

Abstract
The detailed molecular mechanism for the reversible inhibition of mitochondrial respiration by NO has puzzled investigators: The rate constants for the binding of NO and O2 to the reduced binuclear center CuB/a3 of cytochrome oxidase (COX) are similar, and NO is able to dissociate slowly from this center whereas O2 is kinetically trapped, which altogether seems to favor the complex of COX with O2 over the complex of COX with NO. Paradoxically, the inhibition of COX by NO is observed at high ratios of O2 to NO (in the 40–500 range) and is very fast (seconds or faster). In this work, we used simple mathematical models to investigate this paradox and other important biological questions concerning the inhibition of COX by NO. The results showed that all known features of the inhibition of COX by NO can be accounted for by a direct competition between NO and O2 for the reduced binuclear center CuB/a3 of COX. Besides conciliating apparently contradictory data, this work provided an explanation for the so-called excess capacity of COX by showing that the COX activity found in tissues actually is optimized to avoid an excessive inhibition of mitochondrial respiration by NO, allowing a moderate, but not excessive, overlap between the roles of NO in COX inhibition and in cellular signaling. In pathological situations such as COX-deficiency diseases and chronic inflammation, an excessive inhibition of the mitochondrial respiration is predicted.
Keywords: mitochondrial respiration, mathematical model, cytochrome-oxidase-deficiency diseases, inflammation, excess capacity of cytochrome oxidase
The paradigm that the respiratory chain is regulated by ADP and O2 was recently updated to include the reversible inhibition of cytochrome oxidase (COX) by nitric oxide (NO) (1–6). The physiological role and the detailed molecular mechanism of this inhibition, as well as the reason for the apparent excess content of COX compared with other mitochondrial complexes, are fundamental questions of mitochondrial biochemistry that remain unsolved. Concerning the mechanism, the problem is particularly puzzling because the known characteristics of COX inhibition by NO are difficult to conciliate with the available kinetic rate constants for this inhibition. On the one hand, COX is inhibited rapidly, within a time scale of seconds, with half-inhibition of respiration attained at O2/NO ratios in the 40–500 range (4, 7), which apparently indicates that the interaction of COX with NO is stronger than that of COX with O2 and very fast. On the other hand, the rate constants for the binding of O2 and NO with the fully reduced binuclear center (CuB/a3) of COX are similar [1.4 × 108 (8) and 4 × 107 to 1 × 108 M–1·s–1 (9, 10), respectively] and NO dissociates slowly from this center (0.01–0.13 s–1) (11, 12), whereas the apparent dissociation rate constant of O2 with COX is virtually zero because, during the initial steps of reduction of oxygen by COX, oxygen is kinetically trapped (8). Accordingly, several investigators have noticed that the potent reversible inhibition of COX by NO cannot be based on a simple competition between NO and O2 (6, 7, 11, 13), and alternative mechanisms have been proposed: Besides binding to the two-electron-reduced CuB/heme a3 center, NO would bind to a single-electron-reduced CuB/heme a3, either reduced CuB (13) or reduced heme a3 (11), for which O2 has a low affinity (8). Originally, these proposals were supported by mathematical models, but the experimental support is missing. Recently, the use of a bacterial mutant in which the reduction of COX is very slow allowed researchers to observe the binding of NO to the single-electron-reduced CuB/heme a3 center (14), but whether this binding is effective in the wild-type protein remains unknown. Also recently, a mechanism for the inhibition of COX was proposed in which NO reacts with the oxidized form of CuB, in which NO is initially oxidized, yielding nitrite (14–16). However, this inhibition is not competitive with oxygen and is not reversible by light as observed in cells, so it is generally accepted that the inhibition of COX by low physiological levels of NO involves binding, rather than reaction of NO with COX (6).
To establish the molecular mechanism for the inhibition of COX by NO, we analyzed the hypothesis that the potent reversible inhibition of COX by NO cannot be based on a simple competition between NO and O2 for COX by using simple mathematical models. Surprisingly, the results showed that the hypothesis was misleading and that all known aspects of the inhibition of COX by NO can be accounted for by a simple competition between NO and O2 for the two-electron-reduced CuB/heme a3 center. Having set up mathematical models that successfully simulated all known aspects of COX inhibition by NO, we investigated other important biological questions regarding this process, namely: (i) the higher sensitivity to NO inhibition in state 3, during which ADP concentration, rate of ATP production, and rate of respiration are high, as compared with state 4 mitochondrial respiration, during which ADP concentration, rate of ATP production, and rate of respiration are low; (ii) the excessive COX content and activity compared with other respiratory complexes; (iii) the physiological role of the inhibition of COX by NO; and (iv) the pathological significance of COX inhibition by NO in COX-deficiency diseases and inflammation.
The Model
To analyze whether the direct competition between NO and O2 could account for the potent inhibition of COX, a minimal model was set up (Scheme 1, model 1) in which, at the steady state, the rate of electron transfer in the respiratory chain is equal in each of its complexes, and so, by studying the effects of NO on the rate of the catalytic cycle of COX, the effects of NO on the overall respiratory chain also are evaluated. A second model (model 2), which includes the reduction of complex IV by cytochrome c (Scheme 1), also was analyzed.
Scheme 1.

Models describe the inhibition of the respiratory chain by NO in competition with O2. Shown are a minimal model (continuous-line-depicted reactions, model 1) and an extended model (continuous- and dashed-line-depicted reactions, model 2). kNOon and kO2app refer to rate constants describing the combination of NO and O2 with the reduced cytochrome a3–CuB site (Fea32+–CuB+) of COX, respectively. kNOoff refers to the release of NO from the reduced cytochrome a3–CuB site. kIV, first-order rate constant in model 1 and second-order rate constant in model 2, refers to the reduction of the cyt a3–CuB site entailing the arrival to COX of electrons from the other mitochondrial complexes, via cytochrome c (cyt c). kIII, a first-order rate constant, refers to the reduction of cytochrome c in complex III. Fea33+–CuB+–O2 species aggregates the intermediate forms appearing in the COX catalytic cycle. Mass action kinetics were assumed, and simulations were carried out with gepasi (45) and plas (46).
The values for the rate constants and conserved moieties used in the models were taken from the literature as described in the Supporting Text, which is published as supporting information on the PNAS web site.
Results and Discussion
O2 Consumption. Before analyzing the inhibition of COX by NO, we evaluated whether model 1 is able to simulate basic aspects of mitochondrial respiration. At steady state and in the absence of NO, the rate of oxygen consumption predicted by model 1 is a hyperbolic function of the concentration of O2 (Eq. 1, which applies irrespectively of the values used for the rate constants kIV and kO2app) as observed experimentally, conveniently described as K0.5, the O2 concentration that provides the half-maximal rate of O2 uptake. The K0.5 values also are often referred to as KMO2 and [O2]0.5. However, the apparent affinity of respiration for O2, the K0.5 value, is not a constant or a Km (Michaelis–Menten constant) value but is given by the ratio kIV/kO2app. Because kIV, an apparent first-order rate constant that depends on the rate of respiration (see Supporting Text), increases with the rate of respiration, K0.5 also increases with the respiration rate, as is experimentally observed in mitochondrial states 4 and 3 (4, 17, 18). After replacing kIV and kO2app by its values in Eq. 1, K0.5 values for states 3 and 4 were 0.22 μM and 0.022 μM, respectively, in reasonable agreement with the experimental values (0.35–1.7 μM in state 3 and 0.08–0.6 μM in state 4) (4, 17, 18). Accordingly, the model predicts that, at the micromolar levels of oxygen found in tissues, the rate of respiration would be nearly saturated with O2. In conclusion, model 1 accommodates well the known dependencies of mitochondrial respiration on O2.
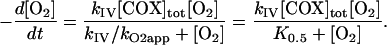 |
[1] |
Inhibition of COX by NO: A Dynamical Analysis. To analyze the inhibition of mitochondrial respiration by NO, we started by simulating typical experiments in which the effect of NO on the rate of O2 consumption by mitochondria is evaluated in a variety of conditions. The main results obtained were as follows: (i) for the same concentration of NO, the degree of inhibition increased when the oxygen concentration decreased (Fig. 1A); (ii) for the same concentration of O2, the degree of inhibition increased with the concentration of NO added (Fig. 1B); (iii) the degree of inhibition caused by the same concentrations of NO and O2 was much more pronounced in state 3 than in state 4 (Fig. 1C); (iv) NO inhibited mitochondrial respiration at high ratios (>1,000) of oxygen to NO (Fig. 1B); and (v) the onset of inhibition by NO was extremely fast, occurring virtually immediately after NO addition (Fig. 1). These general observations in the model are identical to those observed experimentally (1, 4, 19–21).
Fig. 1.

Simulation of typical experiments with isolated mitochondria on the inhibition of respiration by NO. (A) NO was added at 1 μM as indicated by the arrows. (B) NO was added at 4.0, 2.0, 1.0, 0.5, 0.25, or 0.10 μM at 4 min, when O2 concentration was 142 μM. (C) NO at 1 μM was added to respiration with high turnover (state 3-like) and with low turnover (state 4-like). Model 1 was used with the following parameters: COX initial concentration was 0.014 μM (at time 0 it was assumed that all COX was in the form Fea32+–CuB+), which is equivalent to 0.1 mg of mitochondrial protein per ml; O2 initial concentration was 220 μM; kNOon = 4 × 107 M–1·s–1; kNOoff = 0.13 s–1; kO2app = 1.4 × 108 M–1·s–1; kIV = 3 s–1 (to simulate state 4 respiration); and kIV = 30 s–1 (to simulate state 3 respiration). Transition between state 4 and state 3 is indicated by the addition of ADP.
Often, the fast inhibition of mitochondrial respiration by NO is used as an argument against a simple competition between NO and O2 for COX being the basis for NO effects. To evaluate this point in greater depth, the formation of the complex between COX and NO was followed in detail after adding NO. As observed experimentally (11, 13), the formation of this complex occurred within a few seconds after NO addition. In general, the time scale for the inhibition of COX by NO is given by kNOon× [NO]. At a typical [NO] of 1 × 10–8 M, a time scale of ≈1 s was obtained, with higher NO levels giving faster inhibitions, and so the fast inhibition of COX by NO is compatible with a direct competition between NO and O2 for COX.
Inhibition of COX by NO: A Steady-State Analysis. The reproduction of experimental observations by mathematical models is not an end in itself; it is equally important to use the model to improve knowledge about the system under study. In the present case, it is of fundamental importance to understand why, despite binding and dissociation rate constants that seem to favor the complex of COX with O2 over that of COX with NO, NO is able to inhibit mitochondrial respiration at high O2/NO ratios. To address this point, simple equations were deduced that summarize the characteristics of model 1. At the steady state and in the presence of NO, the rate of respiration is given by Eq. 2,
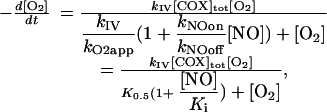 |
[2] |
which describes a simple linear competitive inhibition by NO of COX with an inhibition constant (Ki) given by kNOoff/kNOon. Thus, NO acts by increasing the apparent K0.5, as observed experimentally (1, 4). The ratio of the rate of respiration in the absence of NO to the rate of respiration in the presence of NO, deduced by combining Eqs. 1 and 2, is
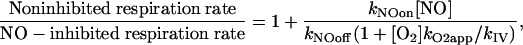 |
[3] |
and the concentration of NO that inhibits mitochondrial respiration by 50% (IC50) is given by Eq. 4:
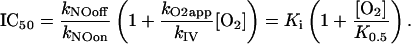 |
[4] |
Eqs. 2, 3, and 4 show that the inhibition caused by NO does not depend only on the [O2]/[NO] ratio and the rate constants for binding and release of these species from COX, as could be intuitively expected. Very importantly, it also depends on the rate constant kIV, i.e., the turnover of COX, which, in the equations deduced, has a role equivalent to a rate constant describing the dissociation of O2 from COX. This finding may be considered surprising, but if the turnover of COX is viewed as the pathway that restores the free enzyme dissociated from O2, it is understandable that kIV has the role of a dissociation rate constant (Scheme 1). This fact constitutes a typical example of an emerging global property of a system that is not expected from the analysis of the individual components. It should be mentioned that if a more complex model were used in which all steps in the catalysis cycle of COX were included, the same result would be obtained. That is, the catalysis cycle would still restore the free enzyme and have a role of a dissociation pathway, and kIV would be replaced by a function of the processes involved in this cycle.
If the dissociation rate constant for NO (in the 0.01–0.13 s–1 range) is compared with the turnover of COX, which operationally affords the dissociation rate constant for O2 and is ≈30 s–1 for active respiration and 3 s–1 for resting respiration, it is understandable why NO can compete with O2 at a very large ratio of O2 to NO. Therefore, the apparent contradiction between the known kinetic data for binding and dissociation of O2 and NO to COX and the strong inhibition of mitochondrial respiration caused by NO is solved.
IC50. Although the most relevant features of the inhibition of mitochondrial respiration by NO were successfully reproduced by model 1, a rigorous quantitative comparison with the degree of inhibition found experimentally is important to further test the validity of the model. IC50 is often obtained in experiments, providing an objective quantitative criterion by which to test the model. As observed in Fig. 2, there is good agreement between the IC50 predicted by the model and the IC50 obtained experimentally for a wide range of O2 concentrations. Notice the large variation in the experimental IC50 (Fig. 2), a situation that is akin to that observed with the apparent affinity of the respiratory chain for O2, which can also vary because of experimental artifacts when measuring O2 (18). Obviously, a different determination of K0.5 also will imply a different determination of IC50 for the inhibition of respiration by NO. The IC50 values predicted by Eq. 4 are a linear function of the oxygen concentration, which is coherent with only one binding site of NO to COX as observed experimentally (22); very importantly, the IC50 values predicted also depend on the turnover of COX.
Fig. 2.

Experimental (♦) and simulated (lines) IC50 values for the inhibition of mitochondrial respiration by NO. IC50 values obtained with model 1 (continuous line) were calculated by using Eq. 4 with kNOon = 4 × 107 M–1·s–1, kNOoff = 0.02 s–1, and kIV = 30 s–1.IC50 values obtained with model 2 (dashed line) were calculated from simulations in which O2 concentration was held constant and NO concentration was varied as a parameter; parameters used were as follows: kNOon = 4 × 107 M–1·s–1, kNOoff = 0.01 s–1, kIV = 1.5 × 106 M–1·s–1, kIII = 23.3 s–1, [cyt c]tot = 200 μM, and [COX]tot = 140 μM. kNOoff in model 2 was half of that in model 1. In both models, the functional dependency of kO2app on the concentration of O2 (see Supporting Text) was explicitly considered. Experimental data were taken from refs. 1, 4, 13, 19, 21, 47, and 48.
One consequence of NO inhibition is an increase of cytochrome c reduction (23, 24). For example, cytochrome c is 50–77% reduced in the presence of NO produced by the specialized NO synthase of the inner mitochondrial membrane, vs. ≈6–15% cytochrome c reduction in the absence of NO (23, 25). It is expected that the increased reduction of cytochrome c upon addition of NO could compensate partially the inhibitory effect of the formation of the complex COX–NO by increasing COX turnover. Therefore, we set up an extended model (Scheme 1, model 2) in which cytochrome c action was included. In this model, as the concentration of NO was increased, the level of reduction of cytochrome c increased as observed experimentally. As expected, the IC50 values obtained with model 2 were higher than those obtained with model 1 for the same set of rate constants, and the experimental IC50 also could be reproduced with model 2 by using plausible values for the rate constants (Fig. 2). In conclusion, the strong inhibition observed with high [O2]/[NO] ratios could be reproduced successfully at a quantitative level by models that assume a simple competition between O2 and NO for the oxygen-binding site of COX.
Effects of NO in State 3 and State 4 Respiration. One of the puzzling questions about NO effects on mitochondria is that state 3 respiration is much more sensitive to NO inhibition than state 4 respiration, a fact that was tentatively attributed to a lower control exerted by COX on the mitochondrial respiration at low rates of respiration (19). This explanation recently found experimental support by the observation that for the same level of COX inhibition by NO the rate of mitochondrial respiration was more inhibited at state 3 than at state 4 (24). However, the difference found was relatively small, and it does not explain the almost complete insensitivity of state 4 observed in some conditions. One feature of the inhibition of state 4 respiration is that it is relatively NO-insensitive until an NO level is reached that lowers the rate of respiration in state 3 to a level similar to that of state 4; after this concentration of NO is attained, state 4 and state 3 respirations are inhibited in a similar way (19). Model 1 was applied with relative success: Although state 4 was less sensitive to inhibition by NO than was state 3 respiration (as predicted from Eqs. 2, 3, and 4 and Fig. 1C), the pattern of inhibition did not follow the one observed experimentally. State 4 respiration was inhibited significantly at levels of NO at which state 3 respiration was significantly higher than state 4 respiration. However, with model 2, the pattern of inhibition of state 3 and state 4 observed experimentally was reproduced successfully (Fig. 3A), showing that cytochrome c counteracts partially the inhibitory effects of NO. This situation is akin to the known relationship between the levels of cytochrome c reduction and oxygen concentration (26, 27).
Fig. 3.

Simulation of the inhibition of state 3 and state 4 mitochondrial respiration by NO in a typical experiment, in vitro (A) ([O2] = 150 μM) and in vivo under normoxia (B) ([O2] = 30 μM) or hypoxia (C) ([O2] = 5 μM) conditions. Model 2 was used with the following parameters: kNOon = 4 × 107 M–1·s–1, kNOoff = 0.01 s–1, kO2app = 1.4 × 108 M–1·s–1, kIV = 1.5 × 106 M–1·s–1, [cyt c]tot = 200 μM, and [COX]tot = 140 μM. Low COX activity in A was simulated by setting [COX]tot at 46.7 μM. State 3 was simulated by setting kIII at 23.3 s–1. State 4 was simulated by setting kIII at 7.77 s–1 (A, to simulate experiment in ref. 19) or 2.33 s–1 (B and C). Notice the difference in the x scales. Arrows indicate range for NO concentration.
Having successfully simulated the experimental pattern of inhibition, we analyzed the reason for the higher sensitivity of state 3 respiration to NO compared with that of state 4 respiration. The pattern of inhibition observed in state 4 with a low COX activity becomes exactly the same as that observed in state 3 with a higher COX activity (Fig. 3A), showing that the cause for higher threshold values for NO in state 4 than in state 3 is the lower fraction of COX bound to NO in state 4, which is the result of a lower COX turnover in this state.
Next, model 2 was applied to predict the state of respiration in vivo under a variety of conditions. At the average O2 concentrations found in tissues (≈30 μM) and for NO levels of 10–50 nM (accepted as the physiological range), the rate of respiration in state 3 was decreased from 8.8 to 5.5 times the rate in state 4, whereas state 4 respiration was insensitive to NO (Fig. 3B). Thus, upon increasing NO concentrations in this range, cells can keep ATP levels by switching a larger fraction of mitochondria from state 4 to state 3. By contrast, in acute or chronic inflammation, in which NO levels in blood and tissues are markedly increased, tissue respiration is decreased despite adequate oxygen supply (28, 29), and the [NO]/[O2] ratio consequently is increased, this switching may not be sufficient to maintain ATP levels. If a 5-fold increase of NO over normal levels (from 50 to 250 nM) is assumed, the rate of state 3 respiration is predicted to be decreased from 5.5 to 1.3 times the rate of state 4 respiration (Fig. 3B), whereas state 4 respiration is predicted to be unaffected by NO. In hypoxic conditions ([O2] = 5 μM, again increasing the ratio [NO]/[O2]), the rate of state 3 is predicted to be severely impaired, decreasing from 4.7 to 1.2 times the rate of state 4 respiration for the physiological 10–50-nM NO range (Fig. 3C); in situations where inflammation is associated with hypoxia, both state 3 and state 4 respirations are predicted to be severely impaired (Fig. 3C). Obviously, the model could be used to simulate the rate of respiration in conditions other than those considered here. It can be concluded that in most in vivo situations, NO is an inhibitor of state 3 respiration but not of state 4 respiration.
Excess Capacity of COX. The molar ratio of COX over complex 1 is ≈8 (30), and the control coefficient of COX in mitochondrial respiration is low (31), indicating that COX activity is excessive. It was suggested that this excess is important in maintaining a high affinity of mitochondrial respiration toward O2 (18), uncoupling acceptor level from electron transfer rates. The results in Fig. 3A provide an alternative explanation that applies at normal oxygen pressures: When COX concentration was decreased to 46.7 μM, the rate of respiration was changed only slightly in the absence of NO but was more susceptible to NO inhibition than it was at a COX concentration of 140 μM, the level found in liver mitochondria. This finding indicates that the excess activity of COX is necessary to avoid an excessive inhibition of NO. To investigate this possibility, we ran a series of simulations in which the total concentration of COX was decreased (Fig. 4). The rate of respiration was not changed in the absence of NO, but in the presence of NO, respiration became very sensitive to the level of COX. For the particular situation simulated, a 5-fold decrease of COX activity implied an increase in the degree of inhibition of mitochondrial respiration by 30 nM NO from 19% to 77% in state 3. It is interesting to note that the curve in the presence of NO starts to plateau in the region of 140 μM, which is the physiological COX level, indicating that the level of COX could have been optimized to decrease the sensitivity of respiration to COX inhibition by NO. In conclusion, the activity of COX cannot be considered excessive in the coexistence of NO as a physiological metabolite.
Fig. 4.

Relationship between activity of COX and NO inhibition of mitochondrial respiration in state 3 in vivo. Model 2 was used with the following parameters: [O2] = 30 μM; [NO] = 0.03 μM, where indicated; kIII = 23.3 s–1; [COX]tot was changed as indicated; and kIV was changed to keep [COX]tot× kIV constant. Other parameters were as in Fig. 3.
Biological Significance. Despite competitive inhibition being a common feature in metabolic control, the physiological and pathological roles of the competitive inhibition of COX by NO are still largely unknown. The finding that the excess COX capacity observed in mammals is necessary to avoid an excessive inhibition of mitochondrial respiration by NO is helpful in elucidating the matter. Liver, heart, and brain mitochondria from mammals show a high affinity of respiration for O2, with K0.5 values for state 3 of 1.1–1.2 μM O2; these values are increased by the endogenous mitochondrial production of NO, as a product of mitochondrial NO synthase activity, to 2.7 μM O2 in liver and heart mitochondria (4, 17) and to 4.7 μM O2 in brain mitochondria (32). Then, endogenous NO and the O2-competitive inhibition of COX decrease the high affinity of mitochondrial respiration for O2 and move from oxyregulation, in which O2 uptake is independent of O2 concentration, to oxyconformity, in which O2 uptake is almost linearly dependent on O2 levels (33). Oxyconformity is classically shown by primitive forms of life, such as sea invertebrates (33), worms (34), snails (35), and reptile eggs (36). However, it has recently been reported that human glioma cells (37) and perfused human placenta (38) behave as oxyconformers. Concerning pathological situations, endogenously generated NO might be involved in the etiology of COX-deficiency diseases, in which there is a 2- to 10-fold decrease in the activity of COX (39), and in chronic inflammation, in which the inducible NO synthase activity is increased severalfold (28, 29). An excessive inhibition of mitochondrial state 3 respiration by NO is predicted in these two situations. Supporting this concept, it has been observed that fibroblasts isolated from Leigh-syndrome patients with COX deficiency show a decreased affinity for O2 (increased K0.5), which can be interpreted as the consequence of the competition between endogenously produced NO and O2 for COX in COX-deficient cells (40); because of the competition between NO and O2 for COX, COX-deficient cells probably show a decreased rate of respiration under normoxic conditions in the presence of NO. However, the existence of an apparent excess capacity of COX can be taken as an indication of the multiple roles of NO. If the primary role of NO in the cell were the regulation of mitochondrial respiration, COX activity could be lower, because the same effect could be attained at lower NO and COX levels. But if other cellular functions of NO are considered, the apparent excess capacity of COX becomes important because it allows a wide cellular range of NO without causing an excessive inhibition of the respiratory chain. For example, the activation of guanylyl cyclase by NO and the NO-induced vascular smooth muscle relaxation that have half-maximal responses at 4–20 nM (41, 42) and 10 nM (43) are possible to attain without inhibition of mitochondrial respiration because of the presence of an apparent excess COX capacity. Supporting this view, in a study in which delivery of NO was rigorously controlled as a steady state, it was found that activation of guanylyl cyclase and the inhibition of COX were nonoverlapping in cerebellar cells, i.e., the maximal degree of guanylyl cyclase activation was reached at the threshold NO level that starts to inhibit COX (42). However, because in this work ADP addition was not mentioned, cells were probably in a state more akin to state 4 respiration than to state 3 respiration; if respiration were measured in state 3, there would probably be some overlap between activation of cGMP production and COX inhibition because of the higher sensitivity of state 3 to NO inhibition. Endogenous mitochondrial NO is able to regulate mitochondrial respiration in vitro; upon supplementation of kidney (44) and brain (32) mitochondria with arginine and with competitive inhibitors of mitochondrial NO synthase, state 3 respiratory rates are decreased and increased, respectively, in the range of –12% to +46% at 150 μM O2, depending on the level of mitochondrial NO synthase activity. The model predicts for this in vitro situation an intramitochondrial NO steady-state concentration facing COX in the range of 100–340 nM NO (Fig. 3A). In conclusion, the physiological role for the NO inhibition of COX remains unknown, although the levels of COX found in cells seem to be optimized to allow a moderate but not excessive overlap between the signaling roles of NO and the inhibition of state 3 mitochondrial respiration by NO.
Supplementary Material
Acknowledgments
This work was supported by Fundação para a Ciência e Tecnologia–Portugal Grant POCTI/BCI/42245/2001 and Fellowship BPD/11487/2002 (to F.A.) and by National Institutes of Health Grants R01-AG16718 and R-01-ES11342 (to E.C.).
Author contributions: F.A., A.B., and E.C. designed research, performed research, analyzed data, and wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: COX, cytochrome oxidase.
References
- 1.Brown, G. C. & Cooper, C. E. (1994) FEBS Lett. 356, 295–298. [DOI] [PubMed] [Google Scholar]
- 2.Cleeter, M. J. W., Cooper, J. M., Darley-Usmar, V. M., Moncada, S. & Schapira, A. H. V. (1994) FEBS Lett. 345, 50–54. [DOI] [PubMed] [Google Scholar]
- 3.Poderoso, J. J., Carreras, M. C., Lisdero, C., Riobo, N., Schopfer, F. & Boveris, A. (1996) Arch. Biochem. Biophys. 328, 85–92. [DOI] [PubMed] [Google Scholar]
- 4.Boveris, A., Costa, L. E., Poderoso, J. J., Carreras, M. C. & Cadenas, E. (2000) Ann. N.Y. Acad. Sci. 899, 121–135. [DOI] [PubMed] [Google Scholar]
- 5.Moncada, S. & Erusalimsky, J. D. (2002) Nat. Rev. Mol. Cell Biol. 3, 214–220. [DOI] [PubMed] [Google Scholar]
- 6.Brown, G. C. & Borutaite, V. (2002) Free Radical Biol. Med. 33, 1440–1450. [DOI] [PubMed] [Google Scholar]
- 7.Brown, G. C. (2001) Biochim. Biophys. Acta 1504, 46–57. [DOI] [PubMed] [Google Scholar]
- 8.Verkhovsky, M. I., Morgan, J. E. & Wikstrom, M. (1994) Biochemistry 33, 3079–3086. [DOI] [PubMed] [Google Scholar]
- 9.Gibson, Q. H. & Greenwood, C. (1963) Biochem. J. 86, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blackmore, R. S., Greenwood, C. & Gibson, Q. H. (1991) J. Biol. Chem. 266, 19245–19249. [PubMed] [Google Scholar]
- 11.Giuffre, A., Sarti, P., D'Itri, E., Buse, G., Soulimane, T. & Brunori, M. (1996) J. Biol. Chem. 271, 33404–33408. [DOI] [PubMed] [Google Scholar]
- 12.Sarti, P., Giuffre, A., Forte, E., Mastronicola, D., Barone, M. C. & Brunori, M. (2000) Biochem. Biophys. Res. Commun. 274, 183–187. [DOI] [PubMed] [Google Scholar]
- 13.Torres, J., Darley-Usmar, V. & Wilson, M. T. (1995) Biochem. J. 312, 169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarti, P., Giuffre, A., Barone, M. C., Forte, E., Mastronicola, D. & Brunori, M. (2003) Free Radical Biol. Med. 34, 509–520. [DOI] [PubMed] [Google Scholar]
- 15.Torres, J., Cooper, C. E. & Wilson, M. T. (1998) J. Biol. Chem. 273, 8756–8766. [DOI] [PubMed] [Google Scholar]
- 16.Torres, J., Sharpe, M. A., Rosquist, A., Cooper, C. E. & Wilson, M. T. (2000) FEBS Lett. 475, 263–266. [DOI] [PubMed] [Google Scholar]
- 17.Costa, L. E., Mendez, G. & Boveris, A. (1997) Am. J. Physiol. 273, C852–C858. [DOI] [PubMed] [Google Scholar]
- 18.Gnaiger, E., Lassnig, B., Kuznetsov, A., Rieger, G. & Margreiter, R. (1998) J. Exp. Biol. 201, 1129–1139. [DOI] [PubMed] [Google Scholar]
- 19.Borutaite, V. & Brown, G. C. (1996) Biochem. J. 315, 295–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishikawa, M., Sato, E. F., Kuroki, T. & Inoue, M. (1997) FEBS Lett. 415, 341–345. [DOI] [PubMed] [Google Scholar]
- 21.Takehara, Y., Kanno, T., Yoshioka, T., Inoue, M. & Utsumi, K. (1995) Arch. Biochem. Biophys. 323, 27–32. [DOI] [PubMed] [Google Scholar]
- 22.Stubauer, G., Giuffre, A., Brunori, M. & Sarti, P. (1998) Biochem. Biophys. Res. Commun. 245, 459–465. [DOI] [PubMed] [Google Scholar]
- 23.Sarkela, T. M., Berthiaume, J., Elfering, S., Gybina, A. A. & Giulivi, C. (2001) J. Biol. Chem. 276, 6945–6949. [DOI] [PubMed] [Google Scholar]
- 24.Brookes, P. S., Kraus, D. W., Shiva, S., Doeller, J. E., Barone, M. C., Patel, R. P., Lancaster, J. R., Jr., & Darley-Usmar, V. M. (2003) J. Biol. Chem. 278, 31603–31609. [DOI] [PubMed] [Google Scholar]
- 25.Poderoso, J. J., Peralta, J. G., Lisdero, C. L., Carreras, M. C., Radisic, M., Schopfer, F., Cadenas, E. & Boveris, A. (1998) Am. J. Physiol. 274, C112–C119. [DOI] [PubMed] [Google Scholar]
- 26.Wilson, D. F., Erecinska, M., Drown, C. & Silver, I. A. (1979) Arch. Biochem. Biophys. 195, 485–493. [DOI] [PubMed] [Google Scholar]
- 27.Korzeniewski, B. & Froncisz, W. (1991) Biochim. Biophys. Acta 1060, 210–223. [DOI] [PubMed] [Google Scholar]
- 28.De Angelo, J. (1999) Expert Opin. Pharmacother. 1, 19–29. [DOI] [PubMed] [Google Scholar]
- 29.Hofseth, L. J., Saito, S., Hussain, S. P., Espey, M. G., Miranda, K. M., Araki, Y., Jhappan, C., Higashimoto, Y., He, P., Linke, S. P., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wikstrom, M. & Saraste, M. (1984) in Bioenergetics, ed. Ernester, L. (Elsevier, Amsterdam), pp. 49–91.
- 31.Brown, G. C. (1992) Biochem. J. 284, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lores-Arnaiz, S., D'Amico, G., Czerniczyniec, A., Bustamante, J. & Boveris, A. (2004) Arch. Biochem. Biophys. 430, 170–177. [DOI] [PubMed] [Google Scholar]
- 33.Abele, D. (2002) Nature 420, 27. [DOI] [PubMed] [Google Scholar]
- 34.Portner, H. O., Heisler, N. & Grieshaber, M. K. (1985) Respir. Physiol. 59, 361–377. [DOI] [PubMed] [Google Scholar]
- 35.Guppy, M., Reeves, D. C., Bishop, T., Withers, P., Buckingham, J. A. & Brand, M. D. (2000) FASEB J. 14, 999–1004. [DOI] [PubMed] [Google Scholar]
- 36.Hastings, D. & Burggren, W. (1995) J. Exp. Biol. 198, 2465–2475. [DOI] [PubMed] [Google Scholar]
- 37.Parliament, M. B., Franko, A. J., Allalunis-Turner, M. J., Mielke, B. W., Santos, C. L., Wolokoff, B. G. & Mercer, J. R. (1997) Br. J. Cancer 75, 311–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mover-Lev, H., Dreval, D., Zakut, H. & Ar, A. (1996) Respir. Physiol. 106, 199–208. [DOI] [PubMed] [Google Scholar]
- 39.Robinson, B. H. (2000) Pediatr. Res. 48, 581–585. [DOI] [PubMed] [Google Scholar]
- 40.Pecina, P., Gnaiger, E., Zeman, J., Pronicka, E. & Houstek, J. (2004) Am. J. Physiol. Cell Physiol. 287, C1384–C1388. [DOI] [PubMed] [Google Scholar]
- 41.Bellamy, T. C., Wood, J., Goodwin, D. A. & Garthwaite, J. (2000) Proc. Natl. Acad. Sci. USA 97, 2928–2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bellamy, T. C., Griffiths, C. & Garthwaite, J. (2002) J. Biol. Chem. 277, 31801–31807. [DOI] [PubMed] [Google Scholar]
- 43.Carter, T. D., Bettache, N. & Ogden, D. (1997) Br. J. Pharmacol. 122, 971–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boveris, A., Valdez, L. B., Alvarez, S., Zaobornyj, T., Boveris, A. D. & Navarro, A. (2003) Antioxid. Redox Signal. 5, 265–271. [DOI] [PubMed] [Google Scholar]
- 45.Mendes, P. (1993) Comput. Appl. Biosci. 9, 563–571. [DOI] [PubMed] [Google Scholar]
- 46.Voit, E. (2000) Computational Analysis of Biochemical Systems: A Practical Guide for Biochemists and Molecular Biologists (Cambridge Univ. Press, Cambridge, U.K.).
- 47.Koivisto, A., Matthias, A., Bronnikov, G. & Nedergaard, J. (1997) FEBS Lett. 417, 75–80. [DOI] [PubMed] [Google Scholar]
- 48.Lizasoain, I., Moro, M. A., Knowles, R. G., Darley-Usmar, V. & Moncada, S. (1996) Biochem. J. 314, 877–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}