer patients. Analysis of miR-193a-3p targets revealed 2 potential binding sites for the miR in the 3′UTR of KRas, suggesting that this miRNA reverses the signature by reducing levels of KRas protein. This decrease in KRas expression led to a reduction in cellular proliferation, G1 arrest and a reduction in colony formation in 3D cultures. Potential therapeutic use of miR-193a-3p was explored using a denuded ex vivo lung model as well as xenograft models, where we demonstrated that miR-193a-3p can reduce growth and metastasis of KRas-mutant lung cancers in vivo, making this a highly clinically relevant miRNA.
Results
miR-193a-3p reverses a KRas signature
KRas mutations occur in 10–30% of lung cancer patients. We identified a protein signature associated with signaling downstream of KRas by analysis of proteins correlated with KRas in lung cancer samples in the TCPA database and literature searches (Figure 1A). The resulting signature (referred to as “our signature”) included phosphorylated forms of members of the canonical MAPK pathway, as well as phospho-YAP1, which has previously been shown to be essential for tumorigenic progression in KRas-mutant pancreatic cancer(14), and CDK1, which has been shown to be regulated by KRas(15, 16). The workflow used to identify miRNA capable of reversing the signature is shown in supplementary figure 1. We initially analyzed lung cancer patient samples to determine miRNA which were significantly negatively correlated with our signature proteins (Figure 1B, red circle and Supp. Fig, 2). Additionally, we analyzed the same samples for miRNA which were negatively correlated with a previously published MAPK signature to identify miRNA which are robustly associated with KRas activity (Figure 1B, blue circle)(17). To further narrow down the number of candidate miRNA, we also determined whether expression of the miRNA was reduced in lung cancer relative to normal lung tissue (Figure 1B, orange circle). We previously published a global miRNA by RPPA screen, in which 879 miRNA were analyzed for their ability to regulate approximately 120 proteins and phospho-proteins(18, 19). To determine whether the 26 miRNA identified here could reverse our KRas signature, we scored their correlation with the signature proteins in this dataset. Additionally, we analyzed publicly available Ago CLIP-seq data to determine which of the miRNA would be able to directly bind to the KRas 3′UTR. Of the 26 identified miRNA, 5 were negatively correlated with the signature in our screen (miR-193a-3p, miR-15b, miR-1292, miR-1306 and miR-484), and only miR-193a-3p was proposed by Ago CLIP-seq studies (20)to bind to the KRas 3′UTR (Figure 1C), suggesting that it could directly regulate the expression of KRas. Using this systems approach of starting with clinical data and integrating a large miRNA-proteomic screen, we identified and chose to further characterize miR-193a-3p to determine its function in mutant KRas driven lung cancer. In support of a role in KRas-mediated tumor pathophysiology, miR-193a-3p is lost in lung cancer (Figure 1D) and colorectal cancer (Supp. Fig. 4), relative to normal lung tissue. We thus sought to determine possible genomic mechanisms of miR-193a-3p expression loss. To this end we analyzed methylation data from lung cancer patients and found that the miR-193a-3p locus is hypermethylated in lung cancer samples relative to normal tissue (Figure 1E), in accordance with a previous study in bladder cancer(21) and the lung cancer cell line A549 (22), suggesting a possible epigenetic mechanism for the decreased expression of miR-193a-3p in cancer. The genomic locus of miR-193a-3p on chromosome 17q11.2 is known to have very rare microdeletions associated with neurofibromatosis-1 but the microdeletions have not been reported in lung cancer. Indeed, we did not observe changes in copy number in the TCGA lung cancer dataset. To verify that miR-193a-3p is capable of reversing our protein signature in vitro, we overexpressed a control miRNA or miR-193a-3p in the KRas mutant lung cancer cell line A549 and analyzed the KRas signature proteins by western blot. As shown in figure 1F, the phosphorylation levels of CRaf, MEK, MAPK, p90RSK, YB1 and YAP1, as well as the total level of CDK1, were all reduced upon expression of miR-193a-3p, showing that this miRNA is capable of reversing the signature downstream of mutant KRas (Figure 1F). Additionally, the KRas signature proteins were analyzed in A549 cells transfected with anti-miR-193a-3p. However, the anti-miR showed negligible effects on the expression of our signature, most likely due to low endogenous levels of miR-193a-3p (Supp Fig 5). This is in accordance with data from the NCI-60 cell line panel, in which miR-193a-3p expression is significantly lower in KRas mutant cells compared to those expressing wild type KRas (Supp Fig 6).
Figure 1.
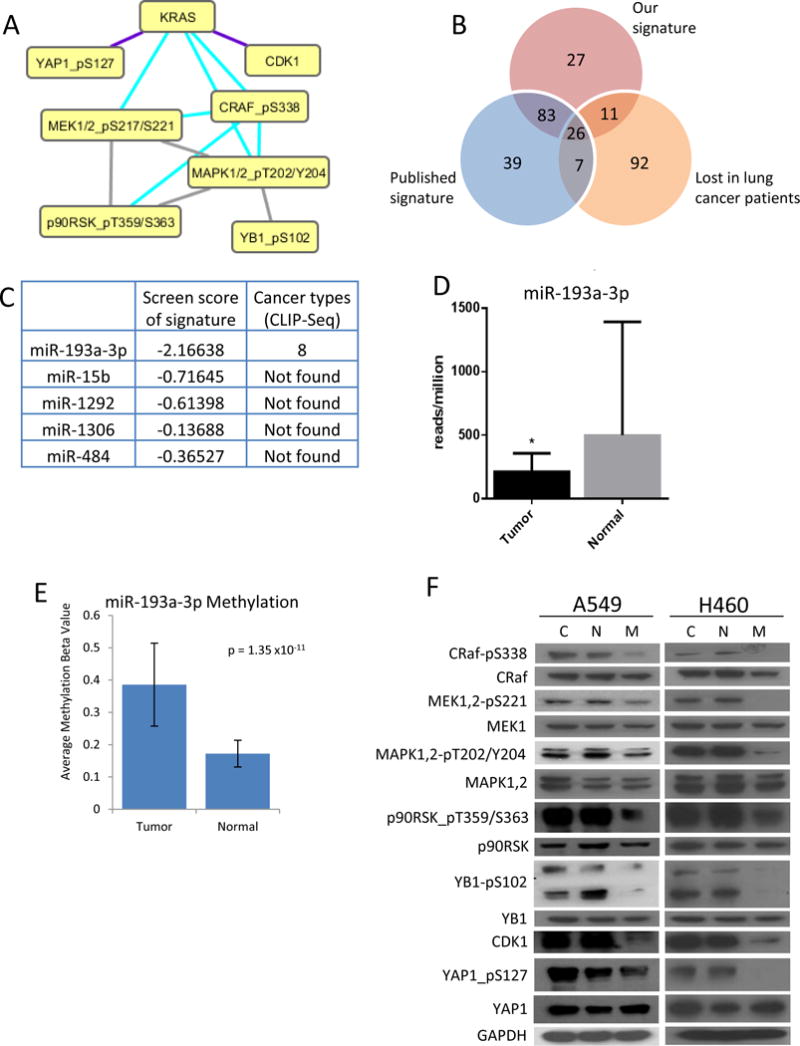
Identification of miR-193a-3p as a clinically significant miRNA. A. Identification of a phospho-protein signature that is activated downstream of Ras signaling. B. miRNA expression in lung cancer patients negatively correlated with our signature (red circle), a previously identified MAPK signature (blue circle), and lost in tumor samples relative to normal tissue (orange circle). C. 5 miRNA negatively regulated the phospho-protein signature in a screen of 879 miRNA, but only miR-193a-3p also binds to the KRas 3′UTR, as identified by Ago-CLIP-seq. D. miR-193a-3p expression is reduced in patients with lung (LUAD) cancer. E. The miR-193a-3p locus shows hypermethylation in patients with lung (LUAD) cancer. F. A549 and H460 cells were transfected with either non-targeting miRNA (N), miR-193a-3p (M) or treated with transfection reagent alone (C) for 48 hours. Western blot analysis confirms the inhibition of signaling through the KRas signature by expression of miR-193a-3p.
miR-193a-3p reduces expression of KRas through two 3′UTR sites
Western blot analysis following the overexpression of miR-193a-3p showed reduced levels of Ras protein (Figure 2A); however, the lack of antibody specificity prevented the direct measurement of individual Ras isoform protein levels. Analysis of the 3′UTR of Ras isoforms revealed two potential miR-193a-3p binding sites in the 3′UTR of KRas (Figure 2B), with no potential binding sites identified in either HRas or NRas, suggesting that miR-193a-3p may be inhibiting Ras signaling by directly targeting and reducing the expression of KRas, in accordance with a previous report by Iliopoulos et al.(23). Additionally, analysis of mRNA expression data from LUAD patients revealed a significant inverse correlation between miR-193a-3p and KRas (r = −0.11, p = 0.049), while neither HRas nor NRas were significantly correlated with miR-193a-3p (r = −0.03, p = 0.57 and r = 0.027, p = 0.62, respectively) (Figure 2C).
Figure 2.
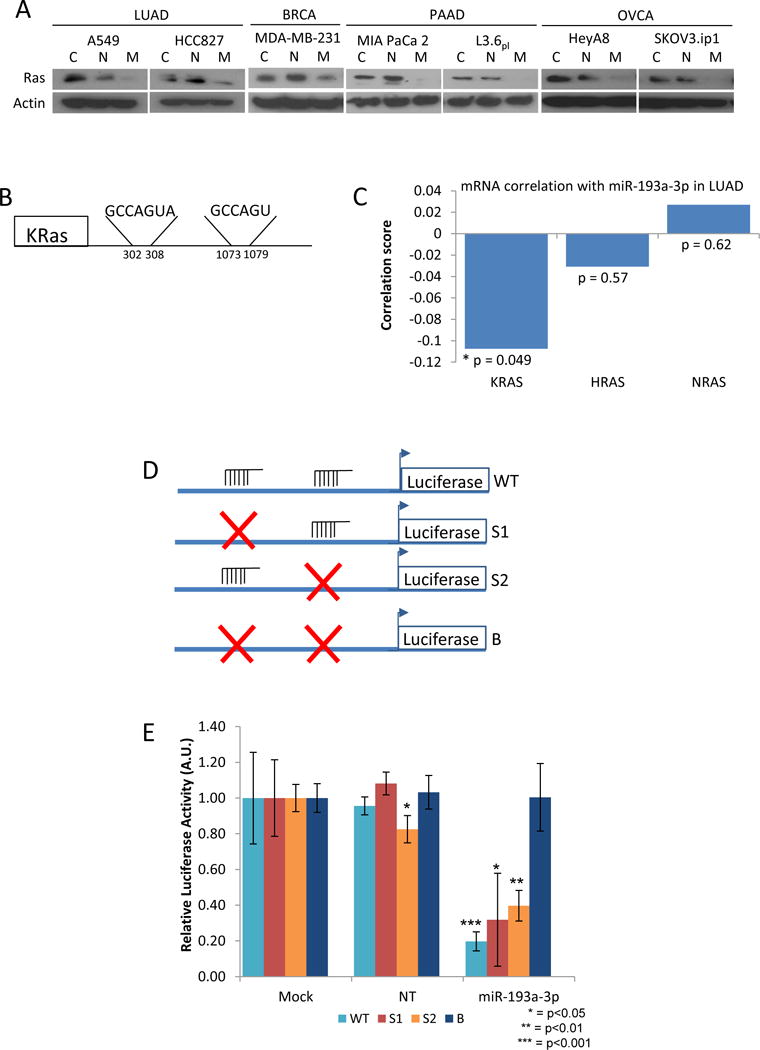
miR-193a-3p targets KRas. A. Cells were transfected with either non-targeting miRNA (N), miR-193a-3p (M) or treated with transfection reagent alone (C) for 48 hours. Expression of Ras was determined by western blot. B. Targetscan identifies 2 independent potential binding sites for miR-193a-3p in the 3′UTR of KRas, with no binding sites identified in either HRas or NRas. C. miR-193a shows significant negative correlation with mRNA levels of KRAS (r = −0.11, p = 0.049), but not with either HRAS (r = −0.03, p = 0.57) or NRAS (r = 0.03, p = 0.62). D. Constructs were generated in which the KRas 3′UTR was mutated (×) to prevent miR-193a-3p binding at either the first (S1), the second (S2) or both (B) sites. E. Expression of miR-193a-3p reduces luciferase activity from either the WT, S1 or S2 constructs in A549 cells, but is unable to block activity when both binding sites are mutated (B).
Having observed two potential miR-193a-3p sites on KRas (nucleotide positions 302–308 and 1073–1079 of the 3′UTR), we wanted to determine the importance of each of the identified sites in targeting KRas. We developed luciferase constructs utilizing the KRas 3′UTR with either no mutations (WT), or the first site (S1), the second site (S2) or both sites mutated (B) (Figure 2D). Expression of these constructs in A549 lung cancer cells in combination with miR-193a-3p showed that the miR is able to target both sites on the KRas 3′UTR suggesting that either of the two miR-193a-3p binding sites is sufficient to target KRas. Only when both sites are mutated did we observe rescue of luciferase activity in the presence of miR-193a-3p (Figure 2E).
miR-193a-3p downregulates a cell cycle network downstream of KRas
Given the multitude of potential miRNA targets, we performed gene expression profiling by microarray to determine global effects of miR-193a-3p. The ratio of gene expression in A549 lung cancer cells transfected with miR-193a-3p was determined relative to the control miRNA, and a network of the top 300 downregulated interactions was determined after edge flux analysis using Netwalker(24, 25) (http://www.netwalker suite.org). To determine the potential for miR-193a-3p to regulate the resulting network through KRas, we intersected the network with that of our KRas signature. Figure 3A shows the downregulated KRas gene network upon transfection with miR-193a-3p, demonstrating that miR-193a-3p regulates multiple genes through the downregulation of KRas and the resulting decrease in signaling through the KRas pathway. Analysis of the GO terms associated with the network revealed that approximately half of the nodes were related to various cell cycle processes, as well as specific cell division-related functions including centrosome organization and DNA packaging (Figure 3A, B). Together, these data suggest a prominent role for miR-193a-3p in regulating the cell cycle through the direct modulation of KRas.
Figure 3.
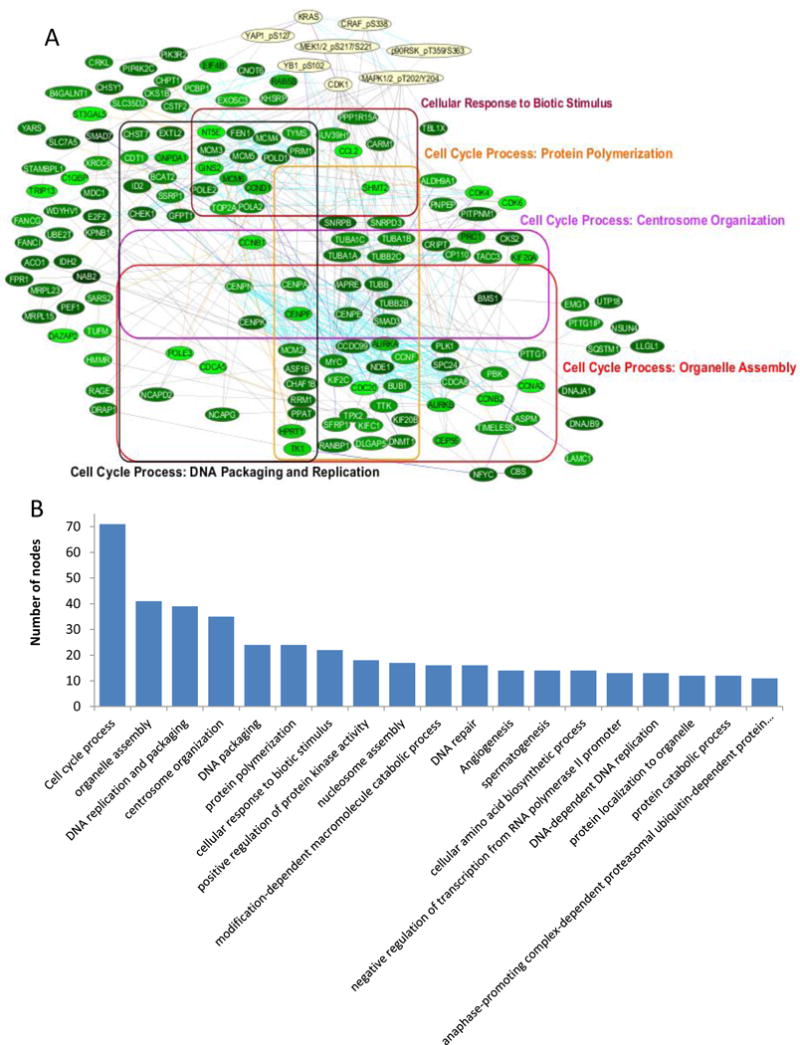
miR-193a-3p regulates a cell cycle-related network downstream of KRas. A. Top 300 downregulated interactions in miR-193a-3p expressing A549 cells relative to control miR expressing cells, after intersection with the KRas signature network. Edge colors indicate interaction type; protein-protein interaction (gray), gene interaction (blue), metabolic interaction (orange) and reactome interaction (cyan). B. GO terms associated with the network after filtering by semantic similarity.
miR-193a-3p functions as a tumor suppressor by inhibiting KRas
Having observed a prominent role of cell cycle regulation by the KRas network in the miR-193a-3p gene array data, we sought to determine the tumor suppressive role for miR-193a-3p in KRas-mutant lung cancer. Analysis showed that miR-193a-3p was able to regulate the cell cycle by inducing a G1 arrest (Figure 4A and Supp Fig 7). To determine the importance of KRas inhibition in the tumor suppressive functions of miR-193a-3p, cell cycle distribution was measured after co-transfection of miR-193a-3p and siRNA directed against KRas. As shown in figure 4B, knockdown of KRas by siRNA caused a similar G1 arrest, with no additional effect of the miR in cells with KRas knockdown (Figure 4B and Supp Fig 8). Furthermore, while miR-193a-3p was able to reduce the proliferation of multiple cell lines (Figure 4C), knockdown of KRas by siRNA reduced the proliferation of the cell lines to the same degree as expression of miR-193a-3p, with no further inhibition occurring following co-transfection with the miR (Figure 4D). Taken together, these data suggest that the effect of miR-193a-3p on the cell cycle and cellular proliferation is mediated through the regulation of KRas expression. Additionally, three dimensional cultures showed that KRas siRNA was able to mimic the effect of miR-193a-3p on both colony size and shape, further supporting the necessity of KRas targeting for the function of miR-193a-3p.
Figure 4.
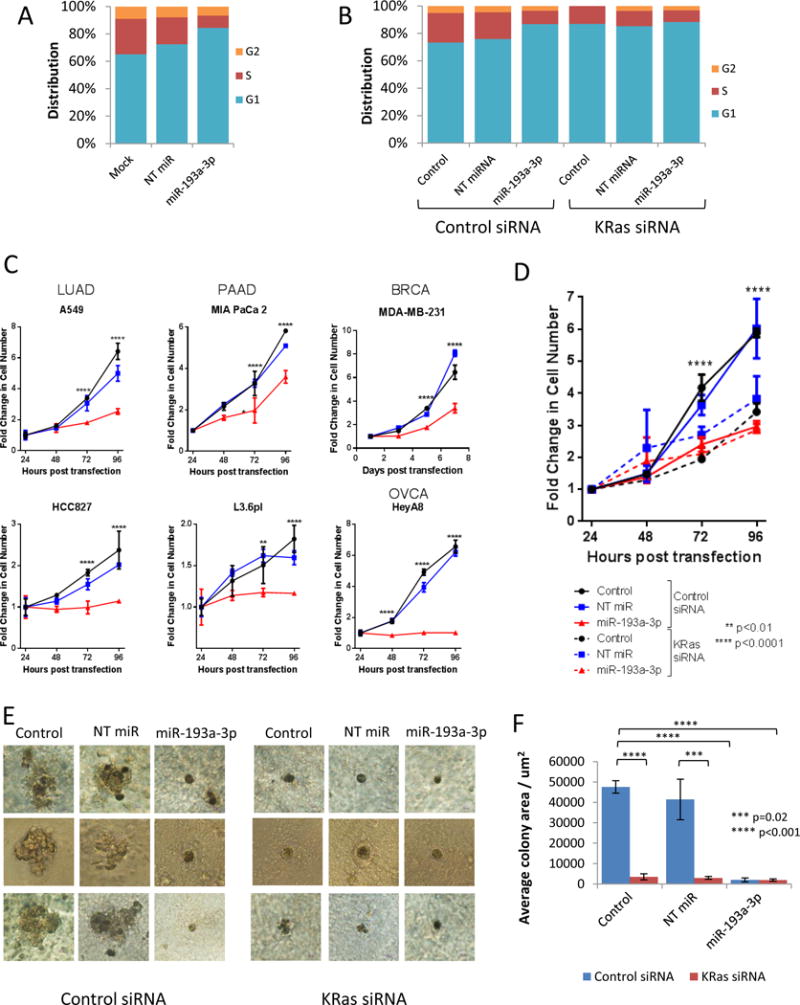
miR-193a-3p functions as a tumor suppressor by inhibiting KRas. A. miR-193a-3p induces G1 arrest. Cell cycle distribution was determined in A549 cells by flow cytometry following staining with propidium iodide. Cyan bars indicate percentage of cells in G1 phase, red bars represent S phase, and orange bars indicate G2 phase. Data shown are representative of three independent experiments. B. miR-193a-3p-induced G1 arrest is dependent of KRas. Cell cycle distribution was determined in A549 cells following transfection of either a control siRNA or KRas-directed siRNA by flow cytometry following staining with propidium iodide. Cyan bars indicate percentage of cells in G1 phase, red bars represent S phase, and orange bars indicate G2 phase. Data shown are representative of three independent experiments. C. Expression of miR-193a-3p reduces proliferation in cell lines from multiple tumor types. Proliferation was analyzed at the indicated time points after transfection by staining with crystal violet and measurement of absorbance at 570nm. Data are normalized to 24 hours to indicate fold change in cell number and statistical significance determined by two-way ANOVA. All cells were plated in triplicate. D. miR-193a-3p inhibition of proliferation is dependent on KRas. Proliferation was analyzed in A549 cells at the indicated time points after transfection including either a control siRNA or KRas-directed siRNA by staining with crystal violet and measurement of absorbance at 570nm. Data are normalized to 24 hours to indicate fold change in cell number and statistical significance determined by two-way ANOVA. All cells were plated in triplicate. E. miR-193a-3p reduces colony formation in 3D. A549 cells were transfected with non-targeting miRNA (NT miR), miR-193a-3p or treated with transfection reagent alone (Control) in combination with either control or KRas-directed siRNA. After plating on matrigel, cells were allowed to grow for 14 days before imaging of colonies. Images are representative of 3 colonies imaged per condition, per experiment. F. Colony area was measured using ImageJ (http://imagej.nih.gov/ij/), with mean and SD calculated based on all imaged colonies.
To further demonstrate that miR-193a-3p-mediated direct targeting of KRas was the mechanism leading to inhibition of cell growth and tumorigenesis, we developed miR-193a-3p-insensitive KRas constructs with targeted mutations in the 3′UTR. To generate the constructs, the 3′UTRs from the luciferase constructs were cloned downstream of a G12V mutant KRas expression construct, resulting in mutant KRas with either the WT, S1, S2 or B 3′UTR sequences (Figure 5A). The co-expression of these constructs with miR-193a-3p in A549 cells revealed that constructs containing at least one functional miR-193a-3p target sites were not able to rescue cells from the effect of the miR. However, the construct with no functional miR-193a-3p sites was able to rescue proliferation (Figure 5B) and the expression of the signature proteins (Figure 5C), confirming that the effect of miR-193a-3p on cellular proliferation and signaling is directly mediated through KRas. Additionally, the expression of mutant KRas with the miR-193a-3p-insensitive 3′UTR (Mut/B) was able to reverse the effects of miR-193a-3p in three dimensional cultures (Figure 5D and E). These data together demonstrate that the oncogenic suppression induced by miR-193a-3p is mediated by direct targeting of KRas.
Figure 5.
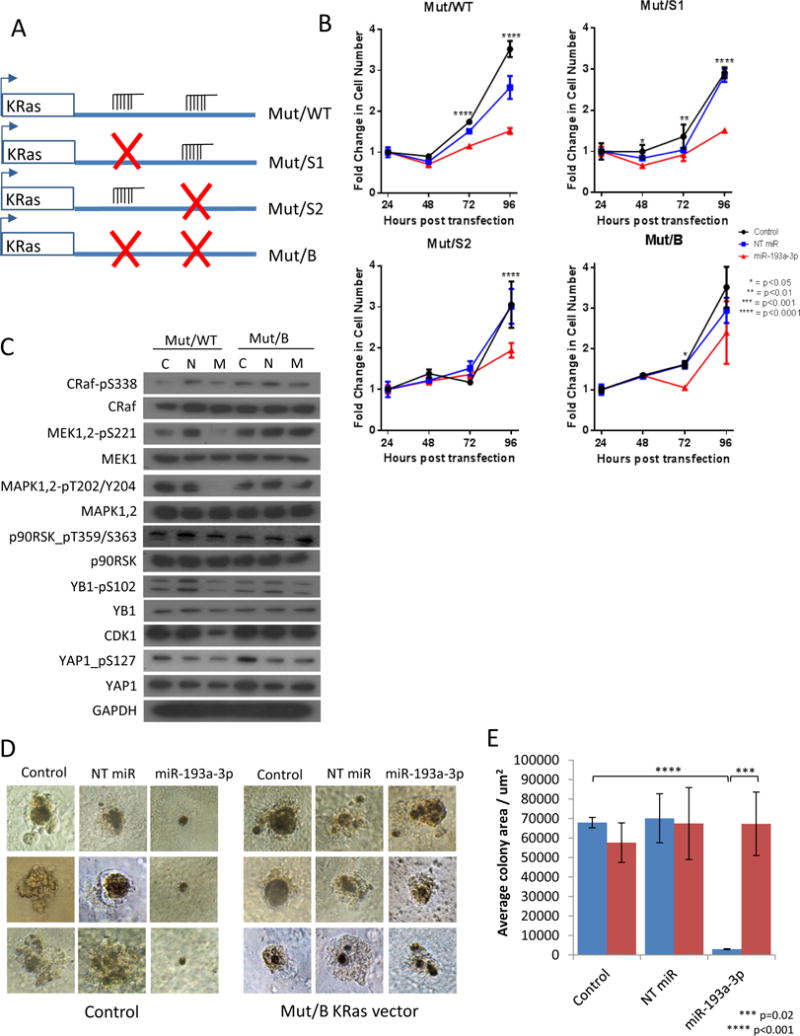
miR-193a-3p effects are rescued by miR-193a-3p-insensitive mutant KRas. A. The 3′UTR constructs were removed from the luciferase vector and cloned downstream of G12V mutant KRas to produce mutant KRas with either no, one or two functional miR-193a-3p target sites. B. miR-193a-3p inhibition of proliferation in A549 cells is rescued by expression of mutant KRas with both miR-193a-3p binding sites mutated (Mut/B), but not by mutant KRas with either the WT 3′UTR (Mut/WT), or with the first (Mut/S1) or second (Mut/S2) binding sites mutated. Data are normalized to 24 hours to indicate fold change in cell number, and statistical significance determined by two-way ANOVA. C. Mut/B KRas is able to rescue the effects of miR-193a-3p on signaling through our KRas signature, as determined by western blot jn A549 cells. D. Mut/B KRas is able to rescue the effects of miR-193a-3p on the size and shape of 3D colonies of A549 cells. Images are representative of 3 colonies imaged per condition, per experiment. E. Colony area was measured using ImageJ (http://imagej.nih.gov/ij/), with mean and SD calculated based on all imaged colonies.
miR-193a-3p inhibits KRas-mutant tumor growth ex vivo and in vivo
In vitro analysis thus far showed that miR-193a-3p directly targets KRas and can block colony formation in a 3D model. To determine if miR-193a-3p could have a beneficial therapeutic effect in inhibiting tumor growth or metastasis, we utilized an ex vivo lung model as well a xenograft animal model. Ex vivo 4D denuded models(26, 27) were seeded with A549 lung cancer cells and treated every 5 days with control (NT) miR or miR-193a-3p incorporated into DOPC nanoliposomes as previously described(10), for 20 days (Figure 6A). H&E staining of the primary tumor showed viable cells on day 20 in 4D models treated with NT miR-DOPC or miR-193a-3p-DOPC (Figure 6A). However, when CTCs were isolated and cultured, those from the 4D model treated with miR-193a-3p-DOPC showed significantly less proliferation compared to those from the 4D model treated with NT miR-DOPC (Figure 6B) suggesting that mir-193a-3p significantly inhibited the viability of CTCs. The presence of tumor cells in the contralateral lobe is indicative of metastasis in this model. We observed that there were significantly fewer tumor cells per high power field in the right lung in the 4D model treated with miR-193a-3p-DOPC compared to the NT miR-DOPC treatment (Figure 6C–D), suggesting that miR-193a-3p significantly inhibited the ability of the tumor cells to form metastases. Both the reduction of viability of CTCs and tumor cells in the contralateral lobe upon treatment with miR-193a-3p suggest that this miRNA targeting of KRas may be of high therapeutic potential. Furthermore, A549 lung cancer cells were implanted orthotopically into athymic mice to study the effects of miR-193a-3p on KRas-mutant tumor growth in vivo. As shown in figure 6, tumor growth was significantly reduced in mice treated with miR-193a-3p relative to those treated with the control miR (Figure 6E–F). Further, the number of metastatic nodules was also significantly reduced, and this was apparent at all locations where metastases were found (Figure 6F). The ex vivo 4D acellular lung and orthotopic xenograft models showed that miR-193a-3p can reduce tumor formation as well as decrease metastasis in KRas-mutant lung carcinoma.
Figure 6.
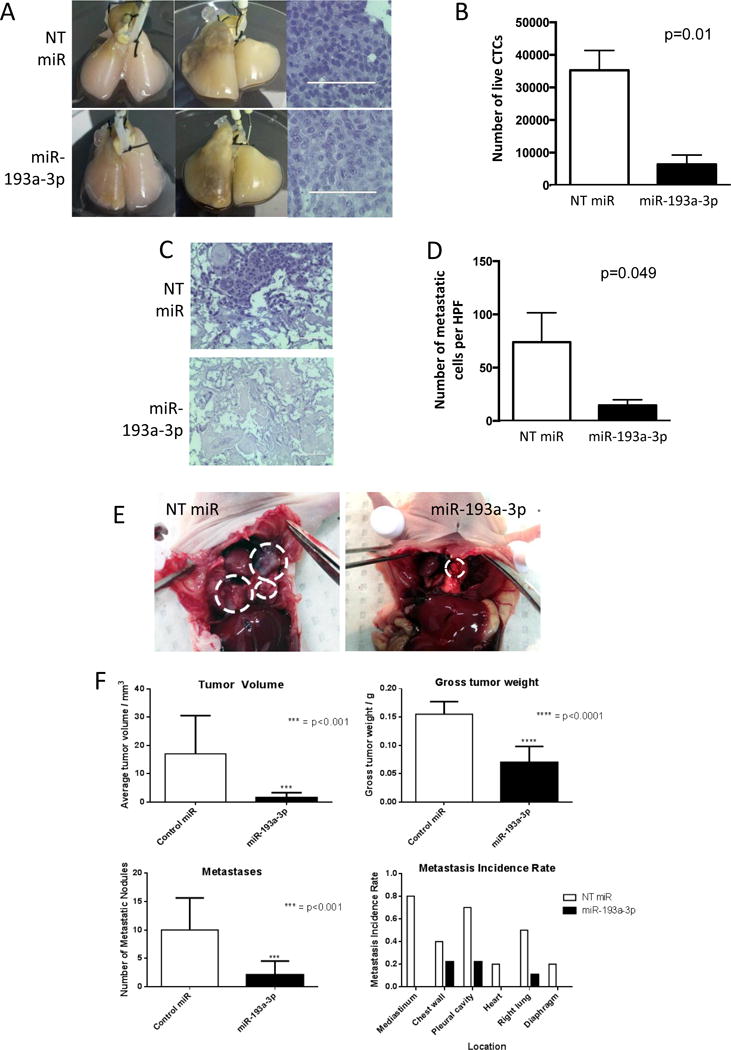
miR-193a-3p functions as a tumor suppressor ex vivo and in vivo. A. A549 cells seeded into acellular rat lungs (left) form primary tumors (middle, right). B. Circulating tumor cells isolated from the miR-193a-3p treatment model show decreased viability in culture (p = 0.01). C. H&E staining of contralateral lobe shows decreased metastasis ex vivo following treatment with miR-193a-3p-DOPC. D. miR-193a-3p-DOPC reduces the number of metastatic cells per HPF (p = 0.049). E. miR-193a-3p-DOPC reduces the growth of A549 cells in in vivo lung xenografts. F. miR-193a-3p-DOPC reduces tumor volume (p = <0.001), gross tumor weight (p = <0.0001) and metastases (p = 0.001) at all locations in vivo. Statistical significance was determined by Student’s t-test.
Discussion
The Ras family of proteins constitutes one of the most widely studied and well characterized oncogene families. While the family includes several members including N, H and KRas, and mutations in KRas are highly correlated with cancer and poor prognosis, this important family of oncogenes is still largely considered to be undruggable. Strategies to target oncogenic Ras using different approaches are essential. The key finding of this study is the ability of miR-193a-3p to directly target KRas, and as such inhibit signaling through its downstream pathway. Here we used a genome-wide miRNA screen to identify miR-193a-3p, for which expression is lost in lung cancer patients, as a key regulator of KRas. We have also determined the details of the mechanism of action, identified the relevant strand as the 3p strand of miR-193a, and identified two target sites in the KRas 3′UTR that are both functional and sufficient to allow miR-193a-3p to downregulate KRas. Significantly the two sites are in regions that exhibit very low genomic alterations as compared to previously characterized KRas targeting miRNA such as let-7(12). Importantly, we have identified KRas as the key target of miR-193a-3p in both 2D and 3D culture systems, and translated the in vitro findings into exciting potential clinical applications by determining the effect of miR-193a-3p on KRas-driven tumor formation and metastasis in ex vivo and in vivo models of lung cancer.
Since Ras mutations result in increased signaling without necessarily being accompanied by a concurrent increase in mRNA or protein expression of Ras itself, downstream pathways from Ras are valuable tools to identify potential therapeutic approaches. Here, we elected to measure KRas activity by studying phosphorylation levels of downstream effector proteins. Utilization of proteomic profiling of patient tumor samples allowed us to rapidly identify such a phospho-protein signature in lung cancer. Such profiling of cancer patients has provided an important step towards personalizing therapy; the hope of which is to identify patients who would benefit from a specific targeted therapy.
Given recent advances in the use of RNAi-based therapeutic approaches(10, 28), including a phase I clinical trial using a derivative of miR-34 to treat liver cancer (http://clinicaltrials.gov/ct2/show/NCT01829971), we elected to screen a library of miRNA to identify those capable of reversing the Ras signature. We identified miR-193a-3p as a candidate therapeutic approach to treat cancers exhibiting the Ras-high protein signature. Analysis of patient data revealed miR-193a-3p as a highly clinically significant miRNA which is lost, potentially due to hypermethylation of the genomic locus. This strongly suggests that miR-193a-3p could have potential as a therapeutic modality in KRas tumors. In vitro characterization demonstrated that miR-193a-3p is capable of modulating multiple cancer-related phenotypes through the inhibition of KRas via two independent 3′UTR target sites. The presence of two independent 3′UTR sites, either of which is sufficient to target KRas, greatly enhances the possibility of utilizing this miRNA as a therapeutic agent. While let-7a has already been characterized as a Ras-targeting miRNA, the presence of a SNP disrupting a single let-7a target site on the KRas 3′UTR (LCS6) is sufficient to increase KRas expression 10 fold relative to KRas containing the WT 3′UTR(12). In contrast, neither of the miR-193a-3p sites has been reported lost or mutated (http://compbio.uthsc.edu/miRSNP). Additionally, our experimental data suggests that even if a SNP was present or acquired in one of the miR-193a-3p target sites, the other site would be sufficient to maintain KRas inhibition.
One of the most often raised issues with utilizing miRNA to target molecules or pathways is the fact that miRNA have pleiotropic effects. Here we show by the development of mutated 3′UTR constructs and targeted knockdown of KRas with siRNA that the effects of miR-193a-3p are primarily due to the direct targeting of KRas. Rescue experiments with KRas mutated 3′UTR constructs demonstrate that if both the 3′UTR sites are disrupted, miR-193a-3p does not alter any of the KRas-driven oncogenic properties assessed. The effect of miR-193a-3p on proliferation, cell cycle and 3D colony growth are all dependent one at least one of the KRas 3′UTR sites being wild type, suggesting that this miRNA may indeed be a potentially valuable therapeutic modality.
Translating in vitro studies including findings from 3D models to show potential applicability to patient use can be a challenge with new molecules including non-coding RNA. The ex vivo 4D lung perfusion model, recently developed by Mishra et al.,(26) combines 3 dimensional aspects of tumor growth with the additional dimension of flow within the endogenous lung matrix and anatomy. Using this system, we developed mutant KRas-driven lung tumors ex vivo and ascertained that the application of miR-193a-3p decreases the viability of circulating tumor cells (CTCs) and decreases metastasis formation on the contralateral lobe, suggesting that application of miR-193a-3p systemically to established tumors could have high therapeutic potential. The ability to measure the impact on different stages of tumor formation using this model greatly enhances our capability to identify high value therapeutic molecules such as miR-193a-3p. Experiments using orthotopic xenograft KRas-mutant lung tumor models targeted with miR-193a-3p encapsulated in DOPC nanoliposome particles showed that miR-193a-3p could significantly reduce tumor volume and growth and more importantly reduce metastasis to various sites.
KRas and its mutations have been well characterized in lung cancers, and the fact that mutant KRas is not only currently untargetable by small molecule therapies, but that it correlates with resistance to multiple modes of therapeutic intervention(4–9), makes miR-193a-3p an exciting prospect for future clinical trials in the treatment of KRas-driven tumors. A systems approach of integrating patient data, genome-wide miRNA proteomic screen as well as in vitro and in vivo models has been essential in identifying a miRNA that directly targets KRas and can block KRas-driven tumor formation and metastasis. Continued work is needed to determine if this miRNA can be utilized as a therapeutic agent to target mutant KRas tumors.
Methods
TCGA data
Level 3 patient RPPA, miRNA expression data, along with genomic methylation data, were downloaded from the TCPA (http://app1.bioinformatics.mdanderson.org/tcpa/_design/basic/index.html)(29) and TCGA (https://tcga-data.nci.nih.gov/tcga/) data portals.
Phospho-protein signature
RPPA and miRNA expression values were standardized to produce z-scores. The mean score of the signature proteins was then correlated with the miRNA expression in each patient to determine miRNA which were significantly negatively correlated with the signature (p<0.05).
miRNA library screen by RPPA
The miRNA library containing 879 miRNA mimics was designed and synthesized by Dharmacon. MDA-MB-231 cells were seeded (3750 cells/well) and transfected with 50 nM miRNA. After 48 hours, cells were lysed and RPPA was carried out as previously described(19). Further details of RPPA technology and data are available from TCGA – RPPA (http://cancergenome.nih.gov/)(30–34). Screen data was normalized for plate variation and with respect to controls as previously described(19).
Ago CLIP-seq data
Ago CLIP-seq data was obtained from the starbase database (http://starbase.sysu.edu.cn/)(20).
Cell culture
All cell lines were obtained from and characterized by ATCC. MDA-MB-231, HeyA8 and SKOV3.ip1 cells were maintained in RPMI 1640 with 5% (v/v) fetal bovine serum (FBS), MCF-7 and MIA PaCa 2 cells were maintained in DMEM with 10% (v/v) FBS, and A549, H460, HCC827 and L3.6pl were maintained in RPMI 1640 with 10% (v/v) FBS. All media was supplemented with 100 I.U./mL penicillin, 100 μg/mL streptomycin and 0.25 μg/mL amphotericin B, and all cells were maintained at 37°C with 5% CO2. Transfections were carried out at approximately 90% confluency using 20 nM miRNA mimics with Lipofectamine 2000 (Life Technologies), according to the manufacturer’s instructions. Where indicated, cells were also transfected with 50 nM KRas siRNA (Dharmacon).
SDS-PAGE and Western blotting
Cells were transfected as described above and incubated for 48 hours. Proteins were extracted in the appropriate volume of lysis buffer, and 30 μg protein was denatured in 0.2 volumes 5× SDS-PAGE loading buffer before loading onto appropriate percentage SDS-PAGE gels. Proteins were transferred to PVDF membrane and blocked for 20 minutes with 3% (w/v) BSA in PBS-T at room temperature. Primary antibody incubations were carried out for 1 hour at room temperature in 3% BSA in PBS-T at the manufacturer’s recommended dilution, following which membranes were washed 5 times for 5 minutes each in PBS-T. HRP-conjugated secondary antibodies (Thermo Scientific) were diluted 1:10,000 in 5% milk in PBS-T and incubated with membranes for 1 hour at room temperature, followed by 5 washes for 5 minutes each in PBS-T. Proteins were visualized using Immobilon Western chemiluminescent HRP substrate (Millipore).
miRNA target prediction
miRNA 3′UTR target predictions were determined using Targetscan v6.2 (http://targetscan.org/).
Luciferase activity
The first 1.5kb of the KRas 3′UTR was cloned upstream of the luciferase gene (Switchgear Genomics). Where indicated, the following mutations were introduced into the miR-193a-3p binding sites; S1: TCGGTCAA, S2: GCCGGTCT, B: TCGGTCAA and GCCGGTCT. Cells were plated into 96 well plates (5000 cells/well) and transfected with miRNA as indicated in combination with a plasmid encoding either the GAPDH 3′UTR or the indicated KRas 3′UTR construct. 24 hours after transfection, cells were lysed and luciferase activity determined using the Lightswitch assay kit (Switchgear Genomics). Activity at the KRas 3′UTR was determined by normalizing the luciferase signal to that from the GAPDH 3′UTR for each condition.
Gene array
A549 cells were transfected with control miRNA or miR-193a-3p as described above. 48 hours after transfection, cells were lysed and prepared for gene expression analysis using the HumanHT-12 v4 Expression BeadChip Kit (Illumina), according to the manufacturer’s instructions. Network analysis was performed using Netwalker (available at https://netwalkersuite.org/). GO terms associated with the network were exported and filtered by semantic similarity using Revigo(35) (http://revigo.irb.hr/revigo.jsp).
Determination of cellular proliferation
Cells were transfected for 6 hours, following which the cells were trypsinised, counted and plated at 1500 cells/well in triplicate in 96 well plates. Where dual transfections are indicated, the miRNA was transfected 16 hours before the siRNA or plasmid containing KRas G12V upstream of the indicated 3′UTR (Epoch Life Sciences), and the cells were plated following a further 6 hours incubation. At the indicated time points, the media was removed and adherent cells were stained with 50 μL crystal violet solution (0.5% crystal violet w/v, 20% methanol). After rinsing to remove excess stain, the wells were dried and the crystals redissolved with 100 μL 20% acetic acid (v/v). Cell density was determined by measuring the absorbance at 570nm using a Vmax kinetic microplate reader (Molecular Devices, Sunnyvale, California). Statistical significance was determined using two-way analysis of variance (ANOVA).
Cell cycle analysis
Cells were plated into 6cm plates and transfected as described above. Where indicated, cells were also transfected with 50 nM control siRNA or 50 nM siRNA targeting KRas (Dharmacon) 6 hours after miRNA transfection. After 48 hours, cells were washed, counted and resuspended in PBS (1×106 to 107 cells in 0.5 mL). Cells were added to 4.5 mL of 70% ethanol in PBS in 12×75mm centrifuge tubes for fixation and kept at −20°C for at least 2 hours. The cells were washed in 2 mL PBS and incubated at 37°C for 15 minutes with 1 mL propidium iodide (0.1% (v/v) Triton X-100 in PBS, 0.2 mg/mL RNase A, 0.02 mg/mL propidium iodide). Cell cycle phase was determined by flow cytometry and analysed with the DNA content histogram deconvolution software Cell Quest Pro by MD Anderson Flow Cytometry core.
3 dimensional cultures
Cells were transfected, trypsinised and counted as described above. 1000 cells/well were plated into 96 well plates containing a thin layer of matrigel, in media supplemented with 2% matrigel, and allowed to grow for 14 days before imaging. Transfections were repeated every 5–6 days to maintain miRNA, siRNA and plasmid expression levels.
Ex vivo 4D lung metastasis model
Experiment protocols were approved by the Institutional Animal Care and Use Committee at the Houston Methodist Research Institute and all experiments were carried out in accordance to guidelines and policies governing the use of laboratory animals in research. Established A549 cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured before seeding on ex vivo 4D model. Lung-heart blocks were harvested from 6–8 week Sprague Dawley rats and decellularised using 0.1% sodium dodecyl sulfate (SDS) and 1% Triton X-100, as previously described(26). The acellular lungs were placed in a bioreactor with an oxygenator and pump with the right main stem ligated with silk suture to create a metastatic ex vivo 4D lung model as previously described(27). The left lung was populated with 25 million A549 cells through the tracheal cannula and perfused with 200 mL of RPMI 1640 supplemented with 10% FBS and antibiotics (Gibco Life Technology, NY USA) through the pulmonary artery cannula daily for 20 days. Models were treated with 20 nM either non-targeting miR or miR-193a-3p on days 0, 4, 8, 12 and 16. The left lung was removed on day 20, and the primary tumor fixed in 10% formalin for H&E staining as described previously(36). The percentage of cells with respective staining was calculated from 10 random images per high power field (HPF, 40×). Circulating tumor cells were collected from the perfused cell culture media present in bioreactor bottle. Every day the 200ml media was centrifuged at 500 ×g for 5 min at room temperature, and the viable CTCS were counted using trypan blue staining.To determine the impact of miR-193a-3p on metastasis, 25,000 circulating tumor cells (CTCs) from the 4D model were plated on 24 well plates in triplicate and cultured for 4 days to assess viability. Additionally, lobectomy of the right lung was performed on day 20 and preserved in 10% formalin for H&E stain followed by examination of 10 random images and counting the number of tumor cells per HPF using Evos XL (Life Technology, USA). Student T-test was performed to compare the number of cells per HPF between the groups and p<0.05 was considered to be significant.
In vivo
Experiment protocols were approved by the Institutional Animal Care and Use Committee at MD Anderson Cancer Center and all experiments were carried out in accordance to guidelines and policies governing the use of laboratory animals in research.10 female athymic nude mice were used per group, all between 8 and 12 weeks of age at the time of injection. For all animal experiments, cells were trypsinised, washed and resuspended in Hanks’ balanced salt solution (HBSS; Gibco, Carlsbad, CA) before injection. 7.5×105 cells in 100 μL of a 1:1 mixture of HBSS and BD Matrigel were injected directly into the lung parenchyma at the left lateral dorsal axillary line, and the incision closed with surgical clips. Mice were assigned by block randomization to groups for treatment with either NT miRNA-DOPC or miR-193a-3p-DOPC. No blinding was performed. Twice weekly treatments of 200 μg miRNA per kg administered by intraperitoneal injection started the day following injection and continued for nearly 4 weeks. Once mice in any group became moribund they were all killed, necropsied and the tumor cells were collected. Tumor weights, number and location of tumor nodules were recorded.
Statistical Analysis
Statistical comparison of patient data was performed using two-tailed student’s T-test with correction for unequal variance, and are stated as mean +/− s.d. All in vitro and ex vivo assays were repeated 3 independent times to provide biological replicates. Where indicated, statistical comparisons were made using two-tailed student’s T-test or two-way ANOVA as appropriate, and are stated as mean +/− s.d. For in vivo assays, 10 animals were used for each group to provide sufficient power for statistical analysis by two-tailed student’s T-test.
Data access
Microarray data is available from GEO under accession number GSE73194.
Supplementary Material
Acknowledgments
All the data reported in the paper are presented. The microarray data are available from GEO under the accession number GSE73194. The work presented here was funded by NIH/NCI ICBP grant U54-CA112970 (Project 2 PI-JWG, Project 4 Co-PI’s PTR and GBM), VS was funded by the CCBTP training grant from the CPRIT, and grants from the NIH (UH2 TR000943), the RGK Foundation, and CPRIT RP110595 (AKS).
References
- 1.Jancik S, Drabek J, Radzioch D, Hajduch M. Clinical relevance of KRAS in human cancers. Journal of biomedicine & biotechnology. 2010;2010:150960. doi: 10.1155/2010/150960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Der CJ, Cooper GM. Altered gene products are associated with activation of cellular rasK genes in human lung and colon carcinomas. Cell. 1983 Jan;32(1):201–8. doi: 10.1016/0092-8674(83)90510-x. [DOI] [PubMed] [Google Scholar]
- 3.Bos JL. ras oncogenes in human cancer: a review. Cancer research. 1989 Sep 1;49(17):4682–9. [PubMed] [Google Scholar]
- 4.Uberall I, Kolar Z, Trojanec R, Berkovcova J, Hajduch M. The status and role of ErbB receptors in human cancer. Experimental and molecular pathology. 2008 Apr;84(2):79–89. doi: 10.1016/j.yexmp.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 5.Katzel JA, Fanucchi MP, Li Z. Recent advances of novel targeted therapy in non-small cell lung cancer. Journal of hematology & oncology. 2009;2:2. doi: 10.1186/1756-8722-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agarwal A, Eide CA, Harlow A, Corbin AS, Mauro MJ, Druker BJ, et al. An activating KRAS mutation in imatinib-resistant chronic myeloid leukemia. Leukemia. 2008 Dec;22(12):2269–72. doi: 10.1038/leu.2008.124. [DOI] [PubMed] [Google Scholar]
- 7.Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008 Jan 20;26(3):374–9. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Gonzalez-Larriba JL, Alberola V, Molina F, Monzo M, Benito D, et al. Single-agent paclitaxel by 3-hour infusion in the treatment of non-small cell lung cancer: links between p53 and K-ras gene status and chemosensitivity. Seminars in oncology. 1995 Dec;22(6 Suppl 14):12–8. [PubMed] [Google Scholar]
- 9.Grana TM, Rusyn EV, Zhou H, Sartor CI, Cox AD. Ras mediates radioresistance through both phosphatidylinositol 3-kinase-dependent and Raf-dependent but mitogen-activated protein kinase/extracellular signal-regulated kinase kinase-independent signaling pathways. Cancer Res. 2002 Jul 15;62(14):4142–50. [PubMed] [Google Scholar]
- 10.Wu SY, Lopez-Berestein G, Calin GA, Sood AK. RNAi therapies: drugging the undruggable. Science translational medicine. 2014 Jun 11;6(240):240ps7. doi: 10.1126/scitranslmed.3008362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiao LR, Frampton AE, Jacob J, Pellegrino L, Krell J, Giamas G, et al. MicroRNAs targeting oncogenes are down-regulated in pancreatic malignant transformation from benign tumors. PloS one. 2012;7(2):e32068. doi: 10.1371/journal.pone.0032068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grechukhina O, Petracco R, Popkhadze S, Massasa E, Paranjape T, Chan E, et al. A polymorphism in a let-7 microRNA binding site of KRAS in women with endometriosis. EMBO molecular medicine. 2012 Mar;4(3):206–17. doi: 10.1002/emmm.201100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, et al. A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nature communications. 2014;5:3887. doi: 10.1038/ncomms4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang W, Nandakumar N, Shi Y, Manzano M, Smith A, Graham G, et al. Downstream of mutant KRAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma. Science signaling. 2014 May 6;7(324):ra42. doi: 10.1126/scisignal.2005049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalinichenko VV, Kalin TV. Is there potential to target FOXM1 for ‘undruggable’ lung cancers? Expert opinion on therapeutic targets. 2015 Jul;19(7):865–7. doi: 10.1517/14728222.2015.1042366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong X, Nguyen HT, Chen Q, Zhang R, Hagman Z, Voorhoeve PM, et al. Opposing activities of the Ras and Hippo pathways converge on regulation of YAP protein turnover. The EMBO journal. 2014 Nov 3;33(21):2447–57. doi: 10.15252/embj.201489385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014 Jul 31;511(7511):543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moss TJ, Luo Z, Seviour EG, Sehgal V, Lu Y, Hill SM, et al. Genome-wide perturbations by miRNAs map onto functional cellular pathways, identifying regulators of chromatin modifiers. Npj Systems Biology And Applications. 2015 Sep 28;1:15001. doi: 10.1038/npjsba.2015.1. online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seviour EG, Sehgal V, Lu Y, Luo Z, Moss T, Zhang F, et al. Functional proteomics identifies miRNAs to target a p27/Myc/phospho-Rb signature in breast and ovarian cancer. Oncogene. 2015 Feb 2; doi: 10.1038/onc.2014.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang JH, Li JH, Shao P, Zhou H, Chen YQ, Qu LH. starBase: a database for exploring microRNA-mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data. Nucleic acids research. 2011 Jan;39:D202–9. doi: 10.1093/nar/gkq1056. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lv L, Deng H, Li Y, Zhang C, Liu X, Liu Q, et al. The DNA methylation-regulated miR-193a-3p dictates the multi-chemoresistance of bladder cancer via repression of SRSF2/PLAU/HIC2 expression. Cell death & disease. 2014;5:e1402. doi: 10.1038/cddis.2014.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heller G, Weinzierl M, Noll C, Babinsky V, Ziegler B, Altenberger C, et al. Genome-wide miRNA expression profiling identifies miR-9-3 and miR-193a as targets for DNA methylation in non-small cell lung cancers. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012 Mar 15;18(6):1619–29. doi: 10.1158/1078-0432.CCR-11-2450. [DOI] [PubMed] [Google Scholar]
- 23.Iliopoulos D, Rotem A, Struhl K. Inhibition of miR-193a expression by Max and RXRalpha activates K-Ras and PLAU to mediate distinct aspects of cellular transformation. Cancer research. 2011 Aug 1;71(15):5144–53. doi: 10.1158/0008-5472.CAN-11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Komurov K, Dursun S, Erdin S, Ram PT. NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics. 2012;13:282. doi: 10.1186/1471-2164-13-282. Epub 2012/06/27. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Komurov K, White M, Ram PT. Use of Data-Biased Random Walks on Graphs for the Retrieval of Context-Specific Networks from Genomic Data. PLoS Comput Biol. 2010;6(8) doi: 10.1371/journal.pcbi.1000889. Epub August 19, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mishra DK, Thrall MJ, Baird BN, Ott HC, Blackmon SH, Kurie JM, et al. Human lung cancer cells grown on acellular rat lung matrix create perfusable tumor nodules. The Annals of thoracic surgery. 2012 Apr;93(4):1075–81. doi: 10.1016/j.athoracsur.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra DK, Creighton CJ, Zhang Y, Chen F, Thrall MJ, Kim MP. Ex vivo four-dimensional lung cancer model mimics metastasis. The Annals of thoracic surgery. 2015 Apr;99(4):1149–56. doi: 10.1016/j.athoracsur.2014.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li CX, Parker A, Menocal E, Xiang S, Borodyansky L, Fruehauf JH. Delivery of RNA interference. Cell cycle. 2006 Sep;5(18):2103–9. doi: 10.4161/cc.5.18.3192. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, et al. TCPA: a resource for cancer functional proteomics data. Nature methods. 2013 Nov;10(11):1046–7. doi: 10.1038/nmeth.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012 Oct 4;490(7418):61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012 Jul 19;487(7407):330–7. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008 Oct 23;455(7216):1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011 Jun 30;474(7353):609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012 Sep 27;489(7417):519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Supek F, Bosnjak M, Skunca N, Smuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PloS one. 2011;6(7):e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra DK, Sakamoto JH, Thrall MJ, Baird BN, Blackmon SH, Ferrari M, et al. Human lung cancer cells grown in an ex vivo 3D lung model produce matrix metalloproteinases not produced in 2D culture. PloS one. 2012;7(9):e45308. doi: 10.1371/journal.pone.0045308. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.