

Abstract
NEK2 is a conserved mitotic regulator critical for cell cycle progression. Aberrant expression of NEK2 has been found in a variety of human cancers, making it an attractive molecular target for the design of novel anticancer therapeutics. In the present study, we have identified a novel compound MBM-5, which was found to bind to NEK2 with high affinity by docking simulations study. MBM-5 potently inhibited NEK2 kinase activity in vitro in a concentration-dependent manner. MBM-5 also suppressed cellular NEK2 kinase activity, as evidenced by the decreased phosphorylation of its substrate Hec1 on S165 in a concentration- and time-dependent manner. This inhibition impeded mitotic progression by inducing chromosome segregation defects and cytokinesis failure; therefore leading to accumulation of cells with ≥4N DNA content, which finally underwent apoptosis. More importantly, MBM-5 treatment effectively suppressed the tumor growth of human gastric and colorectal cancer cells xenografts. Taken together, we demonstrated that MBM-5 effectively inhibited the kinase activity of NEK2 and showed a potential application in anti-cancer treatment regimens.
Keywords: NEK2, mitosis, chromosome misalignment, cytokinesis failure, apoptosis
INTRODUCTION
Never in Mitosis Related Kinase 2 (NEK2) is a member of the Never in Mitosis (NIMA) Related Kinases (NEKs) family that has a broadly structural similarity to the mitotic regulator NIMA of the filamentous fungus Aspergillus nidulans [1]. NEK2 is well recognized as a multifunctional serine/threonine kinase with key roles in cell cycle regulation, particularly with respect to the centrosome cycle. NEK2 localizes to the centrosome and triggers centrosome separation by phosphorylating centrosome cohesion proteins C-Nap1, Rootletin and Cep68 [2–4]. NEK2 also regulates microtubule organization and stabilization through phosphorylation of ninein-like protein (Nlp) [5]. Moreover, NEK2 operates a faithful kinetochore microtubule attachments by phosphorylation of highly expressed in cancer 1 (Hec1) [6, 7]. Through direct interaction with mitotic arrest deficient-like 1 (MAD1) or phosphorylation of Hec1 and Sgo1, NEK2 also modulates chromosome alignment and the spindle assembly checkpoint (SAC) signaling, thus regulating chromosome separation [8–10]. In addition, NEK2B (a splice variant of NEK2) is observed to be required for execution of mitotic exit as NEK2B depleted cells were unable to complete cytokinesis and resulted in the formation of multinucleated cells [11].
NEK2 has been reported to be overexpressed in a wide variety of human cancers, such as gastric cancer [12], colorectal cancer [13, 14], prostate cancer [15] and breast cancer [16, 17]. NEK2 overexpression is associated with tumorigenensis, tumor progression, drug resistance and predicts poor prognosis [18, 19]. Several preclinical studies using RNA interference targeting NEK2 have shown the efficient anti-tumor effect against different type of cancers. Suppression of the NEK2 expression with siRNA inhibited cell proliferation and induced cell death of breast cancer, cholangiocarcinoma, colorectal cancer, multiple myeloma, hepatoma and prostate cancer cells in vitro, and led to a reduction of tumor size in xenograft-nude mouse model [13, 15, 17, 20–23]. Moreover, silencing of NEK2 expression in breast cancer and colorectal cancer cells dramatically increased the susceptibility to chemotherapeutic drugs, such as paclitaxel, doxorubicin and cisplatin [13, 24]. These findings suggest NEK2 as a valuable anticancer target.
Development of NEK2 inhibitors has attracted much attention recently. The sunitinib-like oxindole inhibitor (SU11652), two thiophene-based PLK1 inhibitors and a series of viridian/wortmannin-like compounds were reported to have activity against NEK2, even though their activities against other kinases were greater [25–27]. Then a series of aminopyrazines and benzimidazoles were developed and showed activities against NEK2 kinase with IC50 of micromole range. However, none of them was active in cells because of insufficient membrane permeability [28, 29]. By combining key scaffold of the aminopyrazines and benzimidazoles, the hybrids finally yielded potent compounds with better activity against NEK2 (IC50=0.022-2.680 μM) and capable of modulating the phosphorylation of NEK2 substrates in cells [30]. Besides above reversible compounds, an irreversible inhibitor JH295 with IC50=0.770 μM against NEK2 was reported; however, its antitumor activity against cancer cell lines was not evaluated [31]. Moreover, the in vivo antitumor activity of any compound mentioned above has not been disclosed yet. Therefore, great efforts should be made to develop novel NEK2 inhibitors that are suitable for clinical applications.
By performing molecular docking analysis, we screened five hundred compounds from our in-house compound library to test their affinity to the ATP-site of the NEK2 kinase. Compounds with high docking score were selected to determine their potential activities against NEK2 kinase in vitro. A novel compound named MBM-5 was found to have the best inhibitory activity against NEK2 kinase. Further biologic testing confirmed that MBM-5 potently inhibited cellular NEK2 activity and exhibited antitumor activity in vitro and in vivo.
RESULTS
MBM-5 inhibits NEK2 kinase activity
MBM-5 is a novel compound synthesized in our laboratory (Figure 1A). The molecular docking/dynamic simulation combined with binding free energy calculations were used to study the binding modes of MBM-5 with NEK2. The predicted binding mode of MBM-5 and NEK2 showed that MBM-5 occupied the ATP-binding site and formed three conserved hydrogen bonds with NEK2 (Figure 1B-1D), one was between nitrogen atom of imidazo[1,2-a]pyridine and CYS 89, and the other two were between the amide group and ASP 159. In addition, there was a conserved π-π interaction between MBM-5 and NEK2. The calculated binding free energy between MBM-5 and NEK2 was −9.40 kcal/mol. The HTRF® (Homogeneous Time-Resolved Fluorescence) KinEASE™-STK assay was then carried out to determine the NEK2 kinase inhibition activity of MBM-5. The result showed that the IC50 was 0.340±0.075 μM (Figure 1E). We further determined the inhibitory effect of MBM-5 on NEK2 kinase activity in cancer cells. As an important substrate of NEK2, Hec1 was phosphorylated by NEK2 at S165; therefore the phosphorylation of Hec1 (S165) was determined. MBM-5 effectively inhibited the phosphorylation of Hec1 (S165) in a concentration- and time- dependent manner, indicating MBM-5 was able to enter into cells and inhibit cellular NEK2 kinase activity (Figure 1F). Unexpectedly, the total Hec1 was down-regulated after NEK2 treatment. Considering the similar structure between NEK2 and Aurora A (with 31% structurally identical) [25], we determined the effect of MBM-5 against Aurora A kinase activity by analyzing its phosphorylated protein level. The phosphorylation of Aurora A was not affected by MBM-5 (Figure 1F). These data demonstrated that MBM-5 was able to inhibit NEK2 kinase activity.
Figure 1. MBM-5 inhibits NEK2 kinase activity.
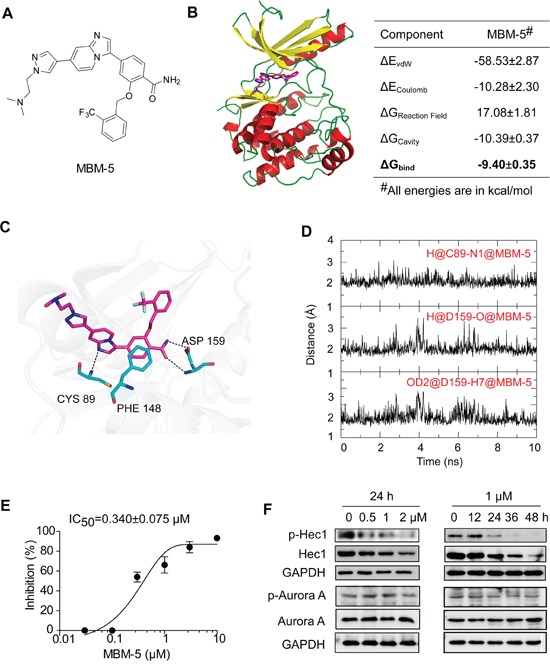
A. Chemical structure of MBM-5. B-D. Computational predicted binding mode of MBM-5 with NEK2. B. Bind free energies of MBM-5 to NEK2 calculated by the SIE method. ΔGbind is the calculated binding free energy; ΔEvdW and ΔECoulomb are contributions of van der Waals and Coulomb interactions to ΔGbind, ΔGReaction Field and ΔGCavity describe differences in the reaction field energy and molecular surface area upon inhibitor binding. C. Proposed binding mode of MBM-5 with NEK2 in the last snapshot of 20 ns molecular dynamic simulations. D. Time evolution of hydrogen bond distance between MBM-5 and NEK2 during MD simulations in the last 10 ns. E. The HTRF® KinEASE™-STK kit was used to measure inhibition of NEK2 by MBM-5. The inhibition rates were determined in reference to the control. Data presented are the mean ± SD of three independent experiments. F. Effects of MBM-5 on phosphorylation of Hec1 and Aurora A in MGC-803 cells. The results are representative of three independent experiments.
MBM-5 shows in vitro antitumor effects in a variety of cancer cells
Because the NEK2 kinase is essential for cell proliferation, we examined the anti-proliferative effects of MBM-5 on a panel of 19 cancer cell lines, including leukemia, gastric, colorectal, prostate, breast and hepatoma cancer cells. Representative concentration-inhibition curves were drawn as shown in Figure 2A, and IC50 values were calculated and listed in Figure 2B and Table 1. The IC50 values ranged from 1 to 10 μM and leukemia, gastric and colorectal cancer cell lines were relatively sensitive to MBM-5 than other cell lines. These data were consistent with the prediction that NEK2 kinase activity is crucial for cellular proliferation, and in line with the findings that NEK2 depletion inhibits cancer cell growth.
Figure 2. MBM-5 has in vitro antitumor activities against cancer cells.
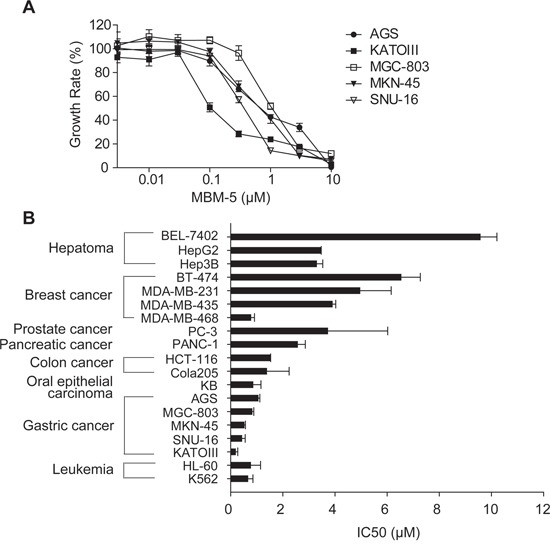
A and B. Growth inhibition assays of MBM-5 in vitro were performed using the MTT assay. The growth rates were determined in reference to the control. The concentration–growth rate curves of MBM-5 on gastric cancer cells were drawn (A), and the IC50 was calculated (B). Data presented are the mean ± SD of three independent experiments.
Table 1. IC50 values for inhibition of cell growth by MBM-5 measured via MTT assay.
| Cell Type | Cell line | IC50 (μM) |
|---|---|---|
| Chronic myeloid leukemia | K562 | 0.65±0.20 |
| Acute myeloid leukemia | HL-60 | 0.76±0.38 |
| Gastric cancer | KATOIII | 0.18±0.09 |
| SNU-16 | 0.42±0.13 | |
| MKN-45 | 0.51±0.05 | |
| MGC-803 | 0.81±0.07 | |
| AGS | 1.05±0.07 | |
| Oral epithelial carcinoma | KB | 0.85±0.31 |
| Colorectal cancer | Cola205 | 1.38±0.87 |
| HCT-116 | 1.51±0.02 | |
| Pancreatic cancer | PANC-1 | 2.56±0.30 |
| Prostate cancer | PC-3 | 3.72±2.30 |
| Breast cancer | MDA-MB-468 | 0.77±0.14 |
| MDA-MB-435 | 3.89±0.14 | |
| MDA-MB-231 | 4.95±1.20 | |
| BT-474 | 6.53±0.74 | |
| Hepatoma | Hep3B | 3.29±0.24 |
| HepG2 | 3.44±0.03 | |
| BEL-7402 | 9.57±0.64 |
MBM-5 induces chromosomal misalignment and triggers mitotic catastrophe
Perturbation of NEK2 function by RNAi or overexpression of kinase-inactive NEK2 leads to mitotic abnormalities represented by spindle configuration changes and chromosome misalignment. We then detected whether MBM-5 treatment would elicit these phenotypes. In contrast to DMSO treatment, MGC-803 cells displayed increased chromosomal misalignment after treated with MBM-5 for 12 h (Figure 3A). The proportion of cells with chromosomal misalignment increased from 0.79% in control group to 7.30% in MBM-5 treated cells (Figure 3B). Multipolar spindle configurations in the mitotic population could be observed, albeit with a very small population (Data not shown). Mitotic progression defects were further evidenced by the appearance of thin chromatin bridges, binucleated and micronucleated interphase cells (Figure 3C).
Figure 3. MBM-5 triggers mitotic catastrophe.
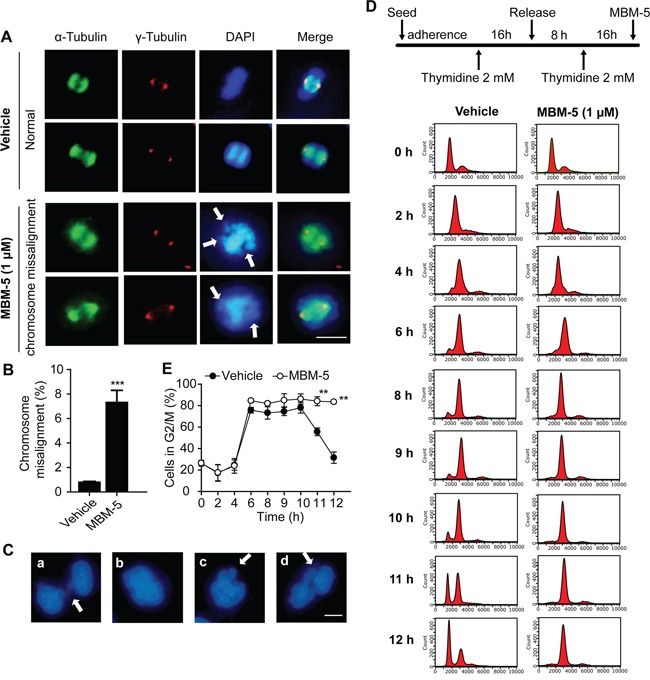
A. Representative images and quantitation of misaligned metaphase chromosomes in MGC-803 cells treated with DMSO and 1 μM of MBM-5 for 12 h. Centrosome, microtubules and chromosome were stained with anti-γ-tubulin, anti-α-tubulin and DAPI, respectively. Scale bar, 10 μm. B. Cells with misaligned metaphase chromosomes in total cells were quantitated. Error bar represent SD, n≥200 per sample. C. Representative images of mitotic progression defects in MGC-803 cells treated with 1 μM of MBM-5 for 12 h. (a) thin chromatin bridges, (b) binucleated cells, (c and d) micronucleated cells. Scale bar, 10 μm. D. Schematic of the protocol used to treat the cells. Following DTB, MGC-803 cells were released into fresh medium in the presence of DMSO or MBM-5 (1 μM). DNA content of cells collected at the indicated time points was assessed by flow cytometric analysis of cells labeled with propidium iodide. The results are representative of three independent experiments. E. The percentages of G2/M phase cells in (D) were quantitated. Data presented are the mean ± SD of three independent experiments.
To determine whether these mitotic defects would lead to cell cycle arrest, cell cycle analysis was performed. MGC-803 cells were first synchronized in G1/S phase by double thymidine block (DTB) treatment, and then were released into MBM-5 in order to better understand the effect of MBM-5 on cell cycle progression. As shown in Figure 3D and 3E, both control and MBM-5 treated cells progressed to G2/M phase by 6 h after DTB release. For control cells, about 80% of cells remained in G2/M phase by 10 h and then gradually progressed from G2/M to G1 phase at 11-12 h. In contrast, cells treated with MBM-5 were still arrested in G2/M phase at least 12 h after DTB release. Together, these observations indicated that MBM-5 induced mitotic catastrophe by inducing chromosomal misalignment and cytokinesis failure, without affecting mitotic entry.
MBM-5 induces cell cycle arrest and polyploid
To fully understand the effect of MBM-5 on mitotic progression, asynchronously-growing MGC-803 and HCT-116 cells were treated with serial dilutions of MBM-5 or with different times. In both cell lines, rising concentrations caused a pronounced enrichment of G2/M phase cells (Figure 4A and 4B). For example, the population of MGC-803 cells at G2/M phase increased from 27.2% to 31.2%, 39.7% and 60.6% after treated with 0.25, 0.5 and 1 μM MBM-5 for 24 h, respectively. Interestingly, an increased population of cells with >4N DNA content was observed after MBM-5 treatment, which was associated with a repeated round of DNA synthesis after cytokinesis failure. We then determined the cell cycle progression after MBM-5 treatment over time. In MGC-803 cells, the population of cells at G2/M phase reached peak level after treated with MBM-5 (0.5 or 1 μM) for 12 h, and then decreased gradually at 24 h and 48 h. Meanwhile, the sub G0 and >4N population increased in a time-dependent manner, suggesting cells arrested in 4N either underwent cell death or entered the G1 phase without cell division. Similar phenomenon was also observed in HCT-116 cells (Figure 4C and 4D).
Figure 4. MBM-5 causes accumulation of cells with ≥4N DNA content.
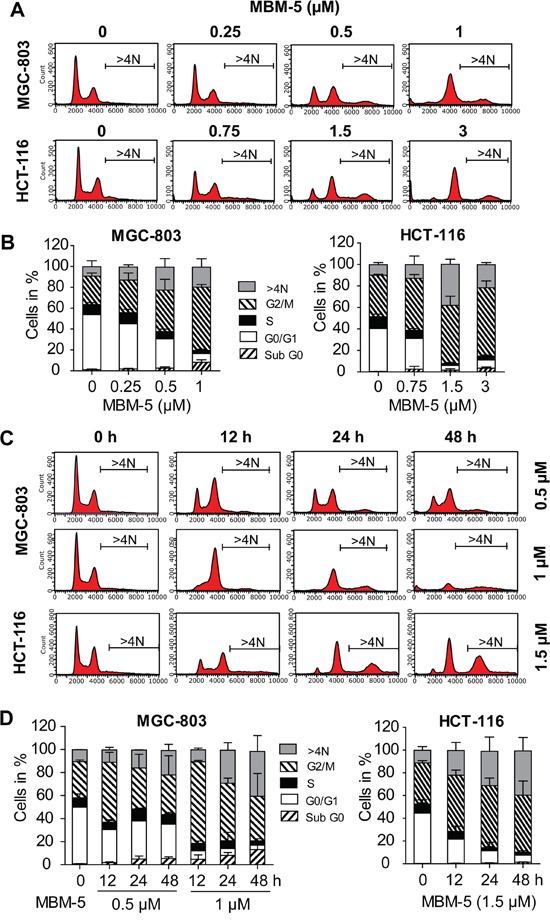
A. MGC-803 and HCT-116 cells were treated with indicated concentration of MBM-5 for 24 h. Cell cycle distribution was assessed by flow cytometric analysis of cells labeled with propidium iodide. Representative data were shown. B. Cell cycle proportion in (A) was quantitated. Data presented are the mean ± SD of three independent experiments. C. MGC-803 and HCT-116 cells were treated with indicated concentration of MBM-5 for different times. Cell cycle distribution was assessed by flow cytometric analysis of cells labeled with propidium iodide. Representative data were shown. D. Cell cycle proportion in (C) was quantitated. Data presented are the mean ± SD of three independent experiments.
In line with the above results, microscopic examination revealed that some cells underwent chromosome decondensation and formation of nuclear envelopes without the normal processes of mitosis. Therefore, parts of cells with 4N DNA content were actually arrested in pseudo-G1 phase with binuclear (Figure 3C). With a repeated round of DNA synthesis in a subsequent S phase, these cells became containing 8N DNA content (Figure 4). In consistence, the multinucleated cells increased from 0.6% to 22.2% after treated with MBM-5 (1 μM) for 24 h, further confirming the perturbed mitotic progression (Figure 5A).
Figure 5. MBM-5 increases formation of multinuclear cells.
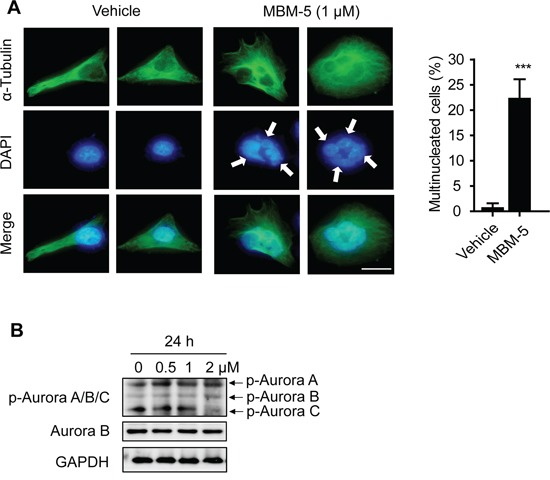
A. Representative images and quantitation of multinuclear MGC-803 cells treated with DMSO and 1 μM of MBM-5 for 24 h. Microtubules and chromosome were stained with anti-α-tubulin and DAPI, respectively. Data show percentage of cells with multinucleation in total cells. Error bar represent SD, n≥200 per sample. Scale bar, 20 μm. B. Effects of MBM-5 on phosphorylation of Aurora A/B/C kinases in MGC-803 cells treated with MBM-5. The results are representative of three independent experiments.
Inhibition of Aurora B kinase activity was reported to cause polyploidy; therefore we speculated whether accumulated cells with >4N DNA content were resulted from inhibition of Aurora B kinase by MBM-5. In our study, we used an antibody that recognizes phospho-Aurora A (T288)/Aurora B (T232)/Aurora C (T198) to determine the activation of Aurora kinases. MBM-5 treatment did not inhibit the phosphorylation of Aurora A/B/C, ruling out all these three kinases as direct targets of MBM-5 (Figure 5B).
MBM-5 induces apoptosis
Given the increased sub G0 population after MBM-5 treatment over time (Figure 4), we then determined whether MBM-5 treatment led to apoptosis or necrosis by performing flow cytometry with Annexin-V/PI staining. As shown in Figure 6A-6D, MBM-5 induced apoptosis of MGC-803 and HCT-116 cells, both in a concentration- and time-dependent manner. For example, the percentage of apoptotic HCT-116 cells increased to 30.0%, 45.6% and 68.4%, after treated with 0.75, 1.5 and 3 μM MBM-5 for 24 h, respectively. Moreover, the apoptotic population reached 91.2% and 85.2% after treated with 1 μM and 1.5 μM MBM-5 for 72 h in MGC-803 and HCT-116 cells, respectively. Western blot results showed that MBM-5 treatment resulted in the cleavage of caspase 3 and PARP-1, indicating that MBM-5 activated the apoptotic pathway (Figure 6E).
Figure 6. MBM-5 induces apoptosis.
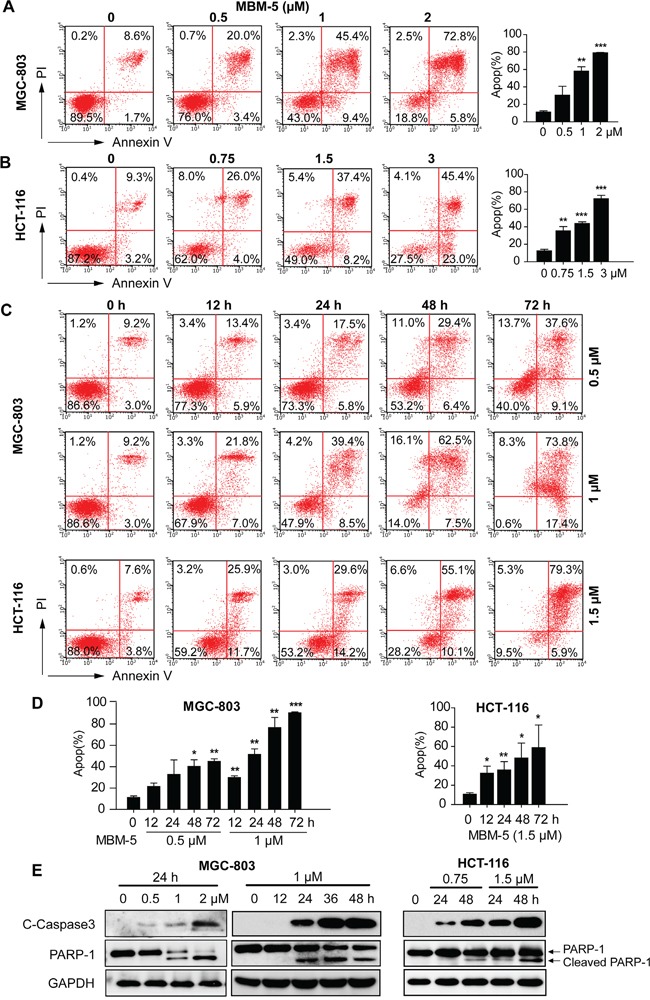
A and B. MGC-803 and HCT-116 cells were treated with indicated concentration of MBM-5 for 24 h. C and D. MGC-803 and HCT-116 cells were treated with indicated concentration of MBM-5 for different times. Apoptosis was assessed by flow cytometric analysis after Annexin V/PI staining. Representative images and quantitation of apoptotic cells were shown. Data presented are the mean ± SD of three independent experiments. E. Effects of MBM-5 on protein expression of cleaved caspase 3 and PARP-1. The results are representative of three independent experiments.
MBM-5 effectively suppresses tumor growth in human gastric and colorectal cancer cells xenografts
To determine the antitumor effect of MBM-5 in vivo, we established a subcutaneous gastric carcinoma model using MGC-803 cells and colorectal cancer model using HCT-116 cells. To improve the water solubility of compound MBM-5, the succinic acid salt of MBM-5 was synthesized for animal studies. And the salt form of MBM-5 was confirmed to have the same anti-cancer activity in vitro (Data not shown). For gastric carcinoma model, tumor growth was suppressed after MBM-5 (30 mg/kg) treatment when compared with vehicle-treated mice. At the end of the experiment, the tumor volume was significantly reduced (p<0.05) with a TGI% of 42.4% (Figure 7A). The average tumor weight of MBM-5-treated mice was 0.54±0.17 g, which was virtually lower than vehicle-treated mice (0.77±0.29 g), albeit without significant difference (Figure 7B). In colorectal cancer model, mice received 20 mg/kg of MBM-5 showed a significant reduction in tumor volume commencing on day 4. At the end of the experiment, the tumor volume was significantly inhibited (p<0.001) compared with control, with a TGI% of 51.1% (Figure 7C). Moreover, the average tumor weight of MBM-5-treated mice was 0.72±0.17 g, which was significantly lower (p<0.01) than vehicle-treated mice (1.31±0.31 g) (Figure 7D). Throughout the experiments, mice treated with MBM-5 displayed neither drug-related death nor remarkable body weight loss of more than 20% (Data not shown).
Figure 7. MBM-5 retards tumor growth in vivo.
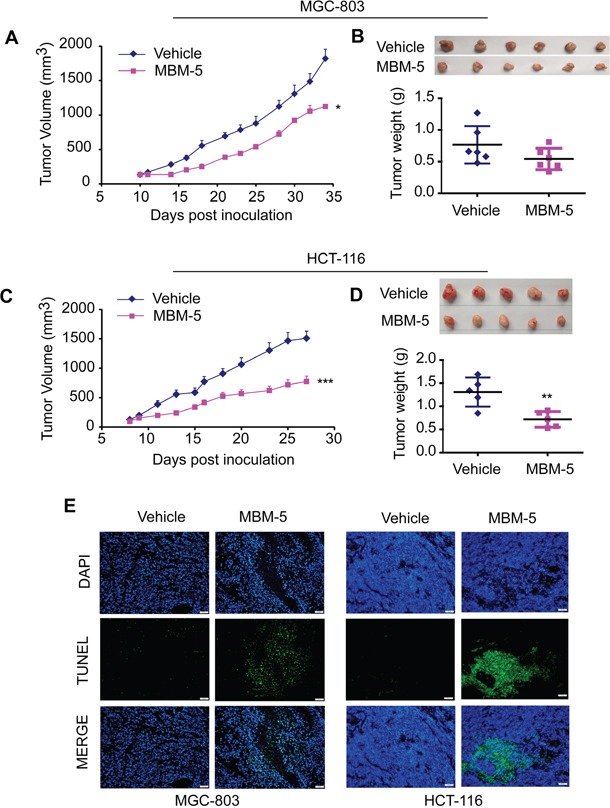
MGC-803 and HCT-116 xenograft bearing nude mice were treated with 30 mg/kg and 20 mg/kg MBM-5 as indicated schedule, respectively. A and C. The tumor volume was recorded and plotted against days post inoculation. Data presented are the mean ± SEM (n=6 in MGC-803 xenograft model, n=5 in HCT-116 xenograft model). B and D. The pictures of tumors and tumor weights. E. Representative images derived from TUNEL staining to evaluate the apoptosis in tumor tissues. Scale bar, 50 μm.
Tumor tissues were assayed for apoptosis using a TUNEL kit which labels apoptotic nuclei with a fluorescent dye. As shown in Figure 7E, tumor tissues from animal receiving MBM-5 treatment showed an obvious increase in apoptosis. These findings coincided with the apoptotic effect of MBM-5 in vitro, highlighting the involvement of apoptosis in the tumor growth inhibitory effects exerted by MBM-5 in vivo.
DISCUSSION
Elevated expression of NEK2 appears to participate in the initiation, maintenance, progression, metastasis of cancer and is positively associated with poor prognosis. RNAi depletion of NEK2 effectively inhibited proliferation of cancer cells and tumor growth in xenograft model [18, 19]. Moreover, compounds (INHs and TAIs) that specifically disrupted the Hec1/NEK2 interaction via direct Hec1 binding and caused NEK2 degradation significantly suppressed tumor growth in vivo without obvious toxicity [19]. Therefore, NEK2 is a promising therapeutic target for cancer treatment. However, the development of NEK2 inhibitors was at the very beginning, as the current reported NEK2 inhibitors was either with low activity or lack of membrane permeability. Great efforts should be made to develop NEK2 inhibitors that are suitable for clinical studies.
In this study, our library's compound MBM-5 was presumed to have potent activity against NEK2 kinase by docking simulation study. The results of the NEK2 kinase assay clearly showed that MBM-5 potently and concentration dependently inhibited NEK2 kinase activity in vitro. We further found that MBM-5 was able to inhibit the cellular kinase activity of NEK2 as evidenced by the decreased phosphorylation of Hec1 (S165). Although the protein level of total Hec1 was down-regulated by MBM-5, it seems not contribute to the decreased phosphorylation of Hec1. Because the phosphorylation of Hec1 was almost undetectable after treated with MBM-5 (0.5 or 1 μM) for 24 h, meanwhile a modest decrease of total Hec1 was observed. A previous study has reported similar phenomenon that the decreased phosphorylation of Hec1 was followed with a down-regulation of Hec1 [32]. Therefore, the phosphorylation of Hec1 might be correlated with the protein level, either affecting its transcription or post-translation modification, which will be studied in our further research. Moreover, MBM-5 prevented proliferation of a diverse range of human cancer cell lines with IC50 values of micromole range. The inhibition was associated with accumulation of G2/M cells, pseudo-G1 cells, and induction of polyploidy cells, as well as apoptosis.
Perturbation of NEK2 function by RNAi or overexpression of kinase-inactive NEK2 was found to cause multipolar spindle configurations [23, 24, 33]. Inhibitors that disrupt NEK2/Hec1 interaction led to aberrant spindles, such as deformed or multipolar spindles [32, 34]. However, this observation was challenged by a study also based on NEK2 RNAi. It reported that NEK2 depleted cells were capable of forming normal bipolar spindles with fully active kinesin motor Eg5 [35]. Therefore, the exact role of NEK2 in bipolar spindle assembly remains controversial. In our experiment, about 90% of mitotic cells exhibited normal bipolar spindle after MBM-5 treatment, suggesting NEK2 might play a supporting role rather than an indispensible role in bipolar spindle assembly.
The spindle assembly checkpoint (SAC) delays sister chromatid separation until all chromosomes are attached to the bipolar spindle. It is able to detect a single unaligned chromosome, causing a prolonged mitotic arrest [36]. Phosphorylation of Hec1 (S165) by NEK2 modulates chromosome alignment and serves as an intricate molecular switch modulating the activity of the SAC. Lack of Hec1 (S165) phosphorylation weakens the SAC signaling and triggers onset of anaphase even with unaligned chromosomes [9]. Here we observed that MBM-5 effectively inhibited the phosphorylation of Hec1 (S165) by NEK2, thus leading to chromosome segregation defects represented by misaligned chromosomes in metaphase cells and thin chromatin bridges in telophase cells.
In both synchronized and asynchronously-growing cells, MBM-5 treatment induced an obvious accumulation of cells with 4N DNA content. Some of these cells were actually in pseudo-G1 phase, as evidenced by the observation of binuclear cells. These might be resulted from mitotic slippage, which means cells escape from mitotic arrest and subsequently enter the G1 phase without cell division. In addition, MBM-5 also increased the proportion of cells with >4N DNA content. Generally speaking, the generation of cells with >4N DNA content could occur by two distinct mechanisms. Cells replicate their DNA without entering mitosis after completion of S phase; or cells proceed through mitosis without dividing, and replicate their DNA in a subsequent S phase [37]. In our experiment, MGC-803 cells released from a G1/S block was able to progress into G2/M phase after treated with MBM-5 for 6 h, suggesting that cells were able to enter mitosis. Therefore MBM-5-induced accumulation of population with >4N DNA content represented cells that exited mitosis without cell division and subsequently experienced a repeated round of DNA synthesis. As a result, MBM-5 treatment increased the level of multinucleated cells. Our study was in consistent with a previous study that some NEK2B depleted cells were unable to complete cytokinesis resulting in the formation of multinucleated cells [11]. Therefore, our study provided a new evidence that NEK2 might regulate cytokinesis.
Cells with prolonged mitotic arrest or continued proliferation with cytokinesis failure subsequently underwent apoptosis. In both MGC-803 and HCT-116 cells, the predominant effect of MBM-5 at 12 h was the accumulation of cells with 4N DNA content. At 24 h, the repeated round of DNA synthesis (represented as >4N DNA content population) occurred in 10-30% cells, and then increased slightly at 48 h. However, no further increase in the degree of polyploidy was observed up to 96 h (Data not shown). Meanwhile, the apoptosis increased significantly with time, indicating MBM-5 triggered apoptosis either during mitotic arrest or after mitotic slippage to a new round of mitotic progression. Moreover, MBM-5 induced apoptosis was also observed in vivo as evidenced by the increased apoptotic nuclei in MBM-5 treated tumor tissues.
Before evaluating the anti-tumor effect of MBM-5 in vivo, we first determined the pharmacokinetic parameters by single i.v. (2 mg/kg) administration of MBM-5 on rats. Using the MBM-5 plasma concentration-time profile curve (Supplementary data 1, Supplementary Figure 1), the pharmacokinetic parameters were determined using Noncompartmental Analysis. The plasma concentration was cleared with a half-life (t1/2) of 1.5 h and clearance (CL) rate of 121 ml/h/kg (Supplementary data 1, Supplementary Table 1). Keeping this in mind, we dosed xenograft mice twice a day. Compared with vehicle treated mice, MBM-5 treatment retarded the tumor growth in both gastric and colorectal cancer cells xenograft models. However, tumor stasis or regression was not observed. Due to the short half-life, more frequent dosing may be needed to enhance the antitumor activity in vivo. Anyway, MBM-5 was the first ATP-competitive NEK2 inhibitor that exhibited the antitumor effect both in vitro and in vivo, therefore was an ideal lead compound worth for further optimization. To improve its kinase inhibitory activity and pharmacokinetic parameters, further chemical modification of MBM-5 is currently in progress.
In conclusion, MBM-5 was identified to be one of the most promising lead compounds of ATP-competitive NEK2 inhibitors. It demonstrated prominent in vitro antitumor activity against a wide variety of cancer cells. MBM-5 interrupted mitotic progression by inducing chromosome segregation defects and cytokinesis failure; therefore leading to accumulation of cells with 4N DNA content (G2/M phase or binuclear) and >4N DNA content (polyploidy or multinuclear), which finally underwent apoptosis. The antitumor potential was further strengthened by its suppressive effect on the growth of MGC-803 gastric and HCT-116 colorectal tumor xenografts in vivo, even though its half-life in vivo was quite short. Together, MBM-5 has been established unequivocally as a lead compound by targeting NEK2 kinase in this study.
MATERIALS AND METHODS
Compounds
MBM-5 (4-(7-(1-(2-(dimethylamino)ethyl)-1H- pyrazol-4-yl)imidazo[1,2-a]pyridin-3-yl)-2-((2-(trifluoromethyl)benzyl)oxy)benzamide), with purity>98% was synthesized by our group. MBM-5 was dissolved in DMSO as 30 mM stock solution for enzymatic assays and in vitro cell based assays. The succinic acid salt of MBM-5, with purity>98% was synthesized and dissolved in ultrapure water for animal studies; the solution was freshly prepared before each use. The information about the purities of MBM-5 and its salt was shown in Supplementary data.
Cell culture and synchronization
All cell lines were purchased from Cell Bank of China Science Academy (Shanghai, China) and maintained in growth medium as recommended at 37°C in a 5% CO2 atmosphere. Penicillin (100 U/ml) and streptomycin (100 μg/ml) were added in the medium. Double thymidine block (DTB) was performed as previously described [38].
Animal treatment
Female BALB/c nu/nu mice (5-6 weeks, 16-18 g) were obtained from Sino-British SIPPR/BK Lab. Animal Ltd, Shanghai, China, with the certification number of 2008001638201. The animals were housed in specific pathogen-free (SPF) conditions at Key Laboratory of Brain Functional Genomics, Ministry of Education, East China Normal University, and were acclimatized for a week prior to use. The use and care of experimental animal was approved by Institutional Animal Care and Use Committee, East China Normal University.
Molecular modeling
X-ray protein structure of NEK2 (PDB ID: 4AFE) was obtained in Protein Data Bank. All water molecules in the crystal structure were removed, and then the structure was used for molecular docking using Sybyl X2. For docked structure, energy minimization and molecular dynamic (MD) simulation were performed using the Sander module in Amber14. A two-step, extensive energy minimization process based on the steepest descent method followed by the conjugate gradient algorithm were carried out to relieve bad contacts and to direct each system toward energetically favorable conformations. After minimization, each system was gently heated from 0 to 300 K in 500 ps at constant volume and equilibrated at 300 K for another 500 ps. Finally, a 20 ns MD simulation without any restrictions was performed at constant pressure, and the coordinates of atoms were saved every 2 ps. During the MD simulation, all bonds involving hydrogen atoms were constrained using the SHAKE algorithm, and a time step of 2 fs was adopted. The temperature was controlled using the Langevin thermostat with a collision frequency of 2.0 ps−1. The particle mesh Ewald (PME) method was applied to treat the long-range electrostatic interactions. The cutoff distances for the long-range electrostatic and van der Waals interactions were set to 12.0 Å. Solvated interaction energy (SIE) method was used to calculate the binding free energies between compounds and NEK2. For the current work, 200 snapshots extracted from the last 10 ns of MD trajectory at an interval of 50 ps were used for the binding free energy analyses.
Kinase assays
The inhibition of MBM-5 on activity of NEK2 kinase (Millipore) was characterized by HTRF® KinEASE™-STK kit according to the manufacturer's instructions (Cisbio Assays).
Cell proliferation assay
Cell proliferation was assessed by the MTT assay [39]. The inhibition rate for each well was calculated as (ODcontrol cells−ODtreated cells)/ODcontrol cells × 100%. The IC50 values were calculated by concentration−response curve fitting using the four parameter method.
Cell cycle analysis
Cell cycle distribution was determined by flow cytometry measurement of DNA content after cells were incubated with RNase A (10 mg/ml) and propidium iodide (50 mg/ml). The cellular DNA content was analyzed on a flow cytometer (Guava EasyCyte 6HT2L). The percentage of each population was measured using the InCyte software. At least 20 000 cells were analyzed for each data point.
Apoptosis analysis
Apoptosis was detected by FITC Annexin V Apoptosis Detection Kit I according to the manufacturer's instructions (Dojindo) to determine the phosphatidyl serine exposure. Apoptotic cells were quantified by a flow cytometer (Guava EasyCyte 6HT2L) using the InCyte software. At least 10 000 cells were analyzed for each data point.
Immunoblotting analysis
Immunoblotting analysis of proteins in cell lysates was performed as previously described [40]. The expression of GAPDH was used as loading control. Primary antibodies used were as follows: anti-NEK2 (#610593) was purchased from BD; anti-p-Hec1(Ser165) (#GTX70013) and anti-Hec1 (#GTX70268) were purchased from Gene Tex; anti-p-Aurora A (#3079), anti-Aurora A (#12100), anti-p-Aurora A/Aurora B/Aurora C (#2914), anti-Aurora B (#3094), and anti-cleaved caspase 3 (#9661) were purchased from Cell Signaling Technology; anti-PARP (#sc-8007) and anti-GAPDH (#sc-25778) were purchased from Santa Cruz.
Immunofluorescence
Cells were grown on 96-well tissue culture plate (Corning, NY) and permeabilized with 0.5% Triton-X100 in PHEM Buffer (80 mM PIPES, 25 mM HEPES, pH 7.2, 10 mM EGTA, 4 mM MgSO4) for 10 min at room temperature, then fixed in PHEM buffer containing 4% paraformaldehyde for 20 min. Cells were blocked with 3% BSA plus 10% FBS in PHEM, and then incubated with primary antibodies: anti-α-tubulin (TU-02) (Santa Cruz) and anti-γ-tubulin (Sigma) in PBS overnight at 4°C. The secondary antibodies Alexa Fluor® 488 goat anti-rabbit IgG (H+L) or Alexa Fluor® 594 goat anti-mouse IgG (H+L) (Invitrogen) were applied respectively for two hours after the plate were washed with TBS thoroughly. 4′, 6-Diamidino-2-phenylindole (DAPI) staining was applied after secondary antibody incubation. Images were captured with a fluorescence microscopy (Olympus IX3).
Antitumor activity in vivo
Human MGC-803 xenografts and HCT-116 xenografts were established by subcutaneously inoculating 5 × 106 cells into nude mice. When the tumors reached a mean group size of 100~150 mm3, the mice were randomized into control and treatment groups to receive treatment accordingly. Mice were treated with vehicle or MBM-5 twice a day via intraperitoneal injection. The tumor growth was recorded with the measurement of length (L) and width (W) by caliper three times a week, and calculated as tumor volume (V) = L × W2/2. The tumor growth inhibition was calculated as TGI%= (1-(mean tumor volume of the treatment group on the first day-mean tumor volume of the treatment group on the end day)/(mean tumor volume of the control group on the first day-mean tumor volume of the control group on the end day)) x100%.
TUNEL histology
To evaluate the apoptotic response in tumor tissue, we applied terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-digoxigenin nick-end labeling technique (TUNEL), to formalin-fixed tumor samples in paraffin blocks, using the commercially available Kit (Promega). The sections (4–5 μm) mounted on glass slides were deparaffinized, rehydrated through graded alcohols to water, treated with 20 μg/ml proteinase K (37°C, 20 min) and then washed in 1× Tris buffer. TUNEL assay was then performed according to the instructions by the manufacturer. Images were captured with a fluorescence microscopy (Olympus IX3).
Statistical analysis
The statistical significance of differences between groups was evaluated by the unpaired Student's t test and indicated with ***P<001, **P<0.01, *P<0.05. All statistical tests were two sided.
SUPPLEMENTARY DATA
Acknowledgments
This work was funded by grants from The Science and Technology Commission of Shanghai Municipality (No. 14431902700), the National Natural Science Foundation of China (No. 81402953 and 21572067), Open Funds of State Key Laboratory of Oncology in South China (HN2016-03), and China Postdoctoral Science Foundation (2015T80416).
Abbreviations
- DTB
double thymidine block
- Hec1
highly expressed in cancer 1
- HTRF
Homogeneous Time-Resolved Fluorescence
- MAD1
mitotic arrest deficient-like 1
- MD
molecular dynamic
- NEK2
Never in Mitosis (NIMA) Related Kinase 2
- NEKs
Never in Mitosis (NIMA) Related Kinases
- Nlp
ninein-like protein
- PME
particle mesh Ewald
- SAC
spindle assembly checkpoint
- SIE
solvated interaction energy
- SPF
specific pathogen-free
- TUNEL
terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-digoxigenin nick-end labeling technique
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Fry AM, O'Regan L, Sabir SR, Bayliss R. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci. 2012;125:4423–33. doi: 10.1242/jcs.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fry AM, Mayor T, Meraldi P, Stierhof YD, Tanaka K, Nigg EA. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J Cell Biol. 1998;141:1563–74. doi: 10.1083/jcb.141.7.1563. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahe S, Stierhof YD, Wilkinson CJ, Leiss F, Nigg EA. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J Cell Biol. 2005;171:27–33. doi: 10.1083/jcb.200504107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Man X, Megraw TL, Lim YP. Cep68 can be regulated by Nek2 and SCF complex. Eur J Cell Biol. 2015;94:162–72. doi: 10.1016/j.ejcb.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Rapley J, Baxter JE, Blot J, Wattam SL, Casenghi M, Meraldi P, Nigg EA, Fry AM. Coordinate regulation of the mother centriole component nlp by nek2 and plk1 protein kinases. Mol Cell Biol. 2005;25:1309–24. doi: 10.1128/MCB.25.4.1309-1324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Riley DJ, Zheng L, Chen PL, Lee WH. Phosphorylation of the mitotic regulator protein Hec1 by Nek2 kinase is essential for faithful chromosome segregation. J Biol Chem. 2002;277:49408–16. doi: 10.1074/jbc.M207069200. [DOI] [PubMed] [Google Scholar]
- 7.Du J, Cai X, Yao J, Ding X, Wu Q, Pei S, Jiang K, Zhang Y, Wang W, Shi Y, Lai Y, Shen J, Teng M, et al. The mitotic checkpoint kinase NEK2A regulates kinetochore microtubule attachment stability. Oncogene. 2008;27:4107–14. doi: 10.1038/onc.2008.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lou Y, Yao J, Zereshki A, Dou Z, Ahmed K, Wang H, Hu J, Wang Y, Yao X. NEK2A interacts with MAD1 and possibly functions as a novel integrator of the spindle checkpoint signaling. J Biol Chem. 2004;279:20049–57. doi: 10.1074/jbc.M314205200. [DOI] [PubMed] [Google Scholar]
- 9.Wei R, Ngo B, Wu G, Lee WH. Phosphorylation of the Ndc80 complex protein, HEC1, by Nek2 kinase modulates chromosome alignment and signaling of the spindle assembly checkpoint. Mol Biol Cell. 2011;22:3584–94. doi: 10.1091/mbc.E11-01-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu G, Ding X, Yuan K, Aikhionbare F, Yao J, Cai X, Jiang K, Yao X. Phosphorylation of human Sgo1 by NEK2A is essential for chromosome congression in mitosis. Cell Res. 2007;17:608–18. doi: 10.1038/cr.2007.55. [DOI] [PubMed] [Google Scholar]
- 11.Fletcher L, Cerniglia GJ, Yen TJ, Muschel RJ. Live cell imaging reveals distinct roles in cell cycle regulation for Nek2A and Nek2B. Biochim Biophys Acta. 2005;1744:89–92. doi: 10.1016/j.bbamcr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Weiss MM, Kuipers EJ, Postma C, Snijders AM, Pinkel D, Meuwissen SG, Albertson D, Meijer GA. Genomic alterations in primary gastric adenocarcinomas correlate with clinicopathological characteristics and survival. Cell Oncol. 2004;26:307–17. doi: 10.1155/2004/454238. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki K, Kokuryo T, Senga T, Yokoyama Y, Nagino M, Hamaguchi M. Novel combination treatment for colorectal cancer using Nek2 siRNA and cisplatin. Cancer Sci. 2010;101:1163–9. doi: 10.1111/j.1349-7006.2010.01504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neal CP, Fry AM, Moreman C, McGregor A, Garcea G, Berry DP, Manson MM. Overexpression of the Nek2 kinase in colorectal cancer correlates with beta-catenin relocalization and shortened cancer-specific survival. J Surg Oncol. 2014;110:828–38. doi: 10.1002/jso.23717. [DOI] [PubMed] [Google Scholar]
- 15.Zeng YR, Han ZD, Wang C, Cai C, Huang YQ, Luo HW, Liu ZZ, Zhuo YJ, Dai QS, Zhao HB, Liang YX, Zhong WD. Overexpression of NIMA-related kinase 2 is associated with progression and poor prognosis of prostate cancer. BMC Urol. 2015;15:90. doi: 10.1186/s12894-015-0085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayward DG, Clarke RB, Faragher AJ, Pillai MR, Hagan IM, Fry AM. The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 2004;64:7370–6. doi: 10.1158/0008-5472.CAN-04-0960. [DOI] [PubMed] [Google Scholar]
- 17.Tsunoda N, Kokuryo T, Oda K, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 2009;100:111–6. doi: 10.1111/j.1349-7006.2008.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia J, Franqui Machin R, Gu Z, Zhan F. Role of NEK2A in human cancer and its therapeutic potentials. Biomed Res Int. 2015;2015:862461. doi: 10.1155/2015/862461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang Y, Zhang X. Targeting NEK2 as a promising therapeutic approach for cancer treatment. Cell Cycle. 2016;15:895–907. doi: 10.1080/15384101.2016.1152430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kokuryo T, Senga T, Yokoyama Y, Nagino M, Nimura Y, Hamaguchi M. Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res. 2007;67:9637–42. doi: 10.1158/0008-5472.CAN-07-1489. [DOI] [PubMed] [Google Scholar]
- 21.Zhou W, Yang Y, Xia J, Wang H, Salama ME, Xiong W, Xu H, Shetty S, Chen T, Zeng Z, Shi L, Zangari M, Miles R, et al. NEK2 induces drug resistance mainly through activation of efflux drug pumps and is associated with poor prognosis in myeloma and other cancers. Cancer Cell. 2013;23:48–62. doi: 10.1016/j.ccr.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang MX, Xu XM, Zhang P, Han NN, Deng JJ, Yu TT, Gan YY, He XQ, Long ZX. Effect of silencing NEK2 on biological behaviors of HepG2 in human hepatoma cells and MAPK signal pathway. Tumour Biol. 2015 doi: 10.1007/s13277-015-3993-y.. [DOI] [PubMed] [Google Scholar]
- 23.Cappello P, Blaser H, Gorrini C, Lin DC, Elia AJ, Wakeham A, Haider S, Boutros PC, Mason JM, Miller NA, Youngson B, Done SJ, Mak TW. Role of Nek2 on centrosome duplication and aneuploidy in breast cancer cells. Oncogene. 2014;33:2375–84. doi: 10.1038/onc.2013.183. [DOI] [PubMed] [Google Scholar]
- 24.Lee J, Gollahon L. Nek2-targeted ASO or siRNA pretreatment enhances anticancer drug sensitivity in triplenegative breast cancer cells. Int J Oncol. 2013;42:839–47. doi: 10.3892/ijo.2013.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rellos P, Ivins FJ, Baxter JE, Pike A, Nott TJ, Parkinson DM, Das S, Howell S, Fedorov O, Shen QY, Fry AM, Knapp S, Smerdon SJ. Structure and regulation of the human Nek2 centrosomal kinase. J Biol Chem. 2007;282:6833–42. doi: 10.1074/jbc.M609721200. [DOI] [PubMed] [Google Scholar]
- 26.Emmitte KA, Adjabeng GM, Andrews CW, Alberti JG, Bambal R, Chamberlain SD, Davis-Ward RG, Dickson HD, Hassler DF, Hornberger KR, Jackson JR, Kuntz KW, Lansing TJ, et al. Design of potent thiophene inhibitors of polo-like kinase 1 with improved solubility and reduced protein binding. Bioorg Med Chem Lett. 2009;19:1694–7. doi: 10.1016/j.bmcl.2009.01.094. [DOI] [PubMed] [Google Scholar]
- 27.Hayward DG, Newbatt Y, Pickard L, Byrne E, Mao G, Burns S, Sahota NK, Workman P, Collins I, Aherne W, Fry AM. Identification by high-throughput screening of viridin analogs as biochemical and cell-based inhibitors of the cell cycle-regulated nek2 kinase. J Biomol Screen. 2010;15:918–27. doi: 10.1177/1087057110376537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whelligan DK, Solanki S, Taylor D, Thomson DW, Cheung KM, Boxall K, Mas-Droux C, Barillari C, Burns S, Grummitt CG, Collins I, van Montfort RL, Aherne GW, et al. Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization. J Med Chem. 2010;53:7682–98. doi: 10.1021/jm1008727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Solanki S, Innocenti P, Mas-Droux C, Boxall K, Barillari C, van Montfort RL, Aherne GW, Bayliss R, Hoelder S. Benzimidazole inhibitors induce a DFG-out conformation of never in mitosis gene A-related kinase 2 (Nek2) without binding to the back pocket and reveal a nonlinear structure-activity relationship. J Med Chem. 2011;54:1626–39. doi: 10.1021/jm1011726. [DOI] [PubMed] [Google Scholar]
- 30.Innocenti P, Cheung KM, Solanki S, Mas-Droux C, Rowan F, Yeoh S, Boxall K, Westlake M, Pickard L, Hardy T, Baxter JE, Aherne GW, Bayliss R, et al. Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: structure-activity relationship, structural biology, and cellular activity. J Med Chem. 2012;55:3228–41. doi: 10.1021/jm201683b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henise JC, Taunton J. Irreversible Nek2 kinase inhibitors with cellular activity. J Med Chem. 2011;54:4133–46. doi: 10.1021/jm200222m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu CM, Zhu J, Guo XE, Chen W, Qiu XL, Ngo B, Chien R, Wang YV, Tsai CY, Wu G, Kim Y, Lopez R, Chamberlin AR, et al. Novel small molecules disrupting Hec1/Nek2 interaction ablate tumor progression by triggering Nek2 degradation through a death-trap mechanism. Oncogene. 2015;34:1220–30. doi: 10.1038/onc.2014.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faragher AJ, Fry AM. Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol Biol Cell. 2003;14:2876–89. doi: 10.1091/mbc.E03-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu G, Qiu XL, Zhou L, Zhu J, Chamberlin R, Lau J, Chen PL, Lee WH. Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 2008;68:8393–9. doi: 10.1158/0008-5472.CAN-08-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mardin BR, Lange C, Baxter JE, Hardy T, Scholz SR, Fry AM, Schiebel E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat Cell Biol. 2010;12:1166–76. doi: 10.1038/ncb2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Musacchio A. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr Biol. 2015;25:R1002–18. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 37.Andreassen PR, Lohez OD, Margolis RL. G2 and spindle assembly checkpoint adaptation, and tetraploidy arrest: implications for intrinsic and chemically induced genomic instability. Mutat Res. 2003;532:245–53. doi: 10.1016/j.mrfmmm.2003.08.020. doi. [DOI] [PubMed] [Google Scholar]
- 38.Jeong AL, Lee S, Park JS, Han S, Jang CY, Lim JS, Lee MS, Yang Y. Cancerous inhibitor of protein phosphatase 2A (CIP2A) protein is involved in centrosome separation through the regulation of NIMA (never in mitosis gene A)-related kinase 2 (NEK2) protein activity. J Biol Chem. 2014;289:28–40. doi: 10.1074/jbc.M113.507954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu H, Luo P, Fu Y, Wang J, Dai J, Shao J, Yang X, Chang L, Weng Q, Yang B, He Q. Dihydromyricetin prevents cardiotoxicity and enhances anticancer activity induced by adriamycin. Oncotarget. 2015;6:3254–67. doi: 10.18632/oncotarget.2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakayama R, Zhang YX, Czaplinski JT, Anatone AJ, Sicinska ET, Fletcher JA, Demetri GD, Wagner AJ. Preclinical activity of selinexor, an inhibitor of XPO1, in sarcoma. Oncotarget. 2016;7:16581–92. doi: 10.18632/oncotarget.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.