

SUMMARY
GpsB regulatory protein and StkP protein kinase have been proposed as molecular switches that balance septal and peripheral (side-wall like) peptidoglycan (PG) synthesis in Streptococcus pneumoniae (pneumococcus); yet, mechanisms of this switching remain unknown. We report that ΔdivIVA mutations are not epistatic to ΔgpsB division-protein mutations in progenitor D39 and related genetic backgrounds; nor is GpsB required for StkP localization or FDAA labeling at septal division rings. However, we confirm that reduction of GpsB amount leads to decreased protein phosphorylation by StkP and report that the essentiality of ΔgpsB mutations is suppressed by inactivation of PhpP protein phosphatase, which concomitantly restores protein phosphorylation levels. ΔgpsB mutations are also suppressed by other classes of mutations, including one that eliminates protein phosphorylation and may alter division. Moreover, ΔgpsB mutations are synthetically lethal with Δpbp1a, but not Δpbp2a or Δpbp1b mutations, suggesting GpsB activation of PBP2a activity. Consistent with this result, co-IP experiments showed that GpsB complexes with EzrA, StkP, PBP2a, PBP2b, and MreC in pneumococcal cells. Furthermore, depletion of GpsB prevents PBP2x migration to septal centers. These results support a model in which GpsB negatively regulates peripheral PG synthesis by PBP2b and positively regulates septal ring closure through its interactions with StkP-PBP2x.
Keywords: pneumococcal cell division, penicillin-binding protein (PBP) regulation, bacterial protein phosphorylation, bacterial protein phosphatases, DivIVA function
Graphical Abstract
Model of GpsB protein interactions and coordination of septal and peripheral peptidoglycan synthesis in Streptococcus pneumoniae, indicating changes in the PhpP protein phosphatase that suppress the requirement for essential GpsB.
ABBREVIATED SUMMARY
GpsB is a small hexameric protein that is essential and acts to balance septal and sidewall-like peptidoglycan synthesis at the midcell of the ovoid-shaped bacterium Streptococcus pneumoniae, which is a serious human respiratory pathogen. Results from genetic, microscopic, physiological, and in vivo co-immunoprecipitation experiments reported herein support a model in which GpsB activates penicillin-binding proteins involved in septal ring closure and inhibits penicillin-binding proteins involved in elongation to maintain normal ovococcus cell shape and size.
INTRODUCTION
GpsB has emerged as a major regulator of peptidoglycan (PG) biosynthesis in low-GC Gram-positive bacteria. GpsB contains a domain that is also found in DivIVA, which is a curvature-binding membrane protein that plays diverse roles in recruiting other proteins to the poles and division septa of rod-shaped bacteria (Claessen et al., 2008, Kaval & Halbedel, 2012, Massidda et al., 1998, Strahl & Hamoen, 2012, Tavares et al., 2008). However, despite this shared region of homology, GpsB plays distinctly different roles from DivIVA in regulating PG biosynthesis in rod-shaped bacteria, including Bacillus subtilis (Bsu) and Listeria monocytogenes (Lmo) (see (Cleverley et al., 2016, Rismondo et al., 2016)). GpsB was initially characterized in Bsu by a screen for mutations that are synthetically lethal with deletion mutations that eliminate EzrA (Claessen et al., 2008), which plays roles in modulating the dynamics of FtsZ-ring formation (see (Cleverley et al., 2014, Land et al., 2014)). In addition, ΔgpsB mutations are synthetically lethal with deletions that eliminate FtsA (Tavares et al., 2008), which anchors the FtsZ ring to cell membranes and recruits downstream proteins required to complete the cell division process (see (Busiek & Margolin, 2015, Ortiz et al., 2016)). Notably, mutations that eliminate GpsB, FtsA, or EzrA are singly not lethal in Bsu, although GpsB is required for growth in high-salt media (Claessen et al., 2008, Tavares et al., 2008).
Localization studies showed that GpsB assembles into the Bsu divisome at ≈ 20% of the cell cycle after FtsZ, FtsA, ZapA, and EzrA (Gamba et al., 2009, Tavares et al., 2008). In both Bsu and Lmo, GpsB is a dynamic protein that cycles between the septum and side wall PG biosynthetic machines (Claessen et al., 2008, Rismondo et al., 2016). Bacterial two-hybrid (B2H) analyses indicated that Bsu GpsB potentially interacts with EzrA and with Class A penicillin-binding protein PBP1 (aPBP1), which catalyzes both transglycosylase (TG) and transpeptidase (TP) activities, as well as the side-wall regulator MreC (Claessen et al., 2008). Consistent with these results, localization studies and characterization of ΔgpsB mutants showed that GpsB is required for normal localization and function of aPBP1 during the cell cycle and in pole maturation (Claessen et al., 2008). Together, these findings led to the model that GpsB acts as a switching protein that shuttles aPBP1 away from poles to carry out PG elongation in the sidewall, whereas EzrA returns the GpsB-aPBP1 complex to division septa (Claessen et al., 2008).
Recent studies have confirmed and further defined roles played by GpsB as a switch between modes of PG septal and elongation synthesis in rod-shaped Gram-positive species (Claessen et al., 2008, Rismondo et al., 2016). In Lmo, GpsB again was observed to dynamically locate between septal and peripheral PG synthesis sites (Rismondo et al., 2016). Lack of Lmo GpsB allowed normal growth at 30°C, retarded growth at 37°C and caused cell elongation, and prevented growth altogether at 42°C (Rismondo et al., 2016). Lmo GpsB is also required for full virulence in animal models of Lmo infection. Combination of ΔgpsB and ΔdivIVA mutations resulted in a synergistic, severe cell morphology defect that was dissimilar to that of either single Lmo mutant. Significantly, a direct interaction between Lmo GpsB and aPBPA1, the homologue of Bsu aPBP1, was inferred from a synthetic-lethal genetic relationship between ΔgpsB and ΔpbpA2, which itself is synthetically lethal with ΔpbpA1 (Rismondo et al., 2016). Direct binding of a negatively charged groove in the structure of the amino-terminal domain of GpsB to the positively charged cytoplasmic amino-terminal domain of aPBP1(A1) was demonstrated by B2H and biochemical assays (Rismondo et al., 2016). Together, these data support the hypothesis that GpsB acts as a switch protein to regulate aPBP1(A1) function and localization in rod-shaped bacteria (Claessen et al., 2008, Rismondo et al., 2016).
Structural studies show that GpsB is a cytoplasmic, membrane-associated hexamer that resembles a tripod structure (Cleverley et al., 2016, Rismondo et al., 2016). The amino terminal domains associate as a trimer of dimers, with each dimer containing amino acids required for domain dimerization, membrane binding, and binding to Bsu aPBP1 and Lmo aPBPA1 (Cleverley et al., 2016, Rismondo et al., 2016). The carboxyl-terminal domains associate as a dimer of homotrimeric, parallel coiled-coils. In Bsu, the carboxyl-terminal domains contain a threonine residue (T75) that is phosphorylated by the PrkC protein kinase, which does not seem to regulate Bsu cell division (Pompeo et al., 2015), but rather plays a role in sporulation (Shah et al., 2008). However, GpsB is required for PrkC kinase activity, and phosphorylation of GpsB may provide a negative feedback loop by reducing PrkC kinase activity (Pompeo et al., 2015).
Streptococcus pneumoniae (Spn) GpsB also likely plays a role in switching between septal and peripheral PG synthesis, which emanate from the midcells of these ovoid cells (Massidda et al., 2013, Land et al., 2013, Fleurie et al., 2014b). In primary, wild-type progenitor strains, such as virulent strains D39 and TIGR4 and isogenic unencapsulated (Δcps) derivatives of D39, GpsB is essential for growth (Land et al., 2013). Depletion of GpsB in D39 Δcps strains causes cultures to stop growing and eventually to lyse. GpsB-depleted cells elongate, enlarge, and contain multiple minimally constricted FtsZ and aPBP1a septal rings. These cell elongation and ring closure defects are consistent with a defect in controlling septal closure and cell elongation when GpsB is depleted (Land et al., 2013). Another study of the requirement for Spn GpsB was performed in unencapsulated laboratory strain R6, which contains at least 81 mutations not found in the D39 progenitor background (Lanie et al., 2007), and strain R800, which was derived from R6 (Fleurie et al., 2014b). In contrast to recently derived D39 strains, GpsB was not essential in the R6 and R800 backgrounds, suggesting accumulation of suppressors in these laboratory strains that allow ΔgpsB mutants to grow.
In addition, Spn R800 ΔgpsB mutants showed several phenotypes that contrasted with phenotypes of Bsu or Lmo ΔgpsB mutants. In the Spn R800 strain, ΔdivIVA mutations are epistatic to ΔgpsB mutations, in that the ΔdivIVA ΔgpsB double mutant shows the same defective cell morphology as the ΔdivIVA mutant compared to the ΔgpsB mutant (Fleurie et al., 2014b). This observation was the basis for a model that GpsB acts as a negative regulator of DivIVA stimulation of cellular elongation, possibly through phosphorylation of DivIVA by the StkP kinase (Fleurie et al., 2014b, Grangeasse, 2016). In contrast, this epistasis was not observed in Lmo, where ΔgpsB, ΔdivIVA, and ΔgpsB ΔdivIVA mutants show distinctively different defects in cell morphology (Rismondo et al., 2016). In Spn R800 GpsB was reported to be required for localization of the StkP Ser/Thr protein kinase into division rings, whereas in Bsu, GpsB was not required for localization of the homologous PrkC protein kinase (Pompeo et al., 2015). On the other hand, GpsB was required for optimal StkP and PrkC protein kinase activity in Spn and Lmo, respectively, suggesting that GpsB may positively regulate the activity of these protein kinases (Fleurie et al., 2014b, Pompeo et al., 2015). However, GpsB is phosphorylated by PrkC in Bsu (Pompeo et al., 2015), whereas phosphorylation of GpsB by StkP has not been detected in Spn (Fleurie et al., 2014b), and replacement of the putative phosphorylated threonine of Lmo GpsB with alanine did not produce a detectable phenotype (Cleverley et al., 2016).
In this study, we tested several of the above hypotheses of GpsB function in an unencapsulated derivative of the D39 progenitor strain and in two other laboratory strains, R6 and Rx1, which were separately derived from the R36A unencapsulated mutant of strain D39 (see (Lanie et al., 2007, Pozzi et al., 1996)). With the exception of a requirement of GpsB for maximal protein phosphorylation, we found phenotypes of R800 ΔgpsB mutants could not be generalized to D39 and the other laboratory strains. In support of an involvement of GpsB in maximizing protein phosphorylation mediated by the StkP protein kinase, we report that lethal ΔgpsB mutations in the D39 progenitor strain are suppressed by mutations that inactivate the cognate PhpP protein phosphatase. This suppression analysis also revealed a new level of control that obviates the requirements for GpsB and for protein phosphorylation. We further show that GpsB activates aPBP2a activity and is required for migration of bPBP2x to the centers of division septa. Co-immunoprecipitation (co-IP) of complexes of proteins crosslinked in cells showed that GpsB resides in complexes with EzrA, StkP, aPBP2a, bPBP2b, and MreC and that StkP is in complexes with bPBP2x, as anticipated from a previous report (Morlot et al., 2013). Together, our results suggest a modified model of GpsB function as a mediator between septal closure and peripheral PG synthesis that accounts for the formation of large, elongated cells with unconstricted septal rings. In this model, GpsB activates StkP-bPBP2x and aPBP2a activities to close septal division rings and acts as a negative regulator of PG elongation by inhibition of bPBP2b and MreC activities.
RESULTS
divIVA mutations are not epistatic to gpsB mutations in pneumococcal strains R6 and D39
It was previously reported in laboratory strain R800 that ΔdivIVA mutations are epistatic to ΔgpsB mutations (Fleurie et al., 2014b). However, GpsB is not essential in unencapsulated laboratory strain R800 (Fleurie et al., 2014b), whereas GpsB is essential in its wild-type, encapsulated, progenitor strain, D39 (Fleurie et al., 2014b, Land et al., 2013). These results suggest that domesticated laboratory strain R800 contains mutations that suppress primary phenotypes caused by ΔgpsB deletions and by mutations in other cell division genes (see (Land et al., 2014, Land & Winkler, 2011, Tsui et al., 2016)). As another example of these strain differences, changes of the single threonine residue in DivIVA (T201A) phosphorylated by StkP result in no morphology phenotype in D39 and two unencapsulated laboratory strains, R6 and Rx1, that were distantly derived from D39 (data not shown; Massidda et al., 2013, Straume et al., 2016), but cause cell elongation and polar bulges in R800 (Fleurie et al., 2012). The normal frequency of transformation and colony morphology observed during construction of divIVA(T201A) mutants in D39 strains are not indicative of suppressor accumulation (data not shown).
Consequently, we tested whether the epistatic genetic relationship between gpsB and divIVA mutations reported in strain R800 is generalizable to progenitor strain D39 and other laboratory strains. We confirmed that laboratory strain R6 readily tolerates a ΔgpsB deletion, similar to R800 (Fleurie et al., 2014b), whereas an Rx1 ΔgpsB mutant grows poorly (Table 1, lines 20 and 21). This result is consistent with accumulation of different combinations of mutations in different lines of laboratory strains derived from the D39 progenitor strain (Lanie et al., 2007). In contrast, encapsulated strain D39 and isogenic unencapsulated D39 Δcps derivatives do not grow when transformed with ΔgpsB mutations (Table 1, lines 1, 9, 13, and 17) (Fleurie et al., 2014b, Land et al., 2013). We next compared the cell morphologies of the R6 parent, the single R6 ΔgpsB or ΔdivIVA mutant, and the R6 ΔgpsB ΔdivIVA double mutant grown exponentially and stained with a fluorescent D-amino acid (FDAA) probe, which indicates regions in the peptidoglycan (PG) of active penicillin-binding protein (PBP) TP activity (Fig. 1A and 1B) (Boersma et al., 2015, Kuru et al., 2012, Kuru et al., 2015). Because septal and peripheral (sidewall-like) PG synthesis are coordinated with FtsZ ring formation in Spn, FDAA-labeled bands serve as a surrogate for FtsZ ring assembly and localization (see (Boersma et al., 2015, Tsui et al., 2014)).
TABLE 1.
Transformation efficiencies with a ΔgpsB<>aad9 amplicon and colony sizes of recipient and transformant strainsa
| Recipient strain and condition |
Genotype of recipient (colony size relative to wild-type (WT) parent strain) |
Number of ΔgpsB<>aad9 transformants at 24 h (colony size after streaking; strain) |
|---|---|---|
| IU1945 (D39 Δcps) genetic background | ||
| 1. IU1945 | WT | 0b |
| 2. IU4846 − fucose | ΔbgaA::PfcsK-gpsB (WT) | <20 (variable; Land et. al, 2013) |
| 3. IU4846 + fucosec | ΔbgaA::PfcsK-gpsB (WT) | >500 (WT; Land et. al, 2013) |
| 4. IU11442 | ΔphpP::Pc-erm (small) | 20–100 (medium; IU11508)d |
| 5. IU7922 | ΔstkP::Pc-[kan-rpsL+] (medium) | 20–100 (medium: IU10109)d |
| 6. IU11460 | ΔstkP::Pc-erm (medium) | 20–100 (medium; IU11546)d |
| 7. IU11462 | Δ[phpP-stkP]::Pc-erm (medium) | 20–100 (medium; IU11512)d |
| 8. K739 | Δ[phpP-stkP]::Pc-[kan-rpsL+] (medium) |
20–100 (medium; IU10107)d |
| IU1824 (D39 Δcps rpsL1) genetic background | ||
| 9. IU1824 | WT | 0 |
| 10. IU7685 |
phpP(G229D) stkP(G10stop) (medium) |
≈ 200 (medium; IU7733) |
| 11. IU10423 | phpP(G229D) (tiny) | ≈ 100 (medium; IU11346) |
| 12. IU11223 | phpP(D192A) (tiny) | >500 (medium; IU11348) |
| IU1690 (D39 cps+) genetic background | ||
| 13. IU1690 | WT | 0e |
| 14. IU11183 | ΔphpP::Pc-erm (small) | 16–20 (medium; IU11350) |
| 15. IU11456 | ΔstkP::Pc-erm (medium) | ≈ 30 (medium; IU11504) |
| 16. IU11458 | Δ[phpP-stkP]::Pc-erm (medium) | ≈ 30 (medium; IU11506) |
| IU1781 (D39 cps+ rpsL1) genetic background | ||
| 17. IU1781 | WT | 0 |
| 18. IU11195 | phpP(G229D) (tiny) | ≈ 30 (small; IU11352) |
| 19. IU11227 | phpP(D192A) (tiny) | ≈ 30 (small; IU11354) |
| R6 or Rx1 genetic background | ||
| 20. R6 (EL59) | WT | >500 (medium; IU8224) |
| 21. Rx1 (IU9256) | WT | ≈ 300 (tiny; IU11574)f |
Recipient strains are described in Table S1. Transformations and visualization of colonies were performed as described in Experimental procedures. The numbers of colonies are normalized to 1 mL of transformation mixture. Similar results were obtained for each D39 Δcps, Rx1, or R6 strain from at least two independent transformation experiments in which multiple isolates were examined. Transformation experiments of D39 cps+ strains was performed once.
0 to <10 colonies were visible after 24 h of incubation from >20 independent transformations. Suppressor strain IU5845 appeared ≈ 24 h after transformation, and IU6441 and IU6442 appeared after ≈ 40 h (see text and Table S1). >500 colonies were obtained for transformations of IU1945 with a ΔpurR<>aad9 control amplicon.
0.8% (wt/vol) L-fucose was added to all steps in the transformation procedure to induce gpsB+ expression in merodiploid strain IU4846 (Land et al., 2013).
Numbers of transformants obtained for ΔphpP or ΔstkP mutants were similar for the ΔgpsB<>aad9 test and ΔpurR<>aad9 control amplicons within experiments, but varied in independent transformations. This variability is consistent with lower and variable transformation efficiency and recovery of ΔphpP or ΔstkP mutants reported previously (Echenique et al., 2004, Saskova et al., 2007).
≈ 80 colonies were obtained for transformation of strain IU1690 with control amplicon ΔhtrA::Pc-erm.
Strain IU11574 (Rx1 ΔgpsB<>aad9) was isolated from a tiny colony 24 h after transformation of strain Rx1.
Fig. 1.

ΔdivIVA mutations are not epistatic to ΔgpsB in pneumococcal strains R6 and D39. A) Representative growth curves of R6 strains. Isogenic strains listed as 1–4 are: 1, R6 (EL59); 2, R6 ΔgpsB (IU8224); 3, R6 ΔdivIVA (IU8371); 4, R6 ΔgpsB ΔdivIVA (IU8369). Average doubling times (±SEM) from 2 independent experiments were calculated for OD620 ≈ 0.015 to 0.2 using a nonlinear regression exponential growth curve program (GraphPad Prism). B) Fluorescent D-amino acid (FDAA) staining and microscopy of live cells labeled with FDAA for 5 min were performed as described in Experimental procedures. The panels shown from left to right are: phase, FDAA, and a phase/FDAA overlay. Genotypes are indicated according to the numbers in panels A and C. Representative images are shown of ≥95% of the cells (n>50 for R6 strains; n>70 for D39 strains) examined manually of each strain. C) Representative growth curves of D39 Δcps strains and D39 Δcps ΔgpsB sup1 strains, which contain a suppressor mutation (phpP(G229D)) of ΔgpsB as described in the text. Isogenic strains listed as 5–8 are: 5, D39 Δcps (IU1945); 6, D39 Δcps ΔgpsB phpP(G229D) (IU6442); 7, D39 Δcps ΔdivIVA (IU8496); 8, D39 Δcps ΔgpsB phpP(G229D) ΔdivIVA (IU11205). Doubling times were calculated as described above. Independent experiments were performed two to three times with similar results. Scale bar = 1 micron.
The R6 ΔgpsB mutant grew at the same rate, but formed larger, elongated cells, often with multiple FDAA-labeled rings, compared to the R6 parent (Fig. 1A and 1B, row 2). The vast majority (≥95%) of FDAA rings were parallel to each other and perpendicular to the long axis in R6 ΔgpsB cells, in contrast to R800 ΔgpsB cells, in which ≈ 25% of FDAA staining was reported in “Z-like” spiral patterns (Fleurie et al., 2014b). The R6 ΔdivIVA mutant formed chains of large compacted, spherical cells containing parallel FDAA-labeled rings (Fig. 1B, row 3), as reported before (Fadda et al., 2003, Fadda et al., 2007). The R6 ΔgpsB ΔdivIVA mutant grew considerably slower and formed large, elongated, misshapen cells with thick rings of FDAA labeling (Fig. 1A and 1B, row 4). We conclude that the ΔgpsB ΔdivIVA mutant has a severe morphological defect distinct from those of either the ΔgpsB or ΔdivIVA mutant and that the ΔdivIVA mutation is not epistatic to the ΔgpsB mutation in laboratory strain R6. In addition, the ΔgpsB mutation did not cause severe FDAA ring mislocalization in the R6 strain, as reported in the R800 strain (Fleurie et al., 2014b).
These conclusions were confirmed by similar experiments in the D39 progenitor genetic background and in the Rx1 laboratory strain, which exhibits a mutator phenotype (Table S1). D39 ΔgpsB and Rx1 ΔgpsB mutants do not grow or grow poorly (Table 1, lines 1, 9, 13, 17, and 21), respectively, and acquire suppressor (sup) mutations, which are described and characterized below (Table 2). In either background, the ΔgpsB (sup) mutants grew similarly to the parent strains and formed cells containing parallel FDAA-labeled rings (below; Fig. 1C and 1B, rows 5 and 6; Fig. S1, rows 1 and 2). The Rx1 ΔgpsB sup4 mutant formed longer cells with multiple division rings compared to the Rx1 parent strain (Fig. S1, rows 1 and 2), and >95% of Rx1 ΔgpsB sup4 cells expressing FtsZ-mCherry from a single chromosomal locus had parallel FtsZ rings perpendicular to their long axis (data not shown), similar to the pattern of FDAA labeling (Fig. S1, row 2). In both the D39 Δcps and Rx1 backgrounds, ΔdivIVA mutants showed the expected phenotype of chains of enlarged, rounded cells (Fig. 1B, row 7 and Fig. S1, row 3). Again, ΔgpsB (sup) ΔdivIVA double mutants of the D39 Δcps and Rx1 strains showed the distinct, severe defects in cell morphology described above for the R6 strain (Fig. 1B, rows 4 and 8; Fig. S1B row 4). Similar to the R6 double mutant, D39 Δcps double mutants had reduced growth rates (Fig. 1C, rows 6–8). Interestingly, the Rx1 double mutant did not have a reduced growth rate compared to the Rx1 single mutants, but was delayed in stationary lysis (Supporting Information Fig S1A, rows 2–4). We conclude that an epistatic genetic relationship between gpsB and divIVA mutations does not exist in the D39 progenitor strain or in laboratory strains, other than R800, and that there is no genetic evidence that GpsB acts as a negative regulator of DivIVA stimulation of cellular elongation (Fleurie et al., 2014b).
Table 2.
Analysis of spontaneous ΔgpsB suppressor mutations that arose in unencapsulated derivatives of strain D39 and Rx1a
| Strain number (suppressor designation) |
Genetic background |
Genotype | Large deletion/ duplication present?b |
Phosphorylation phenotypec |
Other mutations present |
|---|---|---|---|---|---|
| 1. IU6442 (sup1)d |
D39 Δcps ΔgpsB |
phpP(G229D) | No | Normal | None detected; strain reconstructed (see Fig. 4) |
| 2. IU5845 (sup2)e |
Δ[spd_1026– spd_1037] Ω[spd_0889– spd_1026] |
Yes | None |
araD(ΔC at aa 123/224) zmpB(Q456P) miaA(N172K) |
|
| 3. IU6441 (sup3)e |
Δ[spd_1029– spd_1037] Ω[spd_0889– spd_1024] |
Yes | None | None detected | |
| 4. IU9262 (sup4)f |
Rx1 ΔgpsB | phpP(L148S) | No | Normal |
stkP(I102T) Δ[spd_1037– spd_1038] |
| 5. Additional classes of ΔgpsB suppressorsg | |||||
Transformations were performed as described in Experimental procedures. Control transformations with a ΔpurR<>aad9 amplicons gave >500 colonies in 24 h, whereas ΔgpsB<>aad9 transformations gave <10 colonies in 48 h (see Table 1, line 1). Mutations in the sup1–3 suppressors were located by whole-genome sequencing (see Experimental procedures) and are listed in columns 3 and 6. IU6442, IU5845, and IU6441 grew with similar doubling times (≈ 30 min) as the parent strain in BHI broth, but the growth yields of IU5845 and IU6441 were about 25% lower than those of IU6442 and the parent (Fig. 5A; data not shown).
Chromosomal deletions/duplications Δ[spd_1026–spd_1037] (13 genes)/Ω[spd_0889–spd_1026] (134 genes) and Δ[spd_1029–spd_1037]/Ω[spd_0889–spd_1024] are depicted in Figure S3A and 3B.
Detection of proteins phosphorylated at Thr residues was performed by Western blotting using α-pThr antibody as described in Experimental procedures. See Results and Fig. 3 for details.
ΔgpsB suppressor mutants containing phpP(G117D) (IU6444), phpP(T163P) (IU7736), or phpP(R125P) (IU11955) mutations were independently isolated and identified by conventional DNA sequencing of phpP-stkP, but were not further characterized in this study. These mutants contained wild-type stkP+ and lacked deletion of the spd_1034 region, as determined by PCR (Fig. S3).
Deletion in the spd_1034 region was detected by PCR in 15 additional, independently isolated ΔgpsB suppressor mutants that were not characterized further in this study.
sup4(phpP(L148S)) arose spontaneously in Rx1, which also contained the stkP(I102T) and Δ[spd_1037–spd_1038] mutations, which were confirmed by conventional DNA sequencing and PCR, respectively.
>5 independent, spontaneous ΔgpsB suppressors were isolated that are phpP+ stkP+ and lack deletion in the spd_1034 region of the chromosome. These suppressors were not further characterized in this study.
ΔgpsB mutations are suppressed by phpP Ser/Thr phosphatase mutations in strain D39
Transformation of a D39 Δcps strain with a ΔgpsB amplicon infrequently led to the appearance of faster growing suppressor mutants (Table 2, lines 1–3 and 5). We sequenced the genomes of three independently isolated, spontaneous D39 Δcps ΔgpsB suppressor mutants, which had similar growth rates as the parent strain in BHI broth (Table 2, lines 1–3). One of the suppressor mutants contained a single amino acid change (G229D) in phpP, which encodes the only canonical PP2C Ser/Thr phosphatase encoded by Spn (Table 2, line 1; Fig S2A) (Beilharz et al., 2012, Novakova et al., 2005, Osaki et al., 2009). The other two sup2 and sup3 suppressor strains had intact copies of the stkP+ and phpP+ genes, but contained large deletions and adjacent duplications in the spd_1034 region of the chromosome (Fig. S3). Additional independent ΔgpsB suppressors were isolated containing mutations in phpP, including phpP(L148S) in strain Rx1, or deletions in the spd_1034 region of the chromosome, as well as other classes of ΔgpsB suppressor strains that lack the latter mutations (Table 2, lines 4 and 5). This paper focuses primarily on the phpP(G229D) and related suppressor mutations.
The previous characterization of R800 ΔgpsB mutants reported that protein phosphorylation was eliminated in the absence of GpsB (Fleurie et al., 2014b). The isolation of the phpP(G229D) and other putative suppressor mutations in phpP (Table 2, lines 1 and 4) led to the hypothesis that growth of the D39 Δcps ΔgpsB mutant was accompanied by a decrease in protein de-phosphorylation that restored protein phosphorylation levels in the ΔgpsB mutant. Given the major phenotypic differences described above for laboratory strain R800, we first determined the effects of GpsB absence or depletion in the R6 and Rx1 or D39 backgrounds, respectively (Fig. 2). A ΔgpsB mutation reduced phosphorylation of DivIVA or StkP/MapZ(LocZ), which were not resolved on these gels, by ≈3–5× or ≈2–5×, respectively, compared to background levels in the Rx1 and R6 strains (Fig. 2A, lanes 3 and 5; Table S5). Likewise, depletion of GpsB in strain D39 caused ≈ 3–5× reduction in protein phosphorylation (Fig. 2B, lanes 1–2 versus 7–9; Table S6). Consistent with the above hypothesis, protein phosphorylation levels were restored to slightly above the wild-type level in the suppressed D39 ΔgpsB sup1 (phpP(G229D)) mutant (Fig. 3, lane 4; Table S7). Likewise, protein phosphorylation was restored in the fast-growing Rx1 ΔgpsB sup4 suppressed strain (Fig. 2A, lane 2; Table S5), containing the spontaneous phpP(L148S) mutation (Table 2, line 4; Fig. S2A). Moreover, purified PhpP(G229D) and PhpP(L148S) lacked protein phosphatase activity in biochemical assays (Fig. S4), but were properly folded compared to wild-type PhpP based on circular dichroism (CD) spectra and thermal denaturation analyses (data not shown). Unexpectedly, the D39 ΔgpsB sup2 and sup3 deletion mutants (Table 2) completely lacked protein phosphorylation (Fig. 3, lanes 2 and 3), despite encoding an intact phpP+-stkP+ operon.
Fig. 2.
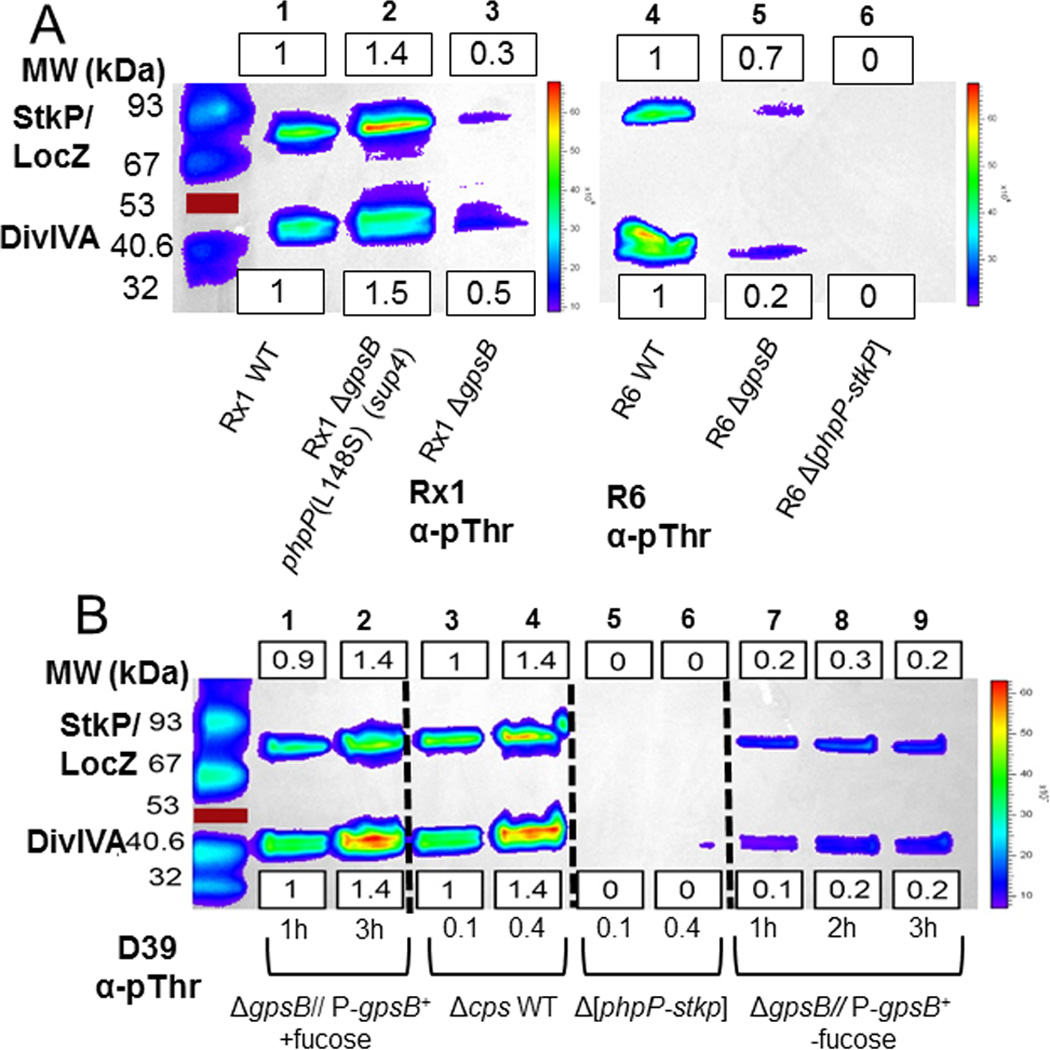
GpsB deletion or depletion results in decreased threonine (Thr) phosphorylation of proteins. Proteins phosphorylated on Thr residues were detected by Western blotting with α-pThr antibody, and blots were imaged and quantitated as described in Experimental procedures. Equal protein loading was confirmed by amido-black staining of membranes after blotting. Representative blots are shown, with background-subtracted luminescence values relative to the wild-type (wt) strain indicated for the StkP/LocZ and DivIVA bands in boxes above or below the blots, respectively. Relative values of band intensities were calculated as described in Experimental procedures, and mean relative intensities (±SEM) are compiled for all experiments in Table S5 and S6. A) ΔgpsB mutants of laboratory strains Rx1 and R6 harvested at OD620 ≈ 0.4. Lane 1, wild-type Rx1 parent (IU9256); lane 2, suppressed Rx1 ΔgpsB phpP(L148S) (IU9262); lane 3, Rx1 ΔgpsB phpP+ stkP+ (IU11574); lane 4 wild-type R6 parent (EL59); lane 5, R6 ΔgpsB (IU8224); and lane 6, R6 Δ[phpP-stkP] control (IU8419). The experiment was performed twice independently with similar results. B) GpsB depletion of D39 Δcps strains as described in Experimental procedures. Lane 1–2, merodiploid strain Δcps ΔgpsB//PfcsK-gpsB+ (IU4888) grown with fucose for the times indicated; lane 3–4, wild-type parent strain D39 Δcps (IU1945) grown to OD620 ≈ 0.1 or 0.4; lanes 5–6, D39 Δcps Δ[phpP-stkP] (E739), and lanes, 7–9, merodiploid strain Δcps ΔgpsB//PfcsK-gpsB+ (IU4888) incubated without fucose for the times indicated. The experiment was performed three times independently with similar results. See text for additional details. Red lines marks a colored 53 kDa standard that did not transfer.
Fig. 3.

phpP(G229D) restores wild-type levels of protein phosphorylation to D39 Δcps ΔgpsB mutants in originally isolated and reconstructed suppressor strains. A representative Western blot of phosphorylated proteins was performed and quantitated as described in Figure 2 and Experimental procedures. Mean relative values (±SEM) of band intensities are compiled for all experiments in Table S7. Strains were harvested at OD620 ≈ 0.4. Lane 1, wild-type parent D39 Δcps (IU1945); lane 2, D39 Δcps ΔgpsB Δ[spd_1026–spd_1037] Ω[spd_0889–spd_1026] (IU5845, sup2) (Table 2, line 2); lane 3, D39 Δcps ΔgpsB Δ[spd_1026–spd_1037] Ω[spd_0889–spd_1026] (IU6441, sup3); (Table 2, line 3); lane 4, D39 Δcps ΔgpsB phpP(G229D) (IU6442, sup1) (Table 2, line 1); Lane 5, D39 Δcps ΔbgaA::Pc-erm (E46) (strain used in reconstruction; Fig. 4B); lane 6, D39 Δcps ΔbgaA::Pc-erm ΔgpsB phpP(G229D) (IU11221) (reconstructed suppressor; Fig. 4B); lane 7, D39 Δcps ΔphpP-Pc-erm (IU11442) (polar mutant with reduced StkP expression); lane 8, D39 Δcps ΔstkP (IU11460) control; and lane 9, D39 Δcps Δ[phpP-stkP] (IU11462) control. The experiment was performed twice independently with similar results. The red line marks a colored 53 kDa standard that did not transfer.
To confirm that the phpP(G229D) mutation is solely responsible for ΔgpsB suppression in the sup1 strain, we reconstructed the suppressed strain by two different genetic schemes (Fig. 4). Conserved G229 points toward R13 and the Mn2+ binding pocket in the predicted active site of PhpP (Rantanen et al., 2007), and purified PhpP(G229D) lacks protein phosphatase activity (Fig. S4). In these schemes, we included another phpP(D192A) allele that inactivates PhpP function by interfering with Mn2+ binding in the active site (Fig. S4 and S5) (Novakova et al., 2005, Rantanen et al., 2007). In the first scheme (Fig. 4A), allele exchange (Sung et al., 2001) was used to replace a kan-rpsL+ Janus cassette inserted between phpP+ and stkP+ (Fig. S2B) with the phpP(G229D) or phpP(D192A) mutation in an unencapsulated derivative of strain D39. Notably, phpP(G229D) stkP+ and phpP(D192A) stkP+ mutants corresponded to tiny-colony transformants, whereas phpP+ stkP+ recombinants or a spontaneous phpP(G229D) stkP(G10stop) mutant formed normal- or medium-size colonies, respectively (Fig. 4A and Table 1, lines 10–12). Transformation of the slow-growing phpP(G229D) stkP+ and phpP(D192A) stkP+ mutants with a ΔgpsB amplicon was tolerated and resulted in faster growing colonies than the phpP mutant strains (Fig. 4A; Table 1, lines 11–12). Similar slow growth and restoration by a ΔgpsB mutation were observed for encapsulated D39 phpP(G229D) stkP+ and phpP(D192A) stkP+ mutants (Table 1, lines 18–19). Finally, the ΔgpsB mutation restored growth to a ΔphpP::Pc-erm mutant (Table 1, lines 4 and 14). The ΔphpP::Pc-erm mutation was polar and considerably reduced the amount of StkP (≈ 10×) (Fig. S7B, last lane), possibly by uncoupling translation between phpP and downstream stkP (Fig. S2A). The decreased amount of StkP without PhpP lowered the amount of phosphorylated StkP/LocZ detected, whereas a wild-type level of phosphorylated DivIVA was maintained in gpsB+ and ΔgpsB strains (Fig. 3, lane 7; data not shown), likely because of repeated catalytic rounds of DivIVA phosphorylation by StkP.
Fig. 4.

Scheme for reconstruction of phpP(G229D) or phpP(D192A) mutants in D39 Δcps rpsL1 gpsB+ and D39 Δcps gpsB+ genetic backgrounds. See Experimental procedures for details. These strains were constructed twice with similar results, and resulting strains are listed in Table S1.
Since non-polar phpP(G229D) stkP+ and phpP(D192A) stkP+ mutants exhibit a severe growth defect, there is the possibility of selecting for additional suppressor mutations before growth is stabilized by transformation with a ΔgpsB mutation. We suspect that the normal growth reported previously for non-polar D39 ΔphpP mutants (Agarwal et al., 2012) was due to suppressor accumulation, because D39 Δcps phpP(G229D) stkP+ or phpP(D192A) stkP+ mutants exhibited improved growth upon repeated culturing (data not shown). To avoid suppressor accumulation, we devised a second genetic scheme to test suppression of a ΔgpsB mutation by the phpP(G229D) or phpP(D192A) mutations (Fig. 4B). In this scheme, a ΔgpsB // PfcsK-gpsB+ merodiploid was depleted for GpsB expression by removing the inducer fucose at the same time as transformation with a phpP(G229D) or phpP(D192A) amplicon. Transformants, which were maintained without fucose to deplete GpsB, each contained only the phpP(G229D) or phpP(D192A) mutation, but not the phpP+ recombinant, and remained stkP+ (Fig. 4B). The resulting strains, which were minimally stressed during construction, were converted to ΔgpsB by removing the ectopic copy of PfcsK-gpsB+. The reconstructed ΔgpsB phpP(G229D) mutant grew similarly to its parent and the original ΔgpsB sup1 mutant (Fig. 5A). We observed that the lengths and widths of these ΔgpsB phpP(G229D) mutant cells were shorter than those of the wild-type parent strains, resulting in small cells with similar aspect ratios as the wild-type parent strains (Fig. 5B and 5C). Western blotting showed that cells of reconstructed ΔgpsB phpP(G229D) and ΔgpsB phpP(D192A) mutants contained approximately the same amounts of PhpP and StkP protein as their isogenic parent strains (Fig. S6). Finally, the reconstructed ΔgpsB phpP(G229D) mutant showed a similar restoration of protein phosphorylation as the original ΔgpsB sup1 (phpP(G229D)) mutant (Fig. 3, lanes 1, 4–6). We conclude that single-amino acid changes that inactivate PhpP protein phosphatase can restore the growth of ΔgpsB mutants, probably by restoring protein phosphorylation decreased by the absence of GpsB.
Fig. 5.

D39 Δcps ΔgpsB phpP(G229D) suppressor strains grow similarly to wild-type parent strains, but form slightly rounder, smaller-sized cells. A) Representative growth curves of the originally isolated ΔgpsB sup1 (phpP(G229D)) (Table 2, line 1) and reconstructed strain (Fig. 4B). Strain 1, D39 Δcps wild-type parent (IU1945); strain 2, original ΔgpsB sup1 strain (D39 Δcps ΔgpsB phpP(G229D); IU6442, Table 2, line 1); strain 3, E46 used in reconstruction (D39 Δcps ΔbgaA::Pc-erm; Fig. 4B); strain 4, reconstructed suppressor (D39 Δcps ΔgpsB phpP(G229D) ΔbgaA::Pc-erm; IU11221; Fig. 4B). Doubling times were calculated as described for Figure 1. B) Representative images of live cells at OD620 ≈ 0.1–0.2 from growth curves in panel A. C) Length, width, aspect ratio, and cell volume relative to the median value of IU1945 for each strain listed in panel A. Over 50 cells were measured per strain in two experimental replicates. Scale bar = 1 micron.
ΔgpsB mutations are suppressed by additional classes of mutations
One class of ΔgpsB suppressors (sup2 and sup3, Table 2, lines 2 and 3) contained wild-type phpP+ and stkP+ genes and large deletion-insertions in the spd_1034 region of the chromosome (Fig. S3); yet, these strains lacked detectable protein phosphorylation (Fig. 3, lanes 2–3), despite expressing nearly wild-type levels of the StkP and PhpP proteins (data not shown). None of the genes in the two large deletions had obvious functions in protein phosphorylation or de-phosphorylation (Fig. S3, Table S8). A constructed Δ[spd_1029–spd_1037] deletion, similar to the one in the ΔgpsB sup3 suppressed strain (Table 2, line 3; Fig. S3), did not alter protein phosphorylation levels compared to the parent strain or restore growth to a ΔgpsB mutant (Fig. S8; data not shown). The large chromosomal duplications adjacent to the deletions (Fig. S3) encode one reading frame annotated as a putative phosphoserine phosphatase (SPD_RS05380) (Table S8). However, overexpression of SPD_RS05380 by itself from a zinc-inducible promoter (strain IU12059, Table S1) did not lead to a detectable change in protein phosphorylation levels or suppress ΔgpsB in the D39 background (data not shown). We conclude that some combination of gene expression changes in these complicated insertion-duplication mutants likely caused the unanticipated lack of protein phosphorylation and compensated for the lack of GpsB in the sup2 and sup3 suppressor strains.
The lack of protein phosphorylation in the ΔgpsB sup2 and ΔgpsB sup3 mutants prompted us to test whether the growth defect of ΔgpsB mutants could be suppressed by the absence of protein phosphorylation in stkP mutants. We could transform ΔgpsB amplicons into ΔstkP, ΔphpP-stkP, and phpP(G229D) stkP(G10stop) mutants in the unencapsulated and encapsulated D39 genetic background (Table 1, lines 5–8, 10, and 15–16). The variable range of transformant recovery in the ΔphpP, ΔstkP, and ΔphpP-stkP mutants (Table 1) was consistent with a previous report (Echenique et al., 2004), and Western-blot controls confirmed the lack of the StkP and PhpP proteins and protein phosphorylation in these mutants (Fig. 2, 3, and S7). However, interpretation of these results was problematic, because the ΔstkP, ΔphpP-stkP, and phpP(G229D) stkP(G10stop) mutants in strain D39 are genetically unstable (see (Massidda et al., 2013)). Upon restreaking and regrowth, these mutants show heterogeneous colony sizes and faster growth properties, indicative of suppressor accumulation (data not shown). Therefore, it is likely that the D39 ΔstkP mutants accumulated additional suppressor mutations that bypassed the requirement for GpsB. In this study, we did not identify these putative suppressors or how they could suppress the requirement for GpsB.
GpsB and StkP have different, but overlapping localization patterns at each division stage, and GpsB is not required for StkP localization in septal rings
To understand the relationship between GpsB and StkP detected in the genetic experiments described above, we performed 2D IFM to localize GpsB-FLAG or GpsB-L-FLAG3 relative to StkP and StkP-HA in the same cells (Fig. 6) Previously we used 2D IFM to localize GpsB-FLAG relative to FtsZ-Myc in pneumococcal D39 cells at different stages of division (Land et al., 2013). The strain expressing GpsB-FLAG exhibited growth (35 min doubling time; equal growth yield) and cell morphology comparable to the parent strain (Fig. 6A and 6B), in contrast to a GFP-GpsB fusion reported in the R800 background (Fleurie et al., 2014b). A further indication that Spn GpsB-FLAG is nearly fully functional is that amino acid changes in the C-terminal domain of GpsB cause loss of GpsB function (unpublished result; Cleverley et al, 2016). The GpsB-L-FLAG3-expressing strain grew like the parent strain, but formed cells that were slightly longer (1.1×) than those of the parent (Fig. 6A and 6C). As reported previously (Land et al., 2013), GpsB localization is somewhat diffuse at early division stages, localizes over the hemispheres of newly divided cells, persists at division septa, and is diffuse around the equators of new daughter cells (Fig. 6B, 6C, 6E, and 6F). This localization pattern overlaps, but is different from that of FtsZ, which exhibits distinct rings in early-division cells, and leaves septa before PBPs and other proteins for the equators of the daughter cells (Land et al., 2013, Tsui et al., 2014), presumably directed by MapZ(LocZ) (Fleurie et al., 2014a, Holečková et al., 2015). Consistent with a previous report (Tsui et al., 2014), StkP localizes differently from either FtsZ or GpsB, and remains at closing septa with PBPs after FtsZ and GpsB have started to move to the equators of the daughter cells (Fig. 6A–6E). We conclude that GpsB and StkP have different localization patterns that overlap at each stage of the cell cycle.
Fig. 6.

GpsB and StkP have different, but overlapping localization patterns at each division stage. Comparison of GpsB and StkP localization by immunofluorescence (IFM) of double- and single-tagged strains and image averaging and quantitation were performed as detailed in Experimental procedures. Averaged IFM images of the indicated number of cells at each division stage (n) and fluorescence intensity traces of protein localization are shown for the following strains: A) IU1945 (D39 Δcps) probed with DAPI and anti-StkP antibody; B) IU5838 (D39 Δcps gpsB-FLAG) probed with anti-FLAG and anti-StkP antibodies; C) IU5458 (D39 Δcps gpsB-L-FLAG3) probed with anti-FLAG and anti-StkP antibodies; D) IU7438 (D39 Δcps stkP-HA) probed with DAPI and anti-HA antibody; E) IU11716 (D39 Δcps gpsB-FLAG stkP-HA) probed with anti-FLAG and anti-HA antibody; and F) IU11412 (D39 Δcps gpsB-L-FLAG3 stkP-HA) probed with anti-FLAG and anti-HA antibodies. See text for additional information.
Consistent with this partial difference in localization, we did not find that GpsB is required for StkP localization to division rings in the R6 and Rx1 laboratory strains and in the progenitor D39 genetic background (Fig. 7 and S9). In the R800 laboratory strain, the absence of GpsB led to diffuse, mislocalization of StkP to cell peripheries instead of to division rings (Fleurie et al., 2014b). In contrast, StkP-FLAG2 localized to rings in a large majority of R6 ΔgpsB cells (Fig. 7, lines 1–2) and in D39 Δcps cells depleted for GpsB (Fig. 7, lines 3–4). Similarly, GFP-StkP localized in defined bands in the majority of Rx1 ΔgpsB cells containing the phpP(L148S) suppressor mutation (Fig. S9B). Thus, StkP still localizes in the majority of cells to rings perpendicular to the long axis of elongated, sometimes irregularly shaped cells lacking or depleted for GpsB. On the other hand, we did notice that GpsB-L-FLAG3, but not GpsB-FLAG, causes some mislocalization of StkP-HA that was not observed when StkP was detected directly with anti-StkP antibody (Fig. 6F compared to Fig. 6B–6E). Moreover, StkP occasionally was detected in aberrant patches away from division rings in elongated cells lacking or depleted for GpsB (Fig. 7, rows 2 and 4; Fig. S9B). Taken together, these results indicate that GpsB is not necessary for StkP ring formation, but GpsB and StkP are likely in a complex together during at least one stage of cell division. This conclusion was supported by in vivo co-IP experiments presented below.
Fig.7.

2D IFM microscopy demonstrates that the absence or depletion of GpsB does not abolish StkP ring formation. 2D IFM was performed as outlined in Experimental procedures. Panels shown from left to right are: phase, FITC antibody labeled FLAG-tagged StkP, and phase/FITC overlay. 1) R6 stkP-FLAG2 (IU8819, sampled at OD620 ≈ 0.2); 2) R6 ΔgpsB stkP-FLAG2 (IU8311, sampled at OD620 ≈ 0.2); 3) D39 Δcps stkP-FLAG2 (IU7434, sampled at OD620 ≈ 0.2); 4) merodiploid strain D39 Δcps stkP-FLAG2 Δcps ΔgpsB//PfcsK-gpsB+ (IU8230) grown for 2 h with fucose addition or without fucose for 2 h or 3 h to deplete GpsB, eventually causing cell lysis. Representative images of each strain are shown for each experiment, which were performed three times independently with similar results. Percentages of cells with StkP rings are based on 100 manually examined cells of each strain. Scale bar = 1 micron.
ΔgpsB and Δpbp1a mutations are synthetically lethal in Spn
Besides possible interactions between GpsB and StkP, recent genetic and biochemical experiments demonstrate that GpsB controls and is required for aPBPA1, but not aPBPA2, activity in Lmo (Introduction) (Cleverley et al., 2016, Rismondo et al., 2016). Similar to Lmo, either Spn aPBP1a or aPBP2a is required for growth, and they cannot be inactivated at the same time (Hoskins et al., 1999, Paik et al., 1999). However, aPBP1a and aPBP2a are not functionally equivalent as illustrated by the markedly smaller cells formed by Δpbp1a compared to Δpbp2a mutants (Land & Winkler, 2011, Tsui et al., 2016). Based on the precedent from Lmo, we tested for a synthetic lethal relationship between GpsB and the three aPBPs of Spn (Table 3). In the R6, Rx1, and the suppressed D39 ΔgpsB phpP(G229D) strains, there is a clear synthetic lethal genetic relationship between ΔgpsB and Δpbp1a mutations, but not Δpbp2a or Δpbp1b mutations (Table 3). Thus, either aPBP1a or aPBP2a is required for viability. Likewise, either aPBP1a or GpsB is required for viability, implying that GpsB is required for aPBP2a activity, since GpsB is not a PBP.
Table 3.
Synthetic lethality between Δpbp1a and ΔgpsB mutations in suppressed strainsa
| Recipient strain |
Genotype | Number of colonies at 20 h after transformation with Δpbp ampliconsb |
||
|---|---|---|---|---|
| Δpbp1a | Δpbp1b | Δpbp2a | ||
| EL59 | R6 | >500 | >500 | >500 |
| IU8224 | R6 ΔgpsB | 0 | >500 | >500 |
| IU1945 | D39 Δcps | >500 | >300 | >300 |
| IU6442c | D39 Δcps ΔgpsB phpP(G229D) |
0 | >300 | >300 |
| IU9256 | Rx1 | >500 | >500 | >500 |
| IU11574 | Rx1 ΔgpsB | 0 | >500 | >500 |
| IU9262 | Rx1 ΔgpsB phpP(L148S) | 0 | >500 | >500 |
Recipient strains were constructed as described in Table S1. Transformations and visualization of colonies from 100 µL or 1 mL of transformation mixture were performed as described in Experimental procedures. Zeros (0) indicate no visible colonies after 40 h of incubation. The same results were obtained for each strain from two independent transformation experiments.
Δpbp1a::Pc-erm, Δpbp1b::Pc-erm, and Δpbp2a::Pc-erm amplicons with ≈ 1 kb flanking sequences were obtained from strains E177, E193, and E180 respectively (Table S1). Numbers of colonies indicated were obtained from 1 mL of transformation mixture.
Control transformations of a ΔgpsB<>aad9 amplicon into strains IU1824 (D39 Δcps rpsL1), IU6741 (IU1824 Δpbp1a), IU1945 (D39 Δcps), E180 (IU1945 Δpbp2a::Pc-erm), or E193 (IU1945 Δpbp1b::Pc-erm) resulted in 0 colonies in 20 h, indicating that D39 Δcps ΔgpsB Δpbp2a or ΔgpsB Δpbp1b strains are only viable when they contain the phpP(G229D) suppressor mutation.
GpsB depletion prevents migration of bPBP2x TP activity to the centers of division septa
Depletion of GpsB in D39-derived strains results in elongated cells containing rings of FtsZ and aPBP1a that fail to contract (Land et al., 2013). Previous surface plasmon resonance (SPR) experiments suggest a potential interaction between GpsB and StkP (Fleurie et al., 2014b). Moreover, StkP binds Class B PBP2x (bPBP2x) (Morlot et al., 2013), which carries out septal ring closure (Giefing et al., 2010, Land et al., 2013, Morlot et al., 2013, Peters et al., 2014, Tsui et al., 2014), although bPBP2x is not known to be phosphorylated by StkP (Morlot et al., 2013). The interactions among GpsB, StkP, and bPBP2x suggests that the defect in ring closure when GpsB is depleted could be caused, in part, by an inability of bPBP2x to move to the centers of division septa during cell division (Tsui et al., 2014). To test this hypothesis, we labeled wild-type and ΔgpsB//PfcsK-gpsB+ merodiploid cells with an FDAA and then examined localization of bPBP2x TP activity, which we showed migrates to the centers of septa of mid-to-late divisional cells, separately from other PBP TP activities (Fig. 8) (Tsui et al., 2014). Consistent with previous results, bPBP2x TP activity accumulates as a dot of FDAA labeling at the centers of septa of wild-type cells and the merodiploid expressing GpsB protein in the presence of the inducer, fucose (arrows, Fig. 8A and 8B). In contrast, merodiploid cells depleted for GpsB (no fucose, Fig. 8C) elongate, contain rings of FDAA labeling that fail to contract, and lack detectable migration of bPBP2x TP activity to the centers of septa. We conclude that GpsB is required for division ring closure and migration of bPBP2x TP activity to the centers of septa.
Fig. 8.

GpsB depletion prevents FDAA labeling of septal centers, indicative of bPBP2x migration. Wild-type parent strain IU1945 (D39 Δcps) and gpsB merodiploid strain IU4888 (D39 Δcps ΔgpsB//PfcsK-gpsB+) were grown and labeled with FDAA (TADA) as described in Experimental procedures. The parent strain and gpsB merodiploid strain grown in fucose to induce GpsB expression were growing exponentially at the time of FDAA labeling (A and B), whereas the gpsB merodiploid switched to medium lacking fucose was depleted for GpsB for 1.5 h or 2.5 h at the time of FDAA labeling (C). White arrows point to the presence of the central septal spot of FDAA labeling within the septal outer ring in growing cells (A and B). Previous work has shown that this central spot corresponds to bPBP2x TP activity (see text). Yellow arrows point to septal outer rings without central septal spots in elongated cells depleted for GpsB (C). A minimum of 100 cells was observed per condition and strain. Wild-type cells and gpsB merodiploid cells grown in fucose had a central septal spot within the septal outer ring ≈ 30% of the time, whereas cells depleted for GpsB for 1.5 h or 2.5 h had a central septal spot within the septal outer ring only 10% or 4% of the time, respectively. The experiment was performed independently twice with similar results. All images are at the same magnification, and scale bar = 1 micron.
GpsB is in complexes with EzrA, StkP, aPBP2a, bPBP2b, and MreC during stages of cell division
Previous B2H assays indicated putative interactions between pneumococcal GpsB and EzrA or DivIVA, but not with FtsZ (Fleurie et al., 2014b). Pairwise binding assays of purified proteins using SPR indicated possible interactions between pneumococcal GpsB and DivIVA (Fleurie et al., 2014b). B2H assays have suggested interactions between Bsu GpsB and EzrA, PrkC (protein kinase), PBP1, or MreC (Claessen et al., 2008, Pompeo et al., 2015). A putative interaction between Bsu GpsB and DivIVA was detected in one study (Pompeo et al., 2015), but not another (Claessen et al., 2008). SPR and B2H assays have defined the interaction between Bsu and Lmo GpsB and the positively charged amino-terminus of aPBP1 and aPBPA1, respectively (Claessen et al., 2008, Rismondo et al., 2016). To date, probing of in vivo complexes containing GpsB have not been reported for any bacterium.
To gain information about in vivo complexes containing pneumococcal GpsB and StkP, we crosslinked exponentially growing cells with formaldehyde and performed pull-down experiments on anti-FLAG magnetic beads of epitope-tagged, bait proteins, GpsB-L-FLAG3, StkP-FLAG2, or EzrA-L-FLAG3, expressed from their normal chromosomal loci (Table 4; Experimental procedures). Control experiments showed that ≈ 55% of the GpsB-L-FLAG3 protein in cell lysates was retained by the beads (data not shown). Following elution from beads, crosslinks were broken by heating, and proteins in complexes were detected pairwise by Western blotting using native antibodies to StkP, MreC, FtsA, FtsZ, or PhpP or to HA or Myc epitope tags attached to prey proteins (Table 4). Strains containing two epitope-tagged proteins exhibited minimal cell morphology or growth defects (Fig. 6E and 6F). Control mock pull downs were run on crosslinked cells that did not express the GpsB-L-FLAG3 bait protein and were used as a background control. Additional control experiments demonstrated that intact PG rings labeled with FDAA and Z rings of FtsZ-GFP were maintained in cells during the crosslinking procedure (data not shown).
Table 4.
Complexes containing GpsB and StkP in non-synchronized exponentially growing cells of unencapsulated derivatives of Spn D39a
| GpsB-L-FLAG3 as bait in co-IP experiments | ||||
| Prey proteins tested |
Primary antibodies in Western blot |
Ratio of prey protein band: GpsB-L-FLAG3 bait to GpsB+ control |
Complex detectedb |
|
| bPBP2b-HA | Anti-HA | 18.3 ± 3.1 (n=2) | + | |
| aPBP2a-HA4 | 4.0 ± 1.2 (n=2) | + | ||
| EzrA-HA | 5.4 ± 1.2 (n=2) | + (+B2H) | ||
| StkP-HA | 22.6 ± 2.4 (n=2) | + (+B2H) | ||
| StkP | Anti-StkP | 25.7 ±2.4 (n=2) | + (+B2H) | |
| MreC | Anti-MreC | 6.2 ± 0.4 (n=2) | + | |
| aPBP1a-HA | Anti-HA | 1.9 ±0.7 (n=2) | ± | |
| bPBP2x-HA | 1.4 ± 0.0 (n=2) | − | ||
| HA-FtsA | 1.0 ± 0.0 (n=2) | − | ||
| FtsA | Anti-FtsA | 1.2 ± 0.2 (n=2) | − | |
| DivIVA-Myc | Anti-Myc | 1.8 ± 0.3 (n=2) | ± (+B2H) | |
| FtsZ-Myc | 1.3 ± 0.4 (n=3) | − (−B2H) | ||
| FtsZ | Anti-FtsZ | 1.3 ± 0.1 (n=2) | − (−B2H) | |
| PhpP | Anti-PhpP | 0.8 ± 0.0 (n=2) | − | |
| StkP-FLAG2 as bait in co-IP experiments | ||||
| Prey proteins tested |
Primary antibodies in Western blot |
Ratio of prey protein band: StkP-FLAG2 bait to StkP+ control |
Complex Detected | |
| bPBP2x-HA | Anti-HA | 3.4 ± 0.3 (n=2) | + | |
| bPBP2b-HA | 8.1 ± 0.3 (n=2) | + | ||
| MreC | Anti-MreC | 8.5 ± 0.1 (n=2) | + | |
| FtsZ | Anti-FtsZ | 1.4 ± 0.4 (n=2) | − | |
| FtsA | Anti-FtsA | 1.6 ± 0.1 (n=2) | − | |
| PhpP | Anti-PhpP | 0.7 ± 0.2 (n=2) | − | |
| EzrA-L-FLAG3 as bait in co-IP experiments | ||||
| Prey proteins | Primary antibody in Western blot |
Ratio of prey protein band: EzrA-L-FLAG3 bait to EzrA+ control |
Complex Detected | |
| FtsZ-Myc | Anti-Myc | 15.01 ± 10.2 (n=2) | + (+B2H) | |
Complexes were detected by pairwise co-IP of the indicated bait and prey proteins from bacterial cells that were crosslinked with paraformaldehyde before lysis as described in Experimental procedures. Detection of prey bands by Western blotting (Fig. 9 and S10–S14) is expressed by the mean ratio (±SEM) of luminescence intensity of the prey band recovered from the co-IP to the bait protein compared to the background area from the control mock co-IP from two or more independent experiments. Brackets indicate testing two different versions of the prey proteins.
+, prey band readily detected; −, prey band not detected; ±, prey band was not visible on blots, or if present, is at the limit of detection by this method; (+B2H), putative direct interaction detected in B2H experiments; (−B2H), direct interaction not detected conclusively in B2H experiment (Fig. S15 and data not shown). B2H assays were performed as described in Experimental procedures.
In a typical experiment (Fig. 9A and S10), Western blots were run on the starting cell lysates before loading onto the beads (Fig. S10A) and on the eluted prey proteins that were in crosslinked complexes with the bait protein (Fig. 9A). Complexes containing GpsB together with bPBP2b, StkP, and/or aPBP2a, but not bPBP2x, were detected in this example. A control Western blot showed that nearly all of the bPBP2x-HA that was loaded onto beads from the cell lysate was recovered in the loading supernates (“flow-through”) fraction, ruling out degradation (data not shown). Recovery of prey proteins was quantitated relative to background areas in the mock control lanes from cells not expressing bait proteins (Table 4), where a 2-fold ratio was the limit of detection for this method. Co-IP experiments with StkP-FLAG2 as bait were also performed and quantitated (Table 4). A complex containing EzrA and FtsZ was also confirmed as part of a study of EzrA function and interactions that will be presented elsewhere (Amilcar Perez, in preparation). Finally, we repeated B2H assays to confirm likely direct interactions between GpsB and EzrA (Table 4; Fig. S15). The in vivo interaction map from these experiments of pneumococcal GpsB and StkP is summarized in Figure 9B and discussed below.
Fig. 9.

A) Pairwise co-IP of GpsB-L-FLAG3 with bPBP2b-HA, StkP-HA, or aPBP2a-HA4, but not with bPBP2x-HA. Co-IP experiments were performed as described in Experimental procedures. Top blot was probed with anti-HA primary antibody for HA-tagged prey proteins, using GpsB-L-FLAG3 as bait protein. 57 µg of each lysate sample were loaded on the input gel, while 20 µL of each elution sample was loaded on to the elution gel, after mixing 1:1 with 2× Laemlli sample buffer. Predicted molecular weight (MW) of bPBP2x-HA, bPBP2b-HA, StkP-HA, and aPBP2a-HA4 are 83.5 kDa, 75.7 kDa, 73.5 kDa, and 85.2 kDa, respectively. Bottom blot was probed with anti-FLAG primary antibody for GpsB-L-FLAG3 (bait). Two major bands are detected by anti-FLAG primary antibody in strains expressing GpsB-L-FLAG3. The bottom band correlates to GpsB-L-FLAG3 monomer (≈ 16.4 kDa), whereas the top band is likely a GpsB-L-FLAG3 trimer based on MW. Lanes shown on blot are as follows (all strains were constructed in the D39 Δcps background, IU1945): lane 1, pbp2x-HA gpsB+ (IU6929); lane 2, gpsB-L-FLAG3 pbp2x-HA (IU11314); lane 3, pbp2b-HA gpsB+ (IU6933); lane 4, gpsB-L-FLAG3 pbp2b-HA (IU11316); lane 5, stkP-HA gpsB+ (IU7438); lane 6, gpsB-L-FLAG3 stkP-HA (IU11412); lane 7, pbp2a-HA4 gpsB+ (IU11560); and lane 8 pbp2a-HA4 gpsB-L-FLAG3 (IU11516). This experiment was performed twice with similar results. B) Map of interactions found by in vivo co-IP that are proposed to coordinate divisome assembly with PBP regulation. GpsB was detected in complexes with EzrA, StkP, aPBP2a, bPBP2b, and/or MreC at stages of the division cycle (above; Fig. S10, S11, and S14). StkP was detected in complexes with bPBP2x, bPBP2b, and MreC (Fig. S13 and S14), although complexes with bPBP2b and MreC could be indirect (blue arrows) via interactions of these proteins with GpsB. EzrA is in complexes with FtsZ and GpsB (Fig. S11 and S12) and other division proteins not shown (Amilcar Perez, in preparation for submission). GpsB did not pull down detectable levels of FtsZ, FtsA, DivIVA, PhpP, bPBP2x, or aPBP1a by this in vivo co-IP method (above; Fig. S10–S12 and S14).
DISCUSSION
Roles of GpsB in modulating septal and peripheral PG synthesis in Spn
Results presented in this paper support a central role for GpsB in mediating a balance between septal and peripheral PG biosynthesis in Spn cell division. Determinations of protein phosphorylation levels and new suppressor analyses (Fig. 2–4; Table 2) support the previous conclusion that GpsB is required for maximal protein phosphorylation in exponentially growing cells (Fleurie et al., 2014b, Grangeasse, 2016). GpsB is also required for optimal protein phosphorylation in Bsu (Pompeo et al., 2015), suggesting that GpsB regulates protein phosphorylation levels in several Gram-positive species. In addition, synthetic-lethality relationships (Table 3), protein localization (Fig. 6 and 8), and in vivo crosslinking-coIP experiments (Fig. 9) show that pneumococcal GpsB activates aPBP2a, is required for septal ring closure by bPBP2x, and is present in complexes with aPBP2a and bPBP2b at stages of the division cycle. These results support and extend previous models based on Bsu and Lmo GpsB that GpsB directly interacts and regulates PBP activities (Claessen et al., 2008, Cleverley et al., 2016, Rismondo et al., 2016).
Combined results from in vivo co-IP experiments indicate that GpsB acts as a central signaling complex poised between the EzrA in the divisome and the PBPs in the septal and peripheral PG machines, as first proposed in Bsu (Claessen et al., 2008) and extended to Spn (Fleurie et al., 2014b). As will be published elsewhere, Spn EzrA is in complexes in vivo with GpsB, FtsZ, FtsA, and other divisome proteins, including DivIVA (Amilcar Perez, in preparation). In contrast, complexes containing EzrA and PBPs have not yet been detected by in vivo co-IP, whereas complexes containing GpsB and three PBPs were readily detected (see below; Fig. 9 and 10). Complex detection reflects relative amounts of proteins and whether interactions are direct or indirect, strong or weak, and frequent or infrequent at different division stages. The interaction map emerging from these studies is consistent with a model in which interactions between EzrA and GpsB play a primary role in linking the dynamics of the divisome to PBP regulation.
Fig. 10.

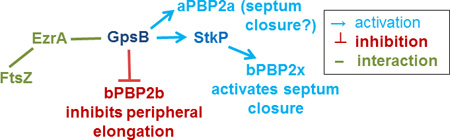
Model of GpsB interactions and coordination of septal and peripheral PG synthesis in Spn strain D39. A) Complexes containing EzrA, which binds to FtsZ, and GpsB link FtsZ-divisome dynamics (which are not shown) with GpsB regulation of downstream functions. Wild-type levels of GpsB mediate the normal protein phosphorylation cycle by StkP kinase and PhpP phosphatase of numerous division proteins, including DivIVA, MapZ(LocZ), whose extracellular E1 and E2 domains are labeled, and other proteins. Septal and peripheral PG synthesis are coordinated by GpsB complexed with aPBP2a, bPBP2b, MreC, and StkP, which interacts with bPBP2x. bPBP2x and possibly aPBP2a catalyze septal ring closure, whereas bPBP2b and MreC catalyze peripheral PG synthesis. Deletion of gpsB is lethal and can be suppressed by non-polar mutations that inactivate the PhpP phosphatase, thereby implicating maintenance of protein phosphorylation levels as an important regulatory function of GpsB; however, the critical phosphorylated protein(s) remain to be determined. B) Genetic scheme of PBP activation by GpsB that can account for the enlarged, elongated cells with unconstricted septa caused by GpsB depletion. According to this scheme, which is based on phenotypes, genetic relationships, microscopy, and interaction maps, GpsB positively regulates septum closure by activating aPBP2a directly and bPBP2x indirectly, via an interaction between GpsB and StkP, whereas GpsB directly or indirectly inhibits bPBP2b/MreC and peripheral PG elongation. See text for additional details.
Epistasis analyses presented here (Figs 1 and S1) do not support a general role for GpsB as a negative regulator of DivIVA activity in peripheral PG synthesis in progenitor Spn strain D39 or laboratory strains except for R800 (Fleurie et al., 2014b). Putative interactions between Spn GpsB and DivIVA are detected by B2H analysis expressed in E. coli (Fig. S15) (Fleurie et al., 2014b), but detection of GpsB in a complex with DivIVA was below the limit of detection of the in vivo co-IP approach used here (Table 4; Fig. S12). The shorter and rounder appearance of Spn ΔdivIVA mutant cells in chains (Fig. 1) suggests a defect in peripheral or in polar PG biosynthesis, as proposed in several papers (Boersma et al., 2015, Fadda et al., 2007, Fleurie et al., 2014b, Massidda et al., 2013, Straume et al., 2016), but the exact function of DivIVA in Spn remains to be determined.
Effects of genetic backgrounds on gpsB mutant phenotypes
The differences in GpsB phenotypes in D39 and laboratory strains undoubtedly reflect the accumulation of bypass suppressors in the domesticated laboratory strains, which have been optimized to study processes, such as competence. Initial sequencing showed that different isolates of encapsulated, virulent D39 strains stored apart for decades have essentially the same genome sequences (Lanie et al., 2007). Encapsulated D39 was the progenitor of unencapsulated mutant R36A, from which laboratory strains R6 and Rx1 were separately derived (Table S1) (Lanie et al., 2007, Pozzi et al., 1996). Laboratory strain R800 was further derived from strain R6 (Lefevre et al., 1979). To complicate things further, the genome sequences of different isolates of the same laboratory strain have diverged. For example, R6 has at least 80 additional mutations than its progenitor, D39 (Lanie et al., 2007), whereas Rx1, which contains a defect in mismatch repair, can contain as many as 600 additional mutations compared to R6 (Yanina Tovpeko and Marco Oggioni, personal communications). Reflecting these different genetic backgrounds, gpsB is essential in the D39 progenitor strain (Fleurie et al., 2014b, Land et al., 2013), nearly essential in strain Rx1 (Table 1), but not at all essential in R800 or R6 (Table 1) (Fleurie et al., 2014b). Epistasis of ΔdivIVA mutations to ΔgpsB mutations (Fleurie et al., 2014b) is confined to the R800 background and was not observed in the D39, R6, and Rx1 strains (Fig. 1 and S1). Likewise, a requirement for GpsB to localize StkP to septal rings is confined to the R800 strain and was not observed for other Spn strains (Fig. 7 and S9). Given that laboratory strains have been mutagenized and selected in culture to optimize specific traits, the chance of characterizing primary cell division mechanisms, rather than bypass mechanisms, is greater in strains derived directly from virulent progenitor strains, such as D39, instead of domesticated laboratory strains containing many additional mutations (Lanie et al., 2007). By starting with bypass mutants, it is difficult to determine the mechanisms that operate in wild-type, virulent Spn strains.
Activation of protein phosphorylation by GpsB
Since gpsB is essential in D39 derivatives, but not laboratory strains, we isolated bypass suppressors in the D39 Δcps background to better understand the role of GpsB in cell division (Table 2). Three categories of ΔgpsB suppressors were found. In this paper, we focus on the mutations that inactivate the phpP protein phosphatase (D192A, G229D, and L148S) (Table 2; Fig. S4) or that abolish protein phosphorylation altogether (sup 2 and sup3, Table 2; Fig. 3 and S3). Western analyses confirmed the previous conclusion (Fleurie et al., 2014b) that GpsB depletion in the D39 Δcps strain or ΔgpsB mutations in R6 or Rx1 significantly reduced the level of protein phosphorylation by the single StkP protein kinase in Spn (Fig. 2). Original or genetically reconstructed D39 Δcps ΔgpsB suppressor strains containing phpP mutations (Fig. 4), which eliminate protein dephosphorylation (Fig. S4) (Novakova et al., 2005), restore the level of protein phosphorylation (Fig. 2 and 3). This result is consistent with the idea that GpsB positively activates protein kinase activity, as proposed previously for Spn StkP and Bsu PrkC (Fleurie et al., 2014b, Pompeo et al., 2015), although it does not rule out the alternative explanation that GpsB negatively regulates PhpP phosphatase activity instead.
Activation of StkP kinase activity by GpsB is implied by several additional findings. In Bsu, phosphorylation of GpsB by PrkC seems to play a role in a negative feedback loop (Pompeo et al., 2015). However, phosphorylation of GpsB by StkP was not detected in Spn (Fleurie et al., 2014b, Pompeo et al., 2015), and we were unable to detect a GpsB~P species by Phos-tag PAGE (see (Zheng et al., 2016)) in the D39 Δcps parent strain or in the D39 Δcps ΔphpP::Pc-erm mutant that contains fully phosphorylated DivIVA~P (Fig. 3; data not shown). Previous binding assays of purified proteins indicate a modest interaction between Spn GpsB and the cytoplasmic kinase domain of StkP (Fleurie et al., 2014b), and a weak interaction between GpsB and StkP was detected by B2H analyses (Table 4; Fig. S15). To date, we have been unable to clone Spn PhpP in the B2H vectors in Eco. Notably, GpsB and StkP were detected together in prominent crosslinked complexes from exponentially growing Spn cells (Table 4; Fig. 9 and S14). It is not yet possible to tell at which stage of cell division GpsB and StkP are in the same complex, because these cells were not synchronized. In contrast, complexes containing GpsB and PhpP were not detected (Table 4; Fig. 9), and there currently is no evidence that GpsB negatively regulates PhpP phosphatase activity. Protein localization and defects in cell morphology also do not definitively distinguish between GpsB acting as a positive or negative regulator of StkP or PhpP activity, respectively. Results presented here show that GpsB and StkP exhibit different, but overlapping localization patterns at each stage of cell division, and epitope-tagged StkP localization was interfered with by epitope-tagged GpsB (Fig. 6F). Previous results show that GFP-PhpP exhibits diffuse localization over entire cells, with some concentration at septa (Beilharz et al., 2012). This pattern more closely matches that of GpsB than StkP (Fig. 6). Finally, both ΔstkP mutants and strains overexpressing PhpP protein phenocopy GpsB depletion (Beilharz et al., 2012, Land et al., 2013, Ulrych et al., 2016). In all three cases, Spn cells elongate, enlarge, and fail to close multiple septa (see Fig. 7 and 8). Additional biochemical assays of purified GpsB and its domains with StkP and PhpP are needed to distinguish whether GpsB activates StkP kinase activity, inhibits PhpP phosphatase activity, or both.
Other classes of ΔgpsB suppressors
Besides numerous mutations in phpP, two other classes of ΔgpsB suppressors were identified (Tables 1 and 2). The sup2 ΔgpsB and sup3 ΔgpsB suppressors contained large deletion/duplications in the spd_1034 region of the chromosome (Fig. S3). Unexpectedly, the sup2 and sup3 suppressors lacked phosphorylated proteins (Fig. 3), despite expressing the StkP+ and PhpP+ proteins. The Δ[spd_1029–spd_1037] deletion from the ΔgpsB sup3 suppressed strain (Table 2, line 3; Fig. S3) did not suppress the ΔgpsB mutation or alter protein phosphorylation (see Results; Fig. S8), and the mechanism of ΔgpsB suppression by these complicated genetic rearrangements remains to be determined. Genetically unstable ΔstkP mutants that likely accumulated suppressor mutations also could suppress ΔgpsB mutations (Table 1; Results). Together, these observations suggest that another, uncharacterized level of regulation of protein phosphorylation, cell division, or both operates in Spn. Additional ΔgpsB suppressor mutations that did not affect protein phosphorylation or the spd_1034 region of the chromosome were found (Table 2) and are being characterized.
Regulation of PBP activities by GpsB
Another phenotype lends support to the conclusion that complexes containing GpsB and StkP control PG biosynthesis in Spn, even though GpsB is not a substrate of StkP kinase activity (Fig. 10). Migration of bPBP2x TP activity to the centers of division septa does not occur when GpsB is depleted (Fig. 8), suggesting some form of regulation of bPBP2x by GpsB. This regulation may be mediated by an interaction between GpsB and StkP (Fig. 10). Previous work demonstrated that StkP interacts with bPBP2x through their extracellular PASTA and pedestal domains, respectively (Morlot et al., 2013), although bPBP2x, like GpsB, is not phosphorylated by StkP in Spn (unpublished results) (Morlot et al., 2013). Consistent with this conclusion, a complex containing StkP and bPBP2x was detected by in vivo co-IP, whereas GpsB did not pull down bPBP2x (Fig. 9 and S13). Altogether, these results support the model (Fig. 10) that an interaction between GpsB and StkP activates bPBP2x to close septal rings, thereby accounting for the multiple unconstricted rings observed in strain D39 depleted for GpsB (Fig. 8) (Land et al., 2013) or deleted for StkP (Beilharz et al., 2012).
This study also suggests that GpsB controls the activities of two other PBPs. Previous structural characterization showed that the dimeric amino terminal domain of GpsB contains a distinctive, negatively charged channel that binds to the cytoplasmic, positively charged amino terminus of Bsu aPBP1 and Lmo aPBPA1 (Claessen et al., 2008, Cleverley et al., 2016, Rismondo et al., 2016). Given that hexameric GpsB is a trimer of these dimers, this arrangement implies that GpsB binds multiple aPBPs, which may coordinate their activities in PG biosynthesis (Cleverley et al., 2016, Rismondo et al., 2016). Genetic analysis demonstrated that suppressed ΔgpsB and Δpbp1a mutations are synthetically lethal in Spn (Table 3), suggesting that GpsB activates aPBP2a activity. Consistent with this notion, GpsB could be detected in complexes with aPBP2a, but not with aPBP1a (Fig. 9 and S11). In addition, a recently discovered recognition sequence (Rick Lewis, personal communication) is present in the positively charged, cytoplasmic amino domain of aPBP2a. Together these data provide strong support for the conclusion that GpsB binding directly activates aPBP2a activity. However, the exact role of aPBP2a in pneumococcal PG synthesis remains largely unknown (see (Land & Winkler, 2011, Tsui et al., 2016)).
In addition, results from the co-IP experiments suggest GpsB directly or indirectly regulates bPBP2b TP activity, which is required for peripheral PG synthesis (Berg et al., 2013, Land et al., 2013, Straume et al., 2016, Tsui et al., 2016). One of the first studies of GpsB showed a potential interaction between Bsu GpsB and MreC by B2H analysis (Claessen et al., 2008). MreC is involved in peripheral PG synthesis in Spn (Fenton et al., 2016, Land & Winkler, 2011, Straume et al., 2016, Tsui et al., 2016). A recent paper claims that mreC is not essential in the D39 genetic background (Straume et al., 2016). However, the original ΔmreC mutation, which was polar on downstream mreD expression, and new ΔmreC mutations that maintain the mreD ribosome binding site indicate that mreC is indeed essential in strain D39 and that ΔmreC mutations can be complemented by ectopically expressed mreC+ (data not shown) (Fenton et al., 2016). Moreover, new Tn-Seq analysis in D39 shows that both mreC and mreD are essential genes (Fenton et al., 2016). It seems likely that ΔmreC mutant that was claimed to be non-essential in D39 (Straume et al., 2016) acquired a pbp1a or another suppressor mutation (Fenton et al., 2016, Land et al., 2013, Tsui et al., 2016).
In support of an interaction between GpsB and MreC, in vivo co-IP experiments revealed that GpsB or StkP pulls down MreC, indicating the three proteins are in a complex together at some stage of cell division (Fig. 9 and S14). This interaction between GpsB and MreC implicates GpsB in interacting with and regulating the peripheral PG synthesis machine that includes MreC, MreD, bPBP2b, RodA, MltG, aPBP1a, RodZ, and CozE (Fenton et al., 2016, Philippe et al., 2014, Straume et al., 2016, Tsui et al., 2016). In support of this conjecture, GpsB or StkP pulls down essential bPBP2b required for peripheral PG synthesis (Fig. 9 and S13). We anticipate that additional in vivo co-IP experiments will reveal complexes containing GpsB and other component proteins of the peripheral PG synthesis machinery listed above.
Model for GpsB function as a mediator of septal and peripheral PG synthesis in Spn
Results and conclusions from this paper can be incorporated into a model that accounts for why GpsB depletion in the D39 progenitor strain causes cells to elongate and enlarge and halts contraction of septal rings (Fig. 9) (Land et al., 2013). According to this model, interactions between EzrA and GpsB coordinate GpsB function with the dynamics of the midcell FtsZ-ring, which mediates both septal and peripheral PG synthesis in ovococcus bacteria (Fig. 10) (see (Land et al., 2013, Massidda et al., 2013, Mura et al., 2016, Tsui et al., 2014)). GpsB functions to balance septal and peripheral PG synthesis by direct or indirect interactions with aPBP2a, bPBP2x, and bPBP2b. GpsB likely activates aPBP2a by a direct interaction between the amino-terminal domains of aPBP2a and GpsB (Fig. 10) (Cleverley et al., 2016, Rismondo et al., 2016). Although the exact function of Spn aPBP2a is unknown, aPBP2a and aPBP1a have a synthetic-lethal relationship, and aPBP1a has been implicated in peripheral PG synthesis (Fenton et al., 2016, Land & Winkler, 2011, Paik et al., 1999); therefore, aPBP2a may play some role in peripheral PG synthesis, especially in the absence of aPBP1a. On the other hand, aPBP1a and aPBP2a may concurrently play roles in septal PG synthesis as well (Fig. 10B) (see (Land et al., 2013, Tsui et al., 2016)).
A GpsB binding motif is absent in the amino terminus of bPBP2x, so it seems likely that GpsB regulation of bPBP2x migration to the center of division septa (Fig. 8) is mediated indirectly through an interaction between GpsB and StkP (Fig. 9), which in turn, interacts with and positively regulates bPBP2x activity (Morlot et al., 2003) (Fig. 10). Reduced bPBP2x activity would account for the failure of septal rings to constrict when GpsB is depleted in D39 strains (Fig. 8) (Land et al., 2013). Like bPBP2x, bPBP2b lacks a GpsB binding motif. GpsB is in complexes with both MreC and bPBP2b at some stage of the cell cycle (Fig. 9 and S14). Since MreC and bPBP2b both mediate peripheral PG synthesis (Fenton et al., 2016, Land et al., 2013, Land & Winkler, 2011, Straume et al., 2016, Tsui et al., 2016), the pronounced elongation of Spn D39 cells upon GpsB depletion could be accounted for if GpsB acts as a negative regulator of bPBP2b TP activity (Fig. 10). By positively regulating septal PG synthesis and negatively regulating peripheral PG synthesis, GpsB would balance the levels of peripheral and septal PG growth at different division stages to maintain normal cell shape and size.
Finally, we do not yet understand the role of phosphorylation in GpsB function. Our data strongly supports the conclusions that GpsB function is required for optimal protein phosphorylation in Spn, that depletion of GpsB severely reduces protein phosphorylation, and that non-polar, non-functional phpP mutations restore growth and phosphorylation levels to a ΔgpsB mutant (see Results). However, how does restoration of protein phosphorylation bypass the requirement for GpsB? In the progenitor D39 background, the role of phosphorylation of single proteins has remained inconclusive (see (Massidda et al., 2013)). DivIVA and MapZ(LocZ) are established substrates of the StkP protein kinase; yet, replacing phosphorylated threonines with alanines and other amino acids does not produce phenotypes indicative of loss of DivIVA function and MapZ(LocZ) function in D39 strains and R6 and Rx1 laboratory strains (Holečková et al., 2015, Massidda et al., 2013). Although GpsB and bPBP2x likely interact with StkP, a phosphorylated form of neither protein can be detected in vivo, even in phpP mutants (see Results). Several explanations could account for bypass of the requirement for GpsB by restoration of protein phosphorylation. The growth defect of gpsB mutants may involve the simultaneous under-phosphorylation of several proteins, instead of the single proteins whose phosphorylated threonines have been changed so far. In addition, new studies show that there are additional StkP-phosphorylated proteins that can be detected in a ΔphpP mutant of laboratory strain Rx1, at least one of which influences cell division (Ulrych et al., 2016). Last, autophosphorylation of StkP may itself regulate interactions between StkP and other proteins. These hypotheses and the model for GpsB mediation of PBP activities will be tested in ongoing and future studies.
EXPERIMENTAL PROCEDURES
Bacterial strains, plasmids, and growth conditions
Bacterial strains and plasmids used in this study are listed in Tables S1–S3. Bacterial strains were derived from IU1945, an unencapsulated derivative of serotype 2 Spn strain D39 (Lanie et al., 2007), from an isolate of the R6 unencapsulated laboratory strain (EL59) (Lanie et al., 2007), or from Rx1, a second unencapsulated laboratory strain (Pozzi et al., 1996). D39 and R6 strains were grown on trypticase soy agar II with 5% (vol/vol) defibrinated sheep blood (TSAII-BA) plates at 37°C in an atmosphere of 5% CO2. Rx1 strains were grown on tryptone soya agar (Oxoid) or columbia agar (Lab Media Servis, SRO) with 5% (vol/vol) defibrinated sheep blood (TSBA and CBA, respectively) plates at 37°C in an atmosphere of 5% CO2. D39 and R6 strains were grown in Becton-Dickinson brain heart infusion (BHI) broth at 37°C in an atmosphere of 5% CO2. Rx1 strains were grown in tryptone soya broth (TSB; Oxoid) or BHI at 37°C in an atmosphere of 5% CO2. When required, chloramphenicol (4.5 µg/mL), erythromycin (0.25-0.3 µg/mL), tetracycline (0.25–2.5 µg/mL), spectinomycin (150 µg/mL) and/or kanamycin (250 µg/mL) were added to culture media.
Antibodies and fluorescent D-amino acid (FDAA)
Antibodies used in 2D-IFM, Western blotting, and co-IP procedures were as follows. Exact concentrations used are specified in individual experimental procedures. Primary antibodies used were monoclonal mouse M2 anti-FLAG (Sigma, F1804), polyclonal rabbit anti-FLAG (Sigma, F7425), polyclonal rabbit anti-HA (Invitrogen, 71–5500), polyclonal rabbit anti-Myc (ThermoFisher Scientific, PA1-981), polyclonal rabbit anti-phosphothreonine (α-pThr) (Cell Signalling, #9381), anti-StkP (Beilharz et al., 2012), anti-PhpP (Beilharz et al., 2012), anti-MreC (Land & Winkler, 2011), anti-FtsZ (Lara et al., 2005), and anti-FtsA (Lara et al., 2005). Secondary antibodies used were polyclonal goat Alexa-Fluor 488 anti-mouse (Life Technologies, A-11029), polyclonal goat Alexa-Fluor 568 anti-rabbit (Life Technologies, A-11011), and ECL anti-rabbit IgG, horseradish peroxidase linked whole antibody (GE healthcare NA93AV). FDAA TADA (tetramethylrhodamine 3-amino-D-alanine) was synthesized as described previously (Kuru et al., 2012, Kuru et al., 2015) and obtained from Michael VanNieuwenhze.
Growth curves and phase-contrast microscopy of strains
For physiological and morphological analyses of strains, cells were inoculated from frozen glycerol stocks into BHI, serial diluted, and incubated for 12–16 hours statically at 37°C in 5% CO2 overnight. The next day, cultures from OD620 ≈ 0.05 to 0.4 were diluted into fresh BHI to OD620 ≈ 0.003 and placed under the same growth conditions. Growth was monitored turbidimetrically every 45 min to 1 hour with a Genesys 2 spectrophotometer (Thermo Scientific). For microscopic analyses, samples (1–2 µl) were taken at OD620 ≈ 0.2 and examined using a Nikon E-400 epifluorescence phase-contrast microscope with a 100× Nikon Plan Apo oil-immersion objective (numerical aperture, 1.40) connected to a CoolSNAP HQ2 charged-coupled device (CCD) camera (Photometrics). Images were processed using NIS-Elements AR software (Nikon), and measurements and calculation of cell width, length, volume, and aspect ratio were performed as described before (Land et al., 2013, Tsui et al., 2016).
Construction of single and double mutants
Transformation of D39, R6, and Rx1 to obtain derivative strains (Table S1) was performed with linear DNA amplicons made by fusion PCR as described previously (Fadda et al., 2003, Tsui et al., 2010). Primers used to construct D39 and R6 derivative strains are listed in Table S2. Primers used to construct Rx1 strains are listed in Table S3. Constructed strains were confirmed by PCR and DNA sequencing of the chromosomal region corresponding to the linear amplicon and surrounding regions.
Rx1 mutant strains were constructed as follows. The Rx1 ΔgpsB phpP(L148S) mutant was obtained using a two-step PCR method as previously described (Fadda et al., 2003) using the three sets of primers listed in Table S3. PCR amplicons were purified using a QIAquick PCR purification kit (Qiagen), mixed, and re-amplified in a second PCR using the external primers. The resulting ΔgpsB::cat construct (2282 bp) was used to transform competent Spn Rx1 cells. A DNA fragment of the corresponding regions not containing the resistance marker was used as a negative control for transformation. Transformants were selected after 24 h growth on TSBA plates containing chloramphenicol (4.5 µg/ml). If no growth was observed, incubation was extended up to 48 h. Eight transformants were verified by PCR for integration of the construct and four of them stored as frozen glycerol stocks for further analysis. One transformant was sequenced in the phpP-stkP region and found to contain a phpP(L148S) mutation (bp T443C). The Rx1 ΔgpsB phpP(L148S) ΔdivIVA double mutant was generated by transforming the ΔdivIVA::erm construct (1844 bp), amplified from chromosomal DNA of a ΔdivIVA mutant (Fadda et al., 2003) using primers LN235/LN236, into Rx1 ΔgpsB phpP(L148S) competent cells. Transformants were selected after 24 h growth on CBA plates containing erythromycin (0.2 µg/ml) and verified for growth on CBA plates containing chloramphenicol and erythromycin, before being stored as frozen glycerol stocks. One transformant, verified for the correct insertion of the construct, was used for further analysis. A clean Rx1 ΔgpsB mutant was obtained via transformation of a ΔgpsB<>aad9 amplicon into the Rx1 parent strain, using methods described previously (Tsui et al., 2010). This strain was sequenced in the phpP-stkP region and determined to have no mutations.
Construction of phpP(D192A)
A phpP(D192A)-Pc-[kanrpsL+] amplicon was constructed by PCR fusion of 5’ and 3’ fragments from IU7673. Primer sets TT546/BR26 and BR25/TT574 were used to obtain the fragments. phpP(D192A) was obtained by PCR from the full-length phpP(D192A)-Pc-[kanrpsL+] amplicon using primers TT546/TT580. phpP(D192A) was then transformed into IU4888 without fucose, resulting in strains IU10180 and IU10191. phpP(D192A) amplicon from IU10180 and IU10191 was then obtained by PCR with primers TT546/TT547 for further experiments.
phpP(G229D) and phpP(D192A) allele exchange and ΔgpsB suppression confirmation
phpP(G229D) amplicon was obtained from IU6442 (D39 Δcps ΔgpsB<>aad9 phpP(G229D)) using primers in Table S2. phpP(D192A) amplicon was obtained as described above. As outlined in Results, two methods were used to confirm that phpP(G229D) or phpP(D192A) could suppress ΔgpsB in reconstructed strains.
In the first method (Fig. 4A), phpP(G229D) or phpP(D192A) amplicons were transformed into strain IU7673 (D39 Δcps rpsL phpP+-Pc-[kan-rpsL+]-stkP+). This transformation yielded both wild-type and tiny sized (1/6th of wild-type) colonies. Tiny colonies were stored and sequenced in the phpP-stkP region. Four out of the six resulting strains from the phpP(G229D) transformation had phpP(G229D), with three being stkP+ (IU8805, IU10423, and IU10424) and one having a mutation at G10 resulting in a stop codon in stkP (IU7685). Two of the resulting strains were wild-type for both phpP+ and stkP+ and were not analyzed further. Two out of the three strains from the phpP(D192A) transformation retained the phpP(D192A) mutation and were stkP+ (IU11223 and IU11240). One of the strains was wild-type for both phpP+ and stkP+. The resulting strains (D39 Δcps rpsL phpP(G229D)-stkP+ or D39 Δcps rpsL phpP(D192A)-stkP+) were then transformed with ΔgpsB<>aad9 from IU4888 or ΔpurR<>aad9 amplicon as a control (Land & Winkler, 2011). This transformation resulted in strains that were phpP(G229D) ΔgpsB (IU11344, IU11346) or phpP(D192A) ΔgpsB (IU11348). Mutations in stored colonies were confirmed by PCR, and sequencing confirmed the presence of ΔgpsB, phpP(G229D) stkP+, or phpP(D192A) stkP+. This construction was performed twice with similar results.
In the second method (Fig. 4B), merodiploid strain IU4888 (D39 Δcps ΔgpsB<>aad9//ΔbgaA::Δcps ΔgpsB//PfcsK-gpsB+), which requires fucose to induce GpsB expression (Land et al., 2013), was grown in the presence of 1.0% (wt/vol) fucose. At OD620 ≈ 0.03, 50 ng of phpP(G229D) or phpP(D192A) amplicon were added to 100 µL of IU4888 and 900 µL of transformation mix without fucose (90% BHI, 10% heat-treated horse serum, 0.18% (wt/vol) glucose, and 100 ng/mL CSP-1), and the mixture was incubated at 37°C in 5% CO2 for 1 h. The transformation mix was plated on TSAII-BA plates without fucose to select for fucose independent transformants. The absence of fucose (absence of GpsB) allowed for selection of phpP(G229D or D192A) cross-in events. Resulting strains [IU10129, IU10138, IU10139, IU10156, and IU10157 for phpP(G229D) and IU10180, IU10191, IU10349, IU10350, IU10363 for phpP(D192A); Table S1] were confirmed for by PCR and sequencing for the presence of the ΔgpsB and phpP(G229D) stkP+ or phpP(D192A) stkP+ mutations. This reconstruction experiment was repeated 3× with similar results. Strains were then transformed with a ΔbgaA::Pc-erm amplicon to replace the ΔbgaA::kan-t1t2-Δcps ΔgpsB//PfcsK-gpsB+ mutation, resulting in strains IU11221 and IU11238. These strains were confirmed by PCR and sequencing for the presence of the ΔgpsB, ΔbgaA::Pc-erm, and phpP(G229D) stkP+ or phpP(D192A) stkP+ mutations.
FDAA short-pulse labeling for 2D epifluorescence microscopy
At OD620 ≈ 0.15-0.2, live cells were subjected to a short pulse of FDAA labeling for 5 min as described before (Boersma et al., 2015). Resulting FDAA labeled live cells were suspended in 10–20 µL ice-cold PBS, and 1–2 µL of the cell suspension was observed using a Nikon E-400 epifluorescence phase-contrast microscope equipped with a mercury lamp, a 100× Nikon Plan Apo oil-immersion objective (numerical aperture, 1.40), and filter block for fluorescence (Alexa 568, EX 532–587, DM 595, and BA 608–683). Images were taken using a CoolSNAP HQ2 charged-coupled device (CCD) camera (Photometrics) and processed with NIS-Elements AR software (Nikon). For FDAA (TADA) pictures, an Alexa 568 filter was used with an exposure time of 50–100 ms to visualize labeling.
2D IFM
Localization of FLAG-tagged proteins, HA-tagged proteins, and native StkP by IFM was performed on exponentially growing cells as described before (Land et al., 2013, Tsui et al., 2011). For strains containing one or two epitope-tagged proteins, the primary antibody used was anti-FLAG mouse monoclonal M2 antibody (1:100 dilution) and/or anti-HA rabbit polyclonal antibody (1:100), and the secondary antibody was Alexa-Fluor 488 goat anti-mouse (1:100 dilution) and/or Alexa-Flour 568 goat anti-rabbit (1:100 dilution). For 2D IFM of native StkP, the primary antibody used was anti-StkP rabbit polyclonal antibody (1:1000 dilution), and secondary antibody used was Alexa-Fluor 568 goat anti-rabbit (1:100 dilution). Cells were mounted using mounting media SlowFade gold antifade reagent with DAPI (ThermoFisher/Life Technologies, S36936) and observed by epifluorescence microscopy as described previously (Land et al., 2013). Control experiments with strains containing untagged proteins and the same antibody setups indicated that non-specific labeling was not detected under these conditions (data not shown). Matlab GUI image analyses of 2D IFM images were performed as described previously (Land et al., 2013).
GpsB depletion experiments
GpsB depletion experiments in D39 Δcps ΔgpsB//PfcsK-gpsB+ merodiploid strain IU4888, which requires fucose addition to induce GpsB expression for growth, were performed as described previously (Land et al., 2013). Bacteria were harvested at OD620 ≈ 0.1 and ≈ 0.4 from cultures induced by fucose addition and after 1, 2, and 3 h from cultures lacking fucose to deplete GpsB for Western blot and microscopic analyses.
FDAA labeling of cells depleted for GpsB for 3D-SIM
Serially diluted overnight cultures of wild-type parent IU1945 (D39 Δcps) and GpsB depletion strain IU4888 (D39 Δcps ΔgpsB//Pfcsk-gpsB+) (grown in fucose) were diluted to OD620 ≈ 0.02 in 3 mL cultures of BHI broth lacking or containing 1.0% (wt/vol) fucose. After 1.5 to 2.5 h of incubation, 500 µL of cultures were centrifuged (16,000×g for 5 min). Cell pellets were resuspended in 250 µL of BHI broth containing the FDAA, TADA (final concentration = 500 µM), and incubated for 5 min at 37 °C. Washing and fixation of TADA-labeled cells were performed as described previously (Tsui et al., 2014). Slides were imaged on a DeltaVision OMX 3D-SIM super-resolution imaging system at Indiana University Bloomington Light Microscopy Imaging Center (http://www.indiana.edu/~lmic/microscopes/OMX.html) as described previously (Tsui et al., 2014) using an exposure time of 5 ms at 31% transmittance.
Western blotting analysis and immunodetection
For Western blot analyses using anti-phosphothreonine (α-pThr), anti-StkP, and anti-PhpP antibodies, bacteria were grown exponentially in 5 mL BHI broth to OD620 ≈ 0.1 to 0.2–0.5. Aliquots of 1.0 to 1.5 mL were centrifuged (5 min, 13,000×g at 25 °C), and cell pellets were put on dry ice. Frozen pellets were resuspended in 20 µL SEDS lysis buffer (0.1% deoxycholate (vol/vol), 150 mM NaCl, 0.2% SDS (vol/vol), 15 mM EDTA pH 8.0) for samples with OD620 ≈ 0.1–0.2 and 40–80 µL for samples with OD620 ≈ 0.4. Samples were incubated at 37°C for 15 min until cells visibly lysed as determined via phase microscopy. The Bio-Rad DC™ protein assay kit I was used to determine total protein concentrations of samples using a standard curve of 0.1 to 3.0 mg/mL of BSA. Absorbance (750 nm) were determined in a 96-well plate reader (Synergy H1 Hybrid Reader, BioTek). Samples were diluted with 2× Laemlli SDS loading buffer (Bio-Rad) and incubated at 95°C for 10 min. 12.5 µg of total protein was loaded per sample onto a 4–15% precast gradient SDS-PAGE gel (Bio-Rad) and subjected to electrophoresis. Proteins were transferred to a nitrocellulose membrane. StkP proteins, PhpP proteins, and threonine-phosphorylated proteins were detected respectively with anti-StkP (1:50,000), anti-PhpP (1:20,000), or α-pThr (1:2000) (Novakova et al., 2010) as primary antibodies, and ECL anti-rabbit IgG horseradish peroxidase linked whole antibody as secondary (1:10,000). Chemiluminescent signals in protein bands were detected with 1–3 min exposures and quantified using an IVIS imaging system as described in previously (Wayne et al., 2010). To determine relative amounts of threonine-phosphorylated proteins and the PhpP and StkP proteins, luminescence values for each band were subtracted by the value of the background (defined as a box enclosing the same area of the blot with no detectable signal or from a Δ[phpP-stkP]). Background-subtracted amounts are expressed relative to amounts in wild-type or bacteria that were not depleted for GpsB. After blotting, membranes were stained with amido black solution to confirm that protein loading was similar between samples.
Co-immunoprecipitation (Co-IP)
Cultures were grown exponentially in 400 mL of BHI to OD620 ≈ 0.25–0.40. Cells were collected by centrifugation (8,000×g for 10 min at 4°C). Cell pellets were washed once with 30 mL of 1X PBS (4°C) and resuspended in 19.8 mL 1X PBS (4°C). 200 µL of 10% paraformaldehyde solution (EMS) were added for crosslinking to a final concentration of 0.1% (vol/vol). Mixtures were incubated at 37°C in an air incubator for 1 h. Cross-linking reactions were quenched by the addition of 4 mL 1.0 M glycine followed by incubation at 25°C for 10 min. Cells were collected by centrifugation (16,500×g for 5 min at 4°C). Pellets were washed once with 20 mL cold 1X PBS (4°C) and resuspended in 2 mL of cold lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X100 (v/v)) with 1 tablet of protease inhibitor (ThermoFisher Scientific, 78429) freshly added per 10 mL of lysis buffer. The suspension was transferred into 2 lysing matrix B tubes (MP Biomedicals) with 1 mL in each tube. Tubes were shaken in FastPrep homogenizer 10× (4×, 5 min on ice, 3×, 5 min on ice, and 3×) with 6.0 M/s for 40 s each at 4°C. Cell debris and lysing matrix from tubes were removed by centrifugation at 16,000×g for 5 min at 4°C. Protein concentration of each sample was determined by Bio-Rad DC× protein assay (Bio-Rad). 1 mL of lysate with similar amounts of total protein (5–7 mg/mL) was added to tubes with 50 µL of anti-FLAG magnetic beads (Sigma, M8823). The same amount of protein was loaded onto the beads for strains expressing FLAG-tagged proteins and the corresponding control strains lacking FLAG-tagged proteins in each experiment. The tubes were rotated for 2 h at 4°C. The beads were washed 3 times with 1 mL of lysis buffer (4°C) with 10 min incubation at 4°C each time. FLAG-tagged protein was eluted from the beads by incubation with 100 µL of FLAG elution solution (150 ng 3× FLAG peptide/µL) (Sigma, F4799) for 30 min at 4°C. 100 µL of the elution and of the original lysate added to magnetic beads (input) were separately mixed with 100 µL 2× Laemmli sample buffer (Bio-Rad) containing 5% (vol/vol) β-mercaptoethanol (Sigma) and heated at 95°C for 1 h to break the cross-links. 50–70 µg of input protein samples and 20 µL of each elution sample were separated by SDS-PAGE on 4–15% precast protein gels (Bio-Rad) in Tris-glycine buffer. Gels were subjected to Western blotting using rabbit anti-FLAG (1:2000), rabbit anti-HA (1:1000), rabbit anti-Myc (1:2000), anti-MreC (1:10,000), anti-FtsZ (1:10,000), anti-FtsA (1:10,000), anti-StkP (1:50,000), and anti-PhpP (1:20,000) as primary antibody. Secondary antibody used was ECL anti-rabbit IgG, horseradish peroxidase linked whole antibody (1:10,000). Chemiluminescent signal in protein bands was quantitated by using an IVIS imaging system as described in (Barendt et al., 2009). A mean ratio of band intensity >2 between FLAG tagged and non-FLAG tagged samples was considered as a positive interaction for proteins, with no background subtraction. Each experiment was performed twice independently.
Bacterial two-hybrid (B2H) experiments
B2H experiments were performed according to (Karimova et al., 2005) and Euromedex manufacturer’s instructions. For plasmid constructions, Spn genes were amplified by PCR from Rx1 chromosomal DNA, using the primers pairs listed in Table S3. PCR fragments were purified, digested with appropriate restriction enzymes (PstI and BamHI for gpsB, divIVA, ezrA, and ftsZ and BamHI and EcoRI for stkP (New England Biolabs)), and subcloned into the corresponding sites of the BACTH vectors pKNT25 and pUT18 to generate the corresponding hybrid proteins fused at the N-terminal ends of the T25 and T18 fragments. Plasmids were transformed into cloning host, E. coli DH5α. After double-strand sequence verification, each pair of plasmids were co-transformed into the E. coli cya− strain BTH101 and co-transformation mixtures were spotted onto LB agar plates supplemented with ampicillin (100 µg/mL), kanamycin (50 µg/mL) and X-Gal (40 µg/mL), with or without 0.5 mM IPTG, followed by incubation at 30°C for 36–40 h, as previously described (Karimova et al., 2005). Interactions (or the lack of them) were additionally confirmed by spotting co-transformation mixtures onto M63-0.2% (wt/vol) maltose or M63-0.2% (wt/vol) glucose agar minimal media, supplemented with ampicillin and kanamycin (50 µg/mL and 25 µg/mL, respectively) and X-Gal (40 µg/mL) and onto MacConkey agar supplemented with the appropriate concentrations of ampicillin and kanamycin and 1% (wt/vol) maltose, with incubation times up to 96 h. Plasmid pairs pKNT25/pUT18 and pKT25-zip/pUT18C-zip were used as negative and positive controls, respectively.
Illumina sequencing
Strains IU5844, IU6441, and IU6442 containing spontaneous suppressor mutations that allowed growth of a D39 Δcps ΔgpsB deletion mutant were isolated as described in Results (see Table 2). Overnight cultures still in exponential phase were diluted into 5 ml of BHI broth to an OD620 ≈ 0.01 and grown to an OD620 ≈ 0.3 to 0.4. Cells were collected by centrifugation (10,000×g for 10 min at 25 °C). Genomic DNA was purified using a MasterPure Gram-positive DNA purification kit (Epicenter Biotechnologies) according to the manufacturer's protocol. DNA library construction, Illumina MiSeq DNA sequencing, and bioinformatics analyses were performed as described previously (Tsui et al., 2016). Visual inspection of the coverage data revealed gaps in genomic sequences corresponding to the expected Δcps Δ[cps2A-cps2H] and ΔgpsB deletion mutations and to the Δ[spd_1026–spd_1037] and Δ[spd_1029–spd_1037] deletions and adjacent chromosomal duplications in strains IU5845 and IU6441, respectively (see Table 2; Fig. S3). The structures of the rearrangements were verified by assembling the genome sequences using newbler (version 2.9). The resulting contig graph was analyzed to determine the organization of the duplications/deletions (see Fig. S3B).
Analysis of the conservation of G229 among bacterial PP2C phosphatases and protein modeling of Spn PhpP and StkP
To determine the conservation of G229 among other bacterial PP2C phosphatases, the NCBI BLASTP v2.2.30+ (Basic Local Alignment Search Tool) interface was used to search for homologous protein sequences among Streptococci and less closely related bacterial species (Altschul et al., 1997, Altschul et al., 2005). The NCBI BLASTP interface was used with preset parameters to search the non-redundant protein sequence database and UniProtKB/Swiss-Prot database for proteins with sequences homologous to the S. pneumoniae D39 PhpP amino acid sequence (Protein Accession YP_816996Pa) (The UniProt Consortium, 2013) (Altschul et al., 1997, Altschul et al., 2005).
Amino acid sequences from other previously identified bacterial PP2C phosphatases were obtained to determine conservation of amino acids in Spn PhpP involved in ΔgpsB suppression. The following phosphatase amino acid sequences were aligned with S. pneumoniae PhpP (Novakova et al., 2005) using Clustal Omega (Sievers et al., 2011) on default settings: S. agalactiae PP2C phosphatase (Rantanen et al., 2007), L. monocytogenes Stp (Archambaud et al., 2005), B. subtilis PrpC (Obuchowski et al., 2000), P. aeruginosa Stp1 (Mukhopadhyay et al., 1999), and S. mutans Pppl (Banu et al., 2010). Amino acid sequences were obtained from the NCBI protein sequence database. The Clustal output file was uploaded to the ConSurf server (Ashkenazy et al., 2016) under the Multiple Sequence Alignment (MSA) section, and default parameters were used to perform a conservation alignment for these sequences only.
To determine how G229D and other changed amino acids in phpP suppressor mutants might affect PhpP structure, BLASTP was used to search the PDB database for related protein structures (Altschul et al., 1997, Altschul et al., 2005, Berman et al., 2000, Madej et al., 2012, Wang et al., 2007). This search method identified the previously solved structure of the PhpP homolog Stp1 in S. agalactiae (PDB ID: 2PK0) (Rantanen et al., 2007) and others listed in Table S4. Structures were then aligned with the Phyre2 generated PhpP threading model (Kelley & Sternberg, 2009) using PyMOL (Schrödinger, LLC). RMSD and Z-scores for each structural alignment were calculated using PyMOL and the DALI server (Schrödinger, LLC, Holm L. and Rosenstrom P) (See Table S4).
Supplementary Material
Acknowledgments
We thank Yana Tovpeko and Kevin Bruce for helpful discussions, Yana Tovpeko and Marco Oggioni for unpublished genome sequences of derivatives of strain Rx1, Gouzel Karimova for strains and plasmids for B2H assays and discussing results, Mike VanNieuwenhze for a sample of FDAA, and James Ford, Kurt Zimmer, and Doug Rusch for assistance with Illumina DNA sequencing analyses. This work was supported by NIH grant 1RO1GM113172 and NIH grant 1R01GM114315 (to M.E.W.), by Czech Science Foundation Grants P302/12/0256 and P207/12/1568 (to P.B.), by LR7/2007 grant CRP2-401 from the Autonomous Region of Sardinia (RAS) (to O.M.), by predoctoral Quantitative and Chemical Biology (QCB) NIH institutional training grant T32 GM109825 (to B.E.R. and A.J.P.), and by a predoctoral scholarship from the Sardinia Regional Government (European Social Fund 2007–2013) (to A. M.).
REFERENCES
- Agarwal S, Agarwal S, Pancholi P, Pancholi V. Strain-specific regulatory role of eukaryote-like serine/threonine phosphatase in pneumococcal adherence. Infect Immun. 2012;80:1361–1372. doi: 10.1128/IAI.06311-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nuc Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272:5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambaud C, Gouin E, Pizarro-Cerda J, Cossart P, Dussurget O. Translation elongation factor EF-Tu is a target for Stp, a serine-threonine phosphatase involved in virulence of Listeria monocytogenes. Molec Microbiol. 2005;56:383–396. doi: 10.1111/j.1365-2958.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, Ben-Tal N. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nuc Acids Res. 2016;44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu LD, Conrads G, Rehrauer H, Hussain H, Allan E, van der Ploeg JR. The Streptococcus mutans serine/threonine kinase, PknB, regulates competence development, bacteriocin production, and cell wall metabolism. Infect Immun. 2010;78:2209–2220. doi: 10.1128/IAI.01167-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barendt SM, Land AD, Sham LT, Ng WL, Tsui HC, Arnold RJ, Winkler ME. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J Bacteriol. 2009;191:3024–3040. doi: 10.1128/JB.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilharz K, Nováková L, Fadda D, Branny P, Massidda O, Veening JW. Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proc Nat Acad Sci USA. 2012;109:E905–E913. doi: 10.1073/pnas.1119172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KH, Stamsas GA, Straume D, Havarstein LS. Effects of low PBP2b levels on cell morphology and peptidoglycan composition in Streptococcus pneumoniae R6. J Bacteriol. 2013;195:4342–4354. doi: 10.1128/JB.00184-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nuc Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma MJ, Kuru E, Rittichier JT, VanNieuwenhze MS, Brun YV, Winkler ME. Minimal Peptidoglycan (PG) Turnover in wild-type and PG hydrolase and cell division mutants of Streptococcus pneumoniae D39 growing planktonically and in host-relevant biofilms. J Bacteriol. 2015;197:3472–3485. doi: 10.1128/JB.00541-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busiek KK, Margolin W. Bacterial actin and tubulin homologs in cell growth and division. Curr Biol. 2015;25:R243–R254. doi: 10.1016/j.cub.2015.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claessen D, Emmins R, Hamoen LW, Daniel RA, Errington J, Edwards DH. Control of the cell elongation-division cycle by shuttling of PBP1 protein in Bacillus subtilis. Molec Microbiol. 2008;68:1029–1046. doi: 10.1111/j.1365-2958.2008.06210.x. [DOI] [PubMed] [Google Scholar]
- Cleverley RM, Barrett JR, Basle A, Bui NK, Hewitt L, Solovyova A, Xu ZQ, Daniel RA, Dixon NE, Harry EJ, Oakley AJ, Vollmer W, Lewis RJ. Structure and function of a spectrin-like regulator of bacterial cytokinesis. Nature Comm. 2014;5:5421. doi: 10.1038/ncomms6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleverley RM, Rismondo J, Lockhart-Cairns MP, Van Bentum PT, Egan AJ, Vollmer W, Halbedel S, Baldock C, Breukink E, Lewis RJ. Subunit arrangement in GpsB, a regulator of cell wall biosynthesis. Microbial Drug Resis. 2016;22:446–460. doi: 10.1089/mdr.2016.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echenique J, Kadioglu A, Romao S, Andrew PW, Trombe MC. Protein serine/threonine kinase StkP positively controls virulence and competence in Streptococcus pneumoniae. Infect Immun. 2004;72:2434–2437. doi: 10.1128/IAI.72.4.2434-2437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda D, Pischedda C, Caldara F, Whalen MB, Anderluzzi D, Domenici E, Massidda O. Characterization of divIVA and other genes located in the chromosomal region downstream of the dcw cluster in Streptococcus pneumoniae. J Bacteriol. 2003;185:6209–6214. doi: 10.1128/JB.185.20.6209-6214.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda D, Santona A, D'Ulisse V, Ghelardini P, Ennas MG, Whalen MB, Massidda O. Streptococcus pneumoniae DivIVA: localization and interactions in a MinCD-free context. J Bacteriol. 2007;189:1288–1298. doi: 10.1128/JB.01168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton AK, El Mortagi L, Lau DTC, Rudner TG, Bernhardt TG. Control of cell elongation by a novel member of the S. pneumoniae MreCD complex. Nature Microbiol. 2016;2:16237. doi: 10.1038/nmicrobiol.2016.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleurie A, Cluzel C, Guiral S, Freton C, Galisson F, Zanella-Cleon I, Di Guilmi AM, Grangeasse C. Mutational dissection of the S/T-kinase StkP reveals crucial roles in cell division of Streptococcus pneumoniae. Molec Microbiol. 2012;83:746–758. doi: 10.1111/j.1365-2958.2011.07962.x. [DOI] [PubMed] [Google Scholar]
- Fleurie A, Lesterlin C, Manuse S, Zhao C, Cluzel C, Lavergne JP, Franz-Wachtel M, Macek B, Combet C, Kuru E, VanNieuwenhze MS, Brun YV, Sherratt D, Grangeasse C. MapZ marks the division sites and positions FtsZ rings in Streptococcus pneumoniae. Nature. 2014a;516:259–262. doi: 10.1038/nature13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, Macek B, Kuru E, VanNieuwenhze MS, Brun YV, Di Guilmi AM, Claverys JP, Galinier A, Grangeasse C. Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet. 2014b;10:e1004275. doi: 10.1371/journal.pgen.1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba P, Veening JW, Saunders NJ, Hamoen LW, Daniel RA. Two-step assembly dynamics of the Bacillus subtilis divisome. J Bacteriol. 2009;191:4186–4194. doi: 10.1128/JB.01758-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giefing C, Jelencsics KE, Gelbmann D, Senn BM, Nagy E. The pneumococcal eukaryotic-type serine/threonine protein kinase StkP co-localizes with the cell division apparatus and interacts with FtsZ in vitro. Microbiol. 2010;156:1697–1707. doi: 10.1099/mic.0.036335-0. [DOI] [PubMed] [Google Scholar]
- Grangeasse C. Rewiring the Pneumococcal cell cycle with serine/threonine- and tyrosine-kinases. Trends Microbiol. 2016;24:713–724. doi: 10.1016/j.tim.2016.04.004. [DOI] [PubMed] [Google Scholar]
- Holečková N, Doubravová L, Massidda O, Molle V, Buriánková K, Benada O, Kofroňová O, Ulrych A, Branny P. LocZ is a new cell division protein involved in proper septum placement in Streptococcus pneumoniae. mBio. 2015;6:e01700–e01714. doi: 10.1128/mBio.01700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins J, Matsushima P, Mullen DL, Tang J, Zhao G, Meier TI, Nicas TI, Jaskunas SR. Gene disruption studies of penicillin-binding proteins 1a, 1b, and 2a in Streptococcus pneumoniae. J Bacteriol. 1999;181:6552–6555. doi: 10.1128/jb.181.20.6552-6555.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Dautin N, Ladant D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J Bacteriol. 2005;187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaval KG, Halbedel S. Architecturally the same, but playing a different game: the diverse species-specific roles of DivIVA proteins. Virulence. 2012;3:406–407. doi: 10.4161/viru.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, de Pedro MA, Brun YV, VanNieuwenhze MS. In Situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl. 2012;51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E, Tekkam S, Hall E, Brun YV, Van Nieuwenhze MS. Synthesis of fluorescent D-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ. Nature Protocols. 2015;10:33–52. doi: 10.1038/nprot.2014.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AD, Luo Q, Levin PA. Functional domain analysis of the cell division inhibitor EzrA. PloS One. 2014;9:e102616. doi: 10.1371/journal.pone.0102616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, Winkler ME. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Molec Microbiol. 2013;90:939–955. doi: 10.1111/mmi.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land AD, Winkler ME. The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. J Bacteriol. 2011;193:4166–4179. doi: 10.1128/JB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol. 2007;189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara B, Rico AI, Petruzzelli S, Santona A, Dumas J, Biton J, Vicente M, Mingorance J, Massidda O. Cell division in cocci: localization and properties of the Streptococcus pneumoniae FtsA protein. Molec Microbiol. 2005;55:699–711. doi: 10.1111/j.1365-2958.2004.04432.x. [DOI] [PubMed] [Google Scholar]
- Lefevre JC, Claverys JP, Sicard AM. Donor deoxyribonucleic acid length and marker effect in pneumococcal transformation. J Bacteriol. 1979;138:80–86. doi: 10.1128/jb.138.1.80-86.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej T, Addess KJ, Fong JH, Geer LY, Geer RC, Lanczycki CJ, Liu C, Lu S, Marchler-Bauer A, Panchenko AR, Chen J, Thiessen PA, Wang Y, Zhang D, Bryant SH. MMDB: 3D structures and macromolecular interactions. Nucl Acids Res. 2012;40:D461–D464. doi: 10.1093/nar/gkr1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massidda O, Anderluzzi D, Friedli L, Feger G. Unconventional organization of the division and cell wall gene cluster of Streptococcus pneumoniae. Microbiol. 1998;144:3069–3078. doi: 10.1099/00221287-144-11-3069. [DOI] [PubMed] [Google Scholar]
- Massidda O, Novakova L, Vollmer W. From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol. 2013;15:3133–3157. doi: 10.1111/1462-2920.12189. [DOI] [PubMed] [Google Scholar]
- Morlot C, Bayle L, Jacq M, Fleurie A, Tourcier G, Galisson F, Vernet T, Grangeasse C, Di Guilmi AM. Interaction of penicillin-binding protein 2× and Ser/Thr protein kinase StkP, two key players in Streptococcus pneumoniae R6 morphogenesis. Molec Microbiol. 2013;90:88–102. doi: 10.1111/mmi.12348. [DOI] [PubMed] [Google Scholar]
- Morlot C, Zapun A, Dideberg O, Vernet T. Growth and division of Streptococcus pneumoniae: localization of the high molecular weight penicillin-binding proteins during the cell cycle. Molec Microbiol. 2003;50:845–855. doi: 10.1046/j.1365-2958.2003.03767.x. [DOI] [PubMed] [Google Scholar]
- Mura A, Fadda D, Perez AJ, Danforth ML, Musu D, Rico AI, Krupka M, Denapaite D, Tsui H-CT, Winkler ME, Branny P, Vicente M, Margolin W, Massidda O. Roles of the essential protein FtsA in cell growth and division in Streptococcus pneumoniae. J. Bacteriol. 2016 doi: 10.1128/JB.00608-16. pii: JB.00608-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S, Kapatral V, Xu W, Chakrabarty AM. Characterization of a Hank's type serine/threonine kinase and serine/threonine phosphoprotein phosphatase in Pseudomonas aeruginosa. J Bacteriol. 1999;181:6615–6622. doi: 10.1128/jb.181.21.6615-6622.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nováková L, Bezousková S, Pompach P, Spidlová P, Sasková L, Weiser J, Branny P. Identification of multiple substrates of the StkP Ser/Thr protein kinase in Streptococcus pneumoniae. J Bacteriol. 2010;192:3629–3638. doi: 10.1128/JB.01564-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nováková L, Sasková L, Pallová P, Janecek J, Novotná J, Ulrych A, Echenique J, Trombe MC, Branny P. Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates. FEBS J. 2005;272:1243–1254. doi: 10.1111/j.1742-4658.2005.04560.x. [DOI] [PubMed] [Google Scholar]
- Obuchowski M, Madec E, Delattre D, Boel G, Iwanicki A, Foulger D, Seror SJ. Characterization of PrpC from Bacillus subtilis, a member of the PPM phosphatase family. J Bacteriol. 2000;182:5634–5638. doi: 10.1128/jb.182.19.5634-5638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz C, Natale P, Cueto L, Vicente M. The keepers of the ring: regulators of FtsZ assembly. FEMS Microbiol Rev. 2016;40:57–67. doi: 10.1093/femsre/fuv040. [DOI] [PubMed] [Google Scholar]
- Osaki M, Arcondeguy T, Bastide A, Touriol C, Prats H, Trombe MC. The StkP/PhpP signaling couple in Streptococcus pneumoniae: cellular organization and physiological characterization. J Bacteriol. 2009;191:4943–4950. doi: 10.1128/JB.00196-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik J, Kern I, Lurz R, Hakenbeck R. Mutational analysis of the Streptococcus pneumoniae bimodular class A penicillin-binding proteins. J Bbacteriol. 1999;181:3852–3856. doi: 10.1128/jb.181.12.3852-3856.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters K, Schweizer I, Beilharz K, Stahlmann C, Veening JW, Hakenbeck R, Denapaite D. Streptococcus pneumoniae PBP2x mid-cell localization requires the C-terminal PASTA domains and is essential for cell shape maintenance. Molec Microbiol. 2014;92:733–755. doi: 10.1111/mmi.12588. [DOI] [PubMed] [Google Scholar]
- Philippe J, Vernet T, Zapun A. The elongation of ovococci. Microbial Drug Resis. 2014;20:215–221. doi: 10.1089/mdr.2014.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompeo F, Foulquier E, Serrano B, Grangeasse C, Galinier A. Phosphorylation of the cell division protein GpsB regulates PrkC kinase activity through a negative feedback loop in Bacillus subtilis. Molec Microbiol. 2015;97:139–150. doi: 10.1111/mmi.13015. [DOI] [PubMed] [Google Scholar]
- Pozzi G, Masala L, Iannelli F, Manganelli R, Havarstein LS, Piccoli L, Simon D, Morrison DA. Competence for genetic transformation in encapsulated strains of Streptococcus pneumoniae: two allelic variants of the peptide pheromone. J Bacteriol. 1996;178:6087–6090. doi: 10.1128/jb.178.20.6087-6090.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantanen MK, Lehtio L, Rajagopal L, Rubens CE, Goldman A. Structure of Streptococcus agalactiae serine/threonine phosphatase. The subdomain conformation is coupled to the binding of a third metal ion. FEBS J. 2007;274:3128–3137. doi: 10.1111/j.1742-4658.2007.05845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismondo J, Cleverley RM, Lane HV, Grosshennig S, Steglich A, Moller L, Mannala GK, Hain T, Lewis RJ, Halbedel S. Structure of the bacterial cell division determinant GpsB and its interaction with penicillin-binding proteins. Molec Microbiol. 2016;99:978–998. doi: 10.1111/mmi.13279. [DOI] [PubMed] [Google Scholar]
- Sasková L, Nováková L, Basler M, Branny P. Eukaryotic-type serine/threonine protein kinase StkP is a global regulator of gene expression in Streptococcus pneumoniae. J Bacteriol. 2007;189:4168–4179. doi: 10.1128/JB.01616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah IM, Laaberki MH, Popham DL, Dworkin J. A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell. 2008;135:486–496. doi: 10.1016/j.cell.2008.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl H, Hamoen LW. Finding the corners in a cell. Curr Opinion Microbiol. 2012;15:731–736. doi: 10.1016/j.mib.2012.10.006. [DOI] [PubMed] [Google Scholar]
- Straume D, Stamsas GA, Berg KH, Salehian Z, Havarstein LS. Identification of pneumococcal proteins that are functionally linked to penicillin-binding protein 2b (PBP2b) Molec Microbiol. 2016;103:99–116. doi: 10.1111/mmi.13543. [DOI] [PubMed] [Google Scholar]
- Sung CK, Li H, Claverys JP, Morrison DA. An rpsL cassette, janus, for gene replacement through negative selection in Streptococcus pneumoniae. App Environ Microbiol. 2001;67:5190–5196. doi: 10.1128/AEM.67.11.5190-5196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavares JR, de Souza RF, Meira GL, Gueiros-Filho FJ. Cytological characterization of YpsB, a novel component of the Bacillus subtilis divisome. J Bacteriol. 2008;190:7096–7107. doi: 10.1128/JB.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, Winkler ME. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Molecular microbiology. 2014;94:21–40. doi: 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Keen SK, Sham LT, Wayne KJ, Winkler ME. Dynamic distribution of the SecA and SecY translocase subunits and septal localization of the HtrA surface chaperone/protease during Streptococcus pneumoniae D39 cell division. mBio. 2011;2:e00202–e00211. doi: 10.1128/mBio.00202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Mukherjee D, Ray VA, Sham LT, Feig AL, Winkler ME. Identification and characterization of noncoding small RNAs in Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol. 2010;192:264–279. doi: 10.1128/JB.01204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui HC, Zheng JJ, Magallon AN, Ryan JD, Yunck R, Rued BE, Bernhardt TG, Winkler ME. Suppression of a deletion mutation in the gene encoding essential PBP2b reveals a new lytic transglycosylase involved in peripheral peptidoglycan synthesis in Streptococcus pneumoniae D39. Molec Microbiol. 2016;100:1039–1065. doi: 10.1111/mmi.13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrych A, Holečková N, Goldová J, Doubravová L, Benada O, Kofroňová O, Halada P, Branny P. Characterization of pneumococcal Ser/Thr protein phosphatase phpP and identification of a novel PhpP substrate, putative RNA binding protein Jag. BMC Microbiol. 2016;16(1):247. doi: 10.1186/s12866-016-0865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Addess KJ, Chen J, Geer LY, He J, He S, Lu S, Madej T, Marchler-Bauer A, Thiessen PA, Zhang N, Bryant SH. MMDB: annotating protein sequences with Entrez's 3D-structure database. Nuc Acids Res. 2007;35:D298–D300. doi: 10.1093/nar/gkl952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne KJ, Sham LT, Tsui HC, Gutu AD, Barendt SM, Keen SK, Winkler ME. Localization and cellular amounts of the WalRKJ (VicRKX) two-component regulatory system proteins in serotype 2 Streptococcus pneumoniae. J Bacteriol. 2010;192:4388–4394. doi: 10.1128/JB.00578-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng JJ, Sinha D, Wayne KJ, Winkler ME. Physiological roles of the dual phosphate transporter systems in low and high phosphate conditions and in capsule maintenance of Streptococcus pneumoniae D39. Front Cell Infect Microbiol. 2016;6:63. doi: 10.3389/fcimb.2016.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.