

Abstract
The potential of the fluorescent protein scaffold to control peptide sequence functionality is illustrated by an exploration of fluorescent proteins as novel probes for targeting integrins. A library of fluorescent mCitrine proteins with RGD motifs incorporated at several positions in loops within the protein main chain was generated and characterized. Amino acid mutations to RGD as well as RGD insertions were evaluated: both led to constructs with typical mCitrine fluorescent properties. Screening experiments against four human integrin receptors revealed two strong‐binding constructs and two selective integrin binders. The effect of the site of RGD incorporation illustrates the importance of the protein scaffold on RGD sequence functionality, leading to fluorescent protein constructs with the potential for selective integrin targeting.
Keywords: fluorescent probes, integrin, protein engineering, protein-protein interactions, RGD
The integrin receptor is the major cell surface receptor responsible for cell–cell and cell–matrix interactions. It is a heteromeric glycoprotein in which each subunit contributes to the ligand binding site.1, 2 Dimeric member are generated by the combination of one 18 α‐ and one 8 β‐subunit in the 24 known mammalian integrins.2 Each integrin has a specific role, as demonstrated by gene knock‐out experiments in mice.3, 4 About half of the integrins bind their ligands with a key tripeptide sequence: arginine‐glycine‐aspartic acid (RGD).2, 5, 6, 7 The interaction between RGD‐containing proteins and their specific integrin receptors regulates a diverse array of fundamental cellular functions, including cell adhesion, migration, differentiation, and signaling.1, 8 Disregulation of these processes is involved in osteoporosis, inflammation, and several types of cancer, thus making integrins an appealing target for anticancer therapy.8, 9 This has resulted in the synthesis of RGD mimics as antagonists, imaging probes, and cell‐adhesion material.1, 10, 11, 12, 13 The relatively low affinity and integrin selectivity of linear RGD sequences can be increased by rigidification strategies, such as the use of cyclic peptides.1, 14, 15, 16 The RGD sequence has also been inserted into protein loops for binding and uptake applications.17, 18
Fluorescent proteins, available in a diverse range of spectral characteristics, are highly attractive for the visualization of biological molecules, functions, and processes.19, 20 A frequently used and brightly fluorescent variant is monomeric Citrine (mCitrine).21 The β‐barrel fold of fluorescent proteins, with the chromophore in the center, is highly stable and amenable to mutations to the β‐strands and the connecting loops.22, 23, 24 As a result, incorporation of peptide epitopes into fluorescent proteins has potential for developing probes for bioimaging applications.25, 26 Fluorescent proteins have many loops as possible insertion sites for RGD motifs. Potentially, the microenvironment of each loop induces a specific conformation of the RGD motif and thus could modulate the affinity and selectivity for several integrins. Here we explore the insertion of the RGD sequences GRGDS into a number of loops of mCitrine and at the termini (Figure 1). We expressed this set of mCitrine variants, and evaluated their expression yield and fluorescence. A simple ELISA‐like integrin‐binding screening protocol was developed and used to evaluate the integrin affinities and specificities of the mCitrine‐RGD probes.
Figure 1.
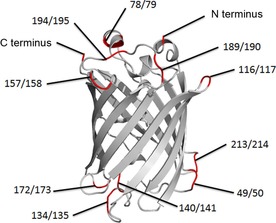
GRGDS insertion sites in mCitrine (PDB ID:1YFP).27
Two strategies were employed for introducing GRGDS at different positions in mCitrine. The amino acid sequence was either inserted into the mCitrine sequence, thus expanding the size of the loop, or replaced a five‐residue loop stretch, thus retaining the size of the loop. Reported mCitrine mutation sites22, 23 were used to introduce the GRGDS sequences (Figure 1), with the aim of retaining the integrity of the β‐barrel fold. The N and C termini were also tested for GRGDS incorporation, as these are non‐structured, exposed protein elements. Site‐directed mutagenesis was used to insert the GRGDS‐encoding sequence in the mCitrine gene. Except for the 190/194 (exchanged) construct, all designed DNA constructs were successfully obtained (Table 1). The constructs were expressed as soluble fusion proteins in Escherichia coli, with an N‐terminal His‐tag for purification and surface immobilization for integrin‐binding studies. The expressed proteins were purified by nickel‐nitrilotriacetic acid (Ni‐NTA) affinity chromatography with stringent washing to obtain mCitrine‐RGD constructs with high purity (Figure S1 in the Supporting Information). Protein yields were between 0.5 and 28 mg per liter of lysogeny broth, thus reflecting the influence of the RGD modifications on mCitrine expression level. Notably, good integrin‐binding constructs (e.g., 140/141 and 194/195; vide infra) expressed with higher yields. The functional integrity of the new protein constructs was assessed by analysis of the fluorescence properties (Table 1). The majority of the proteins exhibited fluorescence intensities and emission maxima similar to those of unmodified mCitrine, thus confirming the tolerance to RGD‐epitope introduction. Nonfluorescent protein constructs were obtained when five residues were exchanged for GRGDS in the amino acid region 116 to 144. Insertion in this region was only problematic for the 134/135 construct. These findings indicate that the loops in this region have an important, probably allosteric, role in maintaining chromophore integrity, so generally do allow insertion of additional amino acids.
Table 1.
mCitrine‐RGD protein constructs and characteristics.
| GRGDS position | Cloning[a] | Fluorescence[b] | ELISA signal intensity [a.u.] | |
|---|---|---|---|---|
| αvβ3 [c] | αvβ5 [c] | |||
| none | + | 398 | 0.058±0.010 | 0.123±0.003 |
| Inserted | ||||
| N terminus | + | n.a. | 0.137±0.014 | 0.133±0.008 |
| 49/50 | + | 445 | 0.238±0.008 | 0.146±0.006 |
| 78/79 | + | 342 | 0.098±0.002 | 0.142±0.006 |
| 116/117 | + | 425 | 0.332±0.013 | 0.249±0.009 |
| 134/135 | + | n.a. | 0.480±0.050 | 0.136±0.013 |
| 140/141 | + | 393 | 1.60±0.05 | 0.98±0.10 |
| 157/158 | + | 372 | 0.164±0.002 | 0.142±0.001 |
| 172/173 | + | 393 | 0.445±0.024 | 0.180±0.010 |
| 189/190 | + | 377 | 0.313±0.008 | 0.195±0.004 |
| 194/195 | + | 240 | 1.90±0.04 | 0.789±0.025 |
| 213/214 | + | 405 | 0.629±0.054 | 0.135±0.006 |
| C terminus | + | 392 | 0.057±0.010 | 0.142±0.005 |
| Exchanged | ||||
| 49/53 | + | 270 | 0.057±0.010 | 0.158±0.003 |
| 78/82 | + | 38 | 0.177±0.006 | 0.483±0.025 |
| 116/121 | + | n.a. | 0.124±0.003 | 0.125±0.003 |
| 135/138 | + | n.a. | 0.64±0.04 | 0.207±0.044 |
| 140/144 | + | n.a. | 0.63±0.02 | 0.439±0.014 |
| 156/160 | + | 479 | 0.244±0.009 | 0.164±0.010 |
| 171/174 | + | 392 | 0.244±0.011 | 0.188±0.030 |
| 190/194 | − | n.d. | n.d. | n.d. |
| 194/198 | + | 235 | 1.158±0.019 | 0.69±0.01 |
| 211/215 | + | 192 | 0.095±0.005 | 0.128±0.009 |
[a]+: successful; −: not successful. [b] Fluorescence intensity (arbitrary units): λ ex=515 nm, λ em=528 nm for 1 μm mCitrine‐RGD in PBS; n.a. not active; n.d. not determined. [c] ELISA signal intensity for two representative integrins. Values are mean±SD (n=3).
A microtiter‐plate‐based integrin‐binding assay was established to simply and rapidly evaluate the binding of the expressed fluorescent mCitrine‐RGD proteins to a set of integrin receptors. The assay was similar to an indirect enzyme‐linked immunosorbent assay (ELISA): integrin receptor was immobilized on a microtiter‐plate, and bound mCitrine‐RGD was detected by its N‐terminal His‐tag (goat anti‐His primary antibody with secondary antibody conjugated to horseradish peroxidase; Figure S2). The assay was initially optimized by using the 140/141 mCitrine‐RGD protein and non‐RGD‐containing mCitrine as a negative control. Optimal signal to background ratios were obtained when using 0.1 % (w/v) milk powder (blocking reagent) and 1:1000 (primary) and 1:10 000 (secondary) antibody dilutions (Figure S3).
All mCitrine‐RGD constructs, including nonfluorescent, were screened against four different human integrin receptors: the RGD binding αvβ3, αvβ5, and α5β1 receptors and the non‐RGD‐binding α1β1 receptor (Figure 2 and Table 1). The screening revealed clear position‐dependent differences in integrin binding. The mCitrine constructs with an additionally integrated GRGDS motif in position 140/141 and 194/195 turned out to be, overall, the strongest binders. For the mCitrine constructs in which five amino acids were exchanged for GRGDS, position 194/198 led to the most potent overall binder. The 78/82‐exchanged construct bound to all integrins tested. However, as the α1β1 integrin typically does not bind its ligand in an RGD‐dependent fashion, the binding of the 78/82 construct to this and the other integrins could result from nonspecific binding. Whereas integrin αvβ5 and especially αvβ3 showed relevant binding to a diverse set of protein constructs, integrins α5β1 and α1β1 basically showed no affinity for the mCitrine‐RGD constructs. In general, integration of the GRGDS motif by insertion led to constructs with higher integrin binding than those generated by exchange. The enhanced binding of the mCitrine mutants with an inserted GRGDS motif is intriguing; it might be due, in part, to the better surface exposure of the RGD motif caused by having five extra amino acids in the protein sequence.
Figure 2.
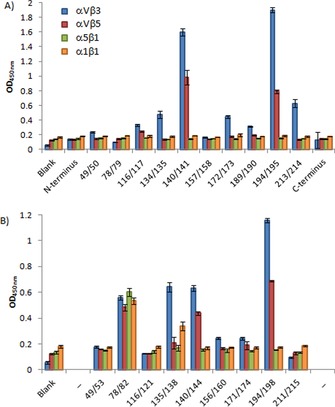
mCitrine‐RGD variants binding to human integrin receptors αvβ3, αvβ5, α5β1, and α1β1. A) GRGDS sequence inserted at the indicted positions. B) GRGDS substituted for five amino acids at the indicted positions.
The importance of the structure of the GRGDS sequence for affinity is illustrated by the absence of binding by the two constructs with the RGD epitope in the flexible N or C terminus (0/1 and 240/241). Both nonfluorescent mutants at amino acid 135 (134/135 and 135/138), and the fluorescent constructs 213/214 and (to a lesser extent) 172/173 displayed selectivity for αvβ3 over αvβ5. The local chemical environment of the loop thus confers binding selectivity.28
The two most prominent integrin‐binding probes, those with insertions at 140/141 and 194/195, were analyzed for integrin‐binding specificity. For this, GRGDS was substituted with GRADS. The single additional methylene group reduced the binding to all four integrins back to the mCitrine background levels (Figure S4), thus confirming the selective binding of the mCitrine‐RGD constructs to αvβ3 and αvβ5. The constrained turns thus orient the side chains with respect to the peptide backbone; this determines binding specificity, as postulated by Kessler and colleagues.8
Fluorescent proteins are versatile tools in chemistry and biology, and incorporation of RGD motifs in the loops provides entry to genetically encoded fluorescent probes for integrins. Our library of 22 mCitrine‐RGD constructs contained strong binders for αvβ3 and αvβ5 as well as integrin subtype‐selective binders. Loop selection was essential, as shown by the inactivity in integrin binding by constructs with the RGD sequence in the flexible termini and in some other loops. The insertion of RGD motifs in mCitrine loops thus imposes a specific structure and microenvironment on the RGD motif; this can facilitate integrin binding and selectivity, and provides a intriguing platform for studies of the structure–function relationship of diverse RGD geometries. The strong‐binding 140/141 and 194/195 constructs have GRGDS at opposite sites of the β‐barrel (Figure 1). The activity and selectivity at these two different positions in mCitrine could be advantageous in homo‐ or heterodimeric sandwich systems, thereby allowing binding to the two sides of mCitrine for integrin targeting and imaging at the cellular level. Constrained peptide motifs enable tissue targeting.5, 8, 9 The mCitrine scaffold also offers the possibility to insert other specific sequences (e.g., NGR29 and YIGSR)30 in combination with GRGDS. Such combinations provide access to specific targeting with fluorescent proteins.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The research was supported by funding from NanoSci‐E+ to the NanoActuate project through STW grant 11022‐NanoActuate and by the Ministry of Education, Culture and Science (Gravity program 024.001.035).
M. H. Sonntag, J. Schill, L. Brunsveld, ChemBioChem 2017, 18, 441.
References
- 1. Ruoslahti E., Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [DOI] [PubMed] [Google Scholar]
- 2. Hynes R. O., Cell 2002, 110, 673–687. [DOI] [PubMed] [Google Scholar]
- 3. De Arcangelis A., Georges-Labouesse E., Trends Genet. 2000, 16, 389–395. [DOI] [PubMed] [Google Scholar]
- 4. Sheppard D., Matrix Biol. 2000, 19, 203–209. [DOI] [PubMed] [Google Scholar]
- 5. Pierschbacher M. D., Ruoslahti E., Nature 1984, 309, 30–33. [DOI] [PubMed] [Google Scholar]
- 6. Pierschbacher M. D., Ruoslahti E., Proc. Natl. Acad. Sci. USA 1984, 81, 5985–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Albelda S. M., Buck C. A., FASEB J. 1990, 4, 2868–2880. [PubMed] [Google Scholar]
- 8. Meyer A., Auernheimer J., Modlinger A., Kessler H., Curr. Pharm. Des. 2006, 12, 2723–2747. [DOI] [PubMed] [Google Scholar]
- 9. Desgrosellier J. S., Cheresh D. A., Nat. Rev. Cancer 2010, 10, 9–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hersel U., Dahmen C., Kessler H., Biomaterials 2003, 24, 4385–4415. [DOI] [PubMed] [Google Scholar]
- 11. Alsibai W., Hahnenkamp A., Eisenblätter M., Riemann B., Schäfers M., Bremer C., Haufe G., Höltke C., J. Med. Chem. 2014, 57, 9971–9982. [DOI] [PubMed] [Google Scholar]
- 12. Pallarola D., Bochen A., Boehm H., Rechenmacher F., Sobahi T. R., Spatz J. P., Kessler H., Adv. Funct. Mater. 2014, 24, 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Marco R., Tolomelli A., Juaristi E., Gentilucci L., Med. Res. Rev. 2016, 36, 389–424. [DOI] [PubMed] [Google Scholar]
- 14. Hautanen A., Gailit J., Mann D. M., Ruoslahti E., J. Biol. Chem. 1989, 264, 1437–1442. [PubMed] [Google Scholar]
- 15. Dechantsreiter M. A., Planker E., Mathä B., Lohof E., Hölzemann G., Jonczyk A., Goodman S. L., Kessler H., J. Med. Chem. 1999, 42, 3033–3040. [DOI] [PubMed] [Google Scholar]
- 16. Haubner R., Schmitt W., Hölzemann G., Goodman S. L., Jonczyk A., Kessler H., J. Am. Chem. Soc. 1996, 118, 7881–7891. [Google Scholar]
- 17. Manning V. A., Hamilton S. M., Karplus P. A., Ciuffetti L. M., Mol. Plant-Microbe Interact. 2008, 21, 315–325. [DOI] [PubMed] [Google Scholar]
- 18. Khan T. A., Wang X., Maynard J. A., Biochemistry 2016, 55, 2078–2090. [DOI] [PubMed] [Google Scholar]
- 19. Tsien R. Y., Annu. Rev. Biochem. 1998, 67, 509–544. [DOI] [PubMed] [Google Scholar]
- 20. Chudakov D. M., Matz M. V., Lukyanov S., Lukyanov K. A., Physiol. Rev. 2010, 90, 1103–1163. [DOI] [PubMed] [Google Scholar]
- 21. Shaner N. C., Steinbach P. A., Tsien R. Y., Nat. Methods 2005, 2, 905–909. [DOI] [PubMed] [Google Scholar]
- 22. Abedi M. R., Caponigro G., Kamb A., Nucleic Acids Res. 1998, 26, 623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baird G. S., Zacharias D. A., Tsien R. Y., Proc. Natl. Acad. Sci. USA 1999, 96, 11241–11246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kadonosono T., Yabe E., Furuta T., Yamano A., Tsubaki T., Sekine T., Kuchimaru T., Sakurai M., Kizaka-Kondoh S., PLoS ONE 2014, 9, e103397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pavoor T. V., Cho Y. K., Shusta E. V., Proc. Natl. Acad. Sci. USA 2009, 106, 11895–11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Viswanathan S., Williams M. E., Bloss E. B., Stasevich T. J., Speer C. M., Nern A., Pfeiffer B. D., Hooks B. M., Li W.-P., English B. P., Tian T., Henry G. L., Macklin J. J., Patel R., Gerfen C. R., Zhuang X., Wang Y., Looger G. M. Rubin L. L., Nat. Methods 2015, 12, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wachter R. M., Elsliger M. A., Kallio K., Hanson G. T., Remington S. J., Structure 1998, 6, 1267–1277. [DOI] [PubMed] [Google Scholar]
- 28. Pfaff M., Tangemann K., Müller B., Gurrath M., Müller G., Kessler H., Timpl R., Engel J., J. Biol. Chem. 1994, 269, 20233–20238. [PubMed] [Google Scholar]
- 29. Corti A., Curnis F., Arap W., Pasqualini R., Blood 2008, 112, 2628–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Graf J., Iwamoto Y., Sasaki M., Martin G. R., Kleinman H. K., Robey F. A., Yamada Y., Cell 1987, 48, 989–996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary