

Significance
The biosynthesis of class I viral membrane fusion proteins as trimers provides the possibility of forming a stable coiled coil core from its transmembrane subunits, on which an effective fusion machinery can be built. Although the coiled coil must engage all three protomers simultaneously, it has remained unclear whether this is also the case with the other steps of their activation pathways. We studied this question with the Moloney murine leukemia virus Env, for which activation can be followed by surface (SU)-transmembrane (TM) intersubunit disulfide isomerization and subsequent SU release. We found that this early activation step occurred sequentially in one protomer after the other, forming asymmetric oligomer intermediates.
Keywords: viral membrane fusion, intermediate, disulfide isomerase, BN-PAGE
Abstract
Viral membrane fusion proteins of class I are trimers in which the protomeric unit is a complex of a surface subunit (SU) and a fusion active transmembrane subunit (TM). Here we have studied how the protomeric units of Moloney murine leukemia virus envelope protein (Env) are activated in relation to each other, sequentially or simultaneously. We followed the isomerization of the SU-TM disulfide and subsequent SU release from Env with biochemical methods and found that this early activation step occurred sequentially in the three protomers, generating two asymmetric oligomer intermediates according to the scheme (SU-TM)3 → (SU-TM)2TM → (SU-TM)TM2 → TM3. This was the case both when activation was triggered in vitro by depleting stabilizing Ca2+ from solubilized Env and when viral Env was receptor triggered on rat XC cells. In the latter case, the activation reaction was too fast for direct observation of the intermediates, but they could be caught by alkylation of the isomerization active thiol.
The viral class I membrane fusion proteins, such as those of the influenza, retro, paramyxo, corona, and Ebola viruses, are typically trimeric transmembrane proteins, where the protomeric unit is composed of a transmembrane subunit (TM) and a peripheral subunit. The former carries the membrane fusion activity, and in most viruses, the latter controls this activity, so that it is triggered at the correct virus entry site in the target cell. Structural analyses of the pre- and postfusion forms of several fusion proteins have revealed a common mechanism of how TM works (1–12). In their prefusion state, part of the polypeptide of the three TM subunits form three central helixes, around which the rest of the fusion protein is organized. On activation, the helices are extended toward the N terminus of TM to form a long coiled coil, which presents the fusion peptide to the cell membrane. The membrane-proximal C-terminal parts of TM are then zippered onto the coiled coil in an antiparallel orientation to form a trimer of TM hairpins. As a result, the viral and the cell membranes are forced together and fuse. TM activation is prevented by its interactions with the peripheral subunit in the native form of the fusion protein. These keep the TM subunits in a metastable conformation, ready to seek their stable trimer-of-hairpin conformation as soon as the peripheral proteins are dissociated by receptor binding and/or low pH (13). An interesting question about the viral fusion protein activation mechanism that has remained unanswered is how much the activation of the three protomeric units depends on each other. Although the coiled coil formation of the TM subunits should be a simultaneous coordinated event, the other steps of the viral fusion protein activation process, before and after coil formation, could be sequential, occurring independently in the three protomeric units. Here we have studied this question using the Moloney murine leukemia retrovirus (Mo-MLV) as a model.
The Mo-MLV fusion protein is called Env (envelope protein). It binds to its receptor (murine cationic amino acid transporter 1) on the cell surface by the N-terminal domain (the receptor binding domain, RBD) of the peripheral or surface subunit (SU) (14–16). The RBD then transmits an activation signal to the TM via the C-terminal SU domain (17, 18). Interestingly, in all type C retroviruses, such as Mo-MLV, the C-terminal domain of SU is linked to TM by a disulfide bond that is rearranged during activation (19–21). The SU Cys residue of the disulfide is part of a disulfide isomerization motif, CXXC, where the other Cys is free. This becomes deprotonated during Env activation and rearranges the intersubunit disulfide to a disulfide isomer within the motif instead. As a result, the SU dissociates, and TM is activated for membrane fusion. If a membrane-impermeable alkylator such as 4-(N-maleimido)benzyl-α-trimethylammonium iodide (MBTA) is present during activation, it will block the free thiol of the CXXC motif before this can attack the intersubunit disulfide and halt the Env activation process in an isomerization-arrested state (21). However, activation can be resumed and the fusion rescued by cleaving the intersubunit disulfide artificially with DTT. It has also been shown that Ca2+ stabilizes the native form of Env, and that its depletion facilitates Env activation on cells. Indeed, it is possible to activate Env in vitro, without the receptor, by Ca2+ depletion, and to generate the alkylation-arrested form (21). These unique features of Env promoted the elucidation of the initial stages of the Env activation process structurally by cryo-EM (22). Analysis of Triton X-100-solubilized native Env showed that the protomeric unit was made up of three protrusions. At the top, there was a bent finger-like protrusion, the tip of which was directed toward the threefold axis, forming interactions with the other protomers. This protrusion corresponded to the RBD, the atomic structure of which had been solved earlier (23). The C-terminal domain of SU, harboring the CXXC motif, formed a lateral protrusion, and the TM a separated Env leg. On activation in vitro to the alkylation-arrested form, there was a clockwise rotation of the RBDs, moving the tips outward. This opened the roof of an apparent central cavity of Env. Most likely, however, the cavity was not real, but the space contained the central triple helix with the fusion peptide, which is typical for class I viral membrane fusion proteins. These structures were probably not resolved in the 18-Å cryo-EM map (22, 24).
In the present study, we have analyzed how the isomerization of the intersubunit disulfide and the subsequent SU release proceed in the protomers of Env: simultaneously in all three, or sequentially in one after the other. Env was triggered both by Ca2+ depletion in vitro and by the receptor on rat XC cells. The dissociation of the Env trimer into free SU and TM trimers was monitored by blue native (BN) PAGE and by ultracentrifugation in 3-(1-pyridinio)-1-propanesulfonate (NDSB-201) density gradients. Using both triggering conditions, we found that this early step of Env activation occurred sequentially in the Env protomers, generating two asymmetric oligomer intermediates.
Results
Env Activation in Vitro.
We first used BN-PAGE analyses to study the time course of Env protomer activation in vitro by Ca2+ depletion from viral Envs during solubilization. If the protomers were activated in a sequential fashion, one would expect a stepwise reduction in the size of the Env oligomer. If, in contrast, the protomers were simultaneously activated, intermediate forms were not expected to appear, only SU monomers and TM trimers. To this end, [35S] Cys-labeled Mo-MLV was solubilized with TX-100 in the presence of 10 mM EDTA and incubated for 0–30 min at 29 °C before analyses by 3–14% (wt/vol) BN-PAGE, as well as 13% (wt/vol) nonreducing SDS/PAGE. At the end of each incubation, before PAGE analyses, 10 mM N-ethyl maleimide (NEM) was added. This effectively prevented any disulfide isomerization after the 29 °C incubation. The SDS/PAGE revealed the isomerization status of the intersubunit disulfide of the SU-TM protomers (Fig. 1A), and the BN-PAGE their oligomeric form or forms (Fig. 1B). In the nonincubated sample, almost all protomeric subunits were covalently linked as SU-TM complexes that were associated into Env trimers, (SU-TM)3 [molecular weight (MW) was about 240 kDa; Fig. 1 A and B, lane 1 and lane 1]. A minor amount of apparently dissociated trimers was also seen as Env monomers, (SU-TM) (Fig. 1B, lane 1). After 30-min incubation, most of the Env subunits had been released by disulfide isomerization into free SU and TM trimers, TM3 (Fig. 1 A and B, lane 7 and lane 7). Most interestingly, the intermediate incubation times showed that the gradual increase of SU-TM disulfide isomerization (Fig. 1A, lanes 2–6) was correlated with a stepwise reduction of the size of the Env complex (Fig. 1B, lanes 2–6) forming two intermediates, I-1 and I-2, before reaching the completely isomerized stage. The precursor/product relationships were confirmed by quantification of the Env-related bands after normalization to the amounts of internal viral proteins (capsid, CA; matrix, MA; and nucleocapsid, NC) in the front band (Fig. 1 C and C′). This result was consistent with a sequential protomer activation model in which the protomers are isomerized and lose their SU one after each other, according to the scheme (SU-TM)3, → (SU-TM)2TM → (SU-TM)TM2 → TM3. However, to demonstrate it, we had to determine the subunit compositions of the intermediate complexes. In particular, we had to show that the intermediates contain noncovalently linked TM.
Fig. 1.

Activation of the Mo-MLV Env in vitro. [35S] Cys-labeled Mo-MLV was solubilized with TX-100 in the presence of 10 mM EDTA, incubated for 0–30 min at 29 °C, and after addition of 10 mM NEM, analyzed by nonreducing SDS/PAGE (A) and BN-PAGE (B). Shown are phosphorimages of the dried gels. The viral proteins or protein complexes are indicated to the right and the migration of standard proteins to the left in each panel. (C and C′) show the quantifications of Env-related proteins and protein complexes in B (after normalization to internal proteins in the front band), with C′ to highlight the less abundant forms. Note the sequential appearance and disappearance of Env trimers, I-1, I-2, SU, and TM3.
To this end, we separated the intermediates by ultracentrifugation and analyzed their content of SU-TM complexes and possible noncovalently associated TM by nonreducing SDS/PAGE. To increase the TM signal, we switched to [35S] Met/Cys-labeled Mo-MLV instead of purely [35S] Cys-labeled virus (Fig. S1). Thus, the double-labeled virus was activated for 15 min at 29 °C during solubilization or left unactivated and then subjected to ultracentrifugation in a 0.4–1.2-M NDSB-201 gradient. BN-PAGE analyses of the fractionated gradient with the activated sample showed that the Env trimers, the I-1 and the I-2 complexes, were partially separated from each other, and that the free SU migrated only slightly slower than the I-2 in the gradient (Fig. 2C). The nonreducing SDS/PAGE indicated that the Env trimers contained only SU-TM complexes, whereas the I-1 and I-2 complexes clearly also contained TM that was noncovalently associated (Fig. 2D, fractions 8–14). Additional TM subunits were present in the upper part of the gradient, apparently as trimers and monomers derived from completely isomerized Env. A quantification of the TM in Fig. 2D and the I-1 and I-2 complexes in Fig. 2C is shown in Fig. 2E. The BN-PAGE analyses of the gradient fractions with the nonactivated sample showed Env mostly as trimers with a minor amount of Env monomers (Fig. 2A). The latter migrated between the I-2 complex and the SU in the activated sample (Fig. S2). Faint bands corresponding to I-1 were also present in the nonactivated sample (Fig. 2A). Corresponding SDS/PAGE analyses showed the SU-TM content of the nonactivated Env trimers, but the absence of noncovalently associated TM (Fig. 2B). Among the internal viral proteins, the CA was found in the upper half of the gradients, apparently as oligomers, whereas the p12 protein migrated with the NC protein only slightly into the gradient, possibly as monomers (Fig. 2 B and D). Interestingly, p12 oligomerized at BN-PAGE conditions and migrated as a diffuse band close to I-2 and SU (Figs. 2 A and C and 3A).
Fig. S1.
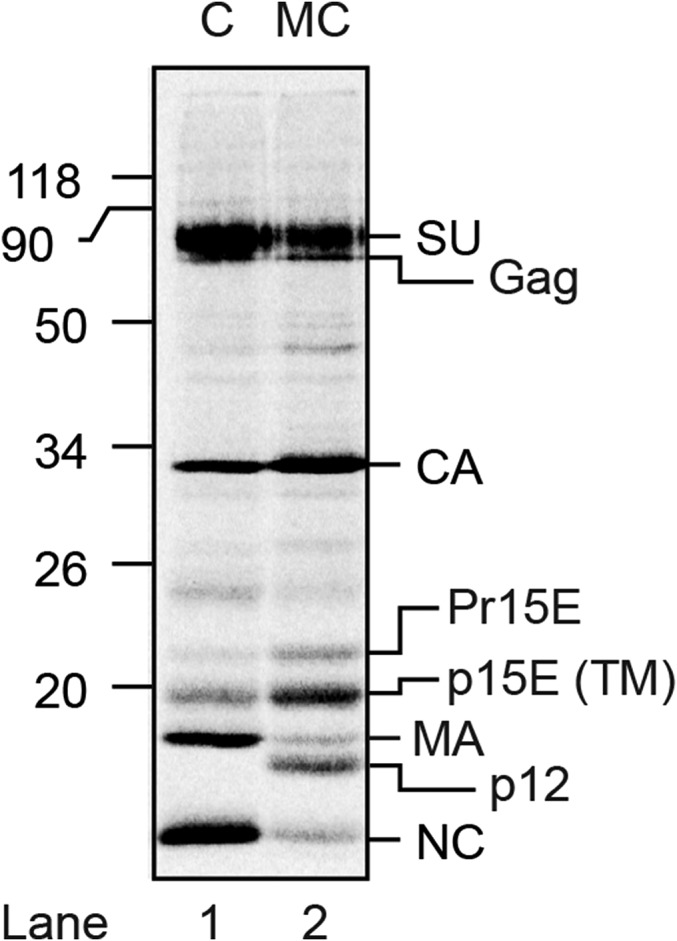
Reducing SDS/PAGE analyses of the proteins in [35S] Cys-labeled (C) and [35S] Met/Cys-labeled (MC) Mo-MLV, respectively. Shown is a phosphorimage of the dried gel. Viral proteins are indicated to the right, and the marker proteins to the left. Gag indicates the uncleaved internal viral precursor protein. Partial Gag cleavage products are also present. Pr15E indicates TM with uncleaved R-peptide (34). Note the more intense labeling of TM in the double-labeled virus. Note also the absence of p12 labeling in the single-labeled virus.
Fig. 2.

Separation of in vitro activated Env intermediates by velocity sedimentation and analyses of their composition. [35S] Met/Cys-labeled Mo-MLV was activated during solubilization for 15 min at 29 °C or left nonactivated and then subjected to ultracentrifugation in a 0.4–1.2-M NDSB-201 gradient. Gradient fractions were analyzed for viral proteins by BN-PAGE (A and C) and nonreducing SDS/PAGE (B and D). A and B show the nonactivated and C and D the activated sample. E shows quantifications of the I-1 and I-2 in C and the TM in D. Note the noncovalent association of TM with the intermediate complexes (fractions 8–14) in addition to TM in apparent trimers (fractions 16–18) and as monomers.
Fig. S2.

Close-by comparison of the migration of proteins and protein complexes of nonactivated and in vitro activated [35S] Met/Cys-labeled Mo-MLV in BN-PAGE. (A) BN-PAGE analyses of fractions 8–15 from the sedimentation of solubilized nonactivated virus shown in Fig. 2A, and B the corresponding analyses from the in vitro activated virus shown in Fig. 2C. Note that the Env monomers (SU-TM) from the nonactivated virus migrate in between the I-2 and SU from the activated virus.
Fig. 3.

2D BN/SDS/PAGE analysis of the compositions of the in vitro activated Env intermediates. [35S] Met/Cys-labeled Mo-MLV was activated as described in Fig. 2 and subjected to BN-PAGE in a first dimension and then to nonreducing SDS/PAGE in a second dimension (A). In the marker lane, to the left, a portion of the sample has been analyzed directly by SDS/PAGE (1D). This provides markers for the migration of the viral proteins in the 2D analysis. The migrations of the viral proteins are indicated to the right, and those of the Env related oligomers in the first dimension BN-PAGE are indicated at the top. The directions of the 2D electrophoreses are indicated. (B) Enlarged cutouts of the SU-TM and TM regions (dotted rectangles) of the 2D gel, at higher contrast. Note the alignment of the two leftmost TM spots with the SU-TM spots of I-1 and I-2. (C) Quantification of the stoichiometric ratio between SU-TM complexes and noncovalently associated TM in I-1 and I-2.
Another way to analyze the composition of the I-1 and I-2 complexes was by 2D BN/SDS/PAGE. In this PAGE system, the solubilized virus sample is first run on a 3–14% (wt/vol) BN-PAGE, the corresponding gel slice cut out, and the contained proteins run in a second dimension in an orthogonal direction on a 13% (wt/vol) nonreducing SDS/PAGE. The SDS/PAGE will dissociate any noncovalent protein–protein interactions of the complexes present in the protein bands of the BN-PAGE gel slice and then separate the proteins. These will arrange as faster and slower migrating spots to the left of a curved front of protein molecules, which have not dissociated. The curved nature of the front is a consequence of the acrylamide gradient in the BN-PAGE gel. Importantly, proteins of dissociated complexes will form spots below each other. Thus, if TM is noncovalently associated with I-1 or I-2, each of these will appear as a spot of SU-TM with a spot of TM straight below (but at the bottom part of the gel) in the 2D analysis. Therefore, a [35S] Met/Cys-labeled virus sample was activated for 15 min at 29 °C and then analyzed by 2D BN/SDS/PAGE (Fig. 3A). A direct SDS/PAGE analysis of the sample was run next to the 2D analysis to provide markers for the viral proteins and their covalent complexes in the 2D gel. The Env trimers, the I-1 and the I-2 complexes, and the Env monomers generated a row of four SU-TM spots from left to right when they moved out from the BN-PAGE into the upper part of the SDS/PAGE in the 2D analysis. Some of the SU-TM complexes of the Env trimers had apparently not become completely dissociated and were left behind as a top spot. Below and to the right of the row of SU-TM spots was the SU spot of completely isomerized protomers. Further down and to the right we identified the CA spot, which smeared considerably to the left, indicating oligomerization. The TM subunits, still lower down in the 2D analysis, formed a row of four spots. The two spots on the right side must represent TM subunits from TM trimer and monomer populations, respectively, of completely isomerized Env (25). In contrast, the two spots on the left side must have a different origin. Remarkably, they can be aligned with the I-1 and I-2 complexes (Fig. 3 A and B). This showed that the intermediate complexes contained noncovalently associated TM, and thus confirmed the results from the gradient analyses described earlier. The possibility that these TM spots were formed by homo oligomerization of TM trimers was ruled out by the absence of these TM spots in 2D analyses of cell receptor-activated Envs that had been completely isomerized (Fig. 5A, bottom cut out). Below the row of TM spots in Fig. 3A, we found the spots of apparent MA monomers, p12 oligomers, and NC monomers.
Fig. 5.

Time-resolved ratio of receptor-activated and MBTA-alkylated Env intermediates. XC cells were spinoculated with [35S] Met/Cys-labeled virus in the cold, shifted to 29 °C, and first incubated for 0–13 min without alkylator (−MBTA) and then in the presence of 0.4 mM MBTA for 0 or 5 min (+MBTA) as indicated, solubilized in the presence of EDTA and NEM, and the virus/cell samples analyzed on 2D BN/SDS/PAGE. (A) Stack of horizontal cutouts from the 2D gels, covering the entire TM region. The positions of I-1, I-2, and TM3, in the first dimension BN-PAGE are indicated at the top. In the cutouts, the noncovalently associated TM of I-1 and I-2 are seen as separate spots to the left of the TM trimer spot. Note the shift in their relative amounts with time of incubation before the alkylation pulse. To the left of the 2D analyses (2D) is TM from samples analyzed by direct SDS/PAGE in the marker lane (1D). (B) Mean relative amount of alkylated I-2, as percentage of the sum of alkylated I-1 and I-2, in samples that have been alkylation pulsed for 5 min at the beginning (after 0 min), in the middle (after 4 min), and at the end (after 8 min) of an activation incubation (± SD; n = 5).
The concentration of the protein radioactivity to defined spots in the 2D analysis facilitated calculation of the stoichiometric ratio of noncovalently associated TM to the SU-TM complexes in the two Env intermediates. We found that I-2 contained about 2 noncovalently associated TM per one SU-TM, and that I-1 contained less than one such TM per two SU-TM (Fig. 3C). The lower-than-expected noncovalent TM content of I-1, according to the sequential activation model, can at least partly be a result of contamination of the I-1 SU-TM spot with that of intact trimers (Fig. 3 A and B). Thus, these results showed that during an in vitro activation of Env, the intersubunit disulfide is isomerized and SU released sequentially from one protomer after the other. An important question was then whether this was also the case during receptor-mediated activation on cells.
Receptor-Triggered Env Activation.
We analyzed Env activation of [35S] Cys-labeled Mo-MLV bound to receptor-positive XC cells in a time course experiment. The virus was spinoculated onto the cells in the cold and incubated for 0–60 min at 24 °C before NEM was added and the samples were solubilized and analyzed by SDS/PAGE and BN-PAGE. Although the nonreducing SDS/PAGE showed that most of the Env protomers had been activated by receptor binding and had isomerized their intersubunit disulfide during the course of the incubation, we could not detect any clear indication of intermediate Env bands in the BN-PAGE, only a gradual increase of uncoupled SU and TM (Fig. S3 A and B). We also tried incubations at 18 °C, 29 °C, and 37 °C, but with the same result. The reason could be that the receptor-activated Envs spent a much shorter time at their intermediate stages than when activated by Ca2+ depletion. Alternatively, the receptor induced all three protomers of Env to isomerize their intersubunit disulfide and release their SU simultaneously, in contrast to what was found in vitro.
Fig. S3.

Nonreducing SDS/PAGE and BN-PAGE analyses of receptor-activated virus. [35S] Cys-labeled Mo-MLV was spinoculated in the cold onto XC cells and incubated at 24 °C for the indicated times. The virus/cell samples were solubilized in the presence of EDTA and NEM and analyzed by nonreducing SDS/PAGE (A) and BN-PAGE (B). Shown are phosphorimages of the dried gels. Viral proteins, protein complexes, and protein standards are indicated to the right and to the left, respectively, in each panel. Note that in this experiment, we have used the [35S] Cys-labeled Mo-MLV, which does not show p12.
To find out, we tried to capture the potential Env activation intermediates on XC cells by alkylation during the incubation (21). This should be possible if the alkylation of the isomerization active thiol in the Env protomers was performed at unsaturating conditions that still allowed partial Env activation and intermediate formation. In this experiment, [35S] Met/Cys-labeled Mo-MLV was bound to XC cells and incubated at 29 °C for 0–13 min in the presence of 0.4 mM of the membrane-impermeable alkylation agent MBTA. This alkylator we used before at higher concentrations to block disulfide isomerization in Env and subsequent membrane fusion (21). The cell samples were then solubilized in the presence of 10 mM EDTA and 10 mM NEM; that is, at conditions where all nonactivated Env protomers, whether in receptor-induced intermediates or untriggered intact Envs, were efficiently isomerization arrested by alkylation. The nonreducing SDS/PAGE analyses showed that isomerization of the SU-TM complexes into SU and TM still took place in the presence of 0.4 mM MBTA (Fig. 4A, lanes 1–5), but less efficiently compared with a sample without the alkylator (Fig. 4A, lane 6). Extending the incubation beyond 13 min did not increase the efficiency. Importantly, the BN-PAGE analysis showed that at these conditions, it was possible to catch apparent I-1 and I-2 intermediates (Fig. 4B). Corresponding bands were clearly identifiable in increasing strength with the incubation. Their ratio appeared constant, which was expected if the Envs are occasionally triggered by the receptor to undergo sequential, but rapid, protomer activation. The SU and the TM trimers also accumulated during the incubation, whereas the Env trimers decreased (Fig. 4 A and B). It should be noted that the Env trimer band contained both the receptor-triggered Env that was alkylation-arrested by MBTA during the incubation and the nontriggered Env, which was NEM-alkylated during solubilization. When different concentrations of MBTA were tested, 0.4 mM was found to be optimal to get a good yield of both intermediates (Fig. 6). Alkylation done at 37 °C also revealed the intermediates. However, for subsequent experiments, we chose 29 °C because samples were easier to process at these conditions.
Fig. 4.

Alkylation capture of receptor-activated Mo-MLV Env intermediates. XC cells were spinoculated with [35S] Met/Cys-labeled Mo-MLV in the cold, incubated with 0.4 mM MBTA for 0–13 min at 29 °C, and solubilized with TX-100 in the presence of EDTA and NEM. The virus/cell samples were analyzed by nonreducing SDS/PAGE (A) and BN-PAGE (B).
Fig. 6.

Reduction of the intersubunit disulfide in alkylation-arrested Env intermediates. [35S] Met/Cys-labeled Mo-MLV was spinoculated in the cold onto XC and DF-1 cells and incubated for 13 min at 29 °C without or with MBTA at the indicated concentrations. A second incubation was done in PBS without or with 20 mM DTT. The virus/cell samples were solubilized with TX-100 in the presence of EDTA and NEM and analyzed by SDS/PAGE (A) and BN-PAGE (B).
The identities of the putative intermediates were confirmed by velocity sedimentation in an NDSB-201 gradient and 2D SDS/BN-PAGE analyses of receptor-activated but alkylation-arrested virus samples. The BN-PAGE analyses of the gradient fractions showed that the sedimentation separated I-1 and I-2 similar to what was found for the in vitro-activated virus (Fig. S4C), and the corresponding nonreducing SDS/PAGE analyses they contained noncovalently associated TM (Fig. S4D), as opposed to the nonactivated virus (Fig. S4 A and B). The 2D gel analyses also showed a noncovalent association of TM with I-1 and I-2 (Fig. S5). The possibility that the I-1 and I-2 were generated nonspecifically was tested by comparing the effect of incubating virus bound to the XC cells in the presence of alkylator to that of virus bound to the receptor-negative chicken DF-1 cells. BN-PAGE analyses revealed the I-1 and I-2 in the virus/XC cell sample, but not in the virus/DF-1 cell sample (Fig. 6). Thus, these results also were consistent with the appearance of the intermediates during the cell receptor–triggered activation process.
Fig. S4.

Separation of receptor-activated and MBTA-alkylated Env intermediates by velocity sedimentation and analyses of their compositions. XC cells were spinoculated with [35S] Met/Cys labeled virus in the cold and left inactivated (A and B) or activated by incubation for 13 min at 29 °C with 0.4 mM MBTA (C and D). Both samples were solubilized in the presence of EDTA and NEM and subjected to ultracentrifugation in 0.4–1.2-M NDSB-201 gradients. Fractions were analyzed by BN-PAGE (A and C) and nonreducing SDS/PAGE (B and D). Shown are phosphorimages of the dried gels. The viral proteins and protein complexes are indicated.
Fig. S5.

2D-BN/SDS/PAGE analyses of the compositions of receptor-activated and MBTA-alkylated Env intermediates. [35S] Met/Cys-labeled Mo-MLV was spinoculated onto XC cells in the cold, shifted to 29 °C, and incubated for 5 min, and then subjected to 2D BN/SDS/PAGE (A). To the left, a sample has been analyzed directly by SDS/PAGE (1D). The migrations of viral proteins are indicated to the right, and those of the Env-related oligomers in the first dimension BN-PAGE are indicated at the top. (B) Cutouts of the SU-TM and TM regions (dotted rectangles) of the 2D gel at higher contrast. Note the alignment of the two leftmost TM spots with the SU-TM spots of I-1 and I-2.
However, an incomplete alkylation of Env undergoing a simultaneous protomer-activation could also yield I-1 and I-2. To investigate this possibility, we followed the formation of the alkylated intermediate Env forms by giving MBTA pulses at different times during an activation incubation of virus with XC cells. We anticipated that a sequential protomer activation process should yield more I-1 relative to I-2 at early points of the incubation than at later ones. This difference could be undetectable with a continuous alkylation protocol, as used earlier, but revealed with MBTA pulses. In contrast, no such shift in intermediate ratio was expected if the protomers were simultaneously activated. To this end, cell-bound virus samples were shifted to 29 °C and either incubated directly for 5 min with MBTA or first incubated without alkylator for 2, 4, 6, or 8 min and then pulsed for 5 min with MBTA. All samples were solubilized in the presence of EDTA and NEM and checked by BN-PAGE. Rather than trying to relate the weak BN-PAGE bands of intermediates that had been alkylated for only 5 min, we used the 2D BN/SDS/PAGE analyses for this purpose. In these gels, the spots of TM that were noncovalently associated in I-1 or I-2 were clearly identified and gave an indication of the intermediate ratio (Fig. 5A). The ratio of I-1- and I-2-derived TM changed from about equal amounts in the early alkylation-pulsed samples to predominantly I-2-derived TM in the later pulsed ones, suggesting a sequential protomer activation. To confirm this, we performed several experiments with 5-min alkylation pulses at the early, middle, or late phase, respectively, of the activation incubation. The mean relative amount of I-2 was calculated from the TM spots in the 2D gels, considering that I-1 contained one and I-2 two noncovalently associated TM subunits. The amount of I-2 was given as percentage of I-1 + I-2 (±SD) and found to increase from 32%, when pulsed early (after 0 min); to 44%, when pulsed in the middle (after 4 min); and to 52%, when pulsed late (after 8 min) (Fig. 5B). The early and late values were compared in a paired one-tailed t test with unequal variance, and the result suggested the increase was significant (P = 0.01).
It remained to be demonstrated that I-1 and I-2 were necessary intermediates in the activation process of the Env. We showed before that the alkylated and isomerization-arrested Env is fusion inhibited, but that the activity could be rescued by reducing the intersubunit disulfide with DTT (21). If the fusion activity was mediated by the I-1 and I-2, we expected that the rescued activity in the above experiment should be linked to a reduction of the intersubunit disulfide bond in the alkylated Env intermediates. To study this possibility, we bound virus to XC cells and incubated them at 29 °C for 13 min in the presence of 0, 0.4, 0.8, or 1.6 mM MBTA before a second incubation for 10 min with or without 20 mM DTT. Corresponding incubations were done with DF-1 cells as controls. All samples were solubilized in the presence of NEM and analyzed by nonreducing SDS/PAGE and BN-PAGE, as before. In the virus/XC cell samples without DTT, we observed the alkylated Env trimers, alkylated I-1 and I-2, free SU, and TM trimers (Fig. 6B, lanes 5, 9, and 13). This ladder of Env-related bands shifted to predominantly I-1 and Env trimers at higher MBTA concentrations. On the receptor-negative DF-1 cells, there was no Env activation, and hence neither I-1 nor I-2 SU or TM trimers were formed (Fig. 6 A and B, lanes 3, 7, 11, and 15). Significantly, the DTT treatment changed the Env band ladders of the XC cell-activated virus, which was obtained at all MBTA concentrations, to that of more I-2 and free TM trimers (Fig. 6B, lanes 6, 10, and 14). The increase of free TM was also seen in the nonreducing SDS/PAGE analyses (Fig. 6A, lanes 5, 6, 9, 10, 13, and 14). This suggested that the DTT treatment reduced the intersubunit disulfide in the alkylation-arrested forms of I-1, I-2, and trimeric Env generating new, later-stage intermediates and TM trimers. It has been shown earlier that DTT treatment does not reduce Envs without receptor triggering (21). This is also shown by the lack of effect of the DTT treatment of Env in virus incubated with DF-1 cells (Fig. 6 A and B, lanes 3, 4, 7, 8, 11, 12, 15, and 16) and of the nontriggered Env fraction of virus incubated without MBTA on XC cells (Fig. 6 A and B, lanes 1 and 2). Thus, these results support the notion that the I-1 and I-2 represent true intermediates of the spike activation process.
Discussion
Here we have studied how an early activation step of Mo-MLV Env proceeds in its three protomeric units: simultaneously in all of them or sequentially in one after the other. We followed the isomerization of the intersubunit disulfide and the subsequent SU release in Env that was triggered in vitro by Ca2+ depletion or in vivo by the receptor on rat XC cells. Our results suggested a sequential activation of the protomers according to the scheme (SU-TM)3 → (SU-TM)2TM → (SU-TM)TM2 → TM3. Thus, in this reaction, the protomers of Env release their SU one after the other, forming asymmetric oligomer intermediates (I-1 and I-2). In contrast, the TM subunits of the isomerized protomers stay noncovalently associated with the partially activated Env. At present, we cannot conclude what controls the sequential protomer activation. One possibility is that it is controlled through sequential receptor interactions. However, a single receptor–protomer interaction might also be sufficient. According to this model, Mo-MLV Env has dynamic features, reversibly exposing structures that are normally hidden in its inactivated form (e.g., the fusion peptide). This has been shown to be the case with the influenza hemagglutinin and the HIV Env (26, 27). The fusion peptide of Mo-MLV could flicker between a hidden and an exposed position, in an uncoordinated manner, in the three Env protomers. When this happens in a protomer that has bound to a receptor, the fusion peptide of this protomer could interact with the cell membrane and become locked in the membrane-embedded position. This might induce isomerization of the intersubunit disulfide and the subsequent release of SU and its bound receptor from Env. However, the resulting Env intermediate will remain membrane bound by virtue of the activated TM subunit, which stays noncovalently associated with Env. This provides the opportunity for the TM subunit of the second protomer to insert its fusion peptide in the cell membrane, isomerize its intersubunit disulfide, and release its SU. Finally, the TM of the third protomer will complete this sequential activation step and initiate coil formation, together with the other TM subunits. Thus, in this model, the sequential feature of the activation process is determined by the independent, uncoordinated, and reversible exposure dynamics of the fusion peptides in the three protomeric units of Env.
This model could also explain why the activation reaction was so much slower in vitro than in vivo. In the in vitro reaction, the fusion peptide cannot be locked in the target membrane and facilitate Env activation in that way. Instead, activation is induced by Ca2+ depletion. It is possible the Ca2+ stabilizes an Env conformation with hidden fusion peptide, and its removal allows fusion peptide exposure and Env activation, but less efficiently.
Earlier, we used single-particle cryo-EM to solve the structures of solubilized native Mo-MLV Env and its Ca2+ depletion-activated, but alkylation-arrested, trimeric form, both at 18 Å resolution (22). We found that the bent finger-like RBD protrusions at the top of Env rotated outward on activation, opening the complex up suggestively for fusion peptide exposure. In view of the present results about sequential protomer activation, the RBD rotation might in an unperturbed situation still control fusion peptide exposure, but occur reversibly in an uncoordinated fashion in the three protomeric units. However, when a fusion peptide has been locked in the cell membrane, the corresponding RBD will remain in the open position, and this protomer will proceed along its activation path. Thus, the all-RBD-open Env that we studied earlier would be generated only by alkylation arrest of the activation in all three protomers.
In this study, we focused on an early step of the Mo-MLV Env activation process. However, the sequential model might not be restricted to early activation steps only. It is possible that the zippering of the C-terminal TM helical and flanking regions onto the central coil also proceeds, if not sequentially, then at least in a less coordinated fashion. This is supported by energy and kinetics measurements of single HIV-1 gp41 trimer-of–hairpins using high-resolution optical tweezers and also by structural dynamics simulations of the conformational transitions occurring during activation of the influenza HA (28, 29).
In contrast, we have shown that the binding of single antigen binding fragment (Fab) of the broadly neutralizing Abs PG9 and PG16 to HIV-1 Env blocked the binding of sCD4 to all three protomers (30). Furthermore, the binding of one sCD4 or one CD4 binding site Ab, b12, Fab to one protomer facilitated the binding of the ligands to the other protomers (30). Clearly, these results support a coordinated, simultaneous activation model of the HIV-1 Env protomers. However, HIV-1 Env activation differs from that of Mo-MLV by initiating the activation process with a primary receptor step, (i.e., CD4 binding) and completing it with a coreceptor step (i.e., binding to CCR5 or CXCR4). It is possible that only the CD4 induced conformational changes (i.e., the bridging sheet formation and the relocations of the V1-V2 and V3 loops in gp120) are coordinated, not the changes mediated by the coreceptor before TM coil formation. These are possibly initiated by insertion of the fusion peptide of one protomer into the cell membrane and could proceed sequentially in the three protomers, equal to the process we here suggest for the Mo-MLV Env.
Materials and Methods
Metabolically labeled Mo-MLV was produced in 293T cells transfected with proviral DNA and isolated by sucrose gradient centrifugation (25, 31). Env was either activated in vitro by Ca2+ depletion during solubilization (10 mM EDTA and 0.3% Triton X-100) for 0–30 min at 29 °C or in vivo by receptor interaction on rat XC cells at 18 °C, 24 °C, 29 °C, or 37 °C for the indicated times. The activation was terminated by addition of NEM, which efficiently alkylated the isomerization active thiol close to the intersubunit disulfide. In some in vivo experiments, the membrane-impermeable alkylator MBTA was included during the activation incubation to capture intermediates, and in some, this was followed by DTT treatment to release intermediates before addition of NEM and Triton X-100. Samples were analyzed by velocity centrifugation in NDSB-201 gradients, nonreducing SDS/PAGE, BN-PAGE, or 2D BN/SDS/PAGE, as indicated. Methodological details are given in SI Materials and Methods.
SI Materials and Methods
Cells and Reagents.
293T, XC, and DF-1 cells were maintained as described (32, 33).
Production of Labeled Mo-MLV.
293T cells were transfected with 20 µg proviral DNA per 175 cm2 bottle, using Ca2+ phosphate precipitation (31). The cultures were labeled with either 0.2 mCi/mL Expre35s35s, [35S] Protein Labeling Mix (NEG072014MC), that is, [35S] Met/Cys, or 0.1 mCi/mL l-[35S] Cysteine (NEG022T005MC), that is, [35S] Cys, both from PerkinElmer, between 24 and 48 h after transfection. The labeling media, DMEM, contained 5 µM unlabeled Met and/or 10 µM unlabeled Cys and 5% (vol/vol) FBS. The supernatants were clarified by low-speed centrifugation, and the virus isolated by sedimentation in a sucrose step gradient, as described (25). Aliquots of the virus in HNC buffer (50 mM Hepes, 100 mM NaCl, 1.8 mM CaCl2 at pH 7.4) and ∼35% (wt/wt) sucrose were frozen in dry ice and stored at −80 °C. Each stock (∼1 mL/175 cm2 transfected 293T) was checked for R-peptide cleavage and SU-TM intersubunit disulfide bond integrity by reducing and nonreducing SDS/PAGE (34). Stocks used for receptor-mediated activation experiments were titrated for optimal activation on XC-cells by spinoculation followed by 13 min at 29 °C (see Receptor-Induced Activation and MBTA Capture of Intermediates).
In Vitro Activation.
Labeled and gradient-purified Mo-MLV was activated by Ca2+ depletion in HN buffer (50 mM Hepes, 100 mM NaCl at pH 7.4) containing 0.3% Triton X-100, 10 mM EDTA, 1 M 3-(1-Pyridinio)-1-propanesulfonate (NDSB-201), and 1% protease-inhibitor mixture for general use, both from Sigma-Aldrich, typically for 0–30 min at 29 °C. At the end of the incubation, the samples were transferred to ice, 10 mM NEM added, and the incubation continued for at least 10 min.
Receptor-Induced Activation and MBTA Capture of Intermediates.
Labeled and purified Mo-MLV was spinoculated at +4 °C (1 h at 1.3 × 103 × g, 2.9 krpm, in a Beckman JS 5.9 rotor) onto receptor carrying XC cells or receptor-negative DF-1 cells in 24-well dishes, in the presence of 0.8 µg/mL polybrene as described (25). The cells were washed twice in ice cold Dulbecco’s PBS with Ca2+ and Mg2+ (PBS), shifted to 29 °C, 350 µL growth medium without or to capture intermediates, with 0.4–1.6 mM of the membrane-impermeable alkylator MBTA (Toronto Research Chemicals Inc) added. The cells were then incubated for the indicated times and temperatures, typically 0–13 min at 29 °C. At the end of the incubation, the cells were washed twice in PBS containing 10 mM NEM and extracted in 50 µL HN buffer containing 0.3% Triton X-100, 10 mM EDTA, 1 M NDSB-201, 1% protease inhibitor mixture for general use, for at least 10 min on ice. Insoluble matter was removed from the virus/cell samples by low-speed centrifugation. In some experiments, the MBTA incubation was preceded by incubations in growth media without alkylator for 2, 4, 6, or 8 min at 29 °C.
DTT Release of Receptor-Induced, MBTA-Captured Intermediates.
Cells were spinoculated and incubated without and with MBTA for 13 min at 29 °C, as earlier, and then washed twice with 0.5 mL PBS containing 20 mM DTT, leaving the last wash on the cells up to a total time of 10 min at 29 °C. Finally, the cells were washed twice with 1 mL PBS containing 20 mM NEM and extracted with Triton X-100 in the presence of NEM and EDTA, as earlier.
Velocity Centrifugation.
Excess sucrose and NDSB-201 were removed from Triton X-100 extracts of virus or virus/cell samples in Zeba desalt spin columns (Pierce), and the samples layered on 0.4–1.2 M NDSB-201 gradients in HNC containing 0.05% Triton X-100 and sedimented for 16 h at 1.6 × 105 × g (36 krpm) at 4 °C in a Beckman SW 55 rotor. Gradients were fractionated from the bottom.
Gel Electrophoresis.
Samples were analyzed by nonreducing SDS/PAGE containing 13% (wt/vol) acrylamide with Pierce prestained protein MW marker 20–120 kDa (catalog number 26612, Thermo Fisher) and by BN-PAGE containing 3–14% (wt/vol) acrylamide gradients, as described, and HMW calibration kit for native electrophoresis, containing thyroglobulin, ferritin, catalase, lactate dehydrogenase, and BSA (catalog number 17-0445-01, VWR International) as markers (25). The cathode buffer contained 0.002% and 0.01% Serva Blue G for virus extracts and virus/cell extracts, respectively. 2D BN/SDS/PAGE was performed as described, with a few important changes (25). A sample was separated on BN-PAGE [3–14% (wt/vol) acrylamide], the middle part of the lane was cut out and directly mounted in an SDS/PAGE well that had been soaked in stacking gel buffer (67 mM Tris, 0.1% SDS at pH 6.8) supplemented with 0.1 M NEM for 2 h at +4 °C. The position of the gel strip was fixed by the addition of 1% agaros (melted and kept at 50 °C before pouring) in SDS/PAGE electrode buffer. The gels [13% (wt/vol) acrylamide, of which 2.6% (wt/vol) comprised bisacrylamide] were run at constant voltage for 20 min at 50 V, and then at 150 V until finished. All SDS gels were fixed in 10% (wt/vol) trichloroacetic acid and 40% (vol/vol) methanol. BN-PAGE gels were fixed in 50% (vol/vol) methanol and 7% (vol/vol) acetic acid and stained with Gel Code Blue staining reagent (Thermo Fisher) to visualize the marker proteins. All gels were dried and exposed to phopohorimage screens (BAS-2025; Fujifilm, Science Imaging Scandinavia).
Quantitations.
Labeled proteins were visualized and quantified using a Molecular Imager FX and the QuantityOne program (Bio-Rad Laboratories). To calculate the stoichiometric ratio of proteins, the radioactivity in individual protein spots were normalized to account for their Cys or Met/Cys content and, where applicable, also for the Met/Cys ratio used in the metabolic labeling. The SU-TM contains 7 Met and 24 Cys, whereas the TM contains 5 Met and 4 Cys. The Met/Cys labeling was carried out in a solution that contained 28.4 Ci/mmole Met and 4.35 Ci/mmole Cys at the activity date specified by PerkinElmer (i.e., 86.7% Met and 13.3% Cys). The resulting weighted radiolabel incorporation (=WRI) was calculated to 9.3 and 4.9 for SU-TM and TM, respectively, using the formula WRIprotein = Metprotein × 0.867 + Cysprotein × 0.133, where Metprotein and Cysprotein are the number of Met or Cys in the protein. Please note that the ratio of radiolabeled Met and Cys does not change with radioactive decay.
Acknowledgments
This work was supported by Swedish Science Foundation Grant 4621 (to H.G.) and Swedish Cancer Foundation Grant 0330 (to H.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1617264114/-/DCSupplemental.
References
- 1.Wilson IA, Skehel JJ, Wiley DC. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature. 1981;289(5796):366–373. doi: 10.1038/289366a0. [DOI] [PubMed] [Google Scholar]
- 2.Bullough PA, Hughson FM, Skehel JJ, Wiley DC. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature. 1994;371(6492):37–43. doi: 10.1038/371037a0. [DOI] [PubMed] [Google Scholar]
- 3.Pancera M, et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature. 2014;514(7523):455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387(6631):426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 5.McLellan JS, et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science. 2013;340(6136):1113–1117. doi: 10.1126/science.1234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McLellan JS, Yang Y, Graham BS, Kwong PD. Structure of respiratory syncytial virus fusion glycoprotein in the postfusion conformation reveals preservation of neutralizing epitopes. J Virol. 2011;85(15):7788–7796. doi: 10.1128/JVI.00555-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature. 2006;439(7072):38–44. doi: 10.1038/nature04322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin HS, Paterson RG, Wen X, Lamb RA, Jardetzky TS. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci USA. 2005;102(26):9288–9293. doi: 10.1073/pnas.0503989102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weissenhorn W, Carfí A, Lee KH, Skehel JJ, Wiley DC. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol Cell. 1998;2(5):605–616. doi: 10.1016/s1097-2765(00)80159-8. [DOI] [PubMed] [Google Scholar]
- 10.Lee JE, et al. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454(7201):177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walls AC, et al. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature. 2016;531(7592):114–117. doi: 10.1038/nature16988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Supekar VM, et al. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc Natl Acad Sci USA. 2004;101(52):17958–17963. doi: 10.1073/pnas.0406128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carr CM, Chaudhry C, Kim PS. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc Natl Acad Sci USA. 1997;94(26):14306–14313. doi: 10.1073/pnas.94.26.14306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albritton LM, Tseng L, Scadden D, Cunningham JM. A putative murine ecotropic retrovirus receptor gene encodes a multiple membrane-spanning protein and confers susceptibility to virus infection. Cell. 1989;57(4):659–666. doi: 10.1016/0092-8674(89)90134-7. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Kavanaugh MP, North RA, Kabat D. Cell-surface receptor for ecotropic murine retroviruses is a basic amino-acid transporter. Nature. 1991;352(6337):729–731. doi: 10.1038/352729a0. [DOI] [PubMed] [Google Scholar]
- 16.Davey RA, Hamson CA, Healey JJ, Cunningham JM. In vitro binding of purified murine ecotropic retrovirus envelope surface protein to its receptor, MCAT-1. J Virol. 1997;71(11):8096–8102. doi: 10.1128/jvi.71.11.8096-8102.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavillette D, Boson B, Russell SJ, Cosset FL. Activation of membrane fusion by murine leukemia viruses is controlled in cis or in trans by interactions between the receptor-binding domain and a conserved disulfide loop of the carboxy terminus of the surface glycoprotein. J Virol. 2001;75(8):3685–3695. doi: 10.1128/JVI.75.8.3685-3695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barnett AL, Davey RA, Cunningham JM. Modular organization of the Friend murine leukemia virus envelope protein underlies the mechanism of infection. Proc Natl Acad Sci USA. 2001;98(7):4113–4118. doi: 10.1073/pnas.071432398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Opstelten DJ, Wallin M, Garoff H. Moloney murine leukemia virus envelope protein subunits, gp70 and Pr15E, form a stable disulfide-linked complex. J Virol. 1998;72(8):6537–6545. doi: 10.1128/jvi.72.8.6537-6545.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinter A, Kopelman R, Li Z, Kayman SC, Sanders DA. Localization of the labile disulfide bond between SU and TM of the murine leukemia virus envelope protein complex to a highly conserved CWLC motif in SU that resembles the active-site sequence of thiol-disulfide exchange enzymes. J Virol. 1997;71(10):8073–8077. doi: 10.1128/jvi.71.10.8073-8077.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallin M, Ekström M, Garoff H. Isomerization of the intersubunit disulphide-bond in Env controls retrovirus fusion. EMBO J. 2004;23(1):54–65. doi: 10.1038/sj.emboj.7600012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu SR, et al. Turning of the receptor-binding domains opens up the murine leukaemia virus Env for membrane fusion. EMBO J. 2008;27(20):2799–2808. doi: 10.1038/emboj.2008.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fass D, Harrison SC, Kim PS. Retrovirus envelope domain at 1.7 angstrom resolution. Nat Struct Biol. 1996;3(5):465–469. doi: 10.1038/nsb0596-465. [DOI] [PubMed] [Google Scholar]
- 24.Bartesaghi A, Merk A, Borgnia MJ, Milne JL, Subramaniam S. Prefusion structure of trimeric HIV-1 envelope glycoprotein determined by cryo-electron microscopy. Nat Struct Mol Biol. 2013;20(12):1352–1357. doi: 10.1038/nsmb.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sjöberg M, Lindqvist B, Garoff H. Stabilization of TM trimer interactions during activation of moloney murine leukemia virus Env. J Virol. 2008;82(5):2358–2366. doi: 10.1128/JVI.01931-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leikina E, Ramos C, Markovic I, Zimmerberg J, Chernomordik LV. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 2002;21(21):5701–5710. doi: 10.1093/emboj/cdf559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Munro JB, et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science. 2014;346(6210):759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiao J, Rebane AA, Ma L, Gao Y, Zhang Y. Kinetically coupled folding of a single HIV-1 glycoprotein 41 complex in viral membrane fusion and inhibition. Proc Natl Acad Sci USA. 2015;112(22):E2855–E2864. doi: 10.1073/pnas.1424995112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin X, et al. Order and disorder control the functional rearrangement of influenza hemagglutinin. Proc Natl Acad Sci USA. 2014;111(33):12049–12054. doi: 10.1073/pnas.1412849111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Löving R, Sjöberg M, Wu S-R, Binley JM, Garoff H. Inhibition of the HIV-1 spike by single-PG9/16-antibody binding suggests a coordinated-activation model for its three protomeric units. J Virol. 2013;87(12):7000–7007. doi: 10.1128/JVI.00530-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Colicelli J, Goff SP. Sequence and spacing requirements of a retrovirus integration site. J Mol Biol. 1988;199(1):47–59. doi: 10.1016/0022-2836(88)90378-6. [DOI] [PubMed] [Google Scholar]
- 32.Löving R, Kronqvist M, Sjöberg M, Garoff H. Cooperative cleavage of the R peptide in the Env trimer of Moloney murine leukemia virus facilitates its maturation for fusion competence. J Virol. 2011;85(7):3262–3269. doi: 10.1128/JVI.02500-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li K, et al. The conserved His8 of the Moloney murine leukemia virus Env SU subunit directs the activity of the SU-TM disulphide bond isomerase. Virology. 2007;361(1):149–160. doi: 10.1016/j.virol.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 34.Rein A, Mirro J, Haynes JG, Ernst SM, Nagashima K. Function of the cytoplasmic domain of a retroviral transmembrane protein: p15E-p2E cleavage activates the membrane fusion capability of the murine leukemia virus Env protein. J Virol. 1994;68(3):1773–1781. doi: 10.1128/jvi.68.3.1773-1781.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]