

Significance
The twin-arginine translocation (Tat) system transports folded proteins across the prokaryotic inner membrane and the thylakoid membrane of plant chloroplasts. Proteins are targeted to the Tat system by signal peptides containing a highly conserved twin arginine motif. We isolated suppressors in the TatB component that allowed a Tat substrate with a defective twin arginine motif to be transported. The strongest of these suppressors, TatB F13Y, resulted in the constitutive assembly of the Tat translocase in the absence of signal peptide binding. These results show that Tat signal peptides have two separable roles: they target their passenger proteins to the Tat machinery but they also trigger the assembly of the active Tat transporter.
Keywords: protein transport, twin arginine signal peptide, Tat pathway, genetic suppressor
Abstract
The twin-arginine protein translocation (Tat) system mediates transport of folded proteins across the cytoplasmic membrane of bacteria and the thylakoid membrane of chloroplasts. The Tat system of Escherichia coli is made up of TatA, TatB, and TatC components. TatBC comprise the substrate receptor complex, and active Tat translocases are formed by the substrate-induced association of TatA oligomers with this receptor. Proteins are targeted to TatBC by signal peptides containing an essential pair of arginine residues. We isolated substitutions, locating to the transmembrane helix of TatB that restored transport activity to Tat signal peptides with inactivating twin arginine substitutions. A subset of these variants also suppressed inactivating substitutions in the signal peptide binding site on TatC. The suppressors did not function by restoring detectable signal peptide binding to the TatBC complex. Instead, site-specific cross-linking experiments indicate that the suppressor substitutions induce conformational change in the complex and movement of the TatB subunit. The TatB F13Y substitution was associated with the strongest suppressing activity, even allowing transport of a Tat substrate lacking a signal peptide. In vivo analysis using a TatA–YFP fusion showed that the TatB F13Y substitution resulted in signal peptide-independent assembly of the Tat translocase. We conclude that Tat signal peptides play roles in substrate targeting and in triggering assembly of the active translocase.
A large proportion of prokaryotic proteins are trafficked into or across the cytoplasmic membrane. Extracytoplasmic proteins are synthesized with cleavable N-terminal signal peptides to address them to export machineries located in the cytoplasmic membrane. Signal peptides are generally between 20 and 30 amino acids in length and have a recognizable tripartite structure comprising a basic n-region, a hydrophobic h-region, and a polar c-region with a signal peptidase cleavage site.
The Sec pathway is the major route of protein export in most prokaryotes, transporting unfolded polypeptides across the cytoplasmic membrane. In bacteria it is comprised of a SecYEG channel complex and a peripheral membrane ATPase, SecA (see refs. 1 and 2 for recent reviews). The initial discovery of Sec components was driven by genetic approaches using Escherichia coli to isolate suppressors of defective Sec substrate proteins and inactive signal peptides (3, 4). These genetic suppressors also contributed significantly to mechanistic understanding of Sec-dependent protein translocation (5–7). More recently, it has been shown that Sec signal peptides have dual roles: they serve to target their passenger proteins to the Sec machinery (3, 8) but also to allosterically activate the SecY channel (9).
The twin-arginine protein translocation (Tat) pathway operates in parallel to the Sec pathway to transport folded proteins across the prokaryotic cytoplasmic membrane and the thylakoid membrane of plant chloroplasts (10, 11). Proteins are targeted to the Tat pathway by N-terminal signal peptides that contain a conserved twin arginine motif at the n-region/h-region boundary (12, 13) (Fig. 1A). The Tat system in the model bacterium E. coli requires three membrane-bound subunits: TatA, TatB, and TatC (14–17). The TatB and TatC proteins form a multivalent complex that binds Tat substrates through their twin arginine signal peptides (18–20). Numerous experiments have shown that the TatC component recognizes the twin arginine motif (21–26), whereas TatB is close to the signal peptide h-region (27, 28). Signal peptides have been shown to penetrate deeply into the TatBC complex (29) and, in thylakoids at least, this deep-binding mode may be modulated by the transmembrane proton electrochemical gradient (PMF) (30).
Fig. 1.

Isolation of signal sequence suppressors in tatB. (A) Schematic representation of a twin arginine signal peptide. The signal peptide sequences of E. coli Tat substrates SufI and AmiA are given underneath, with residues matching the Tat consensus motif in red, the consecutive arginines in red underline, and the signal peptidase cleavage site in black underline. (B and C) An example of screening results. Growth of MCDSSAC ΔtatABC coproducing the indicated TatB variants (with wild-type tatA and tatC) from pTAT101, alongside (B) the HH or (C) RE-substituted signal peptide variants of AmiA, on LB agar supplemented with chloramphenicol and kanamycin, with or without the addition of 2% (wt/vol) SDS as indicated. An 8-μL aliquot of each strain/plasmid combination following aerobic growth to an OD600 of 1.0 was spotted and incubated for 16 h at 37 °C.
It is generally accepted that the TatA protein forms the protein-conducting element of the Tat pathway. TatA oligomers assemble at the substrate-bound TatBC complex, dependent on the PMF (27, 31–35). Current models for Tat transport propose that TatA oligomers either provide form-fitting channels of varying diameter that adapt to the size of the folded passenger domain, or that oligomeric assemblies of TatA cause a localized weakening of the membrane and transient bilayer disruption accompanied by substrate transport (reviewed in refs. 10 and 11). An implicit prediction of the latter model is that transient membrane rupture would be expected to be accompanied by ion leakage.
In this study we have addressed the function of the twin-arginine signal peptide in the Tat transport process by isolating genetic suppressors that either restore transport to signal peptides harboring transport inactivating twin arginine substitutions, or that restore Tat activity to a TatC variant that has an inactive signal peptide binding site. Our results identified a common set of substitutions, primarily located in the transmembrane helix of TatB that can suppress both types of transport defect. Biochemical analysis of Tat translocases harboring these substitutions indicates that at least one of them, TatB F13Y, promotes signal peptide-independent TatA assembly. Our findings show that, like Sec signal peptides, Tat targeting sequences have two separable roles in the transport process.
Results
Single Amino Acid Substitutions in TatB Permit Export of Twin-Arginine Substituted Signal Peptides.
Previous genetic screens using maltose binding protein (21, 22) or GFP (23) fused to the Tat signal sequence of E. coli TorA identified mutations that were able to restore some level of Tat transport to fusions with export-defective twin arginine substitutions. These substitutions were located toward the N-terminal end of the TatB transmembrane helix or within the cytoplasmic loops of TatC. To shed light on potential functions of signal peptides during Tat transport, we initiated an independent genetic screen using a native E. coli Tat substrate, AmiA, as our reporter. AmiA is an N-acetylmuramoyl-l-alanine amidase that remodels the cell wall during growth. In the absence of a functional Tat system, the cell envelope is impaired because of the inability to correctly localize AmiA and the related Tat substrate AmiC, rendering E. coli sensitive to killing by SDS (36). Using a strain lacking chromosomal tat genes and amiA/amiC (37), coproduction of plasmid-encoded AmiA (from the medium copy construct pSUAmiA) (36) alongside a separate plasmid, pTAT1d (producing TatABC from a compatible medium copy plasmid) (38), permits the strain to grow on LB medium containing 2% (wt/vol) SDS (SI Appendix, Fig. S1). We then mutated the consecutive arginines in the Tat signal peptide of plasmid-encoded AmiA to each of the amino acid pairs RD, RE, RH, RN, RQ, KH, KQ, or HH. As expected, each of these substitutions abolished growth on SDS (SI Appendix, Fig. S1).
Next we screened a tatB mutant library, generated in pTAT1d by error-prone PCR, for clones that supported growth of strains carrying AmiA with variant signal peptides (38). Each AmiA twin arginine variant was challenged with this library, screening ∼10,000 clones for each construct. In total across the screening campaign we isolated 30 individual clones. Upon rescreening, 20 of the clones retained the ability to suppress the inactive signal peptide variant of AmiA against which they were originally isolated. These clones are listed in SI Appendix, Table S1. Substitutions appeared to cluster within the transmembrane region of TatB, including L9Q that appeared in seven of the clones and F13Y that occurred in five clones, whereas F6Y, E8K, L9P, and L10P were each found once. It should be noted that E8K, L9P, and L9Q substitutions have previously been identified as suppressors of inactive signal peptides (21, 22). We also found clones, each isolated twice, which contained no substitutions in the transmembrane domain, where the first substitution was in the amphipathic helix of TatB (clones BRQ1, BRQ2, BRQ3, and BRQ5).
We introduced the individual amino acid substitutions F6Y, E8K, L9P, L9Q, L10P, F13Y, K30I, and I36N into TatB encoded within the tatABC operon on the very low copy number plasmid pTAT101 (39) and tested their ability to support export of wild-type AmiA and to suppress each of the RD, RE, RH, RN, RQ, KH, KQ, or HH AmiA signal sequence variants (Fig. 1 B and C, Table 1, and SI Appendix, Fig. S2). Each of the substitutions was able to support transport of wild-type AmiA, but they varied in their ability to permit export of the AmiA signal sequence variants. For example, the F13Y variant of TatB supported growth on SDS for all of the signal peptide variants tested, whereas the L9Q, L10P, K30I, and I36N TatB substitutions could suppress some of the signal sequence variants, but not KH, KQ, and HH substitutions. To determine whether the suppression observed was specific for the AmiA signal peptide, we generated an additional construct where the signal peptide of the Tat substrate SufI was fused to the mature portion of AmiA and the same twin arginine substitutions were introduced (Table 1 and SI Appendix, Fig. S3). The TatB variants generally showed the same pattern of suppression of SufI signal peptide substitutions, although the TatBL10P, K30I, and I36N variants could not suppress RD or RE substitutions in the SufI signal peptide.
Table 1.
Summary of the results obtained from TatB suppressor analysis against the indicated twin-arginine substitutions in the AmiA and SufI signal peptides
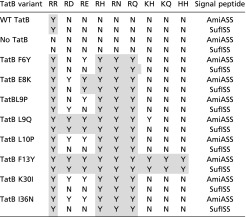 |
N indicates no export; Y indicates export. Gray shading indicates suppression of the equivalent substitutions in both signal peptides.
A Subset of TatB Signal Peptide Suppressors also Suppress TatC Signal Peptide Binding Site Mutants.
In a complementary approach, we asked whether it was possible to select suppressors of defects in the signal peptide binding site on TatC. Residue F94 in E. coli TatC is highly conserved and lies within the signal peptide binding site (25, 40) (Fig. 2A), and substitution to other amino acids is poorly tolerated (41). We constructed substitutions of F94 to each of the small neutral amino acids Ala and Gly, helix-breaking Pro, polar residues Ser and Gln, positively charged Arg and Lys, and negatively charged Asp. These substitutions were introduced into TatC encoded on both the medium copy plasmid pTAT1d and the very low copy plasmid pTAT101 that also carry wild-type tatA and tatB. SI Appendix, Fig. S4 shows that substitutions to Asp, Gln, or Pro resulted in a complete inability of strain DADE (ΔtatABCD, ΔtatE) (42) to grow in the presence of SDS at both medium and very low copy number. We selected the F94Q substitution of TatC and constructed three mutant libraries in the pTAT1d vector by error-prone PCR in an attempt to identify suppressors of this inactivating tatC mutation. LibC1 carried mutations in the first 93 codons of tatC, LibC2 carried mutations in tatC from residue 95 onwards, and LibAB contained mutations in the tatA and tatB genes.
Fig. 2.

Isolation of suppressors of the TatC F94Q inactivating substitution. (A) Model of E. coli TatC (from ref. 47) showing the location of the F94 and E103 residues (in red) that form part of the signal peptide binding site. (B, C, and E) Growth of DADE (ΔtatABCD, ΔtatE) coproducing wild-type TatA alongside; (B and C) F94Q-substituted TatC or (E) E103A-substituted TatC, and the indicated substitution in TatB from plasmid pTAT101 on LB agar or LB agar containing 2% (wt/vol) SDS. (D) Structure of E. coli TatB (from ref. 68) with the locations of the TatCF94Q suppressor substitutions shown in red. (F) Strain MC4100 ΔamiA ΔamiC ΔtatABC coproducing the indicated TatB variants (with wild-type tatA and tatC) from pTAT101, alongside either a signal peptide variant of SufI lacking the h-region fused to the AmiA mature domain (SufIssnoH-AmiA), or the mature AmiA domain alone (mAmiA) on LB agar or LB agar containing 1% SDS. For all growth tests, a single colony of each strain/plasmid combination was resuspended in 30 μL of PBS and an 8 μL aliquot was spotted onto LB agar supplemented with appropriate antibiotics, along with SDS as indicated and incubated for 16 h at 37 °C.
A number of suppressors were identified from screening the LibC1 and LibC2 libraries for growth on SDS plates. However, sequence analysis indicated that for each of these Tat active mutants there was substitution at the tatC F94Q codon to tatC F94Y, W or L codons. In contrast, after screening more than 180,000 clones from the LibAB library, 11 mutants were isolated that were able to rescue the growth defect of TatC F94Q on SDS plates, of which 5 were still able to support growth on SDS following fresh transformation of strain DADE with the isolated plasmid. These clones are listed in SI Appendix, Table S2. Interestingly, each of these suppressors encoded either TatB L10P, F13Y, or I36N, which had been identified in our prior screen for signal sequence suppressors. Introduction of each of these substitutions, individually, into the very low copy number pTAT101CF94Q plasmid supported growth of strain DADE on SDS-containing media (Fig. 2B), indicating that these TatB variants are each able to rescue the Tat-inactivating F94Q TatC substitution.
Because these three TatB substitutions that suppress the TatC F94Q defect were previously isolated as suppressors of inactive Tat signal sequences, we asked whether any of the other tatB signal sequence suppressors we had found could also rescue the TatC F94Q substitution. Fig. 2C shows that in addition to L10P, F13Y, and I36N, L9Q could restore Tat activity to cells producing TatC F94Q. The location of each these residues on a model of TatB is shown in Fig. 2D. We next asked the question whether any of these TatC F94Q suppressors could restore Tat activity to other inactivating substitutions in TatC. Fig. 2E shows that two different inactivating substitutions of E103 in the signal peptide binding site, either E103A or E103K (25, 40, 41) (Fig. 2A), could also be complemented by three of the four suppressors of tatCF94Q (I36N did not suppress these substitutions), but they could not restore Tat activity caused by inactivating TatC substitutions located outside the signal peptide binding site (SI Appendix, Fig. S5). Finally, we tested whether F94Q-suppressing substitutions were additive (i.e., whether when combined they resulted in a stronger suppressing activity). However, SI Appendix, Fig. S6A shows that none of the pairwise combinations we tested gave any suppression of tatCF94Q and, with the exception of the F13Y, I36N substitution that showed some suppression of the RN signal peptide variant, the combined suppressors lost suppressive function of RN or KK substitutions of the SufI signal peptide (SI Appendix, Fig. S6B). We therefore conclude that the suppressor mutations, when combined, have detrimental rather than additive effects on suppression activity.
The TatB F13Y and L9Q Substitutions Support Export of AmiA Lacking a Signal Peptide.
The results above indicate that the TatBF13Y substitution is the strongest suppressor of inactive Tat signal peptides, allowing the E. coli Tat system to recognize all eight of the different twin arginine motif substitutions tested, as well as suppressing the TatC F94Q mutation. We therefore tested whether more severe signal peptide defects could be suppressed by this TatB variant. Fig. 2F shows that, remarkably, even after truncation of the signal peptide by removal of the h-region, or indeed complete removal of the entire signal peptide-coding sequence of AmiA, we could still detect some Tat-dependent translocation of the AmiA passenger domain in the presence of TatBF13Y. The TatB L9Q substitution, which was the strongest suppressor of signal peptide defects after F13Y, also supported some translocation of mature AmiA; however, we were not able to detect export of mature AmiC in the presence of either of these two suppressor substitutions (SI Appendix, Fig. S6C). We conclude that TatB L9Q and F13Y allow at least one Tat substrate to be transported independently of any signal peptide.
Variant TatB Proteins Support Good Transport Activity of a Native Tat Substrate but Much Poorer Transport When the Signal Peptide Is Altered.
We next addressed whether the TatB substitutions were detrimental to the activity of the Tat system. Using overproduced His-tagged, but otherwise native SufI as a substrate, it was seen that mature SufI was clearly detected in the periplasmic fractions of all of the strains tested, although TatBI36N seemed to support only low levels of transport (SI Appendix, Fig. S7A). However, we were unable to detect transport of the RD, RN, or KQ signal peptide variants of SufI in the presence of TatBF13Y (SI Appendix, Fig. S7B) or of a twin-lysine substituted His-tagged CueO in the presence of TatBE8K or F13Y (Fig. 3). It therefore appears that there is very low export efficiency of substrates with variant signal peptides in the suppressor mutant strains.
Fig. 3.

TatB suppressors support export of a Tat substrate with its native signal peptide. E. coli strains producing native levels of TatA, TatC, and the indicated TatB variants, and overproducing His-tagged CueO with a wild-type (Upper) or KK-substituted (Lower) signal peptide were fractionated into whole cell (W), spheroplast (S), and periplasm (P) fractions. Equivalent amounts of each fraction were separated by SDS/PAGE and analyzed by Western blot with antibodies against CueO and the cytosolic marker DnaK.
The TatB Suppressors Do Not Restore Biochemically Detectable Signal Peptide Binding to TatBC.
We subsequently sought to understand the biochemical basis for the action of the TatB suppressors. Our initial hypothesis was that they acted to increase the affinity of the TatBC complex for the variant signal peptides, or to restore binding to complexes containing the TatC F94Q or E103A/E103K substitutions. First we produced His-tagged GFP with variants of the SufI signal peptide at its N terminus and assessed how much TatBC could be copurified with this from detergent-solubilized membrane fractions. Fig. 4A shows that when the wild-type signal peptide was fused to GFP, wild-type TatBC or variants harboring the TatB E8K, F13Y, or I36N substitutions were coeluted with His-tagged SufI-GFP. However, no TatBC was detected when the RR motif in the signal peptide was mutated to RD, RN, or KK, even in the presence of the TatB suppressor substitutions (despite the fact TatBC and GFP were clearly present in all of the input samples) (SI Appendix, Fig. S8 A and B). Very similar behavior was also seen when His-tagged AmiA variants were used as substrate for copurification experiments (SI Appendix, Fig. S9). Thus, TatBC and TatBF13YTatC copurified with the His-tagged wild-type AmiA precursor, but no TatBC was detected when RD, RN, KK, or KQ substitutions were introduced into the signal peptide, or when the AmiA signal peptide was lacking. We conclude that the TatB suppressors do not detectably restore binding of variant signal peptides to the TatBC complex. Because several TatB variants can support transport of substrates with defective signal peptides, but not without signal peptides (Fig. 3), we infer that the defective signal peptides must still weakly interact with the TatBC complex at a level that is not detected by our copurification assay.
Fig. 4.

The TatB suppressors do not restore signal peptide binding to the TatBC complex. (A) C-terminally His-tagged GFP with the wild-type (RR) or twin-arginine substituted SufI signal peptide at its N terminus, as indicated, was purified by magnetic nickel beads from digitonin-treated cell extracts coexpressing TatC along with either wild-type TatB or the E8K, F13Y, or I36N substituted variants. (B) C-terminally His-tagged GFP with the wild-type SufI signal peptide at its N terminus was purified by magnetic nickel beads from digitonin-treated cell extracts coexpressing TatB and TatC with the indicated amino acid substitutions. For both panels the elution fractions from each sample were normalized for GFP fluorescence and an equivalent amount of purified SufIss-GFP-His was loaded onto SDS/PAGE (4–15% Mini-PROTEAN TGX precast gradient gel) followed by Western blot using TatB and TatC mixed antibodies.
We then tested whether any of the four suppressors, TatB L9Q, L10P, F13Y, and I36N, which allow Tat transport in the presence of the TatC F94Q and E103K mutations, acted to restore substrate binding to TatBC complexes containing these signal sequence binding-site substitutions. Although high GFP fluorescence and strong TatBC signals were detected in whole cells (SI Appendix, Fig. S8 C and D), only wild-type TatBC was found to copurify with His-tagged SufIss-GFP (Fig. 4B). Thus, as expected, TatC substitutions F94Q or E103K prevented copurification of the TatBC-substrate complex, consistent with loss of signal peptide binding detected for substitutions at these amino acid positions (25, 40). However, detectable signal peptide binding was not restored by introduction of the individual TatB suppressor substitutions. We conclude that the TatB suppressors do not act by rescuing signal peptide binding.
The TatBC Complexes Harboring TatB Suppressor Substitutions Are Conformationally Altered.
We next investigated whether TatBC complexes could still be detected when any of the TatB L9Q, L10P, F13Y, or I36N substitutions were present in TatB. Membranes harboring wild-type TatA and TatC along with each of these TatB variants were solubilized with digitonin and analyzed by blue native-gel electrophoresis (BN-PAGE). As shown in Fig. 5A, the wild-type TatBC complex solubilized with digitonin migrated close to the 440-kDa marker, as reported previously (e.g., ref. 43). The TatB L9Q, F13Y, and I36N variants were also associated with a complex of apparently identical size to wild-type TatBC, whereas for membranes producing TatB L10P, very little TatBC complex could be detected, even though both proteins were solubilized from the membrane (SI Appendix, Fig. S10). Interestingly, the L9Q and F13Y TatB substitutions also resulted in the appearance of a second band of apparently higher mass that was absent from the sample containing wild-type TatBC (Fig. 5 A and B).
Fig. 5.

A constitutive disulfide cross-link and aberrant BN-PAGE migration induced by a subset of TatB suppressors. (A and B) Membranes from E. coli strain DADE (ΔtatABCD ΔtatE) producing the indicated TatB variants alongside wild-type TatA and TatC from plasmid pTAT1d were solubilized by addition of 2% (wt/vol) digitonin and analyzed by BN-PAGE (4–16% Bis-Tris NativePAGE gels) followed by Western blot with anti-TatA, anti-TatB, or anti-TatC antibodies as indicated. Solubilized membranes (20 μg) were loaded in each lane. (C) Membranes from strain DADE producing the indicated TatB variant alongside wild-type TatC from plasmid pTATBC1d were solubilized and analyzed as in A and B. (D) Whole cells of DADE harboring pTAT101 coproducing either wild-type TatA, the M205C single cysteine variant of TatC and the indicated substitution in TatB, or wild-type TatA, the TatC F94Q M205C variant of TatC, and the indicated substitution in TatB were subjected to oxidizing (O) or reducing (R) conditions. Where indicated the additional plasmid pQE80-CueO (producing His-tagged CueO) was also present. Membranes were prepared from equal quantities of cells following treatment and equivalent amounts of material from each sample were resolved by nonreducing SDS/PAGE (12% acrylamide). TatC was visualized by Western blotting using an anti-TatC antibody and CueO-His with an anti-His antibody.
We wondered whether this additional band might arise because of the presence of excess TatA bound to the variant complexes. However, blotting the BN gels for TatA showed the distinct TatA-laddering pattern reported previously (44, 45) was detectable for all of the samples, but there was no obvious TatA cross-reactive material migrating at the same position as the higher mass TatBC-containing complex (Fig. 5B). To examine whether the presence of TatA was required for these higher molecular weight variant TatBC complexes to form, we repeated the BN-PAGE analysis in the absence of TatA (or its paralog TatE) (16). Surprisingly, this resulted in the apparent aggregation of the variant TatBC complexes, yielding a series of bands of apparent masses well above 440 kDa that were not seen for the wild-type (Fig. 5C). We infer from this finding that there is a conformational alteration in the TatBC complex induced by the presence of the L9Q or F13Y TatB substitutions that in the absence of TatA causes further oligomerization.
Conformational alterations in the TatBC complex have been previously detected by disulfide cross-linking (46, 47). Cléon et al. (47) reported that when a Tat substrate was overproduced, a disulfide cross-link between M205C in transmembrane helix 5 of neighboring TatC proteins could be detected in vivo, suggesting the formation of a transient TatC dimer in response to substrate binding. Fig. 5D confirms that dimerization through TatC M205C is not observed unless cells also harbor an overproduced Tat substrate, in this case CueO. The TatC M205C dimer induced by CueO is almost completely absent when the F94Q substitution is introduced into TatC, again supporting the conclusion that substrate binding promotes TatC dimerization (Fig. 5D). Interestingly, however, when either the TatB L10P or F13Y substitutions were present, a TatC M205C cross-link was detected in the absence of overexpressed substrate. We wondered whether these TatB substitutions rendered the TatBC complex more responsive to the presence of endogenous substrates. To test this, we also introduced the signal peptide binding-defective F94Q substitution into TatC M205C. However, as Fig. 5D shows, the TatC M205C dimer can still be detected in the presence of TatB L10P or F13Y substitutions, even when the F94Q-inactivating substitution is present, and is therefore independent of signal peptide binding. We conclude that at least a subset of the TatB suppressors induces conformational changes in the TatBC complex, and that the TatB L10P and F13Y substitutions potentially mimic the substrate-bound form of the complex.
The TatB F13Y Substitution Promotes Signal Peptide-Independent Oligomerization of TatA in Vivo.
Substrate binding to the TatBC complex is a prerequisite for the assembly of a TatA oligomer. TatA oligomer assembly in vivo can be followed by fluorescence microscopy in cells producing a chromosomally encoded TatA–YFP fusion protein (34). When Tat substrates are present at native level, TatA oligomers are found with low frequency, but this frequency can be significantly increased by overproduction of a Tat substrate protein with a functional signal peptide (34, 35). This finding is confirmed in Fig. 6A, where clusters of TatA–YFP can be seen in cells overproducing AmiA from a plasmid. As expected, introduction of the F94Q codon substitution into chromosomally encoded tatC prevented the AmiA-induced clustering of TatA–YFP, resulting in a halo of delocalized TatA around the cell periphery (Fig. 6A), consistent with the inability of the TatC variant to bind substrates. We next assessed whether any of the TatC F94Q suppressors, TatB L9Q, L10P, F13Y, or I36N (introduced into chromosomal tatB) affected the oligomerization of TatA–YFP (Fig. 6B and SI Appendix, Fig. S11). Remarkably, we found that the presence of the TatB F13Y substitution promoted constitutive assembly of TatA–YFP in the absence of overproduced Tat substrates (Fig. 6B), and the TatA–YFP assemblies persisted even in the presence of the TatC F94Q substitution for this variant (but not for L9Q, L10P, or I36N) (SI Appendix, Fig. S11). Taken together, these results indicate that the TatB F13Y substitution triggers signal peptide-independent assembly of TatA oligomers.
Fig. 6.

The TatB F13Y substitution promotes constitutive oligomerization of TatA in vivo. (A) Fluorescence images of TatA–YFP in representative cells of strains AyBCE or AyBCF94QE (encoding the TatC F94Q substitution in chromosomal tatC) in the presence (pSUAmiA) or absence of plasmid-encoded wild-type AmiA, as indicated. Production of AmiA was induced by addition of 1 mM IPTG to mid-log cell cultures 45 min before harvesting. (B) Fluorescence images of TatA–YFP in representative cells of strains AyBF13YCE (encoding the TatB F13Y substitution in chromosomal tatB) and AyBCF94QE (encoding the TatB F13Y substitution in chromosomal tatB, alongside the TatC F94Q substitution in chromosomal tatC). (Scale bars, 1 μm.)
No Leak Across the Cytoplasmic Membrane When Cells Produce the Tat System Containing TatB F13Y.
One of the current models for Tat transport posits that TatA oligomers facilitate transport of substrates by causing a localized weakening of the bilayer and transient disruption (discussed in refs. 10 and 11). Such a mechanism might be expected to be accompanied by increased permeability of small molecules associated with assembled TatA. The availability of a TatB variant (F13Y) that causes TatA to accumulate in the assembled state provides an experimental tool to investigate this issue.
First, we asked whether overexpression of Tat systems containing the TatB suppressors L9Q, L10P, F13Y, or I36N from an arabinose-inducible promoter had any effect on the growth rate of E. coli. Fig. 7A shows that when production of each of these variant Tat systems was induced by the addition of arabinose, cells grew more slowly than when the wild-type Tat system was overexpressed, with the TatBL9Q substitution having a particularly detrimental effect on growth rate. This finding indicates that some level of toxicity is associated with overproduction of these variants. We next assessed whether the TatB variants facilitated membrane permeability using an osmotic lysis method previously used to monitor solute movement through the Sec protein transport channel (48). Here, spheroplasts containing wild-type or variant Tat translocases were diluted into an iso-osmotic solution of the uncharged sugar xylitol and permeation of xylitol into the cells was assessed by monitoring turbidity associated with osmotically induced spheroplast lysis. Spheroplasts expressing a SecY variant that is known to increase permeability (48, 49) rapidly lysed following dilution into xylitol solution (Fig. 7B). However, no lysis was observed for spheroplasts producing any of the variant Tat translocases, even those harboring the TatA-oligomerizing TatB F13Y variant (Fig. 7B). Western blotting confirmed that the Tat proteins were present in these membranes (Fig. 7C). These results show the TatA assemblies induced by the TatB F13Y substitution do not result in a small molecule leak across the cytoplasmic membrane.
Fig. 7.

No detectable leak of xylitol across the cytoplasmic membrane when cells produce Tat translocases harboring TatB suppressors. (A) Overnight cultures of E. coli strain BW25113 ∆glpF ∆tatABC harboring pBAD24 encoding TatA and TatC-his along with wild type TatB or each of the L9Q, L10P, F13Y, or I36N point substitutions, were subcultured at 1:100 dilution into fresh LB medium containing ampicillin, which was supplemented after 120 min with 0.2% of glucose or arabinose, as indicated. Growth of the strains was followed for a further 6.5 h. Error bars represent SD, n = 3 (biological replicates). (B) The same strain and plasmid combinations as in A, alongside BW25113 ∆glpF harboring pBAD22SecY(∆plug)EG, were subcultured and supplemented with 0.2% of arabinose as described in A and grown for an additional 3 h, after which spheroplasts were prepared and incubated in the presence of xylitol. (C) An aliquot of each sample producing plasmid-encoded Tat proteins was analyzed by SDS/PAGE and Western blotting to confirm expression of TatA, TatB, and TatC-His.
Discussion
In this work, a genetic approach has been taken to shed light on functions of twin arginine signal peptides during Tat transport. Two complementary screens, the first to identify substitutions in TatB permitting export of substrates with inactivating substitutions at the signal peptide arginine pair, and the second to identify rescue mutations of the TatC signal peptide binding site, converged on a similar group of tatB suppressors. Four TatB substitutions were identified: three in the transmembrane domain, L9Q, L10P, and F13Y, and one in the amphipathic helix (I36N) that restored Tat transport in the presence of the inactivating TatC F94Q substitution. The same three substitutions in the transmembrane helix could suppress inactivating substitutions at E103, and also in the signal peptide binding site. Of these three, the F13Y substitution displayed the strongest suppressing activity, allowing export all of the twin arginine substitutions tested, and even allowing some translocation of AmiA completely devoid of a signal sequence.
A combined bioinformatics and mutagenesis approach has shown the TatB transmembrane helix to bind along transmembrane helix 5 of TatC (50), and this is consistent with in vitro disulfide cross-linking studies (25, 39). Signal peptide binding to the TatBC complex is suggested to cause movement of TatB from its resting-state binding site on TatC to a site elsewhere on the protein. This is proposed to prime TatA to occupy the same binding site, which in turn triggers assembly of further TatA molecules to form the active translocase (50) (Fig. 8). After our biochemical experiments revealed that the suppressors did not function by restoring detectable binding of signal peptides to the TatBC complex (Fig. 4 and SI Appendix, Fig. S9), we considered whether the TatB substitutions were mimicking the substrate-driven conformational changes that prime the translocase for TatA recruitment, but in the absence of substrate binding. Our analysis using BN-PAGE showed that the TatB L9Q, L10P, and F13Y substitutions caused conformational alterations in the resting TatBC complex (Fig. 5 A and B). The complex containing the L10P substitution appeared more labile because very little full-sized TatBC complex could be detected, whereas the L9Q and F13Y substitutions yielded a subset of TatBC complexes with apparently increased mass, which may be indicative of altered subunit composition or significant conformational change. Substrate-induced conformational changes in the wild-type complex can also be monitored by appearance of a cross-link between cysteine residues at position 205 at the periplasmic end of TatC transmembrane helix 5. This residue forms part of the TatB resting-state binding site (50), so is occluded from dimerization with a neighboring TatC molecule in the resting state, but has been shown to dimerize in response to overproduction of a Tat substrate (39). Similarly, assembly of TatA-YFP oligomers (indicating assembled translocation sites) that can be monitored by fluorescence microscopy is only seen for the wild-type translocase upon overproduction of Tat substrates (34). For one of the TatB substitutions, F13Y, both TatC M205C dimerization and TatA–YFP assembly were observed not only in the absence exogenous substrates, but also in the presence of a TatC F94Q substitution, which disrupts interaction with signal peptides. We therefore conclude that TatB F13Y has decreased affinity for the “resting” TatC binding site, and an increased affinity for the “activated” binding site, such that it is able to trigger recruitment of TatA and transport of precursors in the absence of signal peptide binding. For the other TatB variants, we propose that each differs in affinity for the resting and activated binding sites, leading to slight differences in conformation for these translocases, and we assume that in these cases, weak residual binding of a signal peptide lacking its twin arginine motif is sufficient to trigger TatA recruitment, whereas in the wild-type system the higher energy of binding of the twin arginine residues is strictly required for this. Hence, the signal peptide plays two distinct roles: in precursor targeting and in translocase activation.
Fig. 8.

Tat translocases containing TatB suppressor variants may more readily transition to the signal peptide-activated state. (Upper) Model for Tat transport. A signal peptide bound through its n-region to the cytoplasmic surface of TatC (step 1) transitions to a deep binding mode (step 2). The deep insertion of the signal peptide displaces TatB from its resting state binding site on TatC (gray arrow). TatB movement allows polymerization of TatA to be nucleated (step 3). The substrate passes across the membrane facilitated by the TatA oligomer (step 4). (Lower) TatB variants that suppress signal sequence defects (represented with an asterisk, *) may be more easily displaced from the resting-state binding site. The TatB variants appear to be on a continuum with TatB F13Y pushing the Tat system into an assembled state (step 4), whereas Tat systems harboring the weaker suppressing variants are more likely to correspond to step 3.
A favored model for Tat-mediated protein translocation is that protein passage across the membrane is facilitated by bilayer disruption arising from TatA oligomerization. Interestingly, it was noted that there was a reduced growth rate associated with overproduction of Tat systems containing TatB suppressors, including F13Y, suggesting apparent toxicity. However, no leak of the small uncharged sugar, xylitol, could be detected in membranes harboring Tat complexes containing TatB F13Y. This may suggest the presence of a substrate precursor is necessary to provide the force required to disrupt the bilayer. Alternatively, it remains possible that the foci of TatA–YFP observed in cells producing TatB F13Y do not correspond to fully assembled translocases and that further recruitment of TatA(–YFP) [for example mediated by the folded mature domain of a Tat substrate (51)] is required. Indeed the export of mature AmiA in the presence of the TatB F13Y substitution might suggest that some Tat substrates have internal targeting information.
In summary, our findings support the notion that Tat signal peptides have two distinct roles. They serve to target their passenger domains to the export machinery, but also to trigger assembly of the active translocase. The isolation of substitutions in the Tat machinery that bypass these steps should prove very useful to dissect the mechanism by which folded protein translocation is achieved.
Materials and Methods
Strain Construction.
The E. coli strains used in this work are listed in SI Appendix, Table S3. Strain JM109 was used for regular cloning and transformation of Quickchange products, and ultracompetent cells of XL10-Gold (Agilent) were used for construction of the random mutagenesis libraries.
Strain DADE [as MC4100, ΔtatABCD, ΔtatE (42)] was used as the background strain for Tat transport activity tests and production of Tat proteins for membrane protein extraction, in vivo disulfide cross-linking, and BN-PAGE, with the exception of Fig. 3, where strain MΔBC [MC4100 ΔtatBC (33)] was used. Strain DADE-P [as DADE, pcnB1 zad-981::Tn10d (Kanr) (52)] was used to coproduce TatB and TatC along with AmiA for copurification experiments. Strain MCDSSAC ΔtatABC (37), in which the 2–33 codons of amiA and 2–32 codons for amiC are deleted and the tatABC operon was replaced with an apramycin-resistance cassette, was used as the background strain for AmiA signal sequence library screening and to analyze transport of AmiA mediated by AmiA or SufI signal peptide variants.
Transport of AmiA mediated by signal sequence truncations was assessed in strain MC4100 ΔamiA ΔamiC ΔtatABC, which was constructed as follows. The ΔamiA:kanr allele from the Keio collection (53) was moved into MC4100 by phage P1 transduction, after which the kanamycin-resistance cassette was eliminated according to Cherepanov and Wackernagel (54). Next, the amiC deletion was introduced and the kanamycin cassette subsequently eliminated using the same approach. Finally, the ΔtatABC::Apra allele was introduced from strain BW25113 ΔtatABC::Apra (54) by P1 transduction. Strain BW25113 ΔglpF ΔtatABC was used in osmotic lysis experiments and was constructed by P1 transduction of the ΔglpF:kanr allele from the Keio collection (53), elimination of the kanamycin resistance, and P1 transduction of the ΔtatABC::Apra allele, as described above.
Strain AyBCE (34), which lacks tatA at the native locus and has a tatA–YFP fusion integrated into the chromosomal att site, was used in fluorescence imaging. Chromosomal point substitutions in tatB and tatC were introduced into this strain via plasmid pMAK-AupBC and its variants using the approach of Hamilton et al. (55).
Strain BL21(DE3) ΔtatABC was used to coproduce TatB and TatC along with SufIss-GFP-His for the copurification experiments. This strain is a derivative of BL21(DE3), where the tatABC genes have been replaced with the apramycin-resistance cassette, and was constructed by recombination as described previously (56).
Plasmid Construction.
The plasmids used and constructed in this work are listed in SI Appendix, Table S4. All point mutations in plasmids, as well as insertion of the flag sequence to create p101C*BCflag, were introduced by Quickchange site-directed mutagenesis (Stratagene) using the primers listed in SI Appendix, Table S5.
Plasmids pTAT101 (39) and pTAT1d (38) were used to express tatABC under the control of the native tatA promoter at very low and medium copy number, respectively. pTAT101 cys-less (47) was used as the backbone to introduce single cys substitutions for in vivo disulfide cross-linking experiments. Plasmid pTATBC1d encodes TatBC and was constructed following amplification of tatBC pTAT1d using primers STIPE-ISH and pT7.5R (SI Appendix, Table S5) was digested using BamHI and PstI and cloned into similarly digested pUNIPROM (57). Plasmid pBADTatABC-His codes for tatABC with a hexa His-tag coding sequence at the 3′ end of tatC in pBAD24 (58). It was constructed following amplification of tatABChis from pUNITAT2 (59), digestion with NcoI and XbaI and cloning into similarly digested pBAD24.
pSUAmiA (36) was used to produce full-length AmiA from a vector specifying chloramphenicol resistance. pSUSufIss-mAmiA was used to produce SufIss-mAmiA and was constructed following separate amplification of DNA encoding the SufI signal sequence, including the sufI ribosome binding site using primers SufIssFE and SufIssR, and the mature region of AmiA (mAmiA) using primers AmiA-mF and AmiA-mRX from the chromosome, and fusing the two fragments by overlap extension PCR according to Heckman and Pease (60). The resultant DNA fragment was then cloned into the pSU18 vector (61) using EcoRI and XbaI sites to generate pSUSufIss-mAmiA plasmid. Plasmids pSUSufIssnoH-mAmiA were used for production of truncated SufIss-mAmiA lacking the signal peptide h-region. It was constructed by removal of codons 11–21 of the SufI signal sequence via Quickchange using pSUSufIss-mAmiA as template with primers SufI-noHF and SufI-noHR. pSUmAmiA was used to produce signaless AmiA and was constructed as follows. A DNA fragment containing the ribosome binding site of amiA and the coding sequence for mature AmiA was amplified using pSUAmiA as template with primers AmiA-nossFE and AmiA-mRX. The DNA fragment was subsequently cloned into pSU18 using EcoRI and XbaI sites to generate pSUmAmiA. To express the mature domains of AmiA or AmiC from pQE70, DNA covering these regions were amplified with primer pairs mAmiA-SphI-F/AmiAnostopBamHI-R or mAmiC-SphI-F/AmiCnostopBamHI-R, respectively, using chromosomal DNA as a template. The DNA fragments were digested with SphI and BamHI and cloned into SphI/BglII digested pQE70 vector (Qiagen). For fractionation experiments, SufI was produced with a C-terminal His-tag from pQE80 (Qiagen). It was cloned by excision of DNA covering a C-terminally His-tagged SufI from pQE60-SufI (62) as a NheI-XhoI fragment and ligation into similarly digested pQE80.
Plasmid pMAK-AupBC was used to introduce the mutations into the AyBCE chromosome and was constructed by amplification of 500 bp of tatA upstream DNA from the chromosome of strain AyBCE using primers TatAup1-XbaI and TatAup2-ClaI, which was cloned into pBluescript KS(+) using XbaI and ClaI sites to give pKS-Aup. Next, a DNA fragment covering the whole of tatBC was amplified from the chromosome of AyBCE strain with primers TatA6B7-ClaI and TatCrev-KpnI, and cloned into pKS-Aup using ClaI and KpnI sites to generate pKS-AupBC. Subsequently the DNA covering the tatA upstream sequence along with tatBC was excised using XbaI and KpnI and cloned into similarly digested pMAK705 (55) to give pMAK-AupBC.
Plasmid pFAT75ΔA-BC, which is a pQE-based plasmid expressing TatB and TatC without a His-tag (19), and pSufIss-GFP-His, which is a pCDFDuet-based plasmid expressing synthetic SufI signal sequence-fused GFP with a His-tag at its C terminus under the control of T7 promoter, were used in copurification experiments. Plasmid pFAT75ΔA-BC-AmiA-His coproduces untagged TatBC along with His-tagged AmiA and was constructed as follows. A DNA fragment covering amiA was amplified from MC4100 genomic DNA using primers AmiAFATApaI-F and AmiAnostopBamHI-R, digested with ApaI and BamHI and cloned into ApaI/BglII digested pFAT75-SufI-His (47). pFAT75ΔA-BC-mAmiA-His was constructed similarly, using primer mAmiAFATApaI-F and AmiAnostopBamHI-R to amply DNA covering the mature region of AmiA.
Mutant Library Construction and Screening.
To screen for Tat signal sequence suppressors, substitutions of the twin arginine sequence of the AmiA signal peptide (to -RD, -RE, -RN, -RQ, -RH, -HH, -KH. and -KQ) were constructed in the pSUAmiA plasmid. The resulting plasmids were individually introduced into strain MCDSSAC ΔtatABC and each resulting strain served as host to screen an existing tatB mutant library (in plasmid pTAT1d, 600,000 individual clones, 0.25% error rate) (38).
To screen for suppressors of the TatC F94Q substitution, three separate mutagenesis libraries, each of which carried the tatC F94Q codon substitution, were constructed that contained random mutations in either tatAB, codons 1–93 of tatC (tatC1) or codons 95–258 of tatC (tatC2), respectively, using a modified MEGAWHOP (megaprimer PCR of whole plasmid) method, as described previously (63). DNA fragments of tatAB, tatC1, and tatC2 containing random mutations were generated using error-prone PCR. Error-prone PCR was carried out in 1× GoTaq buffer, 7 mM MgCl2, 0.2 mM dATP, 0.2 mM dGTP, 1 mM dCTP, 1 mM dTTP, 0.4 μM each primer, 0–0.1 mM MnCl2, 50 ng pTAT1dCF94Q plasmid as template, and 5 U GoTaq DNA Polymerase (Promega) in a total volume of 50 μL using a PCR program: 94 °C for 2 min followed by 20 cycles of incubation at 94 °C for 30 s, 50 °C for 30 s, and 72 °C for 3 min, and a final incubation at 72 °C for 5 min. Primer pairs TatA-FB and TatB-RS were used to amplify tatAB, TatCm6 and TatC93R to amplify tatC1, and TatC95F and TatCR1d were used to amplify tatC2. The DNA fragments were then used as megaprimers to amplify the whole plasmid, which was carried out in a 50-μL mixture containing 1× Herculase II reaction buffer, 0.5 mM each dNTP, 100 ng pTAT1dCF94Q plasmid as template, 500 ng DNA fragment obtained above as megaprimers, and 5 U Herculase II Fusion DNA Polymerase (Agilent) using the PCR program: incubation at 68 °C for 5 min, 95 °C for 2 min, followed by 20 cycles of incubation at 95 °C for 30 s, 55 °C for 30 s, and 68 °C for 6 min. The resultant whole-plasmid PCR products were digested with DpnI to remove the template DNA and incubated with T4 polynucleotide kinase and T4 DNA ligase to repair the nicks. Finally, the whole plasmids were separately transformed into XL10-Gold Ultracompetent Cells (Agilent) resulting in three mutagenesis libraries. Subsequent sequencing of 10 randomly selected colonies from each library revealed an average error rate of ∼2 nucleotides per 1,000 base pairs. Screening of these three libraries was carried out in strain DADE (ΔtatABCD, ΔtatE).
For screening experiments, the libraries were transformed into the respective host strains and subsequently plated onto solid LB medium containing appropriate antibiotics and 2% (wt/vol) SDS for selection. Colonies able to grow under these conditions were isolated, the mutations in the tat genes identified by sequencing and retested for growth in the presence and absence of 2% (wt/vol) SDS. To further verify isolated candidates, individual mutations were introduced into low copy number plasmid pTAT101 by site-directed mutagenesis and the activity was again assessed on 2% (wt/vol) SDS-containing plates.
Protein Methods.
For copurification of TatBC-substrate complexes, cultures of strain BL21(DE3) ΔtatABC harboring pFAT75ΔA (or a point-substituted variant) and pSufI-GFP-His were incubated at 37 °C for 7 h with shaking, after which they were supplemented with 0.2 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and incubated for an additional 17 h at 37 °C. The cells were subsequently harvested and resuspended in 1× PBS, the fluorescence intensity of the suspension was recorded, after which the cells were repelleted, resuspended in 2× lysis buffer (100 mM NaH2PO4 pH 8.0, 600 mM NaCl, 40 mM imidazole, 50 mg lysozyme, 80 U benzonase, and protease inhibitor) and mixed gently at room temperature for 1 h. Cells were snap-frozen at −80 °C, thawed at room temperature, and an equivalent amount of 2.5% (wt/vol) digitonin was added and the sample gently mixed at room temperature for 1 h. Cell debris was pelleted by centrifugation and the supernatant was transferred to a 96-well plate and mixed with 20 μL Ni-NTA Magnetic Agarose Beads (Qiagen) for 1 h. After the beads were washed three times with wash buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 40 mM imidazole, 0.03% digitonin), bound proteins were eluted with 50 μL elution buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 250 mM imidazole, 0.03% digitonin).
In vivo disulfide cross-linking was carried out in strain DADE harboring pTAT101 cys-less CM205C, as described in Cléon et al. (47). BN-PAGE was undertaken according to refs. 47 and 64. Subcellular fractionation was according to Palmer et al. (65). Preparation of membrane fractions was as described previously (39). For analysis of SufI export, E. coli strain DADE harboring wild-type or signal peptide variants of pQE80SufI-His alone or alongside wild-type or TatB variants of pTAT101 was cultured in the presence of 1 mM IPTG until OD600 of 1 was reached. Samples (equivalent to 150 μL of whole cells from an OD600 = 1, or periplasm fractions from the equivalent of 300 μL of cells from an OD600 = 1) were separated by SDS/PAGE and analyzed by Western blot with anti-6× His-tag or anti-RNA polymerase β-subunit antibodies (cytoplasmic control protein). For analysis of CueO export, strain MΔBC harboring wild-type and KK variants of pQE80-CueO alongside wild-type, tatBE8K or tatBF13Y variants of p101C*BCflag were cultured and fractionated as previously described (34). Immunoblotting was according to the methodology of Towbin et al. (66), and antibodies to TatA, TatB, and TatC have been described previously (47, 67). Anti-6× His-tag antibody (HRP-conjugated) was purchased from Abcam (catalog no. ab184607), anti-DnaK mouse monoclonal 8E2/2 antibody was also from Abcam (catalog no. ab69617), and a mouse monoclonal anti-RNA polymerase β-subunit antibody was purchased from NeoClone Biotechnology (catalog no. W0023). Secondary antibodies were goat anti-Rabbit IgG (HRP Conjugate, catalog no. 170-6515) or Goat Anti-Mouse IgG (HRP conjugate, catalog no.170-6516), both from Bio-Rad. Immunoreactive bands were visualized with the Clarity Western ECL Substrate Kit (Bio-Rad) and captured either on light-sensitive film or using the GeneGNOME camera (Syngene).
Cell Permeability Experiments.
Cell permeability experiments were performed according to Park and Rapoport (48). Briefly, cells were grown aerobically at 37 °C with 1:100 inoculation of an overnight culture for 2 h. Production of TatABC harboring TatB variants and of SecY(∆plug)EG was induced by addition of 0.2% arabinose at 37 °C for 3 h. Subsequently, the OD600nm of each sample was normalized using LB, a small volume was withdrawn for Western blotting, and equal volumes of each culture were then harvested and resuspended in fractionation buffer (50 mM Tris⋅HCl buffer, 20% (wt/vol) sucrose, pH 7.5, 5 mM EDTA, 0.6 mg/mL lysozyme) and incubated at room temperature for 20 min to obtain spheroplasts. The spheroplast samples were then adjusted to the same OD600 and a 19-fold excess of 0.616 M xylitol solution was added. The samples were rapidly transferred to a 96-well plate and OD600 was measured every 30 s for 300 s.
Fluorescence Microscopy.
Cells were prepared for fluorescence microscopy and imaged as previously described (47), with the exception that a 550-nm LP emission filter was used.
Supplementary Material
Acknowledgments
We thank Dr. Bérengère Ize for her assistance in constructing pQE70-mAmiA and pQE70-mAmiC; Prof. Ian Collinson for the gift of pBADSecY(Δplug)EG; and Drs. Hajra Basit and Mark Wallace for fluorescence microscopy access and advice. This work was supported by the UK Biotechnology and Biological Sciences Research Council (Grants BB/L002531/1 and BB/N014545/1); the UK Medical Research Council (Grants G1001640, K000721, and MR/L000776/1), the Wellcome Trust through Investigator Award 107929/Z/15/Z (to B.C.B.); a Wellcome Trust PhD studentship (to S.E.R.); and the China Scholarship Council (through a studentship to Q.H.). T.P. and S.M.L. are Wellcome Trust Investigators and T.P. is a Royal Society/Wolfson merit award holder.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1615056114/-/DCSupplemental.
References
- 1.Collinson I, Corey RA, Allen WJ. Channel crossing: How are proteins shipped across the bacterial plasma membrane? Philos Trans R Soc Lond B Biol Sci. 2015;370(1679):20150025. doi: 10.1098/rstb.2015.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denks K, et al. The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol Membr Biol. 2014;31(2-3):58–84. doi: 10.3109/09687688.2014.907455. [DOI] [PubMed] [Google Scholar]
- 3.Emr SD, Hanley-Way S, Silhavy TJ. Suppressor mutations that restore export of a protein with a defective signal sequence. Cell. 1981;23(1):79–88. doi: 10.1016/0092-8674(81)90272-5. [DOI] [PubMed] [Google Scholar]
- 4.Oliver DB, Beckwith J. E. coli mutant pleiotropically defective in the export of secreted proteins. Cell. 1981;25(3):765–772. doi: 10.1016/0092-8674(81)90184-7. [DOI] [PubMed] [Google Scholar]
- 5.Van den Berg B, et al. X-ray structure of a protein-conducting channel. Nature. 2004;427(6969):36–44. doi: 10.1038/nature02218. [DOI] [PubMed] [Google Scholar]
- 6.Smith MA, Clemons WM, Jr, DeMars CJ, Flower AM. Modeling the effects of prl mutations on the Escherichia coli SecY complex. J Bacteriol. 2005;187(18):6454–6465. doi: 10.1128/JB.187.18.6454-6465.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trueman SF, Mandon EC, Gilmore R. Translocation channel gating kinetics balances protein translocation efficiency with signal sequence recognition fidelity. Mol Biol Cell. 2011;22(17):2983–2993. doi: 10.1091/mbc.E11-01-0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emr SD, Schwartz M, Silhavy TJ. Mutations altering the cellular localization of the phage lambda receptor, an Escherichia coli outer membrane protein. Proc Natl Acad Sci USA. 1978;75(12):5802–5806. doi: 10.1073/pnas.75.12.5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gouridis G, Karamanou S, Gelis I, Kalodimos CG, Economou A. Signal peptides are allosteric activators of the protein translocase. Nature. 2009;462(7271):363–367. doi: 10.1038/nature08559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cline K. Mechanistic aspects of folded protein transport by the twin arginine translocase (Tat) J Biol Chem. 2015;290(27):16530–16538. doi: 10.1074/jbc.R114.626820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berks BC. The twin-arginine protein translocation pathway. Annu Rev Biochem. 2015;84:843–864. doi: 10.1146/annurev-biochem-060614-034251. [DOI] [PubMed] [Google Scholar]
- 12.Chaddock AM, et al. A new type of signal peptide: Central role of a twin-arginine motif in transfer signals for the delta pH-dependent thylakoidal protein translocase. EMBO J. 1995;14(12):2715–2722. doi: 10.1002/j.1460-2075.1995.tb07272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berks BC. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol. 1996;22(3):393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- 14.Weiner JH, et al. A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell. 1998;93(1):93–101. doi: 10.1016/s0092-8674(00)81149-6. [DOI] [PubMed] [Google Scholar]
- 15.Bogsch EG, et al. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem. 1998;273(29):18003–18006. doi: 10.1074/jbc.273.29.18003. [DOI] [PubMed] [Google Scholar]
- 16.Sargent F, et al. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 1998;17(13):3640–3650. doi: 10.1093/emboj/17.13.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sargent F, Stanley NR, Berks BC, Palmer T. Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J Biol Chem. 1999;274(51):36073–36082. doi: 10.1074/jbc.274.51.36073. [DOI] [PubMed] [Google Scholar]
- 18.Bolhuis A, Mathers JE, Thomas JD, Barrett CM, Robinson C. TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J Biol Chem. 2001;276(23):20213–20219. doi: 10.1074/jbc.M100682200. [DOI] [PubMed] [Google Scholar]
- 19.Tarry MJ, et al. Structural analysis of substrate binding by the TatBC component of the twin-arginine protein transport system. Proc Natl Acad Sci USA. 2009;106(32):13284–13289. doi: 10.1073/pnas.0901566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma X, Cline K. Multiple precursor proteins bind individual Tat receptor complexes and are collectively transported. EMBO J. 2010;29(9):1477–1488. doi: 10.1038/emboj.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreutzenbeck P, et al. Escherichia coli twin arginine (Tat) mutant translocases possessing relaxed signal peptide recognition specificities. J Biol Chem. 2007;282(11):7903–7911. doi: 10.1074/jbc.M610126200. [DOI] [PubMed] [Google Scholar]
- 22.Lausberg F, et al. Genetic evidence for a tight cooperation of TatB and TatC during productive recognition of twin-arginine (Tat) signal peptides in Escherichia coli. PLoS One. 2012;7(6):e39867. doi: 10.1371/journal.pone.0039867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strauch EM, Georgiou G. Escherichia coli tatC mutations that suppress defective twin-arginine transporter signal peptides. J Mol Biol. 2007;374(2):283–291. doi: 10.1016/j.jmb.2007.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoufaly S, et al. Mapping precursor-binding site on TatC subunit of twin arginine-specific protein translocase by site-specific photo cross-linking. J Biol Chem. 2012;287(16):13430–13441. doi: 10.1074/jbc.M112.343798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rollauer SE, et al. Structure of the TatC core of the twin-arginine protein transport system. Nature. 2012;492(7428):210–214. doi: 10.1038/nature11683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma X, Cline K. Mapping the signal peptide binding and oligomer contact sites of the core subunit of the pea twin arginine protein translocase. Plant Cell. 2013;25(3):999–1015. doi: 10.1105/tpc.112.107409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alami M, et al. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell. 2003;12(4):937–946. doi: 10.1016/s1097-2765(03)00398-8. [DOI] [PubMed] [Google Scholar]
- 28.Gérard F, Cline K. Efficient twin arginine translocation (Tat) pathway transport of a precursor protein covalently anchored to its initial cpTatC binding site. J Biol Chem. 2006;281(10):6130–6135. doi: 10.1074/jbc.M512733200. [DOI] [PubMed] [Google Scholar]
- 29.Blümmel AS, Haag LA, Eimer E, Müller M, Fröbel J. Initial assembly steps of a translocase for folded proteins. Nat Commun. 2015;6:7234. doi: 10.1038/ncomms8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gérard F, Cline K. The thylakoid proton gradient promotes an advanced stage of signal peptide binding deep within the Tat pathway receptor complex. J Biol Chem. 2007;282(8):5263–5272. doi: 10.1074/jbc.M610337200. [DOI] [PubMed] [Google Scholar]
- 31.Mori H, Cline K. A twin arginine signal peptide and the pH gradient trigger reversible assembly of the thylakoid [Delta]pH/Tat translocase. J Cell Biol. 2002;157(2):205–210. doi: 10.1083/jcb.200202048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dabney-Smith C, Mori H, Cline K. Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J Biol Chem. 2006;281(9):5476–5483. doi: 10.1074/jbc.M512453200. [DOI] [PubMed] [Google Scholar]
- 33.Dabney-Smith C, Cline K. Clustering of C-terminal stromal domains of Tha4 homo-oligomers during translocation by the Tat protein transport system. Mol Biol Cell. 2009;20(7):2060–2069. doi: 10.1091/mbc.E08-12-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alcock F, et al. Live cell imaging shows reversible assembly of the TatA component of the twin-arginine protein transport system. Proc Natl Acad Sci USA. 2013;110(38):E3650–E3659. doi: 10.1073/pnas.1306738110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rose P, Fröbel J, Graumann PL, Müller M. Substrate-dependent assembly of the Tat translocase as observed in live Escherichia coli cells. PLoS One. 2013;8(8):e69488. doi: 10.1371/journal.pone.0069488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ize B, Stanley NR, Buchanan G, Palmer T. Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol Microbiol. 2003;48(5):1183–1193. doi: 10.1046/j.1365-2958.2003.03504.x. [DOI] [PubMed] [Google Scholar]
- 37.Keller R, de Keyzer J, Driessen AJ, Palmer T. Co-operation between different targeting pathways during integration of a membrane protein. J Cell Biol. 2012;199(2):303–315. doi: 10.1083/jcb.201204149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maldonado B, et al. Characterisation of the membrane-extrinsic domain of the TatB component of the twin arginine protein translocase. FEBS Lett. 2011;585(3):478–484. doi: 10.1016/j.febslet.2011.01.016. [DOI] [PubMed] [Google Scholar]
- 39.Kneuper H, et al. Molecular dissection of TatC defines critical regions essential for protein transport and a TatB-TatC contact site. Mol Microbiol. 2012;85(5):945–961. doi: 10.1111/j.1365-2958.2012.08151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holzapfel E, et al. The entire N-terminal half of TatC is involved in twin-arginine precursor binding. Biochemistry. 2007;46(10):2892–2898. doi: 10.1021/bi062205b. [DOI] [PubMed] [Google Scholar]
- 41.Buchanan G, et al. Functional complexity of the twin-arginine translocase TatC component revealed by site-directed mutagenesis. Mol Microbiol. 2002;43(6):1457–1470. doi: 10.1046/j.1365-2958.2002.02853.x. [DOI] [PubMed] [Google Scholar]
- 42.Wexler M, et al. TatD is a cytoplasmic protein with DNase activity. No requirement for TatD family proteins in sec-independent protein export. J Biol Chem. 2000;275(22):16717–16722. doi: 10.1074/jbc.M000800200. [DOI] [PubMed] [Google Scholar]
- 43.Richter S, Brüser T. Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. J Biol Chem. 2005;280(52):42723–42730. doi: 10.1074/jbc.M509570200. [DOI] [PubMed] [Google Scholar]
- 44.Oates J, et al. The Escherichia coli twin-arginine translocation apparatus incorporates a distinct form of TatABC complex, spectrum of modular TatA complexes and minor TatAB complex. J Mol Biol. 2005;346(1):295–305. doi: 10.1016/j.jmb.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 45.Gohlke U, et al. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA. 2005;102(30):10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aldridge C, Ma X, Gerard F, Cline K. Substrate-gated docking of pore subunit Tha4 in the TatC cavity initiates Tat translocase assembly. J Cell Biol. 2014;205(1):51–65. doi: 10.1083/jcb.201311057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cléon F, et al. The TatC component of the twin-arginine protein translocase functions as an obligate oligomer. Mol Microbiol. 2015;98(1):111–129. doi: 10.1111/mmi.13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park E, Rapoport TA. Preserving the membrane barrier for small molecules during bacterial protein translocation. Nature. 2011;473(7346):239–242. doi: 10.1038/nature10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dalal K, Duong F. The SecY complex forms a channel capable of ionic discrimination. EMBO Rep. 2009;10(7):762–768. doi: 10.1038/embor.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alcock F, et al. Assembling the Tat protein translocase. eLife. 2016;5:e20718. doi: 10.7554/eLife.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taubert J, et al. TatBC-independent TatA/Tat substrate interactions contribute to transport efficiency. PLoS One. 2015;10(3):e0119761. doi: 10.1371/journal.pone.0119761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee PA, et al. Cysteine-scanning mutagenesis and disulfide mapping studies of the conserved domain of the twin-arginine translocase TatB component. J Biol Chem. 2006;281(45):34072–34085. doi: 10.1074/jbc.M607295200. [DOI] [PubMed] [Google Scholar]
- 53.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158(1):9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- 55.Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol. 1989;171(9):4617–4622. doi: 10.1128/jb.171.9.4617-4622.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orriss GL, et al. TatBC, TatB, and TatC form structurally autonomous units within the twin arginine protein transport system of Escherichia coli. FEBS Lett. 2007;581(21):4091–4097. doi: 10.1016/j.febslet.2007.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jack RL, et al. Coordinating assembly and export of complex bacterial proteins. EMBO J. 2004;23(20):3962–3972. doi: 10.1038/sj.emboj.7600409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177(14):4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDevitt CA, Hicks MG, Palmer T, Berks BC. Characterisation of Tat protein transport complexes carrying inactivating mutations. Biochem Biophys Res Commun. 2005;329(2):693–698. doi: 10.1016/j.bbrc.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 60.Heckman KL, Pease LR. Gene splicing and mutagenesis by PCR-driven overlap extension. Nat Protoc. 2007;2(4):924–932. doi: 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 61.Bartolomé B, Jubete Y, Martínez E, de la Cruz F. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene. 1991;102(1):75–78. doi: 10.1016/0378-1119(91)90541-i. [DOI] [PubMed] [Google Scholar]
- 62.Tarry M, et al. The Escherichia coli cell division protein and model Tat substrate SufI (FtsP) localizes to the septal ring and has a multicopper oxidase-like structure. J Mol Biol. 2009;386(2):504–519. doi: 10.1016/j.jmb.2008.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miyazaki K, Takenouchi M. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechni. 2002;33(5):1033-1034–1036-1038. doi: 10.2144/02335st03. [DOI] [PubMed] [Google Scholar]
- 64.Wittig I, Braun HP, Schägger H. Blue native PAGE. Nat Protoc. 2006;1(1):418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 65.Palmer T, Berks BC, Sargent F. Analysis of Tat targeting function and twin-arginine signal peptide activity in Escherichia coli. Methods Mol Biol. 2010;619:191–216. doi: 10.1007/978-1-60327-412-8_12. [DOI] [PubMed] [Google Scholar]
- 66.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76(9):4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sargent F, et al. Purified components of the Escherichia coli Tat protein transport system form a double-layered ring structure. Eur J Biochem. 2001;268(12):3361–3367. doi: 10.1046/j.1432-1327.2001.02263.x. [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Wang L, Hu Y, Jin C. Solution structure of the TatB component of the twin-arginine translocation system. Biochim Biophys Acta. 2014;1838(7):1881–1888. doi: 10.1016/j.bbamem.2014.03.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.