

Abstract
Among pregnant women ibuprofen is one of the most frequently used pharmaceutical compounds with up to 28% reporting use. Regardless of this, it remains unknown whether ibuprofen could act as an endocrine disruptor as reported for fellow analgesics paracetamol and aspirin. To investigate this, we exposed human fetal testes (7–17 gestational weeks (GW)) to ibuprofen using ex vivo culture and xenograft systems. Ibuprofen suppressed testosterone and Leydig cell hormone INSL3 during culture of 8–9 GW fetal testes with concomitant reduction in expression of the steroidogenic enzymes CYP11A1, CYP17A1 and HSD17B3, and of INSL3. Testosterone was not suppressed in testes from fetuses younger than 8 GW, older than 10–12 GW, or in second trimester xenografted testes (14–17 GW). Ex vivo, ibuprofen also affected Sertoli cell by suppressing AMH production and mRNA expression of AMH, SOX9, DHH, and COL2A1. While PGE2 production was suppressed by ibuprofen, PGD2 production was not. Germ cell transcripts POU5F1, TFAP2C, LIN28A, ALPP and KIT were also reduced by ibuprofen. We conclude that, at concentrations relevant to human exposure and within a particular narrow ‘early window’ of sensitivity within first trimester, ibuprofen causes direct endocrine disturbances in the human fetal testis and alteration of the germ cell biology.
Analgesics, including paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin and ibuprofen, are among the most widely used and environmentally prevalent pharmaceutical drugs in the world1. They are regarded as extremely effective medications and are widely used for self-medication, including by pregnant women during early pregnancy2,3,4. During pregnancy, analgesics are generally taken to relieve migraine, pain, and fever, but are also used in inflammatory conditions and are frequently used during preterm labor1,5,6,7,8. These medications are known to cross the placenta and to be present in meconium, neonatal urine, and in breast milk, indicating painkiller transmission from the mother to the fetus and to neonates9,10,11,12,13,14,15. Attention has recently been focused on the associations between analgesic use during the second and third trimesters of pregnancy and a number of unwanted effects in children. These include low birthweight, risk of premature closure of the ductus arteriosus, cardiac defects, decreased fetal and neonatal renal function, and asthma16,17,18,19,20. Concerns have also been raised recently about the use of over-the-counter painkillers during the first and second trimesters of pregnancy and an association with congenital cryptorchidism, the most frequent congenital reproductive disorder in newborn boys7,8,21,22, as well as with shorter anogenital distance (AGD) in male infants23,24, the latter being a biomarker for androgen action during fetal life. A series of studies undertaken in utero in the rat and ex vivo in rat and human fetal testes and in xenografted mouse models have shown that paracetamol and the NSAIDs aspirin and indomethacin can disrupt the testicular endocrine system. These disruptive endocrine effects may highlight analgesic involvement in fetal maldescended testes1,7,25,26,27,28.
Ibuprofen is the only of the 3 most common over-the-counter painkiller with paracetamol and aspirin, whose endocrine disruptive potential has not yet been investigated ex vivo. This is despite the fact that its consumption has continued to increase even though aspirin consumption has decreased1,6. The percentage of pregnant women reporting ibuprofen use averages 10%, but this varies widely across studies, ranging from 0.5% to over 28%3,6,7,16,29,30,31,32,33,34. In addition to cryptorchidism7, ibuprofen use by pregnant women has also been associated with hypospadias35, another congenital abnormality featuring a midline fusion defect of the male ventral urethra. The latter association was not found in another study36. Cryptorchidism and hypospadias are associated disorders and although some cases arise from genetic syndromes, most cases remain idiopathic37. However, both cryptorchidism and hypospadias most probably reflect subnormal levels of androgens during the development of the male urogenital tract38,39.
In this study, we investigated whether ibuprofen disrupts the endocrine system and the differentiation of the human fetal testis during the first and second trimesters of pregnancy. We used a combination of an organotypic culture system (Fetal Gonad Assay; FEGA) based on culturing human fetal testes fragments, and a human fetal testes xenograft system. These approaches, have been separately used for the study of endocrine-disrupting substances26,27,40,41, but never integrated in the same series of experiments. In the FEGA system, after 1 to 3 days of exposure to ibuprofen at 10−7–10−4 M, we assessed the gross morphology, endocrine function, and gene expression for the main cell types of the first trimester human fetal testis ex vivo, whilst the xenograft system was used to determine the effect of prolonged (7 day) ibuprofen exposure on the endocrine function of the second trimester testis.
Results
Ibuprofen and Leydig cell morphology and function
No apparent changes in general morphology or Leydig cell marker expression was seen in the ibuprofen-exposed FEGA explants as compared to the non-exposed (Fig. 1A–C).
Figure 1. Ibuprofen and Leydig cell steroidogenic function.

(A–C) Representative images of CYP11A1 immunostaining in explants of a 7.9 gestational week (GW) human fetal testis cultured with DMSO or ibuprofen. CYP11A1 appears brown (3,3′-diaminobenzidine tetrahydrochloride staining), and sections were counterstained with hematoxylin. Scale bar = 100 μm. (D) Testosterone production as a function of culture duration in 7–7.9 GW human fetal testes in the presence of DMSO (Control) or 10−5 M of ibuprofen. Results are expressed as fold change from the respective control testis (% of Control). Values are means ± SEM of 6 testes from 6 different fetuses. Repeated measures analysis of variance (ANOVA) on ranks was performed (p = 0.172). (E–G) Testosterone production after culture of 8-9.9 GW (E,F) and 10–12 GW human fetal testes (G) in the presence of DMSO (Control) or 10−7–10−4 M of ibuprofen for 72 h (E,G) and for 24, 48 and 72 h (F). Results are expressed as fold change from the first day of culture (FC to D0). Values are means ± SEM of 14–37 testes from 14–37 fetuses for the 8–9.9 GW group, 6–14 testes from 6–14 fetuses for the 8–9.9 GW at 24, 48 and 72 h group, and 13–21 testes from 13–21 fetuses for 10–12 GW. ANOVAs with a random fetus effect were performed using unstructured covariance matrices. A Wilcoxon test was also performed for pairwise comparisons (**p < 0.01, ***p < 0.001). (H) Seminal vesicle weight and (I) plasma testosterone production in host mice carrying human fetal testis xenografts (14–17 GW; n = 4 fetuses) after 7 days (7d) of exposure to vehicle (Corn Oil; open circles) or ibuprofen (10 mg/kg 3 times daily; closed circles) with overall mean ± SEM for vehicle (white bars) and ibuprofen (black bars). Values are means ± SEM from 4 fetuses. Data analyzed by two-way ANOVA.
In the 7–7.9 gestational week (GW) 10−5 M ibuprofen-treated testes, no significant effect was observed on testosterone levels when compared to the non-treated testes (Fig. 1D). By contrast, when the 8–9.9 GW testes were exposed ex vivo to ibuprofen, a significant dose-dependent testosterone decrease was seen after 72 h of exposure (slope β = −0.076, p < 0.0001); the pairwise comparisons versus the control condition evidenced an effect with 10−5 M (−32.3%; p < 0.001 and with 10−4 M (−35.6%; p < 0.01) (Fig. 1E). In the 8–9.9 GW testes, we also observed time- and dose-dependent effects of ibuprofen (after 24 h, slope β = −0.052, p < 0.0001; after 48 h, slope β = −0.081, p < 0.0001; after 72 h, slope β = −0.085, p < 0.0001), with the decrease of testosterone occurring after only 24 h of exposure: −12.8% with 10−5 M (p < 0.01); and −24.6% with 10−4 M (p < 0.001). This dose-dependent decrease was amplified after 48 h (−19.8 and −49%, respectively; both p < 0.001), and further amplified after 72 h (−29.3% and −55.3%, respectively; both p < 0.001) (Fig. 1F). In contrast, no effect was observed ex vivo on testosterone levels in the 10–12 GW testes for doses of ibuprofen of 10−7–10−4 M (Fig. 1G).
Given the apparent specific time window for testosterone reduction during the mid-late first trimester, we then investigated the effect of ibuprofen exposure during the second trimester (14–17 GW) using the xenograft system. Exposure to a 7 day therapeutic regimen of ibuprofen (10 mg/kg, 3 times daily) did not affect testosterone production from the human fetal testis as determined by both host mouse seminal vesicle weight (14.63 mg versus 13.21 mg; p = 0.62; n = 4 Fig. 1H) and plasma testosterone (0.84 versus 0.82 ng/ml; p = 0.85; n = 4; Fig. 1I) in host mice. Similarly, plasma 5-dihydrotestosterone (5α-DHT) was unaffected by exposure to ibuprofen (3.26 versus 2.44 ng/ml; p = 0.56; n = 4; Fig. S1). Mean plasma ibuprofen concentration 1 hour after the final dose in ibuprofen-exposed host mice was 2.78 ± 0.55 μg/ml and undetectable in vehicle-exposed controls (Fig. S1)
Using gas chromatography-tandem mass spectrometry (GC/MS-MS), we then measured the entire complement of detectable steroids both in the FEGA testis explants themselves and in the media after 48 h of exposure to 10−5 M of ibuprofen in the 8–9.9 GW testes (Fig. 2A,B). Our results reveal that in this fetal age-range, only 3 steroids were found to be above the threshold of the GC/MS-MS technique in the non-treated fetal testis: pregnenolone (1.39 ± 0.7 ng/mg of testicular tissue); dehydroepiandrosterone (DHEA) (1.7 ± 1.1 ng/mg of testicular tissue); and testosterone (54.4 ± 25.7 ng/mg of testicular tissue). Levels of these 3 steroids in the explants were not significantly affected by ibuprofen at 10−5 M (data not shown). Parallel investigation in the corresponding culture media allowed us to detect, for the first time in humans, 6 steroids in the control (i.e. unexposed) condition in both the Δ5 and Δ4 steroid pathways: - Δ5, 17-hydroxy-pregnenolone (17-OH-preg) (4.3 ± 1.9 ng/mL); DHEA (21.3 ± 4.7 ng/mL) (Fig. 2A); - Δ4, 17-hydroxy-progesterone (17-OH-prog) (9.6 ± 5.9 ng/mL); androstenedione (183.7 ± 97.0 ng/mL); testosterone (1597.8 ± 722.5 ng/mL); and 5-dihydrotestosterone (5α-DHT) (7.1 ± 8.0 ng/mL of culture media) (Fig. 2B). Of these, the potent androgens testosterone and α-DHT were found to be significantly inhibited by 10−5 M of ibuprofen (36.8 and 70.2%, respectively at p < 0.01). The levels of the other androgen androstenedione were also reduced (−41.2%), but this was not statistically significant at p < 0.05 (Fig. 2B).
Figure 2. Ibuprofen and global steroidogenesis.

(A,B) Global analysis of ibuprofen effects on the Δ5 (A) and Δ4 pathways (B) of testosterone production using GC-MS/MS. Human fetal testis explants (8–9.9 GW) were incubated for 48 h with DMSO (Control) or 10−5 M of ibuprofen. Steroid precursors of the Δ5 and Δ4 pathways were measured by GC-MS/MS in the media. Values are mean ± SEM of 8 fetuses pooled in 5 independent experiments, and are expressed as the percentage of variation from the control. **p < 0.01 by non-parametric signed rank Wilcoxon test on paired data. (C) Quantitative RT-PCR of BZRP, STAR, CYP11A1, CYP17A1, HSD17B3, HSD3B2 and SRD5A3 was performed on control testes (white bars) and testes treated with 10−5 M of ibuprofen (grey bars) for 24 and 48 h. Each column shows a pool of 11–15 fetal testes. Each bar represents the mean ± SEM of the fold change in target gene expression relative to the reference gene RPLP0. A non-parametric signed rank Wilcoxon test on paired data was performed (*p < 0.05, **p < 0.01, ***p < 0.001).
After 48 h of exposure to ibuprofen, the expression of 3 genes that encode for steroidogenic enzymes was markedly decreased: by 48.6% (p < 0.05) for CYP11A1, by 82% (p < 0.001) for CYP17A1, and by 70.4% (p < 0.05) for HSD17B3 (Fig. 2C). By contrast, the mRNA levels of HSD3B2 and SRD5A3, 2 other genes which encode enzymes of the steroidogenic pathway, as well as the mRNA levels of BZRP and STAR, the 2 genes encoding proteins involved in cholesterol transport to the mitochondria, were not affected by exposure to ibuprofen (Fig. 2C).
The other key fetal Leydig cell-produced hormone investigated was INSL3. For explants from 8–12 week gestation fetuses, INSL3 levels were dose-dependently and significantly decreased after 72 h of exposure to ibuprofen at doses ranging from 10−7–10−4 M, (slope β = −0.155, p = 0.007) (Fig. 3A). At 10−5 M the decrease of INSL3 (−39%) was significant (p < 0.05), but at 10−4 M no significant difference was observed. To assess whether a window of sensitivity exists, we again divided the fetuses into 2 age groups, 8–9.9 GW and 10–12 GW. In fact, there was a significant dose-response decrease in INSL3 levels for the 10–12 GW age group (slope β = −0.181, p = 0.03) (Fig. 3B,C). In addition to decreased INSL3 production, after 48 h of culture with 10−5 M of ibuprofen, INSL3 transcripts were also repressed by 57.1% (p < 0.001) (Fig. 3D). As in previous studies26,42, INSL3 production appears to have substantial interindividual variability.
Figure 3. Ibuprofen and production of INSL3 by Leydig cells.

(A–C) Ibuprofen and INSL3 production after exposure to ibuprofen during 72 h of human fetal testis culture at (A) 8–12 gestational weeks (GW), (B) 8–9.9 GW, and (C) 10–12 GW. Results are expressed as fold change from the first day of culture (FC to D0). Values are means ± SEM (n = 12–31 testes from 12–31 fetuses). ANOVAs with a random fetus effect were performed using unstructured covariance matrices. A Wilcoxon test was performed for pairwise comparisons. (*p < 0.05). (D) Quantitative RT-PCR for INSL3 was performed on control testes (white bars) and testes treated with 10−5 M of ibuprofen (grey bars) for 24 and 48 h. Each column is made up of a pool of 11–15 fetal testes. Each bar represents the mean ± SEM of the fold change in target gene expression relative to RPLP0 reference gene. A non-parametric signed rank Wilcoxon test on paired data was performed (***p < 0.001).
Ibuprofen and Sertoli cell morphology and function
No apparent change in Sertoli cell number or topographical organization was observed in FEGA explants after exposure to 10−5 M or 10−4 M of ibuprofen, based on expression of AMH or cytokeratin 18 (KRT18) a marker of the immature Sertoli cell intermediate filaments, (Fig. 4A–F). This was further demonstrated by an unchanged Sertoli cell density after 72 h of exposure to 10−5 M of ibuprofen (Fig. 4G).
Figure 4. Ibuprofen and Sertoli cell function.

(A–F) Representative images of KRT18 (A–C) and AMH (D–F) immunostaining in cultured explants of 7–7.9-gestational week (GW) human fetal testis. KRT18 and AMH appear brown (3,3-diaminobenzidine tetrahydrochloride staining), and sections were counterstained with hematoxylin. Scale bar = 100 μm. (G) Sertoli cell numbers were determined by counting AMH-positive cells in control testes (white bars) and testes treated with 10−5 M (grey bars) and 10−4 M (black bars) of ibuprofen for 72 h. Data are presented as the number of cells per surface area unit (0.01 mm2) (means ± SEM) based on 1 or 2 explants per treatment in 7 fetuses. A Wilcoxon test was performed for pairwise comparisons. (H) AMH production after 24, 48, and 72 h of exposure to DMSO (Control) or 10−5 M of ibuprofen in 7–7.9 GW human fetal testes. Results are expressed as fold changes from the control testis (% of Ctrl). Values are means ± SEM for 7 testes from 7 fetuses. Repeated measures analysis of variance (ANOVA) on ranks was performed (*p < 0.05, ****p < 0.0001). (I–K) AMH production after culture of 8–12 GW (I); 8–9.9 GW (J); and 10–12 GW (K) human fetal testes in the presence of the control solvent DMSO (Control) or 10−7–10−4 M of ibuprofen. Results are expressed as fold change from the first day of culture (FC to D0). Values are means ± SEM of 6–17 testes from 6–17 fetuses for the 8–9.9 GW, and of 6–7 testes from 6–7 fetuses for the 10–12 GW. ANOVAs with a random fetus effect were performed using unstructured covariance matrices. A Wilcoxon test was performed for pairwise comparisons (*p < 0.05; ***p < 0.001). (L) Plasma AMH in individual host mice carrying human fetal testis xenografts (14–17 GW; n = 4 fetuses) after 7d exposure to vehicle (Corn Oil; open circles) or ibuprofen (10 mg/kg 3 times daily; closed circles) with overall mean ± SEM for vehicle (white bars) and ibuprofen (black bars). Data analyzed by two-way ANOVA.
When the testes from the youngest human fetuses (7–7.9 GW) were exposed to 10−5 M of ibuprofen, a significant decrease in AMH was seen from 48 h of culture onwards (−31.1% at 48 h (p < 0.05), −53.0% at 72 h (p < 0.0001), and −38.1% at 96 h (p < 0.05)) (Fig. 4H). When fetuses of 8–12 GWs were analyzed together, a dose-dependent suppression of AMH production was observed (slope β = −0.294, p < 0.0001), with −38.4% (p < 0.001) at 10−5 M and −76.2% (p < 0.001) at 10−4 M (Fig. 4I). The dose-dependent decrease of AMH levels was also seen both in the 8–9.9 GW group (slope β = −0.295, p < 0.0001), with −31.0% (p < 0.05) at 10−7 M, −54.8% (p < 0.001) at 10−5 M and −66.7% (p < 0.001) with 10−4 M (Fig. 4J) and in the 10–12 GW group (slope β = −0.291, p = 0.003) (Fig. 4K). In accordance with these findings, the expression of AMH mRNA was found to be suppressed by ibuprofen exposure in hFEGA: −42.1% at 24 h and −89.9% (p < 0.001) at 48 h (Fig. 5). As with AMH mRNA, the mRNA of SOX9, a specific marker of the differentiation of Sertoli cells, was repressed after ibuprofen exposure: −43.3% (p < 0.001) at 24 h; and −59.7% (p < 0.001) at 48 h. The mRNA expression of the 2 SOX9 targets investigated DHH and COL2A1 was decreased by −51.3% (p < 0.01) and −61.7% (p < 0.01) at 24 h; and −79.8% (p < 0.001) and 79.6% (p < 0.001), respectively (Fig. 5).
Figure 5. Ibuprofen and Sertoli cell markers.

Quantitative RT-PCR for SOX9, AMH, DHH and COL2A1 were performed on control testes (white bars) and testes treated with 10−5 M of ibuprofen (grey bars) at 24 or 48 h of culture. Each column represents a pool of 11–15 fetal testes. Each bar shows the mean ± SEM of the fold change in target gene expression relative to the RPLP0 reference gene. A non-parametric signed rank Wilcoxon test on paired data was performed (*p < 0.05; **p < 0.01; ***p < 0.001).
We also investigated the effect of ibuprofen exposure on AMH production in the second trimester human fetal testis using the xenograft system. In contrast to the results in the first trimester testis there was no significant difference in AMH production in second trimester xenografts exposed to ibuprofen compared to vehicle-exposed controls (0.75 versus 1.34 ng/ml; p = 0.038; Fig. 4L).
Ibuprofen alters germ cell markers
Neither the morphology nor the density of the germ cells appeared altered after 72 h of exposure to 10−5 and 10−4 M of ibuprofen (Fig. 6A–D). This contrasted with the expression of 5 germ cell genes which were repressed after 48 h of ibuprofen exposure: POU5F1, a transcription factor essential for the pluripotency maintenance in embryonic stem cells43, was significantly decreased by 57.2% (p < 0.01); TFAP2C, a factor believed to regulate the expression of several genes involved in cell growth and differentiation44, was also significantly decreased by 67.9% (p < 0.001); LIN28A, which regulates the germ cell pool45, was significantly decreased by 32.9% (p < 0.05); ALPP and KIT, which are typical markers of germ cells46,47, were significantly decreased by 91.8% (p < 0.05) and 72.7% (p < 0.05), respectively (Fig. 6E).
Figure 6. Ibuprofen and germ cells.

(A–C) Representative images of M2A immunostaining in cultured explants of an 8.6-gestational week (GW) human fetal testis. M2A appears brown (3,3′-diaminobenzidine tetrahydrochloride staining), and sections were counterstained with hematoxylin. Scale bar = 100 μm. (D) Numbers of germ cells were determined by counting AMH-negative cells after culturing human fetal testis (n = 7 testes from 7 fetuses) in the presence of 0.01% of DMSO (Control, white bars) or 10−5 M (grey bars) or 10−4 M (black bars) of ibuprofen. Data are presented as the number of cells per surface area unit (0.01 mm2) (means ± SEM) based on 1 or 2 explants per treatment from 7 fetuses. A non-parametric signed rank Wilcoxon test on paired data was performed. (E) Quantitative RT-PCR for POU5F1, TFAP2C, LIN28A, ALPP and KIT was performed on control testes (white bars) and testes treated with 10−5 M of ibuprofen (grey bars) at 24 or 48 h of culture. Each column shows a pool of 11 or 15 fetal testes. Each bar represents the mean ± SEM of the fold change in target gene expression relative to the RPLP0 reference gene. A non-parametric signed rank Wilcoxon test on paired data was performed (*p < 0.05, **p < 0.01; ***p < 0.001).
Ibuprofen suppresses the production of prostaglandin PGE2, but not that of PGD2
Cyclooxygenase enzymes (COX1 and COX2) are responsible for catalyzing the formation of prostaglandins from arachidonic acid, and ibuprofen is classically known to inhibit these enzymes48,49. We therefore assayed the prostaglandins PGD2 and PGE2, both previously found to be produced by the human fetal testis26. At all ages investigated, ibuprofen did not have a significant effect on PGD2 levels (Fig. 7A–C). In contrast, an ibuprofen-induced dose-dependent inhibition of PGE2 levels was demonstrated (slope β = −0.109, p = 0.03). Furthermore, significant inhibitory effects on PGE2 levels were observed in the 8–12 GW fetal testes after 72 h of exposure to 10−5 and 10−4 M of ibuprofen (−31.6%, p < 0.01 at 10−5 M; and −32.7%, p < 0.01 at 10−4 M) (Fig. 7D). This ibuprofen-suppressive effect was more pronounced in the 8–9.9 GW fetal testes under the same conditions: −29.7% at 10−7 M; −36.5% at 10−6 M; −51.4% (p < 0.01) at 10−5 M; and −42.0% (p < 0.05) at 10−4 M (Fig. 7E), again with a significant dose-dependent suppression of PGE2 production (slope β = −0.122, p = 0.02). In the 10–12 GW testes, no significant inhibition of PGE2 was seen (Fig. 7F). However, high levels of PGE2 were observed when the 10–12 GW testes were exposed to the lowest doses of ibuprofen (+128.1% at 10−7 M, p < 0.05 and +78.1% at 10−6 M, p < 0.01).
Figure 7. Effect of ibuprofen on human fetal testis PGD2 and PGE2 production.

(A–C) PGD2 concentrations after 72 h of culture of 8 to 12 gestational week (GW) (A), 8–9.9 GW (B), and 10–12 GW (C) human fetal testes in the presence of the solvent DMSO (Control) or 10−7–10−4 M of ibuprofen. (D–F) PGE2 concentrations after 72 h of culture of 8–12 GW (D), 8–9.9 GW (E), and 10–12 GW (F) human fetal testes in the presence of DMSO (Control) or 10−7–10−4 M of ibuprofen. Results are expressed as fold change from the first day of culture (FC to D0) for both PGD2 and PGE2 productions. Data are means ± SEM (n = 4–10 testes from 4–10 fetuses for PGD2; n = 6−18 testes from 6–18 fetuses for PGE2). ANOVAs with a random embryo effect were performed using unstructured covariance matrices. A Wilcoxon test was performed for pairwise comparisons (*p < 0.05, **p < 0.01).
Discussion
The 3 most frequently consumed mild analgesics during the first trimester of pregnancy are paracetamol (also known as acetaminophen) and the NSAIDs aspirin and ibuprofen1. Several recent epidemiological studies have reported that the consumption of ibuprofen during the first trimester is associated with an increased risk of cryptorchidism and/or hypospadias7,8,33. In animals and humans, these congenital abnormalities are considered complex disorders involving either rare genetic traits, or abnormal endocrine activity or production in fetal testes37,38,39,50,51,52. Since the in utero assessment of ibuprofen’s effects on the development and function of human fetal testes is impossible for obvious ethical reasons, we developed an ex vivo model system which has already proven to be useful for studying the effects of paracetamol, NSAIDs (aspirin and indomethacin) and environmental chemicals (bisphenol A) on the human fetal testis during the first trimester26,40. In addition, as a complementary tool of investigation, we utilized a xenograft system which has been shown to model the effects of prolonged exposure to therapeutic doses of analgesics in the human testis during the second trimester27.
The present study demonstrates, through the use of the FEGA, that concentrations which are equivalent to or even lower than peak plasma levels of ibuprofen53,54 markedly affect the biology of the 2 major human fetal testicular somatic cell populations, as well as of the germ cells, and that these effects occur during specific periods of human fetal testis development. This occurred without any obvious changes in the number of these cells or in the general morphology of the testis at any age. Thus, ibuprofen markedly decreased testosterone levels and steroidogenesis at large. This inhibitory effect was observed using both the immunoassay of testosterone and GC/MS-MS analysis, and was found to be dose-, time- and age-dependent. Of note is that this appeared restricted to the 8–9.9 GW fetal testes. Androgens play an essential role in the masculinization of the urogenital tract55. This narrow window of ibuprofen sensitivity corresponding to the younger fetal ages investigated is similar to that previously observed for aspirin and indomethacin26. This likely reflects the fact that Leydig cells go through markedly different steps of differentiation and thus biological states56, some of which appear to be resistant to ibuprofen’s anti-androgenic effects. Indeed, we were able to demonstrate that testosterone production was not reduced by exposure to ibuprofen in late first trimester human fetal testis (10–12 GW) in the ex vivo culture system, nor in second trimester human fetal testis (14–17 GW) using the xenograft approach. The ibuprofen-induced testosterone inhibition during the specific 8–99 GW time window contrasts greatly with the effects observed in previous studies in aspirin- and indomethacin-treated fetal testes ex vivo. In fact, we found that the latter NSAIDs actually stimulate human testis testosterone levels26, rather than suppressing them as in the case of ibuprofen. Another striking difference is that, while paracetamol was previously found to reduce both plasma levels of testosterone and the weight of androgen-dependent seminal vesicles in the xenograft model-system27, ibuprofen had no effect in the xenograft-system model as revealed here. These differences are likely to reflect the different nature and thus mechanisms of action of the two analgesics considered, ibuprofen being a NSAID while paracetamol is not.
The present work also reveals that at least 3 steps of steroidogenesis are significantly affected ex vivo by ibuprofen, as it suppressed the transcripts encoding CYP11A1, CYP17A1, and HSD17B3. CYP11A1 is important as it catalyzes the first reaction of steroidogenesis (i.e. the conversion of cholesterol to pregnenolone). CYP17A1 catalyzes 17α-hydroxylation of progesterone and pregnenolone, and also converts 17α-hydroxypregnenolone to DHEA and 17α-hydroxyprogesterone to androstenedione. CYP17A1 is typically regarded as a key determinant of testosterone production, susceptible to both androgen production regulation and perturbation57,58. HSD17B3 catalyzes the ultimate conversion of androstenedione to testosterone, androstenedione being the first androgen produced in the downstream chain line of steroidogenesis. The present study represents the first GC/MS-MS global analysis of fetal testicular steroidogenesis demonstrating that, in addition to suppression of testosterone, the effect of ibuprofen on the expression of CYP17A1 also negatively impacted androstenedione and to a greater extent 5α-DHT. Further supporting evidence for ibuprofen being an anti-androgenic compound is the observation that low doses of this NSAID (5–6 mg/kg/day for 35 days) inhibited plasma levels of testosterone in adult male mice59. Ibuprofen has also been shown to inhibit the human UDP-glucuronosyltransferases UGT2B15 and UGT2B17, and the latter is very important for glucuronidation and testosterone excretion60.
It is important to note that not only androgens were impacted by ibuprofen but also INSL3, which is the peptidic hormone produced by Leydig cells and which was dose-dependently inhibited by ibuprofen. In accordance with this, INSL3 expression levels were also suppressed by ibuprofen. Insl3 knock-out mice are cryptorchid50,61 and some mutations of the human INSL3 gene have been associated with cases of cryptorchidism38. The first phase of testicular descent which occurs in humans between 8 and 17 GW is controlled by INSL328,38,51,62. In a previous study, 8–12 GW human fetal testes treated with paracetamol had reduced INSL3 production26, and in the present study ibuprofen exposure in testis explants at the same gestational age for 72 h induced significant alterations in INSL3 production. These findings support the hypothesis that ibuprofen-induced suppression of INSL3 expression and production could be related to the increased risk of cryptorchidism in boys whose mothers were exposed to ibuprofen during pregnancy7.
Interestingly, ibuprofen also induced a dose-dependent suppression of AMH production in the human fetal testis ex vivo. To the best of our knowledge, ibuprofen is the first endocrine disruptor which is found to display direct suppressive action on the endocrine function of the human (and other mammalian) fetal Sertoli cells. However, the reduced AMH production following ibuprofen exposure was restricted to the first trimester testis during a specific period from 7–9.9 GW. Exposure in late first trimester (10–12 GW) explants and in second trimester (14–17 GW) xenografts did not result in a significant change in AMH production although the variability between samples clearly increased in the late first trimester. In addition, there was a large variability in AMH production between fetuses for the second trimester xenografts which was accounted for using two-factor analysis as previously described27. The difference observed here between the FEGA ex vivo (i.e. decreased levels of AMH) and the xenograft model system (i.e. no change in AMH levels) could result from the age-difference of the fetal testes used; it also cannot be excluded, however, that the intrinsic difference of the two assay-systems discussed before28 could also account for it. Taken together, the results obtained ex vivo indicate that there exist specific ‘windows of sensitivity’ during which certain Sertoli cell function may be affected by exposure to ibuprofen. It is likely that the ibuprofen suppression of SOX9 mRNA expression evidenced in this study along with several of its known targets (AMH, DHH, COL2A1), represents the basis by which ibuprofen also suppresses AMH.
Our study reveals not only that ibuprofen disturbs the endocrine homeostasis of the testis, in both the Sertoli and Leydig cells, but also that exposure to this NSAID markedly alters the expression of the germ cells genes POU5F1, TFAP2C, LIN28A, ALPP, and KIT. These genes are expressed in a population of mitotic fetal germ cells named gonocytes. NSAIDs have been shown to promote cell cycle arrest and apoptosis thereby decreasing the initiation and/or progression of various cancers (colorectal, bladder, skin, esophageal adenocarcinoma, ovarian)63,64,65,66. Whether ibuprofen effects occur by similar mechanisms remains to be established. In addition, whether ibuprofen-induced alteration of germ cell gene expression results from a direct effect of this NSAID on the human gonocytes, or if it is secondary to the marked suppression of the endocrine function of both Sertoli and Leydig cells is also unknown.
PGD2 and PGE2, the 2 prostaglandins investigated in this study, are thought to be involved in the differentiation of the fetal testis. In fact, exposing mouse testis explants (11, 12, and 14 days post-coitum) to PGE2 prevented the differentiation of testis cords67. Furthermore, mice deficient in Ptgds, which encodes the lipocalin-type prostaglandin D2 synthase enzyme responsible for PGD2 synthesis, display unilateral cryptorchidism68. However, here, we show that exposure to ibuprofen had no significant effect on PGD2 production. In contrast, ibuprofen induced marked changes in PGE2 levels. It is interesting to note that, as in our previous study on paracetamol, aspirin, and indomethacin, human fetal testis PGD2 production appears to be much less sensitive to the effects of ibuprofen than PGE226. The precise mechanism of action underlying the inhibitory effect of ibuprofen on PGE2 production remains to be explored. PGE2 is known to modulate immune and inflammatory responses through its receptors EP1 to EP469, and EP2 has recently been detected within the rat fetal testis70. It also remains to be determined what the consequences of ibuprofen-induced suppression of PGE2 might be on the human fetal testis biology and development. It would be important to understand which testicular cell type(s) is/are responsible for PGE2 production and which ones express the receptor for PGE2 within the human fetal testis.
Of all the analgesics that we have tested so far using our ex vivo human fetal testis system26, ibuprofen appears to be the agent which alters the human fetal endocrine balance with the widest range of effects as it affects all the major testicular cell types. Interestingly, paracetamol, aspirin, indomethacin, and ibuprofen each display their own endocrine-disrupting signatures (Table 1). The only unequivocally shared effect observed with these 4 analgesics is their inhibitory action on prostaglandin E2 production in 8–9.9 GW fetuses. However, whereas paracetamol and aspirin did not have any effect on testosterone production at this stage of gestation, the same group of testes treated with ibuprofen had lower testosterone production. Several other differences between analgesic effects also exist: ibuprofen - unlike aspirin but similar to paracetamol - suppresses INSL3 production. Furthermore, aspirin increased AMH production whereas paracetamol had no such effect, and ibuprofen showed a dose-response inhibition of this Sertoli cell produced hormone (Table 1). Taken together, these observations indicate that each drug has its own mechanism of action within the human fetal testis. Due to the different analgesic-induced biological signatures induced within the fetal testis, it is likely that this explains why when pregnant women simultaneously use more than one analgesic (paracetamol, aspirin, and ibuprofen), the risk of cryptorchidism observed is almost 10 times higher (odds ratio 2 fold versus 17 fold) than when a single painkiller is used7. Likewise, simultaneous use of both paracetamol and NSAIDs was found to be associated with significantly shorter AGD in boys, while exposure to paracetamol only was not significantly associated with shorter AGD24. In any case, the different therapeutic indications and contra-indications of these medications, i.e. paracetamol and of the NSAIDs, also reflect their different structures and properties. The present data complement our previous findings, which show that analgesics can behave as endocrine disruptors during key stages of the development of the urogenital tract26. This study provides new mechanistic explanations for the increased risk of cryptorchidism and hypospadias observed after in utero exposure to these medications by different groups.
Table 1. Summary of the endocrine-disrupting signatures of each tested analgesic ex vivo.
| Cell type | Hormone | Paracetamol | Ibuprofen | Aspirin | Indomethacin |
|---|---|---|---|---|---|
| Leydig cell | Testosterone | ⇔ |
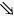 1 1
|
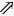 |
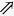 |
| INSL3 | 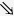 |
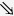 |
⇔ | ⇔ | |
| Sertoli cell | AMH | ⇔ | 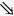 |
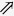 |
⇔ |
| Unknown | PGD2 | ⇔ | ⇔ | ⇔ | ⇔ |
| PGE2 | 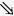 |
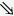 or or 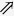 2 2
|
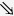 |
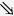 |
Data for paracetamol, aspirin and indomethacin come from26.
1After 24, 48 and 72 h of exposure.
2 In the 8–9.9 GW and 8–12 GW testes at 10−5 and 10−4 M;
In the 8–9.9 GW and 8–12 GW testes at 10−5 and 10−4 M;  in the 10–12 GW testes at 10−7 and 10−6 M.
in the 10–12 GW testes at 10−7 and 10−6 M.
In conclusion we show that exposure to therapeutic levels of ibuprofen during specific ‘windows of sensitivity’ can result in multiple effects on Sertoli-, Leydig- and germ-cell development and function in the human fetal testis. This includes effects on production of several testicular hormones during the first trimester. These findings are based on the results of 2 different model systems of human fetal testis development. Whilst they cannot be directly translated into recommendations for the use of ibuprofen in humans they provide experimental support to the epidemiological association between analgesic use and the development of male reproductive disorders, evidence that would support the avoidance of ibuprofen use during the first trimester where practicable.
Materials and Methods
Ethics statement
First trimester (7–12 gestational week) and second trimester (14–17 gestational week) human fetuses were obtained from abortions performed at the Rennes Sud Hospital in Rennes, France and the Royal Infirmary of Edinburgh, Edinburgh, UK, respectively. None of the pregnancy terminations were due to fetal abnormalities. Women gave written informed consent as per the legal procedures put in place by the French national biomedical research agency (authorization #PFS09-011; Agence de la Biomédecine) and the Declaration of Helsinki – Ethical Principles for Medical Research Involving Human Subjects. All experiments were performed in accordance with relevant guidelines and regulations. Ethical approval for the study was obtained from Rennes Sud Hospital local ethics committee (notice #11-48) and South East Scotland Research Ethics Committee (reference number: LREC08/S1101/1).
Human fetal testis collection
First-trimester human fetal testes were recovered from the abortion aspiration products using a binocular microscope (Olympus SZX7, Lille, France). They were immediately placed in ice-cold phosphate-buffered saline (PBS) solution. Second trimester human fetal testes were recovered and placed into ice-cold ‘xenograft media’ containing Liebowitz L-15 with glutamine, 10% fetal bovine serum, 1% penicillin/streptomycin and 1% non-essential amino acids (all Sigma, Poole, UK).
Culture and xenograft procedures
The testes were cut into explants of less than 1 mm3 according to a standardized protocol previously described26,40. For the testes younger than 8 GW, 2 wells were designed to accommodate 1 testis and either control or 10−5 M of ibuprofen treatment. These testes were immediately exposed to treatments. For the 8 to 10 GW group, 4 wells were prepared for 4 different culture conditions (1 control and 3 ibuprofen concentrations). These wells were each half a testis in length and contained 2 to 3 pieces of a single testis. For the older testes (10 to 12 GW), 6 wells for 6 different culture conditions (1 control and 5 ibuprofen concentrations) were created, each containing 1 or 2 testis pieces from a single testis. The explants were cultured in inserts (0.4 μm pores; Falcon, Becton-Dickinson, Le Pont de Claix, France) placed in 24-well companion culture plates (Becton-Dickinson). Human chorionic gonadotrophin (Sigma Aldrich, Saint-Quentin, France) was added at a concentration of 0.1 IU/mL71, and the cultures were incubated at 37 °C for 96 h in a humidified atmosphere of 95% air and 5% CO2. The medium was removed every 24 h and divided into at least 2 aliquots that were immediately snap-frozen on dry ice and stored at −80 °C. To assess dose-response effects, after the first 24 h of culture (D0) the explants were exposed to either the control, DMSO at a final concentration of 0.1%, or to the ibuprofen treatment, with concentrations of 10−7 M to 10−4 M added to the medium.
For xenograft studies, human fetal testes were grafted into castrate host mice as previously described27. Briefly, small pieces (1 mm3 approx; 4–6 per mouse) of testis tissue were placed subcutaneously, either side of the midline, under the dorsal skin of the mice using a 13 G cancer implant needle (Popper and Sons, US). In general, 3–6 mice were xenografted with tissue from each fetus, and mice were maintained for 7 days to ensure vascularisation before any host treatments commenced. One week after grafting, host mice commenced treatment with subcutaneous injection of human chorionic gonadotropin (20 IU hCG every 72 hours; Pregnyl, Organon Laboratories) to mimic the human in utero environment72. Host mice were also randomly allocated to receive a therapeutic regimen of either ibuprofen (10 mg/kg three times daily), or vehicle (corn oil) by daily oral administration with analysis 1 hour after the final dose. Host mice were sacrificed by cervical dislocation, and blood was obtained by cardiac puncture for assessment of plasma testosterone, AMH and ibuprofen. Testosterone production and action was assessed by measuring plasma testosterone and seminal vesicle weight. No differences were observed between vehicle- and ibuprofen-exposed host mice in terms of body weight, number of grafts retrieved or total graft weight (Fig. S1).
Immunostaining and stereology
Immunohistochemistry was performed on 4% paraformaldehyde-PBS and Bouin solution-fixed, paraffin-embedded tissues, as previously described26. The Sertoli cells were labeled with an AMH goat primary antibody (1:100; Santa Cruz Biotechnology, CA, USA). Leydig cells were stained with a rabbit anti-cytochrome P450, family 11, subfamily A, polypeptide 1 (CYP11A1) antibody (1:250; Sigma Aldrich). A mouse M2A primary antibody (1:100; Abcam, Paris, France) was used for gonocyte immunolabeling. For the AMH and M2A antibodies, antigens were retrieved for 40 min at 80 °C in 10 mM citrate buffer, pH 6.
A NanoZoomer 2.0-RS scanner (Hamamatsu, Tokyo, Japan) was used to capture pictures of the whole slides at 40x magnification. The surface area of 5 to 10 sections randomly selected within the whole explant were calculated with NDP.view software (Hamamatsu). ImageJ software (US National Institutes of Health, Bethesda, MD, USA) was used to perform the stereological cell counting. Germ and Sertoli cells were identified and counted as intra-cordal AMH-negative and AMH-positive cells, respectively.
Hormone assays
Testosterone was assayed using a specific radioimmunoassay (RIA): a direct testosterone RIA with an intra-assay coefficient of variation (CV) ≤ 8.6% and an inter-assay CV of 11.9% (Immunotech, Beckman Coulter, Villepinte, France). A specific RIA was also used for insulin-like 3: a human INSL3/RLF RIA kit with intra- and inter-assay CVs of ≤15 and 7%, respectively (Phoenix Pharmaceuticals, Strasbourg, France). AMH levels were assayed using an AMH/MIS Enzyme-Linked Immunosorbent Assay (ELISA) kit with an intra-assay CV of 3.2 to 12.3% and inter-assay CV of 5.8 to 14.2% (Immunotech, Marseille, France). PGD2 was measured with ELISA using a prostaglandin D2-MOX EIA Kit having an intra-assay CV of 8 to 15% and an inter-assay CV of 10 to 17% (Cayman Chemical, Ann Arbor, MI, USA). PGE2 was also assayed using a prostaglandin E2 EIA Kit (intra-assay CV 3.7 to 30.4% and inter-assay CV 6.4 to 35%) (Cayman Chemical).
The hormone assay data from the culture system using the testes of the fetuses younger than 8 GW were expressed as hormone production fold change as compared to the control testis. Since the older testes were cut into explants, the first 24 hours of culture (day 0 without treatment, or D0) served as the baseline for normalization of hormone production in each testicular sample after 24, 48, and 72 h of exposure to DMSO or ibuprofen. Depending on fetal age, we established cut-offs for normal relative testosterone production in the control experiments, calculated at D3 normalized to the D0 result in the same culture well. These were >0.7 for the 8–9.8 GW explants, and ≥0.1 for the 10–12 ones.
Global analysis of steroids
The procedures for applied sample preparation and GC-MS/MS measurements have been previously described73,74. Tissue samples were homogenized after addition of 50:50 v/v methanol and water (300 μl/10 mg tissue). Prior to extraction for quantification according to isotopic dilution, tissue homogenates, and cell culture media (200 μl) were spiked with internal standards (17β-estradiol-16,16,17-d3; 5α-androstan-3α,17β-diol-16,16,17-d3; etiocholanolone-2,2,3β,4,4-d5; 17β-testosterone-16,16,17-d3; 4-androsten-3,17-dione-19,19,19-d3; 5α-androstan-17β-ol-3-one-16,16,17-d3; and 17-methyltestosterone-d3). Non-polar compounds were extracted from the aqueous phase (pH 5.2) with diethyl ether, followed by a liquid/liquid partitioning to separate androgens and estrogens. Both fractions were purified on 200 mg Upti-Clean SI-S cartridges (Interchim, Montluçon, France), then derivatized with a silylation reagent as described by Courant et al.74. Two micro-liters of each final extract were injected into the GC-MS/MS system. The detection and quantification of the targeted compounds were performed on a spectrometer (Bruker, Fremont, CA, USA). Electron impact (70 eV) was the ionization mode used for androgens, while negative chemical ionization (80 μA filament current) with methane reactant gas was applied for estrogens. The source temperature was set to 250 °C and the transfer line to 300 °C. The chromatographic conditions were previously described by Courant et al.74. The mass spectrometer was operated in multiple reaction monitoring (MRM) acquisition mode, and argon was used as collision gas at 2.0 mTorr. The diagnostic signals were monitored for the targeted compounds have been reported elsewhere73,74.
For xenografts, testosterone and ibuprofen were extracted from mouse plasma by solid phase extraction using 10 mg HLB Oasis cartridges (Waters, UK). Briefly, 50 μL plasma was enriched with 1 ng 13C3-Testosterone (QMX, UK) as internal standard. Cartridges were conditioned with methanol and water, the sample loaded and washed with water, 5% methanol and then eluted with 1 mL methanol. The eluate was reduced to dryness under nitrogen at 40 °C and reconstituted in mobile phase (70 μL water/acetonitrile (70:30, v/v)). Chromatographic separation was achieved by injecting 30 μL sample onto an Acquity UPLC system with an ACE Excel C18-AR column (150 × 2.1 mm; 2 μm) protected by a Kinetex KrudKatcher (Phenomenex) and operated at 30 °C. The mobile phase consisted of 0.1% formic acid (Sigma Aldrich) in water and 0.1% formic acid in acetonitrile (B) at a flow rate of 0.5 mL/min. Gradient elution from 30–100% B was achieved, with a total run time of 9 mins. Testosterone, and Ibuprofen eluted at 5.40 and 5.7 min respectively. Following separation, testosterone and ibuprofen were detected on a QTrap 5500 triple quadrupole mass spectrometer (Sciex, Warrington, UK) operated in positive ion electrospray mode (5.5 kV, 550 °C, ion source gas 60/40). Transitions monitored were m/z 289.1 → 97.0, and m/z 156.1 → 114.1 for testosterone and ibuprofen, respectively. The ratio of the peak area of each analyte to the internal standard was used to generate a calibration curve and using linear regression analysis the amount of each analyte was calculated.
RNA extraction and quantitative Polymerase Chain Reaction (qPCR)
RNA extractions from testes were done using a NucleoSpin XS kit (Macherey-Nagel, Hoerdt, France) according to the manufacturer’s instructions. Total RNA (250 ng) was reverse transcribed with random primers and Moloney murine leukemia virus reverse transcriptase (Invitrogen, ThermoFisher, Courtaboeuf, France). Quantitative PCR was performed according to the manufacturer’s instructions using the SYBR Green master mix (Applied Biosystems, ThermoFisher) with a 0.5 μl cDNA template in an Applied Biosystems 7500 Real-Time PCR system. The amplification program was as follows: a 2 min holding stage at 50 °C; initial denaturation of 10 min at 95 °C; 40 cycles of 15 sec denaturation at 95 °C; and 1 min at 60 °C for annealing and extension. Dissociation curves were produced using a thermal melting profile performed after the last PCR cycle. To avoid amplification of contaminating genomic DNA, primer pairs were selected on either side of an intron. RPLP0 and RPS20 mRNA were used as internal controls for normalization75 (Table 2). Results were calculated by the ΔΔCT method as n-fold differences in target gene expression with respect to the reference gene and the calibration sample (made of an equal mix of each of the samples tested).
Table 2. Primers used for qPCR.
| Gene | Upstream primer | Downstream primer | Product length (bp) | Annealing temp °C |
|---|---|---|---|---|
| ALPP | TCTGGGTACTCAGGGTCTGG | ATCGCTACGCAGCTCATCTC | 101 | 62 |
| AMH | CGCCTGGTGGTCCTACAC | GAACCTCAGCGAGGGTGTT | 162 | 67 |
| BZRP | GGCTTCACAGAGAAGGCTGT | ACTGACCAGCAGGAGATCCA | 87 | 66 |
| COL2A1 | GGCAATAGCAGGTTCACGTACA | CGATAACAGTCTTGCCCCACTT | 57 | 53 |
| CYP11A1 | AGACCTGGAAGGACCATGTG | TCCTCGAAGGACATCTTGCT | 435 | 65 |
| CYP17A1 | GTGGAGACCACCACCTCTGT | GCTGAAACCCACATTCTGGT | 108 | 67 |
| DHH | TGATGACCGAGCGTTGTAAG | GCCAGCAACCCATACTTGTT | 196 | 67 |
| HSD3B2 | GCCTGTTGGTGGAAGAGAAG | GCAGGCTCTTTTCAGGAATG | 158 | 62 |
| HSD17B3 | TCCTTGGCCTCTCTACTCCA | AGACAGCATATGGGGTCAGC | 125 | 62 |
| INSL3 | CCCAGAGATGCGTGAGAAGT | CCAGCCACTGTAGCAACTCA | 229 | 68 |
| KIT | TTCTTACCAGGTGGCAAAGG | AAATGCTTTCAGGTGCCATC | 209 | 62 |
| LIN28A | AAGCTGCACATGGAAGGGTT | CCGCCTCTCACTCCCAATAC | 138 | 67 |
| POU5F1 | TACTCCTCGGTCCCTTTCC | CAAAAACCCTGGCACAAACT | 131 | 67 |
| RPLP0 | TCTACAACCCTGAAGTGCTTGAT | CAATCTGCAGACAGACACTGG | 167 | 66 |
| RPS20 | AACAAGCCGCAACGTAAAA | ACGATCCCACGTCTTAGAA | 96 | 67 |
| SOX9 | AACGCCTTCATGGTGTGG | TCTCGCTCTCGTTCAGAAGTC | 124 | 59 |
| SRD5A3 | AGGAATGCCTACATAACAGGGAA | CTCCAAATGGGATCCTGTGGT | 181 | 62 |
| STAR | GGCTGGCATGGCCACAGACT | TTGGGCAGCCACCCCTTGA | 162 | 77 |
| TFAP2C | CCGGTCCTTGCGGGAGAAGTT | CTGGTTTACTAGGAAATTCGGCTTCACA | 164 | 75 |
Statistical analysis
Hormone measurements (means ± SEM) are expressed either as percentages of the values as compared to the control testes (7 to 7.9 GW fetuses), as the fold change from the respective untreated first day of culture (D0; 8–12 GW fetuses), or as absolute values for serum in xenografted mice.
Testosterone and AMH production as a function of culture duration in 7–7.9 GW testes were analyzed using repeated measures analysis of variance (ANOVA) on ranks. Ibuprofen dose-response relationships were assessed for testosterone, INSL3, AMH, PGD2, and PGE2 using variance analyses, with the treatment as explanatory variable and a random fetus effect. To normalize the data, logarithm transformations were used for INSL3, AMH, PGD2 and PGE2 data. The dose was transformed into a categorical variable (ranging from 1 for the lowest dose to 4 for the highest one) then introduced into the models as continuous variables. The corresponding slope β therefore represents the change in hormone secretion for each supplementary dose unit. Pairwise differences between the ibuprofen treatments and controls were analyzed using the non-parametric Wilcoxon signed-rank test. Quantitative PCR data, GC-MS/MS data, and cell-counts were also analyzed with the Wilcoxon test. For analysis of AMH, testosterone and ibuprofen in xenografts, a two-way ANOVA was performed. Significance was defined as a confidence level of p < 0.05. Statistical analyses were performed using SAS software (SAS/STAT version 9.3; SAS Institute Inc., Cary, NC) and SigmaPlot 12.0 software (Systat Software, San Jose, CA, USA).
Additional Information
How to cite this article: Ben Maamar, M. et al. Ibuprofen results in alterations of human fetal testis development. Sci. Rep. 7, 44184; doi: 10.1038/srep44184 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
We thank all the staff of the Department of Obstetrics and Gynecology and the Department of Pediatric Surgery of the Rennes Sud Hospital (Rennes, France) and the participating women, without whom this study would not have been possible. We also thank the nursing staff of the Bruntsfield Ward at the Royal Infirmary of Edinburgh and the Human Developmental Biology Resource (www.hdbr.org) for providing some of the fetal material (Joint MRC/Wellcome Trust; Grant No: 099175/Z/12/Z). We also thank Natalie Homer from the Edinburgh CRF Mass Spectrometry Core for her assistance. This study was funded by Inserm, University of Rennes 1, EHESP – School of Public Health, by grants from the Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM; AAP-2012–037), and the Danish Council for independent Research (Medical Sciences), and the Wellcome Trust (Grant No: 098522).
Footnotes
The authors declare no competing financial interests.
Author Contributions M.B.M. performed experiments (organ collection, FEGA, hormone measurements, Qt RT-PCR and histology), analyzed data and wrote the manuscript draft. J-P.A., B.L.B. and K.H. performed the global steroid analyses and wrote the corresponding section of the manuscript. L.L. performed cell counts, statistics and contributed to the writing of the manuscript. C.D.L. contributed to hormone measurements and Qt RT-PCR validations. K.K. performed xenografting experiments. I.C. contributed to organ collection and tissue culture. A.D.R. designed and performed Qt-RT-PCR. C.C. performed multivariate analyses. N.D.R. and D.M.K. contributed to critical discussions. V.L. supervised the collection of the first trimester human fetal testis samples. R.T.M. designed and conducted the xenograft experiments, analyzed data, and contributed to the writing of the manuscript. S.M.G. designed and performed experiments (organ collection, FEGA, Qt RT-PCR and histology), analyzed data, and contributed to writing of the manuscript. B.J. designed the study, analyzed data, and contributed to the writing of the manuscript. All authors approved the final version.
References
- Kristensen D. M. et al. Analgesic use - prevalence, biomonitoring and endocrine and reproductive effects. Nat. Rev. Endocrinol. doi: 10.1038/nrendo.2016.55 (2016). [DOI] [PubMed] [Google Scholar]
- Verlicchi P., Al Aukidy M., Galletti A., Petrovic M. & Barceló D. Hospital effluent: investigation of the concentrations and distribution of pharmaceuticals and environmental risk assessment. Sci. Total Environ. 430, 109–18 (2012). [DOI] [PubMed] [Google Scholar]
- Kassaw C. & Wabe N. T. Pregnant women and non-steroidal anti-inflammatory drugs: knowledge, perception and drug consumption pattern during pregnancy in ethiopia. N. Am. J. Med. Sci. 4, 72–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassoun-Barhamji R., Raia Barjat T. & Chauleur C. In the Era Of Self-Medication, What Do Pregnant Women Know about Anti-Inflammatory Drugs? Therapie 70, 369–76 (2015). [DOI] [PubMed] [Google Scholar]
- Ertresvåg J. M., Zwart J.-A., Helde G., Johnsen H.-J. & Bovim G. Headache and transient focal neurological symptoms during pregnancy, a prospective cohort. Acta Neurol. Scand. 111, 233–7 (2005). [DOI] [PubMed] [Google Scholar]
- Werler M. M., Mitchell A. A., Hernandez-Diaz S. & Honein M. A. Use of over-the-counter medications during pregnancy. Am. J. Obstet. Gynecol. 193, 771–7 (2005). [DOI] [PubMed] [Google Scholar]
- Kristensen D. M. et al. Intrauterine exposure to mild analgesics is a risk factor for development of male reproductive disorders in human and rat. Hum. Reprod. 26, 235–44 (2011). [DOI] [PubMed] [Google Scholar]
- Snijder C. A. et al. Intrauterine exposure to mild analgesics during pregnancy and the occurrence of cryptorchidism and hypospadia in the offspring: the Generation R Study. Hum. Reprod. 27, 1191–201 (2012). [DOI] [PubMed] [Google Scholar]
- Alano M. A., Ngougmna E., Ostrea E. M. & Konduri G. G. Analysis of nonsteroidal antiinflammatory drugs in meconium and its relation to persistent pulmonary hypertension of the newborn. Pediatrics 107, 519–23 (2001). [DOI] [PubMed] [Google Scholar]
- Berlin C. M., Yaffe S. J. & Ragni M. Disposition of acetaminophen in milk, saliva, and plasma of lactating women. Pediatr. Pharmacol. (New York). 1, 135–41 (1980). [PubMed] [Google Scholar]
- Bitzén P. O., Gustafsson B., Jostell K. G., Melander A. & Wåhlin-Boll E. Excretion of paracetamol in human breast milk. Eur. J. Clin. Pharmacol. 20, 123–5 (1981). [DOI] [PubMed] [Google Scholar]
- Ellfolk M. & Hultzsch S. In Drugs During Pregnancy and Lactation: Treatment options and risk assessment. (eds Schaeffer C., Peters P. & Miller R.) 4.1, 653–670 (Elsevier, 2015).
- Levy G., Garrettson L. K. & Soda D. M. Letter: Evidence of placental transfer of acetaminophen. Pediatrics 55, 895 (1975). [PubMed] [Google Scholar]
- Naga Rani M. A., Joseph T. & Narayanan R. Placental transfer of paracetamol. J. Indian Med. Assoc. 87, 182–3 (1989). [PubMed] [Google Scholar]
- Weigand U. W., Chou R. C., Maulik D. & Levy G. Assessment of biotransformation during transfer of propoxyphene and acetaminophen across the isolated perfused human placenta. Pediatr. Pharmacol. (New York). 4, 145–53 (1984). [PubMed] [Google Scholar]
- Nezvalová-Henriksen K., Spigset O. & Nordeng H. Effects of ibuprofen, diclofenac, naproxen, and piroxicam on the course of pregnancy and pregnancy outcome: a prospective cohort study. BJOG 120, 948–59 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerman C. & Kaplan M. Patent ductus arteriousus in the premature neonate: current concepts in pharmacological management. Paediatr. Drugs 1, 81–92 (1999). [DOI] [PubMed] [Google Scholar]
- Marsh C. A., Cragan J. D., Alverson C. J. & Correa A. Case-control analysis of maternal prenatal analgesic use and cardiovascular malformations: Baltimore–Washington Infant Study. Am. J. Obstet. Gynecol. 211, 404.e1–404.e9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieux R., Fresson J., Guillemin F. & Hascoet J. M. Perinatal drug exposure and renal function in very preterm infants. Arch. Dis. Child. Fetal Neonatal Ed. 96, F290–5 (2011). [DOI] [PubMed] [Google Scholar]
- Sordillo J. E. et al. Prenatal and infant exposure to acetaminophen and ibuprofen and the risk for wheeze and asthma in children. J. Allergy Clin. Immunol. 135, 441–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz G. S. & Lapinski R. H. Risk factors for cryptorchidism: a nested case-control study. Paediatr. Perinat. Epidemiol. 10, 39–51 (1996). [DOI] [PubMed] [Google Scholar]
- Jensen M. S. et al. Maternal use of acetaminophen, ibuprofen, and acetylsalicylic acid during pregnancy and risk of cryptorchidism. Epidemiology 21, 779–85 (2010). [DOI] [PubMed] [Google Scholar]
- Fisher B. G. et al. Prenatal paracetamol exposure is associated with shorter anogenital distance in male infants. Hum. Reprod. 31, 2642–2650 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind D. V. et al. Maternal use of mild analgesics during pregnancy associated with reduced anogenital distance in sons: a cohort study of 1027 mother-child pairs. Hum. Reprod. 32, 223–231 (2017). [DOI] [PubMed] [Google Scholar]
- Kristensen D. M. et al. Paracetamol (acetaminophen), aspirin (acetylsalicylic acid) and indomethacin are anti-androgenic in the rat foetal testis. Int. J. Androl. 35, 377–84 (2012). [DOI] [PubMed] [Google Scholar]
- Mazaud-Guittot S. et al. Paracetamol, aspirin, and indomethacin induce endocrine disturbances in the human fetal testis capable of interfering with testicular descent. J. Clin. Endocrinol. Metab. 98, E1757–67 (2013). [DOI] [PubMed] [Google Scholar]
- van den Driesche S. et al. Prolonged exposure to acetaminophen reduces testosterone production by the human fetal testis in a xenograft model. Sci. Transl. Med. 7, 288ra80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jégou B. Paracetamol-induced endocrine disruption in human fetal testes. Nat. Rev. Endocrinol. 11, 453–4 (2015). [DOI] [PubMed] [Google Scholar]
- Glover D. D., Amonkar M., Rybeck B. F. & Tracy T. S. Prescription, over-the-counter, and herbal medicine use in a rural, obstetric population. Am. J. Obstet. Gynecol. 188, 1039–45 (2003). [DOI] [PubMed] [Google Scholar]
- Wen S. W. et al. Patterns of pregnancy exposure to prescription FDA C, D and X drugs in a Canadian population. J. Perinatol. 28, 324–9 (2008). [DOI] [PubMed] [Google Scholar]
- Damase-Michel C., Christaud J., Berrebi A., Lacroix I. & Montastruc J.-L. What do pregnant women know about non-steroidal anti-inflammatory drugs? Pharmacoepidemiol. Drug Saf. 18, 1034–8 (2009). [DOI] [PubMed] [Google Scholar]
- Stephansson O. et al. Drug use during pregnancy in Sweden - assessed by the Prescribed Drug Register and the Medical Birth Register. Clin. Epidemiol. 3, 43–50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel S. et al. Fetal exposure to nonsteroidal anti-inflammatory drugs and spontaneous abortions. CMAJ 186, E177–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Marter L. J., Hernandez-Diaz S., Werler M. M., Louik C. & Mitchell A. A. Nonsteroidal antiinflammatory drugs in late pregnancy and persistent pulmonary hypertension of the newborn. Pediatrics 131, 79–87 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind J. N. et al. Maternal medication and herbal use and risk for hypospadias: data from the National Birth Defects Prevention Study, 1997-2007. Pharmacoepidemiol. Drug Saf. 22, 783–93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez R. K., Werler M. M., Romitti P., Sun L. & Anderka M. Nonsteroidal antiinflammatory drug use among women and the risk of birth defects. Am. J. Obstet. Gynecol. 206, 228.e1–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akre O. & Richiardi L. Does a testicular dysgenesis syndrome exist? Hum. Reprod. 24, 2053–60 (2009). [DOI] [PubMed] [Google Scholar]
- Foresta C., Zuccarello D., Garolla A. & Ferlin A. Role of hormones, genes, and environment in human cryptorchidism. Endocr. Rev. 29, 560–80 (2008). [DOI] [PubMed] [Google Scholar]
- Thorup J., Nordenskjöld A. & Hutson J. M. Genetic and environmental origins of hypospadias. Curr. Opin. Endocrinol. Diabetes. Obes. 21, 227–32 (2014). [DOI] [PubMed] [Google Scholar]
- Ben Maamar M. et al. An investigation of the endocrine-disruptive effects of bisphenol a in human and rat fetal testes. PLoS One 10, e0117226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell R. T. et al. Do phthalates affect steroidogenesis by the human fetal testis? Exposure of human fetal testis xenografts to di-n-butyl phthalate. J. Clin. Endocrinol. Metab. 97, E341–8 (2012). [DOI] [PubMed] [Google Scholar]
- Anand-Ivell R., Ivell R., Driscoll D. & Manson J. Insulin-like factor 3 levels in amniotic fluid of human male fetuses. Hum. Reprod. 23, 1180–6 (2008). [DOI] [PubMed] [Google Scholar]
- Rajpert-De Meyts E. et al. Developmental expression of POU5F1 (OCT-3/4) in normal and dysgenetic human gonads. Hum. Reprod. 19, 1338–44 (2004). [DOI] [PubMed] [Google Scholar]
- Hoei-Hansen C. E. et al. Transcription factor AP-2gamma is a developmentally regulated marker of testicular carcinoma in situ and germ cell tumors. Clin. Cancer Res. 10, 8521–30 (2004). [DOI] [PubMed] [Google Scholar]
- Shinoda G. et al. Lin28a regulates germ cell pool size and fertility. Stem Cells 31, 1001–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen N. et al. Expression of immunohistochemical markers for testicular carcinoma in situ by normal human fetal germ cells. Lab. Invest. 72, 223–31 (1995). [PubMed] [Google Scholar]
- Robinson L. L., Gaskell T. L., Saunders P. T. & Anderson R. A. Germ cell specific expression of c-kit in the human fetal gonad. Mol. Hum. Reprod. 7, 845–52 (2001). [DOI] [PubMed] [Google Scholar]
- Mitchell J. A., Akarasereenont P., Thiemermann C., Flower R. J. & Vane J. R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 90, 11693–7 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D. L., Botting R. M. & Hla T. Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 56, 387–437 (2004). [DOI] [PubMed] [Google Scholar]
- Nef S. & Parada L. F. Cryptorchidism in mice mutant for Insl3. Nat. Genet. 22, 295–9 (1999). [DOI] [PubMed] [Google Scholar]
- Hutson J. M., Balic A., Nation T. & Southwell B. Cryptorchidism. Semin. Pediatr. Surg. 19, 215–24 (2010). [DOI] [PubMed] [Google Scholar]
- Serrano T., Chevrier C., Multigner L., Cordier S. & Jegou B. International geographic correlation study of the prevalence of disorders of male reproductive health. Hum. Reprod. 28, 1974–1986 (2013). [DOI] [PubMed] [Google Scholar]
- Källström E., Heikinheimo M. & Quiding H. Bioavailability of three commercial preparations of ibuprofen 600 mg. J. Int. Med. Res. 16, 44–9 (1988). [DOI] [PubMed] [Google Scholar]
- Karttunen P., Saano V., Paronen P., Peura P. & Vidgren M. Pharmacokinetics of ibuprofen in man: a single-dose comparison of two over-the-counter, 200 mg preparations. Int. J. Clin. Pharmacol. Ther. Toxicol. 28, 251–5 (1990). [PubMed] [Google Scholar]
- Jost A. A new look at the mechanisms controlling sex differentiation in mammals. Johns Hopkins Med. J. 130, 38–53 (1972). [PubMed] [Google Scholar]
- Pelliniemi L. J. & Niemi M. Fine structure of the human foetal testis. I. The interstitial tissue. Zeitschrift für Zellforsch. und mikroskopische Anat. (Vienna, Austria 1948) 99, 507–22 (1969). [DOI] [PubMed] [Google Scholar]
- Chauvigné F. et al. Mono-(2-ethylhexyl) phthalate directly alters the expression of Leydig cell genes and CYP17 lyase activity in cultured rat fetal testis. PLoS One 6, e27172 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke J. A. et al. A novel communication role for CYP17A1 in the progression of castration-resistant prostate cancer. Prostate 69, 928–37 (2009). [DOI] [PubMed] [Google Scholar]
- Stutz G. et al. Functional activity of mouse sperm was not affected by low doses of aspirin-like drugs. Arch. Androl. 44, 117–28 (2000). [DOI] [PubMed] [Google Scholar]
- Sten T., Finel M., Ask B., Rane A. & Ekström L. Non-steroidal anti-inflammatory drugs interact with testosterone glucuronidation. Steroids 74, 971–7 (2009). [DOI] [PubMed] [Google Scholar]
- Zimmermann S. et al. Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol. Endocrinol. 13, 681–91 (1999). [DOI] [PubMed] [Google Scholar]
- Bay K. & Andersson A.-M. Human testicular insulin-like factor 3: in relation to development, reproductive hormones and andrological disorders. Int. J. Androl. 34, 97–109 (2011). [DOI] [PubMed] [Google Scholar]
- Fischer S. M., Hawk E. T. & Lubet R. A. Coxibs and other nonsteroidal anti-inflammatory drugs in animal models of cancer chemoprevention. Cancer Prev. Res. (Phila). 4, 1728–35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santander S. et al. Cyclooxygenase inhibitors decrease the growth and induce regression of human esophageal adenocarcinoma xenografts in nude mice. Int. J. Oncol. 40, 527–34 (2012). [DOI] [PubMed] [Google Scholar]
- Rodríguez-Burford C. et al. Effects of nonsteroidal anti-inflammatory agents (NSAIDs) on ovarian carcinoma cell lines: preclinical evaluation of NSAIDs as chemopreventive agents. Clin. Cancer Res. 8, 202–9 (2002). [PubMed] [Google Scholar]
- Valle B. L. et al. Non-steroidal anti-inflammatory drugs decrease E2F1 expression and inhibit cell growth in ovarian cancer cells. PLoS One 8, e61836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taketo T., Thau R. B., Adeyemo O. & Koide S. S. Influence of adenosine 3′:5′-cyclic monophosphate analogues on testicular organization of fetal mouse gonads in vitro. Biol. Reprod. 30, 189–98 (1984). [DOI] [PubMed] [Google Scholar]
- Philibert P. et al. Unilateral cryptorchidism in mice mutant for Ptgds. Hum. Mutat. 34, 278–82 (2013). [DOI] [PubMed] [Google Scholar]
- Hirata T. & Narumiya S. Prostanoids as regulators of innate and adaptive immunity. Adv. Immunol. 116, 143–74 (2012). [DOI] [PubMed] [Google Scholar]
- Dean A. et al. Analgesic exposure in pregnant rats affects fetal germ cell development with inter-generational reproductive consequences. Sci. Rep. 6, 19789 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmark N. et al. Effects of monobutyl and di(n-butyl) phthalate in vitro on steroidogenesis and Leydig cell aggregation in fetal testis explants from the rat: comparison with effects in vivo in the fetal rat and neonatal marmoset and in vitro in the human. Environ. Health Perspect. 115, 390–6 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell R. T. et al. Xenografting of human fetal testis tissue: a new approach to study fetal testis development and germ cell differentiation. Hum. Reprod. 25, 2405–14 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courant F. et al. Exposure assessment of prepubertal children to steroid endocrine disrupters 1. Analytical strategy for estrogens measurement in plasma at ultra-trace level. Anal. Chim. Acta 586, 105–14 (2007). [DOI] [PubMed] [Google Scholar]
- Courant F. et al. Assessment of circulating sex steroid levels in prepubertal and pubertal boys and girls by a novel ultrasensitive gas chromatography-tandem mass spectrometry method. J. Clin. Endocrinol. Metab. 95, 82–92 (2010). [DOI] [PubMed] [Google Scholar]
- Svingen T., Jørgensen A. & Rajpert-De Meyts E. Validation of endogenous normalizing genes for expression analyses in adult human testis and germ cell neoplasms. Mol. Hum. Reprod. 20, 709–18 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.