

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disorder worldwide. The disease is characterized by renal cysts and progressive renal failure due to progressive enlargement of cysts and renal fibrosis. An estimated 45% to 70% of patients with ADPKD progress to end-stage renal disease by age 65 years. Although both targeted and nontargeted therapies have been tested in patients with ADPKD, tolvaptan is currently the only pharmacological therapy approved in Canada for the treatment of ADPKD. The purpose of this consensus recommendation is to develop an evidence-informed recommendation for the optimal management of adult patients with ADPKD. This document focuses on the role of genetic testing, the role of renal imaging, predicting the risk of disease progression, and pharmacological treatment options for ADPKD. These areas of focus were derived from 2 national surveys that were disseminated to nephrologists and patients with ADPKD with the aim of identifying unmet needs in the management of ADPKD in Canada. Specific recommendations are provided for the treatment of ADPKD with tolvaptan.
Keywords: kidney, autosomal dominant polycystic kidney disease, ADPKD, Canadian, consensus
Abrégé
La polykystose rénale autosomique dominante (PKRAD) est le trouble rénal héréditaire le plus fréquent dans le monde. La maladie est caractérisée par la présence de kystes rénaux et par une insuffisance rénale progressive provoquée par l’élargissement progressif des kystes et par une fibrose rénale. Environ 45 à 70% des patients atteints de PKRAD verront leur état évoluer vers l’insuffisance rénale terminale avant l’âge de 65 ans. Bien que les thérapies ciblées et non ciblées aient été testées chez des patients atteints de PKRAD, le tolvaptan est le seul médicament approuvé au Canada pour le traitement de la PKRAD. L’objectif de cette recommandation consensuelle est l’élaboration de recommandations fondées sur des données probantes pour une prise en charge optimale des patients adultes atteints de PKRAD. Ce document met l’accent sur le rôle du dépistage génétique et de l’imagerie rénale, sur les façons de prédire le risque de progression de la maladie et sur les options de traitement pharmacologique de la PKRAD. Ces domaines d’action dérivent de deux enquêtes nationales diffusées aux néphrologues et aux patients canadiens atteints de PKRAD, et qui avaient pour but d’identifier les besoins non satisfaits dans la prise en charge le la PKRAD au Canada. Des recommandations spécifiques sont fournies pour le traitement de la PKD avec le tolvaptan.
Why is this review important
This consensus review was done to develop evidence-informed recommendations for Canadian nephrologists to guide optimal management of adult patients with ADPKD.
What are the key messages
Based on existing data, genetic testing is only required for a subset of patients with ADPKD, and it is not needed prior to initiating treatment options.
Although the gold standard for imaging is MRI, other less costly and more readily available imaging modalities should be used in routine clinical practice for diagnosis and determination of risk of progression.
Blood pressure targets and ADPKD-specific treatment options are recommended for patients who fulfill specific criteria.
We recommend treatment with tolvaptan for patients who fulfill the enrollment criteria of the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes (TEMPO) 3:4 study: 18 to 50 years of age, Cockcroft-Gault GFR >60 mL/min, and TKV >750 mL. In the absence of Cockcroft-Gault GFR, Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) >45 mL/min may be used, and in the absence of TKV, ultrasound (US) kidney length (KL) >16.5 cm may be used.
We suggest treatment with tolvaptan for patients who, according to the Mayo Classification, are classified as 1D or 1E with eGFR in CKD stage 3 or higher. Treatment with tolvaptan should be considered for patients who are classified as 1C and are younger than 50 years or have other risk factors for rapid progression.
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited renal disorder, with a prevalence of 1:500 to 1:1000.1 ADPKD accounts for 7% to 11% of patients on renal replacement therapy in Europe and approximately 5% of patients requiring dialysis in the United States.2 ADPKD is characterized by bilateral renal cysts and may be associated with kidney pain, urinary tract infection, hematuria, nephrolithiasis, hypertension, and progressive renal failure due to progressive enlargement of cysts and fibrosis.3-5 Cyst growth displaces and destroys normal kidney tissue, culminating in fibrosis, renal architectural derangement, and ultimately kidney failure.6,7 An estimated 45% to 70% of patients with ADPKD progress to end-stage renal disease (ESRD) by age 65 years.8
Several targeted pharmacological therapies have been tested in patients with ADPKD, including mammalian target of rapamycin (mTOR) inhibitors, somatostatin analogues, and the vasopressin V2-receptor antagonist tolvaptan2; however, tolvaptan is the only approved therapy in Canada for the treatment of ADPKD.9 The purpose of this consensus recommendation is to develop an evidence-informed recommendation for the optimal management of adult patients with ADPKD. The focus will be on the role of genetic testing, the role of renal imaging, risk prediction of disease progression, and pharmacological treatment options. These areas of focus were derived from 2 national surveys that were disseminated to nephrologists and patients with ADPKD.
Nephrologists’ Survey
With the goal of identifying the top needs in the management of ADPKD from the perspective of nephrologists, a survey was disseminated to members of the Canadian Society of Nephrology. A total of 73 physicians completed the survey. The survey results are summarized in Table 1.
Table 1.
Results of the survey disseminated to members of the Canadian Society of Nephrology.
| What treatment guidelines/algorithm, if any, do you use to treat your patients with ADPKD? |
| 82% None |
| 11% Clinical guidelines developed by the Caring for Australians with Renal Impairment |
| 7% Workshop results in Kidney International, literature-guided therapy, evidence-based literature, control of BP, and encouragement of increased water consumption |
| What screening tools do you use to diagnose ADPKD? |
| 92% Ultrasonography |
| 3% Magnetic resonance imaging |
| 3% Molecular genetic testing |
| 1% Computed tomography |
| 1% None |
| 0% Urinary biomarkers |
| What treatments have you used for your patients with ADPKD? |
| 42% Surgery |
| 29% Tolvaptan |
| 24% Other types of therapy (eg, BP monitoring, interventional radiology) |
| 3% Mammalian target of rapamycin inhibitors (eg, everolimus, sirolimus) |
| 1% Somatostatin analogues (eg, octreotide, lanreotide, pasireotide) |
| What do you perceive to be the top needs in the management of ADPKD in Canada? |
| Lack of general practitioners’ awareness of ADPKD |
| Lack of management guidelines and care pathways |
| Lack of knowledge of comprehensive integrated and accepted guidelines for the evaluation of extrarenal manifestations of ADPKD |
| Lack of established diagnostic algorithms integrating clinical signs and symptoms with kidney imaging and genetic testing |
| Lack of consensus on the optimal approach to predict prognosis |
| Lack of therapeutic options |
| Lack of clarity on subgroups of patients that may benefit from existing treatment |
Note. ADPKD = autosomal dominant polycystic kidney disease; BP = blood pressure.
Patients’ Survey
A second survey was disseminated to patient members of the PKD Foundation of Canada. A total of 88 patients completed the survey that was designed to assess what symptoms of ADPKD have the greatest effect on their lives, and what they perceive to be the greatest unmet needs in the management of ADPKD. The results of the survey are summarized in Table 2.
Table 2.
Results of the Survey Disseminated to Patient Members of the PKD Foundation of Canada.
| What symptoms bother you the most? |
| Pain |
| Fatigue |
| Enlarged abdomen/bulky organs/bloating |
| Edema |
| Hypertension |
| Side effects of therapy |
| In your opinion, what should the priorities be for research on ADPKD? |
| Finding a cure |
| Finding therapy that will stop/slow disease progression/cyst growth |
| Finding therapy that could reduce cysts to a manageable size or prevent cyst development |
| Identifying nonpharmacological therapies that could be used to delay the initiation of pharmacological therapies |
| Providing better access to transplantation |
| Improving the longevity of transplants |
| Finding treatments with improved efficacy and safety |
| Identifying molecular-based therapies that could correct the gene defect |
| Identifying modifiable lifestyle factors |
Note. ADPKD = autosomal dominant polycystic kidney disease.
Methods
The present consensus recommendations are based on the experience and opinions of the authors, and on a literature search conducted in PubMed, the Cochrane Library, and Google Scholar using the search terms ADPKD or polycystic kidney in combination with the following terms: CKD or chronic kidney disease or diagnosis or end-stage renal disease or ESRD or gene or imaging or management or mTOR inhibitor or risk or pharmacological or screening or somatostatin or surgery or TKV or total kidney volume or height adjusted TKV or tolvaptan or transplantation or treatment. We selected publications that were published in the past 10 years but did not exclude highly regarded older publications. In addition, we searched the reference lists of articles identified by this search strategy and selected additional relevant references. For topics that were beyond the scope of this consensus document, review articles are cited to guide readers to sources with more details. The authors reached a consensus on the recommendations published herein. The following aspects of the disease are addressed: genetic testing, renal imaging, predicting disease progression, and pharmacological treatment options.
Genetic Testing in ADPKD
ADPKD is genetically heterogeneous with 2 genes identified: PKD1 (chromosome 16p13.3) and PKD2 (4q21).10 Mutations in the PKD1 gene occur in 85% to 90% of cases of ADPKD, whereas mutations in the PKD2 gene account for the remainder of cases.11,12 The PKD1 and PKD2 genes encode 2 proteins, polycystin-1 and polycystin-2, that constitute the transient receptor potential polycystin subfamily of transient receptor potential channels.10 Genic, allelic, and gene modifier effects contribute to the high phenotypic variability of ADPKD, and truncating PKD1 mutations are associated with more severe disease and earlier decline in kidney function compared with nontruncating PKD1 mutations.13 According to the PKD Foundation of Canada, there are currently 2323 known mutations in the PKD1 gene and 278 known mutations in the PKD2 gene (Autosomal Dominant Polycystic Kidney Disease Mutation Database [PKDB], http://pkdb.mayo.edu/).
Genetic testing for ADPKD can be carried out using DNA linkage analysis, gene-based mutation screening (also referred to as Sanger sequencing), or, in the near future, next-generation sequencing (NGS). Up-to-date information regarding laboratories currently offering genetic testing for ADPKD can be obtained from GeneTests (www.genetests.org), a valuable web-based resource funded by the National Institutes of Health.14 The Web site provides a comprehensive list of academic and commercial facilities worldwide that offer testing for PKD1 or PKD2 mutations on a clinical or research basis.
DNA linkage analysis seeks to identify the presence of a segment of the chromosome at either the PKD1 or PKD2 locus that completely segregates with the disease. Thus, there is no need to identify the exact ADPKD mutation as the presence of these markers and not the mutations themselves is being tracked. There are currently 15 microsatellite markers for PKD1 and 8 for PKD2.15 DNA linkage analysis, however, is useful only in familial cases and requires a large family with at least 4 affected members in at least 2 generations, with radiological studies in both affected and unaffected individuals, for conclusive results. Results must be interpreted with caution if there are de novo mutations, mosaicisms, and hypomorphic alleles.
Gene-based mutation screening is the most commonly used method for genetic diagnosis of ADPKD. This approach seeks to identify the exact mutation in the PKD1 and PKD2 genes. Because most mutations are unique to a single family with no clear “hot spots,” exon-by-exon screening of these genes is necessary to ensure high sensitivity in detecting disease-causing mutations in PKD1 and PKD2.16 Challenges of gene-based mutation screening include the difficulty in differentiating disease-causing missense mutations from benign variants, the detection of a definitive mutation in no more than 65% to 75% of patients tested, and the lack of a confirmed pathogenic mutation in approximately 8% of patients with ADPKD.13
NGS, also known as high-throughput sequencing, refers to a number of different modern sequencing technologies that can sequence millions of small fragments of DNA in parallel and use bioinformatic analyses to piece together these fragments and provide accurate data on genetic mutations.17 Compared with gene-based mutation screening, NGS offers the benefits of high fidelity, high throughput, and high speed.18 NGS was validated in a cohort of 25 patients who had previously undergone genetic testing using gene-based mutation screening.19 NGS identified 250 genetic variants in the PKD1 and PKD2 genes, including all 16 pathogenic mutations and 3 novel mutations that gene-based mutation screening did not identify. In this study, NGS showed sensitivity of 99.2% and specificity of 99.9%, with cost and turnaround time reduced by approximately 70% compared with gene-based mutation screening sequencing.
Although genetic testing for ADPKD mutations is indicated in some patients, it is not indicated for all patients. Genetic testing is not needed when a firm positive or negative diagnosis can be made by imaging alone or, as in the case of a patient with suspected ADPKD, when a diagnosis can be made based on the imaging results of the patient’s parents or based on the presence of extrarenal manifestations. Genetic testing should be considered in potential living related donors to confirm the absence of any mutations for ADPKD, in patients without a family history of ADPKD (especially if radiographic presentation is atypical, if renal disease is mild, if extrarenal symptoms are atypical, or if prognostic information is required), in families with atypical radiographic patterns of kidney cysts to possibly exclude other cystic kidney diseases, in families affected by early-onset polycystic disease, and in patients who want a prenatal or preimplantation diagnosis.15,20
Family history can be highly predictive of the genetic mutation.21 A family history of having at least one family member with early-onset ESRD ≤55 to 58 years of age has a positive predictive value (PPV) of 100% for the presence of a mutation in the PKD1 gene. In contrast, a family history of having at least one family member who remained renal sufficient or developed ESRD ≥68 to 70 years of age had a PPV of 100% for a mutation in the PKD2 gene. Applying these 2 criteria can correctly predict the PKD1 or PKD2 mutation in approximately 75% of cases.
Recommendations
Based on existing data, genetic testing is not necessary for selecting treatment options for patients with a confirmed diagnosis of ADPKD.
If genetic testing is to be done, it should be done by a Clinical Laboratory Improvement Amendments (CLIA)-certified center and interpreted by those familiar with ADPKD genetics.
Renal Imaging
Several imaging modalities are currently available to diagnose and evaluate ADPKD, including abdominal ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI). In terms of diagnosis, our nephrologist survey indicated that 92% of clinicians are using US as their diagnostic modality. US has robust performance in this setting; age-based criteria and diagnostic performance have been previously published and serve as good criteria for imaging-based diagnosis.22 MRI and high-resolution US have greater sensitivity for imaging-based diagnosis at younger ages than conventional US,23 but the availability of these tests in some centers may be limited.
For size determination, MRI appears to be the preferred imaging modality; it has greater accuracy and precision when compared with US, and although CT performs well, it requires radiation exposure.24,25 US is a more practical and more cost-effective approach; however, there are concerns that, compared with CT or MRI, US is more user-dependent and has higher variability making it difficult to obtain accurate and reproducible results.26 Emerging US techniques, including 3-dimensional and high-resolution US, have some data in the diagnosis of ADPKD,23 but these modalities are not widely available, and their performance in size determination has not been evaluated.27
In ADPKD, kidney volume can be divided into cystic and noncystic components; however, changes in overall total kidney volume (TKV) are less variable than changes in either of these components individually, and TKV is easier to obtain.28 For these reasons, TKV is the more commonly used measure, and in most cases, it is unnecessary to divide the volume into cystic and noncystic components.
TKV is typically obtained either by stereology or various formulae that estimate volume from a more limited set of measurements.28,29 Stereology can be quite labor-intensive but is presently considered the gold standard for the measurement of TKV.28 The most common method of estimation is the ellipsoid equation, which has been shown to approximate the stereological approach accurately and is less labor-intensive.29 Another estimation formula based on a single midcoronal measurement has also been shown to yield volumes that are highly correlated with stereology measures.30 More recently, automated methods of TKV determination have been reported to yield results similar to stereology; for these measures, patients are classified based on height-adjusted (ht) TKV and age.31,32
The nephrologist survey indicated that US is the first imaging modality in the majority of cases; the information extracted from these already available US images should be maximized, but it is important to consider the performance of this test. When measuring TKV, US tends to be more variable than MRI and tends to overestimate volume. Therefore, it may be useful to group patients into broad categories of kidney size.26 Of the component measurements, US kidney length has less variability than the other dimensions.26 US kidney length was recently found to correlate well with htTKV measured by MRI and seemed to predict the development of stage 3 chronic kidney disease (CKD) in a similar manner.33
Before interpreting TKV measurements, patients should be categorized according to the recently published Mayo Clinic Classification (Figure 1)29 (http://www.mayo.edu/research/documents/pkd-center-adpkd-classification/doc-20094754). According to this system, patients with typical symmetric, bilateral, diffuse cyst distribution are categorized as class 1 (approximately 90% of patients), whereas patients with atypical, asymmetric, or segmental cyst distribution are categorized as class 2 (Table 3). Class 1 patients can be further divided into subclasses A through E by integrating htTKV with age. Classes 1C, 1D, and 1E show the highest propensity for developing early-onset renal disease. There are subtypes of class 2 (atypical) enlargement, and class 2 patients are generally not thought to be at risk of rapid renal progression, although the original studies had too few class 2 patients to definitively comment on renal progression in this group.29
Figure 1.
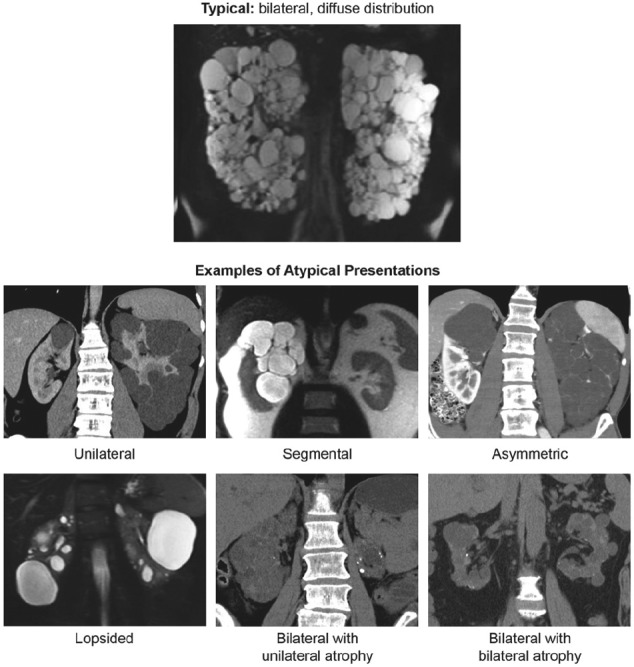
Mayo clinic classification of autosomal dominant polycystic kidney disease.
Source. Republished from Irazabal et al29 with permission of the American Society of Nephrology; permission conveyed through Copyright Clearance Center, Inc.
Table 3.
Classification of ADPKD Based on Imaging Characteristics According to the Mayo Clinic Classification.
| Class, subclass, and term | Description |
|---|---|
| 1. Typical ADPKD | Bilateral and diffuse distribution, with mild, moderate, or severe replacement of kidney tissue by cysts, where all cysts contribute similarly to TKV |
| 2. Atypical ADPKD | |
| Unilateral | Diffuse cystic involvement of one kidney causing marked renal enlargement with a normal contralateral kidney, defined by a normal kidney volume (<275 mL in men; <244 mL in women) and having 0-2 cysts |
| Segmental | Cystic disease involving only one pole of one or both kidneys and sparing the remaining renal tissue |
| Asymmetric | Diffuse cystic involvement of one kidney causing marked renal enlargement with mild segmental or minimal diffuse involvement of the contralateral kidney, defined by a small number of cysts (>2 but <10) and volume accounting for <30% of TKV |
| Lopsided | Bilateral distribution of renal cysts with mild replacement of kidney tissue with atypical cysts where ≤5 cysts account for ≥50% TKV (the largest cyst diameter is used to estimate individual cyst volume) |
| Bilateral presentation with acquired unilateral atrophy | Diffuse cystic involvement of one kidney causing moderate to severe renal enlargement with contralateral acquired atrophy |
| Bilateral presentation with bilateral kidney atrophy | Impaired renal function (serum creatinine ≥1.5 mg/dL) without significant enlargement of the kidneys, defined by an average length <14.5 cm, and replacement of kidney tissue by cysts with atrophy of the parenchyma |
Source. Republished from Irazabal et al29 with permission of the American Society of Nephrology; permission conveyed through Copyright Clearance Center, Inc.
Note. ADPKD = autosomal dominant polycystic kidney disease; TKV = total kidney volume.
There are limited data on the role of repeated imaging.24,34 In patients where serial measurements are taken, an increase of >5% per year in TKV, corresponding to the threshold for class 1D, appears to correlate well with predicting rapid renal progression.29 Recent recommendations by the European Renal Association-European Dialysis and Transplant Association (ERA-EDTA) and the Japanese regulatory authorities also proposed that patients with an increase in TKV of >5% annually should be placed in a higher risk category for renal disease progression.35,36 If repeated measurements are obtained by the clinician, the performance of these tests should be considered. With MRI, differences in TKV can be detected with as little as a 6-month interval between measurements.37 Conversely, although US can detect differences in kidney size over many years, it is not suitable for short-term follow-up as the inherent inaccuracy in US measurements is approximately the same as the annual rate of growth.38
Recommendations
1. We recommend that before quantifying the size of the kidneys, patients should be classified according to the Mayo Clinic classification for typical versus atypical morphology with renal imaging.
2.1. We recommend that a baseline assessment of renal size be undertaken in patients with ADPKD. The objective of these measurements is to determine which patients are suitable candidates to be considered for therapeutic intervention based on their risk of progression.
2.2. Although the gold standard for measuring TKV is MRI stereology, we recommend the use of ellipsoid TKV or US to determine TKV in routine clinical practice. We suggest that MRI or CT htTKV is currently the most accurate method of assessing renal size in patients with ADPKD.
2.3. In the absence of MRI, imaging by CT may be used to determine TKV. In situations where an MRI or CT is not easily obtainable, we suggest US-measured KL as a suitable surrogate. US can be used to determine TKV; however, TKV obtained using US may introduce error and does not provide an advantage over KL.
3. We recommend that routine assessment of TKV or KL should not exceed a frequency of once yearly.
Predicting Disease Progression
Prognostic factors related to disease progression in ADPKD include TKV, the type of genetic mutation, vasopressin activity, uric acid, and the presence of certain urine biomarkers, as summarized in Table 4.39 Disease progression in ADPKD is characterized by loss of renal function, defined as a decline of ≥5 mL/min/1.73 m2 in 1 year, or average decline of ≥2.5 mL/min/1.73 m2 over 5 years, measured by creatinine clearance using the Cockcroft-Gault equation or by estimated glomerular filtration rate (eGFR) using the CKD-EPI equation.29,40 However, in the early stages of disease, there is little change in renal function yet detectable changes in TKV. As such, TKV is a more sensitive measure of disease progression.28
Table 4.
Prognostic Factors Related to Disease Progression in ADPKD.
| Imaging-based prognostic factors |
| ● TKV shows a strong inverse association with the slope of GFR28,41 |
| ● Height-adjusted TKV shows a good correlation with GFR at baseline (r = 0.22), and an even stronger correlation after 3 years and 8 years (r = 0.44 and r = 0.65, respectively)42 |
| Genetic prognostic factors |
| ● PKD1 mutations are associated with an earlier onset of ESRD compared with PKD2 mutations43 |
| ● Truncating PKD1 mutations are associated with an average onset of ESRD at 55 years of age, whereas nontruncating PKD1 mutations and PKD2 mutations are associated with an average onset of ESRD at 67 and 79 years of age, respectively44 |
| ● Hypomorphic alleles are associated with milder disease as polycystin activity is not completely abrogated; if these alleles are coupled with another mutation, a more severe disease progression may develop45 |
| Urinary biomarkers |
| ● Urinary neutrophil gelatinase–associated lipocalin and interleukin-18 levels increased over 3 years in the CRISP study; however, the increases in these 2 urine biomarkers did not correlate with changes in TKV or kidney function46 |
| Other prognostic factors |
| ● Higher levels of vasopressin activity (measured using 24-h urine osmolality as a surrogate marker) were associated with greater declines in GFR from year 1 to 6 in the CRISP study47 |
| ● Increased vasopressin activity (measured using copeptin levels as a surrogate marker) was associated with higher morning urine osmolality, higher BP, increased TKV, and decreased GFR in the CRISP study48 |
| ● Elevated serum uric acid levels are associated with disease progression; a 5.8% increase in TKV and a 4.1% increase in TKV/body surface area for every 1-mg/dL increase in uric acid have been demonstrated49 |
Note. ADPKD = autosomal dominant polycystic kidney disease; TKV = total kidney volume; GFR = glomerular filtration rate; ESRD = end-stage renal disease; CRISP = Consortium of Renal Imaging Studies in Polycystic Kidney Disease; BP = blood pressure.
Risk Prediction Using PROPKD Score
Cornec-Le Gall et al developed the PROPKD score as a prognostic model to predict renal outcomes in patients with ADPKD on the basis of genetic and clinical data from 1341 patients from the Genkyst cohort.50 The scoring system assigns points as shown in Table 5. Thus, an individual’s PROPKD score can range from 0 to 9 points. Three risk categories of progression to ESRD were subsequently defined: low (0-3 points), intermediate (4-6 points), and high (7-9 points). The predicted median age of onset for ESRD and predicted disease progression for these 3 risk categories are listed in Table 6. Of note, the PROPKD scoring system cannot be applied to patients with no history of urological events or hypertension and has not been widely validated in independent cohorts.
Table 5.
The PROPKD Scoring System.
| Factor | Points |
|---|---|
| Male | 1 |
| Hypertension before age 35 y | 2 |
| First urological event before age 35 y | 2 |
| PKD2 mutation | 0 |
| Nontruncating PKD1 mutation | 2 |
| Truncating PKD1 mutation | 4 |
Table 6.
Predicted Median Age of Onset of ESRD and Predicted Disease Progression by PROPKD Risk Category.
| PROPKD risk category for progression to ESRD |
|||
|---|---|---|---|
| Low risk (0-3 points) | Intermediate risk (4-6 points) | High risk (7-9 points) | |
| Predicted median age of onset for ESRD, y | 70.6 | 56.9 | 49.0 |
| Predicted disease progression | Excludes progression to ESRD before 60 years of age (negative predictive value of 81.4%) | Prognosis is unclear | Rapid progression to ESRD before 60 years of age (positive predictive value of 90.9%) |
Note. ESRD = end-stage renal disease.
Risk Prediction Using Genetic Scoring
For some patients, such as those younger than 35 years and those missing clinical data, the Predicting Renal Outcome in Polycystic Kidney Disease (PROPKD) score cannot be applied.50 In these cases, genetic scoring may be carried out, comprising only genetic data and gender. In this scoring system, patients fall into 1 of 4 prognostic groups:
patients with PKD2 mutations (1 point)
patients with nontruncating PKD1 mutations (2 points)
women with truncating PKD1 mutations (3 points)
men with truncating PKD1 mutations (4 points)
Patients with a genetic score ≥2 points have a predicted onset of ESRD before age 65 years. Although genetic scoring is less accurate than the PROPKD score, it offers good prediction of ESRD.
Risk Prediction Using Mayo Classification
The proposed Mayo Classification defines groups of patients with different risks for eGFR decline.29 As shown in Figure 2A, class 1 patients are categorized into subclasses 1A through 1E based on htTKV and age at baseline, which, in turn, predicts decline in eGFR, as shown in Figure 2B. The Mayo Classification, although a useful clinical tool, was developed with the aim of identifying patients eligible to participate in clinical trials.
Figure 2.
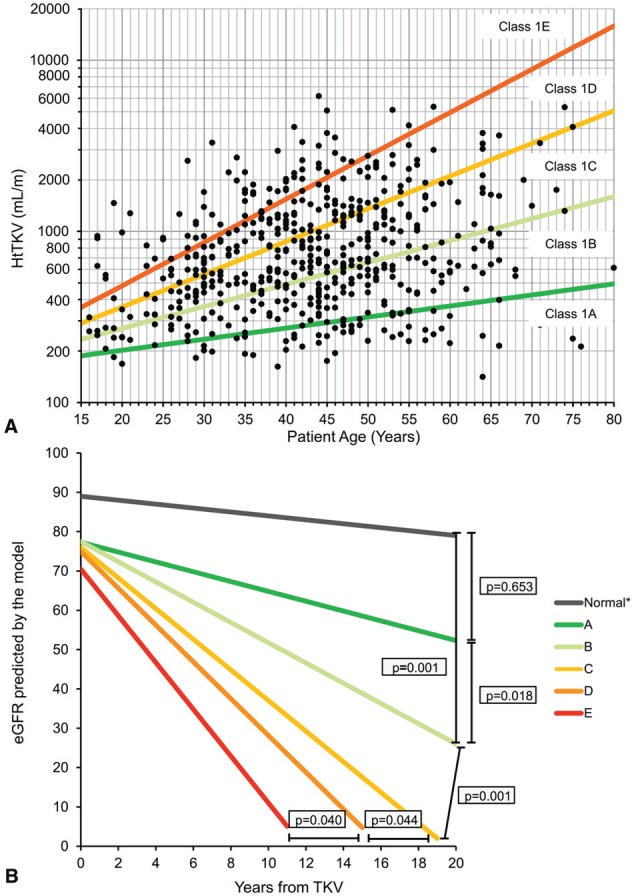
A, Subclassification of patients with class 1 autosomal dominant polycystic kidney disease at baseline based on htTKV and age at baseline. B, Predicted change in eGFR over time in class 1 patients (slopes shown are those for men).
Source. Republished from Irazabal et al29 with permission of the American Society of Nephrology; permission conveyed through Copyright Clearance Center, Inc.
Note. htTKV = height-adjusted total kidney volume; eGFR = estimated glomerular filtration rate; TKV = total kidney volume.
Recommendations
We recommend that in current clinical practice, patients with a TKV measurement be categorized in terms of their risk of progression as per the Mayo Clinic classification or other validated clinical tools. We highlight that the application of the Mayo Classification to clinical practice has not yet been delineated; however, it appears to be the most robust clinical prediction tool as it pertains to the important marker of htTKV.
Currently available TKV-based prognostication tools should not be applied to class 2 (atypical morphology) patients, as we suggest that these patients are unlikely to be rapid progressors. Certain patients may require further clinical evaluation.
We suggest that patients who are classified as Mayo class 1C, D, or E be considered to be at risk of rapid progression of their ADPKD renal disease.
We recommend that patients who demonstrate a sequential increase of >5% annually in TKV on imaging should be considered at risk of rapid progression of their ADPKD-related renal disease.
We recommend that patients with an US KL of >16.5 cm bilaterally should be considered at high risk of progression of their ADPKD-related renal disease. A KL >16.5 cm has been shown to correlate with a TKV of 750 mL; however, direct measurement of TKV would be required if more accurate assessment is needed.
We suggest that baseline TKV and KL are important determinants of renal progression of ADPKD; however, serial TKV and KL measurements have not been established as markers to monitor response to therapy.
Nontargeted Treatment Options
Nontargeted treatment options for ADPKD include protein restriction, increased fluid intake, and blood pressure (BP) control. To date, no study has been able to demonstrate the benefit of protein restriction in patients with ADPKD.51
Increased fluid intake has received a great deal of attention as a therapeutic approach to improving disease progression in ADPKD; however, there are currently no compelling data to support increased water intake as a treatment option to prevent disease progression in ADPKD. A recent study demonstrated no benefit on disease progression in ADPKD among patients in the high water intake group compared with the free water intake group.52
Rigorous BP control (95/60-110/75 mm Hg) was associated with a significantly lower annual rate of increase in TKV compared with a standard BP target (120/70-130/80 mm Hg): 5.6% versus 6.5%; P = .006.53 Patients in the tighter BP control group also experienced a reduction in urinary albumin excretion per year (−3.8%) versus an increase (2.4%) in the standard BP group (P < .001), but there was no significant difference between the 2 groups in annual change in eGFR (−2.9 mL/min/1.73 m2 vs −3.0 mL/min/1.73 m2, respectively; P = .55). Similarly, a post hoc analysis of the early ADPKD population in the Halt Polycystic Kidney Disease (HALT-PKD) Study A demonstrated a stronger benefit of rigorous BP control on TKV increase, as well as a stronger benefit on eGFR decline, in the subgroup of patients with severe disease (classes 1D and E).54
Recommendation
We recommend that patients with ADPKD who are younger than 50 years with eGFR >60 mL/min/1.73 m2 and without significant cardiovascular comorbidities should have a target BP of ≤110/75 mm Hg, realizing that in some patients an individual target may be needed.
ADPKD-Specific Treatment Options
A recent meta-analysis of 4 randomized, controlled trials of the mTOR inhibitor sirolimus in adults with ADPKD showed a positive impact on TKV but not on eGFR.55 Similar results have been reported with the mTOR inhibitor everolimus.56 Thus, sirolimus and everolimus are effective in reducing the increase in TKV in patients with ADPKD but have not been shown to slow or improve loss of renal function.
Treatment with the somatostatin analogue octreotide in its standard or long-acting formulation inhibits or slows renal enlargement in patients with ADPKD but has not been shown to improve loss of renal function.57-59 The efficacy of pravastatin in the treatment of ADPKD has been demonstrated in pediatric patients.60 At the end of 3 years of treatment with pravastatin, a significant decrease in percent change in htTKV was observed when adjusted for age, sex, and hypertension status, compared with placebo (23 ± 3% vs 31 ± 3%, respectively, P = .02). Further studies are required to assess efficacy in adults.
Tolvaptan, a selective vasopressin V2-receptor antagonist approved by Health Canada in 2015, is indicated to slow the progression of kidney enlargement in patients with ADPKD.9,61 Tolvaptan received approval based on the results of the phase 3, double-blind TEMPO 3:4 trial.62 In this 3-year trial, 1445 ADPKD patients aged 18 to 50 years with a TKV ≥750 mL and a creatinine clearance of ≥60 mL/min, as estimated by the Cockroft-Gault formula, were randomized to either tolvaptan (highest of 3 doses based on tolerability) or placebo. The annual rate of change in TKV (primary endpoint) was 2.8% with tolvaptan, compared with 5.5% with placebo (P < .0001). The rate of growth was reduced by 2.7 percentage points per year with tolvaptan, and the ratio of the geometric means of growth rate was 0.97 (P < .001). Loss in kidney function, determined as the reciprocal of the serum creatinine level, from the end of dose escalation to month 36, was significantly reduced with tolvaptan (slope of −2.61 [mg/mL]−1 per year) compared with placebo (slope of −3.81 [mg/mL]−1 per year).62 The overall treatment effect was an increase of 1.20 (mg/mL)−1 per year (P < .001). Analysis of the annual estimated GFR slope (which gave results similar to those of the slopes of the reciprocal of the serum creatinine level) showed an estimated GFR slope of −2.72 mL per minute per 1.73 m2 per year in the tolvaptan group versus −3.70 in the placebo group (treatment effect, an increase of 0.98 mL per minute per 1.73 m2 per year; 95% confidence interval, 0.60 to 1.36; P < .001).63 Figure 3 shows the effect of tolvaptan on TKV growth and eGFR stratified by CKD stage.63 Tolvaptan also significantly reduced the occurrence of clinically significant kidney pain—defined as pain necessitating medical leave, pharmacological treatment (opioid or last-resort analgesic agents), or invasive intervention—compared with placebo (hazard ratio [HR] 0.64; P = .007).62
Figure 3.

Effect of tolvaptan on eGFR (left panel) and TKV (right panel) by CKD stage in the TEMPO 3:4 trial.
Source. Republished from Torres et al63 with permission of the American Society of Nephrology; permission conveyed through Copyright Clearance Center, Inc.
Note. eGFR = estimated glomerular filtration rate; TKV = total kidney volume; CKD = chronic kidney disease.
A post hoc analysis of TEMPO 3:4 clinical data was carried out using Mayo Classification to exclude 10% of the original patient population who had a lesser risk for progression (classes 1A-B and 2), resulting in a patient population enriched in categories 1C-E.64 A comparison of the enriched population to the original cohort showed that the effect of tolvaptan on TKV and eGFR slopes increased in classes 1C to E: TKV was significantly lower with tolvaptan versus placebo (5.8% vs 2.9%; P < .001; 2.8% in TEMPO 3:4) and reduced the decline in the eGFR slope (−3.93 mL/min/1.73 m2 per year to −2.82 mL/min/1.73 m2 per year; P < .001; −2.78 mL/min/1.73 m2 per year in TEMPO 3:4), and significantly reduced the risk of clinical progression (HR 0.84; P = .0032).
Cost-effectiveness of Tolvaptan
There have been no published manuscripts addressing the cost-effectiveness of tolvaptan. The Canadian Agency for Drugs and Technologies in Health (CADTH) recently recommended that tolvaptan not be listed on provincial formularies to slow the progression of kidney enlargement in patients with ADPKD.65 The manufacturer submitted a cost-utility analysis comparing tolvaptan with the standard of care that suggested a base-case incremental cost utility ratio (ICUR) of $244 402 per quality-adjusted life year (QALY).66 From their conclusions, it appears that the model is sensitive to varying assumptions around rate of disease progression and lower drug efficacy, which inflate the ICUR considerably. Although we are unable to comment directly on how sensitive the model is to pricing of the drug, it is logical that the model would be exquisitely sensitive to this parameter. If we assume a liberal willingness-to-pay threshold of $100 000 per QALY to fund new health interventions, further negotiation on pricing may influence decisions to list this medication on provincial formularies moving forward. Therefore, at the time of writing this article, tolvaptan is only available through private insurance, and our recommendations are predicated on patients having private health care insurance that will cover the cost of the drug.
Recommendations
We suggest that all patients be referred to a nephrologist for initial assessment to determine what treatment should be initiated, in particular to initiate tolvaptan as soon as possible in patients determined to be appropriate candidates who would benefit from this therapy.
We recommend treatment with tolvaptan for patients who fulfill the enrollment criteria of the TEMPO 3:4 study: 18 to 50 years of age, Cockcroft-Gault GFR >60 mL/min, and TKV >750 mL. In the absence of Cockcroft-Gault GFR, CKD-EPI >45 mL/min may be used, and in the absence of TKV, US KL >16.5 cm may be used.
We suggest treatment with tolvaptan for patients who, according to the Mayo Classification, are classified as 1D or 1E with eGFR in CKD stage 3 or higher. Treatment with tolvaptan should be considered for patients who are classified as 1C and are younger than 50 years or have other risk factors for rapid progression. We do not recommend tolvaptan for patients classified as 1A or 1B.
We suggest that treatment with tolvaptan be stopped when the patient develops ESRD. In the predialysis setting, there are no data to guide when treatment with tolvaptan should be stopped.
Acknowledgments
The authors gratefully acknowledge the contribution of Angela Styhler in the drafting of the manuscript.
Footnotes
Declaration of Conflicting Interests: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Steven Soroka reports receiving honoraria for lecturing on autosomal dominant polycystic kidney disease (ADPKD), and developing educational material and participating in advisory boards from Otsuka Canada. Dr Daniel G. Bichet reports receiving honoraria for lectures on ADPKD from Otsuka Canada. Dr Ahsan Alam reports receiving honoraria for consultancy and lecturing from Otsuka Canada and Amgen. Dr Louis-Philippe Girard reports receiving honoraria for his involvement in continuing medical education and his participation in advisory boards from Otsuka Canada. Dr Philip McFarlane reports receiving honoraria for his participation in advisory boards from Otsuka Canada. Dr Paul Tam reports receiving a research grant from Janssen and honoraria for his participation in advisory boards from Amgen. Drs Micheli Bevilacqua, Paul Komenda, Sanjaya Pandeya, and Rolf Loertscher have no disclosures to report regarding their contributions to this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Otsuka, the funding sponsor, offered unrestricted support to the development of these recommendations and did not have any part in creating this document. Funding sponsor representatives were not present at the meeting. The meeting that produced the recommendations presented was organized by SNELL Medical Communication. Honoraria were provided to the participants to create and present slides to generate discussion. The funding also provided the authors with the services of an experienced and qualified medical writer to ensure a professional manuscript. The medical writer, solely under the direction and outline of the authors, assisted in researching the topic and preparing a first draft. At no time did the medical writer have any involvement in determining the content of the manuscript.
References
- 1. Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-1301. [DOI] [PubMed] [Google Scholar]
- 2. Akoh JA. Current management of autosomal dominant polycystic kidney disease. World J Nephrol. 2015;4(4):468-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Paul BM, Vanden Heuvel GB. Kidney: polycystic kidney disease. Wiley Interdiscip Rev Dev Biol. 2014;3(6):465-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Romão EA, Moysés Neto M, Teixeira SR, Muglia VF, Vieira-Neto OM, Dantas M. Renal and extrarenal manifestations of autosomal dominant polycystic kidney disease. Braz J Med Biol Res. 2006;39(4):533-538. [DOI] [PubMed] [Google Scholar]
- 5. Thong KM, Ong AC. The natural history of autosomal dominant polycystic kidney disease: 30-year experience from a single centre. QJM. 2013;106(7):639-646. [DOI] [PubMed] [Google Scholar]
- 6. Belibi FA, Reif G, Wallace DP, et al. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004;66(3):964-973. [DOI] [PubMed] [Google Scholar]
- 7. Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lentine KL, Xiao H, Machnicki G, Gheorghian A, Schnitzler MA. Renal function and healthcare costs in patients with polycystic kidney disease. Clin J Am Soc Nephrol. 2010;5(8):1471-1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. JINARC (tolvaptan). Product Monograph. Saint-Laurent, Quebec: Otsuka Canada Pharmaceutical Inc.; 2015. [Google Scholar]
- 10. Mochizuki T, Wu G, Hayashi T, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272(5266):1339-1342. [DOI] [PubMed] [Google Scholar]
- 11. Cornec-Le Gall E, Audrézet MP, Le Meur Y, Chen JM, Férec C. Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum Mutat. 2014;35(12):1393-1406. [DOI] [PubMed] [Google Scholar]
- 12. Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13(9):2384-2398. [DOI] [PubMed] [Google Scholar]
- 13. Heyer CM, Sundsbak JL, Abebe KZ, et al. Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27:2872-2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pei Y, Watnick T. Diagnosis and screening of autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17(2):140-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Torra-Balcells R, Ars-Criach E. Molecular diagnosis of autosomal dominant polycystic kidney disease. Nefrologia. 2011;31(1):35-43. [DOI] [PubMed] [Google Scholar]
- 16. Pei Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2006;1(5):1108-1114. [DOI] [PubMed] [Google Scholar]
- 17. Behjati S, Tarpey PS. What is next generation sequencing? Arch Dis Child Educ Pract Ed. 2013;98(6):236-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rossetti S, Hopp K, Sikkink RA, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol. 2012;23(5):915-933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tan AY, Michaeel A, Liu G, et al. Molecular diagnosis of autosomal dominant polycystic kidney disease using next-generation sequencing. J Mol Diagn. 2014;16(2):216-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harris PC, Rossetti S. Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2010;6(4):197-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barua M, Cil O, Paterson AD, et al. Family history of renal disease severity predicts the mutated gene in ADPKD. J Am Soc Nephrol. 2009;20(8):1833-1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barua M, Pei Y. Diagnosis of autosomal-dominant polycystic kidney disease: an integrated approach. Semin Nephrol. 2010;30(4):356-365. [DOI] [PubMed] [Google Scholar]
- 23. Pei Y, Hwang Y-H, Conklin J, et al. Imaging-based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2015;26(3):746-753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bae KT, Grantham JJ. Imaging for the prognosis of autosomal dominant polycystic kidney disease. Nat Rev Nephrol. 2010;6(2):96-106. [DOI] [PubMed] [Google Scholar]
- 25. Sise C, Kusaka M, Wetzel LH, et al. Volumetric determination of progression in autosomal dominant polycystic kidney disease by computed tomography. Kidney Int. 2000;58(6):2492-2501. [DOI] [PubMed] [Google Scholar]
- 26. O’Neill WC, Robbin ML, Bae KT, et al. Sonographic assessment of the severity and progression of autosomal dominant polycystic kidney disease: the Consortium of Renal Imaging Studies in Polycystic Kidney Disease (CRISP). Am J Kidney Dis. 2005;46(6):1058-1064. [DOI] [PubMed] [Google Scholar]
- 27. Alam A, Dahl NK, Lipschutz JH, et al. Total kidney volume in autosomal dominant polycystic kidney disease: a biomarker of disease progression and therapeutic efficacy. Am J Kidney Dis. 2015;66(4):564-576. [DOI] [PubMed] [Google Scholar]
- 28. Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354(20):2122-2130. [DOI] [PubMed] [Google Scholar]
- 29. Irazabal MV, Rangel LJ, Bergstralh EJ, et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J Am Soc Nephrol. 2015;26(1):160-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bae KT, Tao C, Wang J, et al. Novel approach to estimate kidney and cyst volumes using mid-slice magnetic resonance images in polycystic kidney disease. Am J Nephrol. 2013;38(4):333-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim Y, Ge Y, Tao C, et al. Automated segmentation of kidneys from MR images in patients with autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2016;11(4):576-584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kline TL, Korfiatis P, Edwards ME, et al. Automatic total kidney volume measurement on follow-up magnetic resonance images to facilitate monitoring of autosomal dominant polycystic kidney disease progression. Nephrol Dial Transplant. 2016;31(2):241-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhutani H, Smith V, Rahbari-Oskoui F, et al. A comparison of ultrasound and magnetic resonance imaging shows that kidney length predicts chronic kidney disease in autosomal dominant polycystic kidney disease. Kidney Int. 2015;88(1):146-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spithoven EM, van Gastel MD, Messchendorp AL, et al. Estimation of total kidney volume in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2015;66(5):792-801. [DOI] [PubMed] [Google Scholar]
- 35. Clinical Guidelines for Polycystic Kidney Disease 2014 Advisory Committee. Evidence-based clinical practice guidelines for polycystic kidney disease 2014. Japan. Clin Exp Nephrol. 2016;20:493-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gansevoort RT, Arici M, Benzing T, et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol Dial Transplant. 2016;31(3):337-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kistler AD, Poster D, Krauer F, et al. Increases in kidney volume in autosomal dominant polycystic kidney disease can be detected within 6 months. Kidney Int. 2009;75(2):235-241. [DOI] [PubMed] [Google Scholar]
- 38. Fick-Brosnahan GM, Belz MM, McFann KK, Johnson AM, Schrier RW. Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease: a longitudinal study. Am J Kidney Dis. 2002;39(6):1127-1134. [DOI] [PubMed] [Google Scholar]
- 39. Alam A. Risk factors for progression in ADPKD. Curr Opin Nephrol Hypertens. 2015;24(3):290-294. [DOI] [PubMed] [Google Scholar]
- 40. Eknoyan G, Lameire N, Eckardt KU, et al. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. 2013;3(1):5-14. [DOI] [PubMed] [Google Scholar]
- 41. Chapman AB, Guay-Woodford LM, Grantham JJ, et al. Renal structure in early autosomal-dominant polycystic kidney disease (ADPKD): the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP) cohort. Kidney Int. 2003;64(3):1035-1045. [DOI] [PubMed] [Google Scholar]
- 42. Chapman AB, Bost JE, Torres VE, et al. Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2012;7(3):479-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hateboer N, Dijk MA, Bogdanova N, et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study Group. Lancet. 1999;353(9147):103-107. [DOI] [PubMed] [Google Scholar]
- 44. Cornec-Le Gall E, Audrézet MP, Chen JM, et al. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24(6):1006-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rossetti S, Kubly VJ, Consugar MB, et al. Incompletely penetrant PKD1 alleles suggest a role for gene dosage in cyst initiation in polycystic kidney disease. Kidney Int. 2009;75(8):848-855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Parikh CR, Dahl NK, Chapman AB, et al. Evaluation of urine biomarkers of kidney injury in polycystic kidney disease. Kidney Int. 2012;81(8):784-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Torres VE, Grantham JJ, Chapman AB, et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6(3):640-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boertien WE, Meijer E, Li J, et al. Relationship of copeptin, a surrogate marker for arginine vasopressin, with change in total kidney volume and GFR decline in autosomal dominant polycystic kidney disease: results from the CRISP cohort. Am J Kidney Dis. 2013;61(3):420-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Helal I, McFann K, Reed B, et al. Serum uric acid, kidney volume and progression in autosomal-dominant polycystic kidney disease. Nephrol Dial Transplant. 2013;28(2):380-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cornec-Le Gall E, Audrézet MP, Rousseau A, et al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2016;27(3):942-951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Levey AS, Greene T, Beck GJ, et al. Dietary protein restriction and the progression of chronic renal disease: what have all of the results of the MDRD study shown? Modification of Diet in Renal Disease Study group. J Am Soc Nephrol. 1999;10(11):2426-2439. [DOI] [PubMed] [Google Scholar]
- 52. Higashihara E, Nutahara K, Tanbo M, et al. Does increased water intake prevent disease progression in autosomal dominant polycystic kidney disease? Nephrol Dial Transplant. 2014;29(9):1710-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schrier RW, Abebe KZ, Perrone RD, et al. Blood pressure in early autosomal dominant polycystic kidney disease. N Engl J Med. 2014;371(24):2255-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Irazabal MV, Abebe KZ, Bae KT, et al. Prognostic enrichment design in clinical trials for autosomal dominant polycystic kidney disease: the HALT-PKD clinical trial [published online ahead of print August 2, 2016]. Nephrol Dial Transplant. doi: 10.1093/ndt/gfw294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu YM, Shao YQ, He Q. Sirolimus for treatment of autosomal-dominant polycystic kidney disease: a meta-analysis of randomized controlled trials. Transplant Proc. 2014;46(1):66-74. [DOI] [PubMed] [Google Scholar]
- 56. Walz G, Budde K, Mannaa M, et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2010;363(9):830-840. [DOI] [PubMed] [Google Scholar]
- 57. Caroli A, Perico N, Perna A, et al. Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet. 2013;382(9903):1485-1495. [DOI] [PubMed] [Google Scholar]
- 58. Hogan MC, Masyuk TV, Page LJ, et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J Am Soc Nephrol. 2010;21(6):1052-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hogan MC, Masyuk TV, Page L, et al. Somatostatin analog therapy for severe polycystic liver disease: results after 2 years. Nephrol Dial Transplant. 2012;27(9):3532-3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cadnapaphornchai MA, George DM, McFann K, et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2014;9(5):889-896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Health Canada. Jinarc Notice of Compliance (NOC) information. https://health-products.canada.ca/noc-ac/info.do?no=16640&lang=en. Accessed February 20, 2017.
- 62. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407-2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Torres VE, Higashihara E, Devuyst O, et al. Effect of tolvaptan in autosomal dominant polycystic kidney disease by CKD stage: results from the TEMPO 3:4 trial. Clin J Am Soc Nephrol. 2016;11(5):803-811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Irazabal MV, Blais J, Perrone RD, et al. Prognostic enrichment in the TEMPO 3:4 ADPKD clinical trial. J Am Soc Nephrol. 2015;26(Abstract suppl):831.25145932 [Google Scholar]
- 65. Canadian Agency for Drugs and Technologies in Health. Reports: tolvaptan. https://www.cadth.ca/tolvaptan-7. Published May 4, 2015. Accessed on August 11, 2016.
- 66. CADTH (2016). CADTH Canadian drug expert committee final recommendation for Jinarc (tolvaptan). Available online at: https://www.cadth.ca/sites/default/files/cdr/complete/SR0435_complete_Jinarc-Feb_26_16_e.pdf. Accessed February 20, 2017.