

Abstract
The efflux transporter P-glycoprotein (P-gp) plays a vital role in the transport of molecules across cell membranes and has been shown to interact with a panoply of functionally and structurally unrelated compounds. How human P-gp interacts with this large number of drugs has not been well understood, although structural flexibility has been implicated. To gain insight into this transporter's broad substrate specificity and to assess its ability to accommodate a variety of molecular and structural changes, we generated human-mouse P-gp chimeras by the exchange of homologous transmembrane and nucleotide-binding domains. High-level expression of these chimeras by BacMam- and baculovirus-mediated transduction in mammalian (HeLa) and insect cells, respectively, was achieved. There were no detectable differences between wild-type and chimeric P-gp in terms of cell surface expression, ability to efflux the P-gp substrates rhodamine 123, calcein-AM, and JC-1, or to be inhibited by the substrate cyclosporine A and the inhibitors tariquidar and elacridar. Additionally, expression of chimeric P-gp was able to confer a paclitaxel-resistant phenotype to HeLa cells characteristic of P-gp-mediated drug resistance. P-gp ATPase assays and photo-crosslinking with [125I]-Iodoarylazidoprazosin confirmed that transport and biochemical properties of P-gp chimeras were similar to those of wild-type P-gp, although differences in drug-binding were detected when human and mouse transmembrane domains were combined. Overall, chimeras with one or two mouse P-gp domains were deemed functionally equivalent to human wild-type P-gp, demonstrating the ability of human P-gp to tolerate major structural changes.
Introduction
For nearly 40 years, the ATP-Binding Cassette (ABC) transporter P-glycoprotein (P-gp, ABCB1, MDR1) has been extensively studied due to its ability to recognize and transport an array of structurally diverse molecules. P-gp functions physiologically in both excretory and/or protective capacities by regulating concentration gradients of xenobiotics across biological membranes.1 Endogenously, P-gp is expressed at various physiological barriers, such as the blood-brain, blood-placental, and blood-testis barriers, where it prevents entry of exogenous molecules from the blood or lumen into organs. P-gp is also expressed in the liver, kidneys, lungs, and gastrointestinal tract to efflux exogenous compounds and their metabolites into the bile, urine, mucus, and feces, respectively.2 Thus, a consequence of P-gp's broad substrate specificity and its expression in various organs is that P-gp can affect drug absorption, distribution, metabolism and excretion.3 Additionally, in vitro expression of P-gp in cancer cells is notoriously associated with multidrug resistance (MDR). 1, 4
Structurally, P-gp is typified by a four-domain architecture consisting of two cytoplasmic nucleotide-binding domains (NBDs) that bind and hydrolyze ATP and two transmembrane domains (TMDs) that recognize and transport substrates. TMDs are embedded in the lipid bilayer with NBDs located in the cytosol, thus having access to cellular stores of ATP that can fuel the export of substrates. Mammalian P-gp homologs arise from the folding of a single polypeptide chain that is transcribed and translated in the order: (N-term) TMD1-NBD1-TMD2-NBD2 (C-term). P-gp folds to form two almost symmetrical halves, each consisting of a TMD containing six α-helices and a NBD. The halves are connected via a flexible linker region of approximately 75 amino acids in length, joining NBD1 to TMD2.5
A defining feature of P-gp is its ability to transport a panoply of structurally unrelated compounds. Studies by multiple laboratories have utilized a variety of biochemical approaches to investigate P-gp's polyspecificity, revealing that the drug-binding pocket of P-gp consists of multiple overlapping drug-binding sites.6 Despite these advances, the residues that comprise the drug-binding site(s) remain unknown. Additionally, understanding how P-gp interacts with drugs has been severely limited by the lack of high-resolution structures of human P-gp. Mouse P-gp (87% homologous to human P-gp) has been crystallized and several structures have been recently reported.7-9 However, while human and mouse P-gps are highly homologous, both subtle and profound functional differences between homologs have been reported. 10-13 For example, one study found cells expressing mouse P-gp were approximately three- and 22-fold more resistant to actinomycin D and colchicine than cells expressing comparable amounts of human P-gp.12 These findings indicate that the profile of substrates effluxed by mouse and human P-gp are overlapping but not identical.
The generation of protein chimeras has been used as a strategy to study numerous mechanistic, biochemical, and structural properties of ABC transporters.14-22 Chimeras generated by the exchange of homologous domains between mouse and human P-gp can be used to investigate the structural flexibility of the protein and probe for residues that are essential for normal protein function. Additionally, human-mouse chimeras can determine whether protein domains between species are functionally homologous. This study reports the generation of chimeras of mouse and human P-gp homologs by a domain swapping approach. We describe a comprehensive functional characterization of P-gp chimeras to ascertain if, despite sequence differences, the various domains have comparable function. Expression by BacMam- and baculovirus-mediated transduction was used to attain high-level expression in mammalian and insect cells, respectively. The ability of chimeras to interact with and transport various substrates, bind and hydrolyze ATP, and confer a MDR phenotype was investigated.
Experimental Procedures
Materials
All chemicals were sourced from Sigma Aldrich (St. Lois, MO) unless otherwise stated.
Bacterial Transformation and Purification of Plasmid DNA
Chemically competent One Shot Top10 (Life Technologies, Carlsbad, CA) E. coli cells (genotype: F- mcrA Δ(mrr-hdRMS-mcrBC) Φ80lacZΔM15 Δ lacX74 recA1 araD139 Δ(araleu)7697 galU galK rpsL (StrR) endA1 nupG) were transformed with plasmid DNA according to the manufacturer's protocol and cultured on LB agar plates containing 50 μg/mL kanamycin for 24 h at 34-37°C to yield single colonies. The mdr1a plasmid was kindly supplied by A. Schinkel (Netherlands Cancer Institute) and P. Gros (McGill University). Single colonies of transformed bacteria were inoculated into either 5 mL or 500 mL of LB media (50 μg/mL kanamycin) for small- and large-scale culturing and subsequent plasmid purification, respectively. For small-scale plasmid purification, 2-5 mL of culture was purified using the Wizard Plus SV Miniprep DNA Purification system (Promega, Madison, WI) according to the manufacturer's protocol. For large-scale plasmid purification, 200-500 mL of culture was purified using the HiSpeed Plasmid Maxi Kit (Qiagen, Limburg, Netherlands) according to the manufacturer's protocol. Purified plasmid DNA was eluted in sterile water and stored at -20°C. DNA was quantified using the ThermoScientific Nanodrop ND1000 Spectrophotometer.
Restriction Enzyme Digests and Ligations of DNA Fragments
All digests were performed using New England Biolabs Ltd. (Hitchen, UK) restriction enzymes according to the manufacturer's instructions. DNA fragments to be ligated were incubated with T4 DNA ligase and 1× ligation buffer in approximately a 1:7 vector to insert ratio for a minimum of 2 h at 18°C. Ligations were transformed into One Shot Top10 Chemically Competent E. coli cells as described above.
Agarose Gel Purification and Extraction of DNA Fragments
DNA to be gel purified was electrophoresed on a 0.7% agarose gel at 70 volts for 2 h to allow for optimal band separation. DNA was visualized using a hand-held UV light (365 nm). DNA bands of interest were excised using a sterile scalpel and purified using the QIAEXII Gel Extraction Kit (Qiagen, Limburg, Netherlands) according to the manufacturer's protocol. Purified DNA fragments were eluted with nuclease-free water and stored at -20°C.
Sequencing and Sequence Analysis of cDNA
Sequencing was carried out using Big Dye Terminator sequencing kits (Life Technologies, Carlsbad, CA). Analysis of sequencing chromatograms was carried out using MacVector alignment software (MacVector, Cary, NC). Sequencing chromatograms of wild-type mdr1a and MDR1 were aligned to reference sequences from Genbank identifications NM_011076.2 (mdr1a) and NM_000927.4 (MDR1). mdr1a cDNA carrying a M107L mutation was used for all cloning work carried out. Mouse P-gp carrying M107L is functionally equivalent to wild-type mouse P-gp. 23
Codon Optimization of mdr1a and MDR1 cDNA
Codon optimization for P. pastoris expression was performed using GeneOptimizer software and completed by GeneArt Gene Synthesis services (Life Technologies, USA).
Mammalian Cell Culture
All mammalian cell lines were grown at 37°C in 5% CO2 and cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% fetal bovine serum, 5 mM L-glutamine, 50 units/mL penicillin, and 50 μg/mL streptomycin. Additionally, the human and mouse P-gp-expressing cell lines KB-8-5-11 and C3M were cultured in 100 ng/mL and 1 μg/mL colchicine, respectively, to maintain P-gp expression.24, 25
Generation of Recombinant BacMam and Baculovirus
The Bac-to-Bac Baculovirus Expression System (Life Technologies, Carlsbad, CA) was used according to the manufacturer's protocol to generate recombinant baculovirus and BacMam baculovirus. Briefly, Gateway cloning was used to transfer wild-type and chimeric cDNA from bacterial cloning plasmids to the pDest-625 and pDest-008 expression vectors for BacMam and baculovirus expression, respectively. Expression vectors were transformed into E. coli DH10Bac cells where site-specific transposition of the gene of interest into a baculovirus shuttle vector (bacmid) occurred. Blue-white screening was carried out, white colonies were selected, and bacmid DNA was purified by alkaline lysis. Bacmid DNA was transfected into High-Five insect cells to generate recombinant baculovirus and BacMam baculovirus.
Transduction of HeLa Cells with BacMam Baculovirus
BacMam transductions were carried out according to a previously optimized protocol.26 In short, HeLa cells were counted, centrifuged, and resuspended to a density of 2.5 × 106 cells in 3 mL of DMEM media. BacMam virus was added to HeLa cells at a ratio of 50 virus particles per cell, and incubated at 37°C for 1 h, followed by addition of 23 mL of DMEM media and incubation for a further 3 h. Sodium butyrate (NaBT) was added to a final concentration of 10 mM and cells were incubated for 20 to 24 h at 37°C with 5% CO2 before harvesting for assays.
Baculovirus-Mediated Protein Expression in High-Five Insect Cells
Protein expression in insect cells was carried out using a previously optimized protocol.27 Briefly, a 1 L culture of High-Five cells was infected (MOI of 10) with recombinant baculovirus containing the P-gp construct to be expressed. Approximately 60-72 h post-infection, cells were examined for signs of baculovirus infection by trypan blue viability staining. After approximately 30 to 40% cell death was observed, cells were harvested, centrifuged, washed once with PBS containing 1% aprotinin, and stored at -80°C.
Western Blotting
Samples were prepared by combining the desired amount of protein with 1x SDS sample buffer and loaded into each well of a 7% Tris-Acetate NuPAGE gel (Life Technologies, Carlsbad, CA) and electrophoresed at 150 V. Protein was transferred to a PVDF membrane using the iBlot Dry Blotting System (Life Technologies, Carlsbad, CA). Membranes were blocked in 10% non-fat milk for at least 1 h. After blocking, membranes were incubated with either the C219 (Fujirebio Diagnostics, Malvern, PA), PEPG-13 (Laboratory of Cell Biology, National Institutes of Health, MD), or GAPDH (Life Technologies, Carlsbad, CA) primary antibody for 1 h. Membranes were washed and incubated with either the anti-mouse IgG HRP-linked (for C219 and GAPDH antibodies) or anti-rabbit IgG HRP-linked (for PEPG-13 antibody) secondary antibodies for an additional hour before addition of chemiluminescent horseradish peroxidase (HRP) antibody detection reagent and exposure to chemiluminescence film. The C219 and GAPDH antibodies were used at a 1:10,000 dilution and the PEPG-13 antibody was used at a 1:2000 dilution.
ATPase Assay
ATPase assays were carried out using membranes prepared from baculovirus-infected High-Five insect cells as previously described 27 and ATPase assays were performed as previously reported.28 Membrane vesicles (10-20 μg of protein/100 μL) were incubated with varying concentrations of the drug to be tested in the presence and absence of 0.3 mM sodium orthovanadate in ATPase 2x assay buffer (100 mM MES-Tris buffer (pH 6.8), 100 mM KCl, 10 mM sodium azide, 2 mM EGTA, 2 mM ouabain, 20 mM MgCl2, and 4 mM DTT) for 5 min. The reaction was started by the addition of 5 mM ATP. Each assay condition was allowed to incubate at 37°C for 20 mins before the addition of 100 μL of 5% SDS. To determine the release of inorganic phosphate, 500 μL of deionized water, 400 μL of Pi reagent (composed of 1% ammonium molybdate in 2.5 M sulfuric acid and 0.014% potassium antimonyl tartrate trihydrate) and 200 μL of 1% ascorbic acid were added into each tube. Color was allowed to develop for 10 mins before the absorbance was measured spectrophotometrically at 880 nm. Absorbance values were used to calculate the rate of ATPase specific activity (nmoles Pi/min/mg protein).
Transport of Fluorescent Substrates by Flow Cytometry
HeLa cells were trypsinized, counted, and 2 × 105 cells per assay conditions were transferred into polystyrene flow cytometry tubes. Cells were incubated in Iscove's Modified Dulbecco's Medium (IMDM) containing either 1.5 μM rhodamine 123 (Rh123), 10 μM calcein-AM (CAM) or 10 μM JC-1 with or without P-gp modulators at 37°C for 45 mins. After incubation, cells were centrifuged, supernatant was removed, and resuspended in 200 μL of cold PBS and placed on ice to stop transporter activity. The efflux rate of P-gp and chimeras was determined by depletion of cellular ATP by incubation in PBS containing 20 mM 2-deoxy-d-glucose (2-DG) and 5 mM sodium azide for 20 mins. After incubation, 2-DG and sodium azide were removed and cells were incubated on ice in the presence of Rh123 for an additional 20 mins. Transport of Rh123 by P-gp was initiated when PBS containing 50 mM glucose was added to cells and the incubation temperature was adjusted to 37°C. A total of 10,000 individual cell-counting events were recorded using a FACSCanto II flow cytometer. A 488 nm blue laser was used to excite all fluorophores and emission was measured either at 530/30 nm (Rh123 and CAM) or 575/26 nm (JC-1). FACS data were analyzed using FlowJo Single Cell Analysis software (FlowJo, Ashland, OR).
Determination of Cell Surface Expression of P-gp Chimeras Using MRK-16 Antibody
Cells were trypsinized, counted, and 2 × 105 cells per assay condition were transferred into polystyrene flow cytometry tubes. Cells were centrifuged, supernatant removed, and resuspended in 1 mL of IMDM containing either 1 μg human P-gp-specific MRK-16 primary antibody or the control primary antibody isotype control (mouse IgG2a Kappa) and incubated at 37°C for 30 mins. After incubation, cells were centrifuged and supernatant containing MRK-16 and control antibody was removed. One mL of IMDM media containing 1 μg secondary antibody (FITC-labeled anti-mouse IgG2a) was added to tubes and incubated in the dark at 37°C for an additional 20 mins. After incubation, flow cytometry was carried out as described above.
Cytotoxicity Assay
Cytotoxicity assays were carried out using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI). Cells were trypsinized and counted, and plated into 96-well, white-walled plates at a density of 4,000 cells per well in 100 μL of media. After a 24-h incubation, cells were dosed with the compound to be tested for toxicity. Cells were incubated with drug for an additional 48 h. Viability was assessed by the removal of cell culture media, and the addition of 50 μL of CellTiter-Glo Reagent. Viability was assessed by the quantification of luminescent signal using a Tecan Infinite M200 pro Luminometer.
Immunohistochemistry
Immunohistochemistry was performed using the anti-P-gp antibody C219 (1:200, BioLegend, San Diego, CA) on a Ventana Ultra automated immunostainer with the ultraView Universal DAB Detection Kit and hematoxylin counterstain (Roche, Indianapolis, IN).
Photo-Crosslinking With [125I]-Iodoarylazidoprazosin (IAAP)
The crude membranes of P-gp-expressing High-Five insect cells containing 0.5-0.7 μg/μL protein (100 μL volume) were incubated in 50 mM MES-Tris buffer (pH 6.8), in the absence (control) and presence of compounds at indicated concentration, for 10 mins at 37°C. The samples were then transferred to a water bath at room temperature under subdued light for the addition of 2-4 nM of [125I]IAAP (2,000 mCi/mmol, PerkinElmer, Boston, MA), and were subsequently photo-crosslinked upon exposure to UV light using a 365 nm UV-lamp for 10 mins. Proteins were separated by electrophoresis and [125I]IAAP incorporation into P-gp bands was quantified as described previously.29
Statistical Analysis
Curve-fitting and all statistical analysis was performed using GraphPad Prism 6.0. Multiple groups were analyzed using one-way analysis of variance (ANOVA) where significance was defined as p < 0.05. Data from dose-response curves of ATPase assay on crude membranes were fitted with the Michaelis-Menten curve from which the Km and Vmax values were calculated.
Results
Generation of P-gp Chimeras
Human-mouse P-gp chimeras were generated by the insertion of unique restriction enzyme sites into mdr1a (mouse) and MDR1 (human) cDNA and subsequent digestion and ligation of individual domains. All inserted restriction enzyme sites were created by synonymous mutations to mdr1a and MDR1 cDNA, and thus did not affect the final amino acid sequence of mouse and human P-gp. Human and mouse cDNA containing unique restriction enzyme sites at the domain boundaries were designed in silico and synthesized by GeneArt Gene Synthesis services (Life Technologies). Additionally, gene synthesis allowed for codon optimization of mdr1a and MDR1 cDNA for expression in the methylotrophic yeast Pichia pastoris. P. pastoris is commonly used for heterologous protein expression of ABC transporters, and has been used to obtain large amounts of purified mouse P-gp for crystallographic studies.7, 30, 31
While a total of 14 chimeras could be generated by the abovementioned domain swapping approach, four chimeras were constucted and characterized in this study as a “proof-of-principle” to test if human-mouse chimeras are useful for functional and biochemical analysis that may elucidate mechanistic aspects of the transporter. Chimeras generated were termed HHHM (consisting of human TMD1/NBD1/TMD2 and mouse NBD2), HMHH (consisting of human TMD1/TMD2/NBD2 and mouse NBD1), HMHM (consisting of human TMD1/TMD2 and mouse NBD1/NBD2), and HHMM (consisting of human TMD1/NBD1 and mouse TMD2/NBD2) (Figure 1). Human P-gp expressed from wild-type MDR1 cDNA and mouse P-gp expressed from wild-type mdr1a were termed HHHH_WT and MMMM, respectively. Human P-gp expressed from MDR1 codon-optimized for expression in P. pastoris was termed HHHH_Opti. The HHMM chimera was constructed from wild-type mdr1a and MDR1 cDNA while the HHHM, HMHH, and HMHM chimeras were constructed from codon-optimized cDNA.
Figure 1.
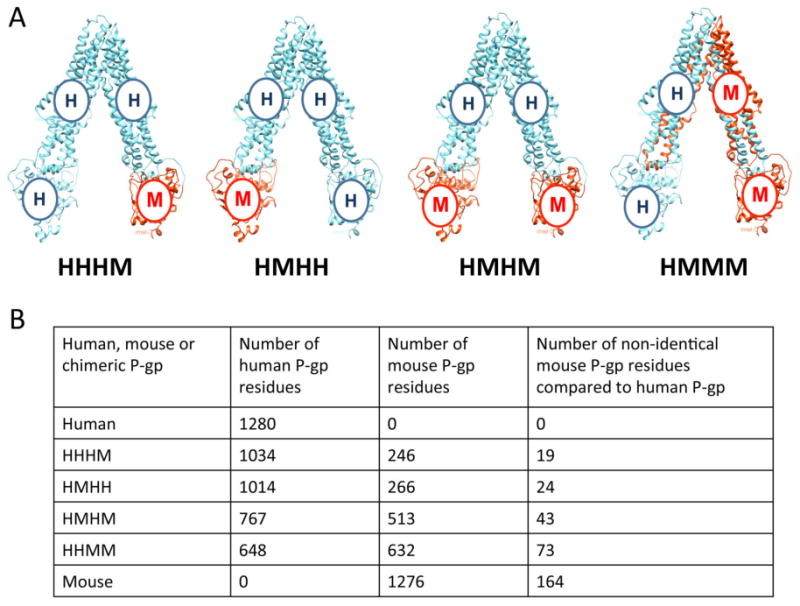
Representation of human-mouse P-gp chimeras. (A) Human (cyan) and mouse (orange) P-gp consist of two TMDs and two NBDs formed by the folding of a single polypeptide chain that is transcribed and translated in the order: (N-term) TMD1-NBD1-TMD2-NBD2 (C-term). A nomenclature to describe chimeras was devised by attributing the first letter from the cDNA of the species of origin, H or M for human or mouse P-gp, respectively, in a particular domain to the order that it is present in the polypeptide. The four chimeras generated in this study are depicted (human and mouse wild-type P-gp are not shown). (B) Table depicting the number of human and mouse residues present in each P-gp chimera. The last column specifies the number of mouse P-gp residues in the chimera that differ from those in human P-gp.
BacMam- and Baculovirus-Mediated Expression of P-gp Chimeras in Human and Insect Cells
Transient expression of P-gp chimeras was achieved using two well-characterized systems: baculovirus-mediated expression in insect cells and BacMam-mediated expression in HeLa cells.26, 32 Expression of chimeric, codon-optimized and wild-type human P-gp in HeLa cells was assessed by Western blotting (Figure 2A). The C219 antibody binds to a 14-residue epitope in the ATP-binding regions of NBDs, consisting of the sequence VVQEALDKAREGRT, that is highly conserved among P-gp homologs.33 C219 recognizes all mammalian P-gps, including mouse and human P-gp. Thus, it was expected that C219 would bind to all P-gp chimeras generated irrespective of whether they contained the mouse or human P-gp epitope. Two cell lines known to express either human or mouse P-gp were also included as positive controls: the human KB-8-5-11 cell line and the mouse C3M cell line. These cell lines have been previously characterized and are known to possess a MDR phenotype due to P-gp expression.24, 34
Figure 2.
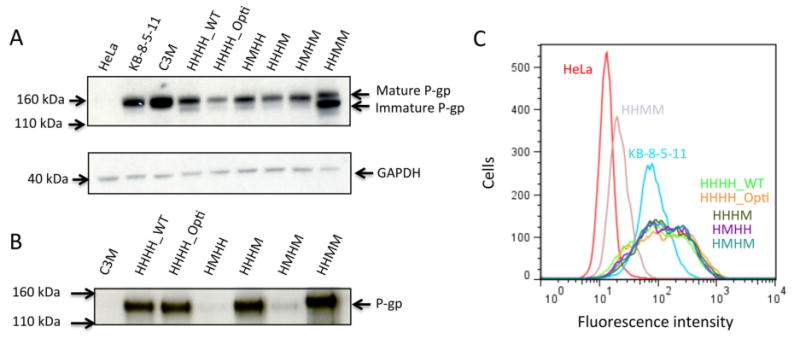
Detection of expression of chimeric P-gps by immunoblotting and flow cytometry with C219, PEPG-13, and MRK-16 antibodies. (A) SDS-PAGE was performed with non-transduced HeLa cells and HeLa cells transduced with wild-type human P-gp (HHHH_WT), codon-optimized P-gp (HHHH_Opti), and the HHHM, HMHH, HMHM, and HHMM chimeras (5.0 μg protein/lane) and Western blotting with the C219 monoclonal antibody was carried out. Additionally, the cell lysates of drug-selected cell lines C3M and KB-8-5-11-expressing wild-type mouse and human P-gp, respectively, were included. Size markers from a protein standard (Novex Sharp Pre-stained Protein Standard, Life Technologies) were used to assess molecular weight. Fully glycosylated P-gp (indicated as mature P-gp) is a 170-kDa protein and non-glycosylated P-gp is 140 kDa (indicated as immature P-gp). GAPDH was used as a loading control (37 kDa). (B) Verification of domains of human and mouse origin in chimeras using the human P-gp-specific antibody PEPG-13. Crude membranes were isolated from insect cells infected with recombinant baculovirus expressing P-gp chimeras as described in the Experimental Procedures. Crude membranes (1.0 μg protein/lane) were subjected to SDS-PAGE and subsequent immunoblotting with human PEPG-13 antibody at a 1:2000 dilution as previously described.36 Blots shown are representative of three individual experiments. (C) Cell surface expression of chimeras as detected by the human P-gp-specific monoclonal antibody MRK-16. Non-transduced and virus-transduced HeLa cells were incubated with MRK-16 antibody (1 μg/200,000 cells) for 30 mins at 37°C followed by incubation with FITC-conjugated anti-mouse IgG2a secondary antibody as described in Experimental Procedures. Trace colors correspond to the following key: non-transduced HeLa (red), KB-8-5-11 (cyan), HHHH_WT-transduced (light green), HHHH_Opti-transduced (orange), HHHM-transduced (dark green), HMHH-transduced (purple), HMHM-transduced (turquoise), and HHMM-transduced (lavender). Non-transduced HeLa cells and drug-selected KB-8-5-11 cells were used as negative and positive controls for MRK-16 binding, respectively. Each trace is representative of three independent experiments.
Several differences were observed in the expression levels and molecular weights of the P-gp chimeras expressed in HeLa cells (Figure 2A). At least two distinct bands between 140 and 170 kDa were present for chimeric, codon-optimized, and wild-type human P-gp when transduced in HeLa cells. Wild-type mouse and human P-gp contain three N-linked glycosylation sites in the first extracellular loop of TMD1. Fully glycosylated P-gp has a molecular weight of 170 kDa, while non-glycosylated P-gp is approximately 140 kDa.35 Previous studies have shown that the degree of glycosylation does not affect P-gp transport function.35 It is likely that the two distinct bands appearing at 170 and 140 kDa correspond to glycosylated (often referred to as “mature”) and non-glycosylated (“immature”) P-gp, respectively. While all transduced HeLa cells express a dominant glycosylated form migrating at 170 kDa and a secondary form migrating at 140 kDa, the HHMM chimera does not show this glycosylation pattern, having a large amount of non-glycosylated P-gp. Both wild-type mouse or human P-gp from the C3M or KB-5-8-11 drug-selected cell lines appear to contain fully glycosylated P-gp, lacking the secondary band that is present in all transduction conditions (P-gp was not detected in untransfected HeLa cells). It is not known why BacMam baculovirus transductions yield P-gp with variable glycosylation. However, previous reports using BacMam for the expression of human P-gp have indicated the presence of both glycosylated and non-glycosylated P-gp, and this was therefore not pursued further here.26 Levels of total P-gp expression also differ between chimeras. Codon-optimized P-gp (HHHH_Opti) and chimeras generated using codon-optimized cDNA (HHHM, HMHH, and HMHM) showed lower expression than wild-type P-gp (HHHH_WT). This could indicate that the codon optimization process for P. pastoris reduces P-gp expression in mammalian cells.
The PEPG-13 antibody was used to confirm the chimeric cDNA was accurately sub-cloned from the cloning vectors into baculovirus expression vectors (Figure 2B). PEPG-13 recognizes an epitope in NBD1 between amino acid residues 592 and 636, and is specific to human P-gp.28, 36 Given this, the PEPG-13 antibody would be expected to recognize human wild-type and codon-optimized P-gp and the HHHM and HHMM chimeras. As the HMHH and HMHM chimeras contain mouse NBD1, reactivity with PEPG-13 would not be expected. Western blotting of wild-type, codon-optimized and chimeric P-gp generated the expected pattern of PEPG-13 antibody reactivity. Faint bands are observed in the HMHH and HMHM lanes of the gel, which indicates a possible slight recognition of mouse P-gp by the PEPG-13 antibody. Note glycosylation in wild-type, chimeric, and codon-optimized P-gp are not observed, as insect cells do not glycosylate human or mouse P-gp.37
P-gp Chimeras Are Expressed at the Cell Surface to Similar Levels as Human Wild-Type P-gp
Subtle mutations in the primary amino acid sequence of proteins can potentially influence protein folding, trafficking, and degradation. The human-specific P-gp antibody MRK-16 was used to determine the cell surface expression in transduced HeLa cells (Figure 2C). MRK-16 is a conformationally-sensitive antibody that recognizes an external epitope composed of the extracellular loops of P-gp,38 and it is also specific to human P-gp. It was unknown if MRK-16 would bind to P-gp with chimerized TMDs, such as the HHMM chimera.
Wild-type P-gp (HHHH_WT), codon-optimized human P-gp, and all chimeras containing both human TMDs (HHHM, HMHH, and HMHM chimeras) were recognized by MRK-16 (Figure 2C). This can be appreciated by the approximately 10-fold increase in mean fluorescence intensity indicating MRK-16 binding compared with non-transduced HeLa cells. The HHMM chimera, which contains chimerized TMDs and therefore a mixture of extracellular loops from both mouse and human P-gp, showed only an approximately 2-fold increase in fluorescence as compared with non-transduced HeLa cells, potentially indicating lower cell-surface expression. However, immunohistochemistry performed with the C219 antibody displayed equivalent cell surface expression, indicating the reduced fluorescence intensity observed in flow cytometry may be due to alteration of the MRK-16 epitope in the HHMM chimera (Figure S1). There were no noteworthy differences in MRK-16 binding between the HHHH_WT control construct and the HHHH_Opti, HHHM, HMHH, and HMHM chimeras, indicating that both wild-type human P-gp and chimeras recognized by MRK-16 were expressed to a similar extent on the cell surface. It can be concluded that the chimerization process did not greatly affect cell surface expression for the chimeras that showed reactivity with MRK-16.
Steady-State Rhodamine 123, Calcein-AM, and JC-1 Efflux Assays
Rhodamine 123 (Rh123), calcein-AM (CAM), and JC-1 efflux assays were used to establish chimera functionality (Figure 3). All fluorophores are membrane-permeable and substrates of wild-type mouse and human P-gp. Rh123, CAM, and JC-1 are structurally diverse compounds. While there is no detailed understanding of where each substrate interacts inside the drug-binding pocket, it is probable that, due to the structural diversity of these compounds, they interact with distinct sites within the pocket.39 Therefore, the use of Rh123, CAM, and JC-1 may yield insight into distinct substrate profiles of the chimeras versus wild-type P-gp homologs. Additionally, non-functional EQ (Walker B glutamate substitution; E556Q/E1207Q) mutant P-gp, with no ATPase activity, was included to ensure that differences in fluorophore accumulation were independent of the transduction process.40
Figure 3.
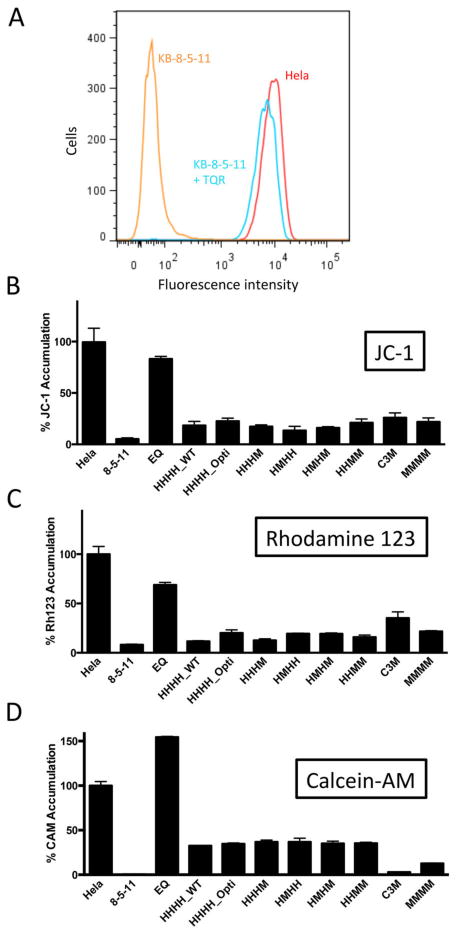
Efflux of the fluorescent substrates Rh123, JC-1 and CAM by chimeric P-gp. (A) A representative histogram exhibiting P-gp-mediated efflux of the fluorescent substrate Rh123 by P-gp-expressing KB-8-5-11 cells (orange) compared to non-P-gp-expressing HeLa cells (red) or KB-8-5-11 cells with inhibited P-gp (KB-8-5-11 +TQR, cyan). HeLa cells or KB-8-5-11 cells were incubated with 1.5 μM Rh123 in the presence or absence of 100 nM TQR, a P-gp inhibitor, for 45 mins and transport of fluorescent substrates by flow cytometry was carried out as described under Experimental Procedures. (B-D) HeLa cells were transduced with wild-type (HHHH_WT), codon-optimized (HHHH_Opti), HHHM, HMHH, HMHM, HHMM, MMMM, and non-functional EQ P-gp as described above. Transduced cells and the drug-selected KB-8-5-11 and C3M cell lines were incubated with substrates (B) JC-1, (C) Rh123, and (D) CAM as described in Experimental Procedures to determine functionality. All chimeras were functional and able to transport the fluorescent substrates tested, as seen by the significant reduction in fluorophore accumulation in chimera-expressing HeLa cells compared to non-transduced HeLa cells (p≤ 0.0001). Data were normalized to 100% accumulation in non-transduced HeLa cells. Data represent the mean fluorescence intensity and standard deviation of three independent flow cytometry experiments. Multiple groups were analyzed using one-way ANOVA where significance was defined as p < 0.05 in GraphPad Prism.
All chimeras were functional and able to efflux Rh123, CAM, and JC-1 compared with parental HeLa cells (Figure 3; p ≤ 0.0001). There were no significant differences in the extent of efflux of these compounds between chimeras and the HHHH_WT P-gp at steady state. HeLa cells transduced with non-functional EQ P-gp accumulated levels of intracellular fluorescence comparable to those of non-transduced HeLa cells. Additionally, the codon optimization process did not influence fluorophore transport, and there were no differences between HHHH_WT and HHHH_Opti efflux. Wild-type human and mouse P-gp in the drug-selected cell lines KB-8-5-11 and C3M were also able to efflux all fluorophores efficiently. While it is tempting to draw conclusions regarding species differences between mouse and human P-gp from these data (for example, C3M cells appear less efficient at Rh123 efflux than KB-8-5-11 cells), species differences in fluorophore affinity cannot be deduced from these assays. This is due to variable P-gp expression levels between cell lines and differences in cellular backgrounds; the C3M cell line is derived from the mouse embryonic fibroblast cell line NIH/3T3, while the KB-8-5-11 cell line is derived from a sub-line of the human adenocarcinoma HeLa cell line that may affect baseline fluorophore accumulation in cells.
Modulators Tariquidar, Cyclosporine A, and Elacridar Inhibit Transport by Chimeric P-gps
P-gp is known to contain multiple drug-binding sites within its central cavity. Therefore, it was unknown if or how the chimerization process would affect the ability of inhibitors to interact with P-gp chimeras. Additionally, inhibitors can be used to confirm that decreased fluorophore accumulation is attributable to P-gp expression, as the presence of P-gp inhibitors during the course of the assay should fully inhibit fluorophore efflux and thus restore fluorescence intensities to levels of non-transduced HeLa cells. The P-gp inhibitors tariquidar (TQR), cyclosporine A (CsA), and elacridar were utilized for their capacity to inhibit P-gp and allow for Rh123 accumulation within transduced cells.
TQR, CsA, and elacridar were all able to significantly increase Rh123 accumulation compared with uninhibited treatments for all chimeras (Figure 4; p≤ 0.0001). The presence of inhibitors did not significantly change Rh123 accumulation in cells transduced with the EQ mutant, non-transporting P-gp. It can be appreciated that in some instances the presence of an inhibitor results in the intracellular accumulation of Rh123 above 100%. For example, various chimeras and P-gp homologs inhibited by CsA accumulate Rh123 to a greater level than non-transduced HeLa cells. This effect seems to be independent of P-gp expression, as even non-transduced Hela cells that do not express P-gp accumulate higher levels of intracellular Rh123 in the presence of CsA. This effect has been previously observed, but it is unclear why some inhibitors can increase levels of intracellular fluorescence. This effect may be attributed to off-target effects of the inhibitor at concentrations used in this assay that have yet to be elucidated.
Figure 4.
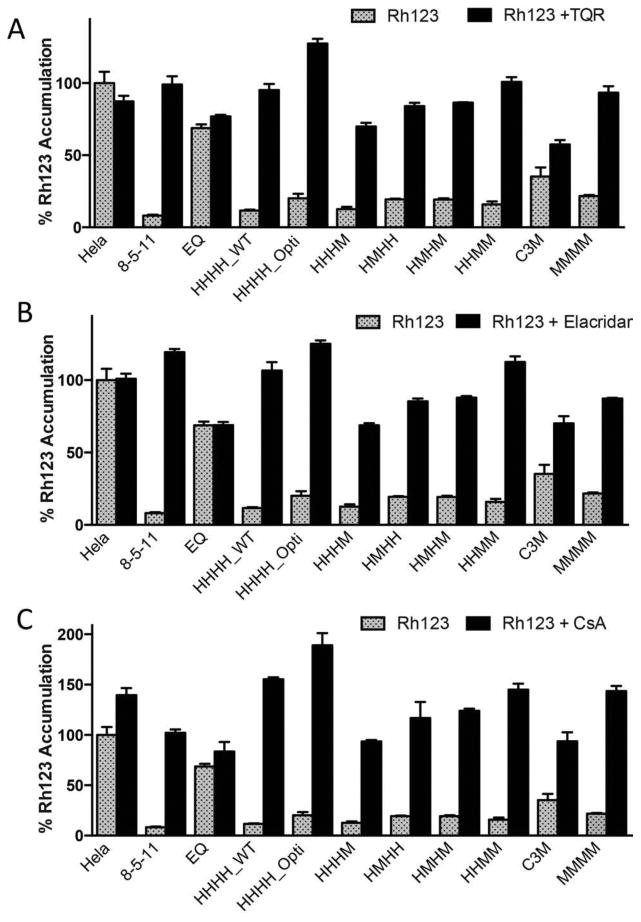
Inhibition of Rh123 efflux by chimeras with tariquidar, cyclosporine A, and elacridar. HeLa cells were transduced with wild-type (HHHH_WT), codon-optimized (HHHH_Opti), HHHM, HMHH, HMHM, HHMM, MMMM and non-functional EQ P-gp as described above. Transduced cells and the drug-selected KB-8-5-11 and C3M cell lines were incubated with 1.5 μM Rh123 either alone (gray bars) or in combination with the P-gp modulators. (A) TQR (100 nM), (B) elacridar (100 nM), and (C) CsA (10 μM) (black bars). All data were normalized to 100% accumulation in non-transduced HeLa cells. Data are presented as the mean fluorescence intensity and standard deviation of three individual flow cytometry events. Multiple groups were analyzed using one-way analysis of variance (ANOVA) where significance was defined as p < 0.05 in GraphPad Prism.
The Rate of Efflux of Rhodamine 123 is Not Affected by Chimerization of P-gp
Flow cytometry time-course experiments were undertaken to determine if chimeras showed differences in the rate of Rh123 efflux (Figure 5). There were no noteworthy differences in the rate of Rh123 efflux between codon-optimized (HHHH_Opti), chimeric (HHHM, HMHH, HMHM, HHMM), and wild-type (HHHH_WT) P-gp. All chimeras reached a steady state of efflux within 10 mins. Rh123 efflux for up to 45 mins was also measured. However, no changes in Rh123 efflux occurred after 15 mins (data not shown). Non-transduced HeLa cells and HeLa cells expressing the EQ mutant P-gp were also assessed for Rh123 efflux and did not exhibit efflux activity (data not shown). Additionally, the time required for P-gp chimeras and homologs to efflux 50% of intracellular Rh123 (T1/2) was calculated. Slight variations in T1/2 were observed with the HHHM and HMHH chimeras having a 1.8-fold decrease in efflux rate compared to HHHH_WT P-gp. While this difference may indicate that the HMHH and HHHM are slightly less efficient at Rh123 efflux, differences in efflux appear to be small.
Figure 5.
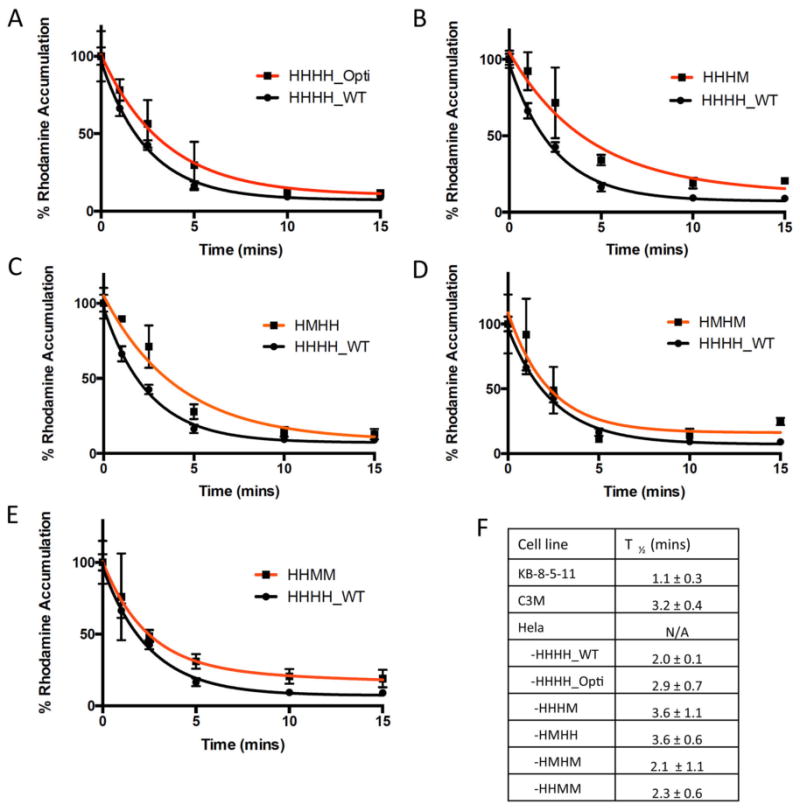
Time-course of Rh123 efflux by HeLa cells expressing WT and chimeric P-gps. Rh123 efflux of HeLa cells transduced with BacMam constructs expressing (A) HHHH_Opti, (B) HHHM, (C) HMHH, (D) HMHM and (E) HHMM as compared with wild-type P-gp (HHHH_WT). 200,000 cells were de-energized before loading with Rh123 as described under Experimental Procedures. The efflux of the Rh123 was initiated by resuspending cells in PBS with 50 mm glucose at 37°C and was monitored over time by flow cytometry. The accumulation of Rh123 at 0 min is taken as 100%, and the values are calculated as the percentage of remaining fluorescence inside the cells. Each time point represents the average and standard deviation of triplicate assays. (F) T1/2 values for all chimeras, and wild-type human and mouse P-gp from KB-8-5-11 and C3M drug-selected cell lines (mean ± SD).
Chimeric P-gp is Able to Confer a Drug-Resistant Phenotype to HeLa Cells
In order to assess if chimeric P-gps were able to confer a drug-resistant phenotype on HeLa cells, transductions were completed in the absence of the histone deacetylase inhibitor sodium butyrate (NaBT) due to its confounding cytotoxic effect. Transductions completed without NaBT result in reduced, but detectable, levels of cell surface P-gp expression (Figure S2). Resistance of transduced cells can be attributed to expression of P-gp by abrogation of resistance when co-incubated with the P-gp inhibitor TQR during the course of the cytotoxicity assay. The degree of P-gp-mediated resistance can be expressed as the ratio of IC50 values with and without the inhibitor TQR. This ratio is often referred to as a resistance ratio, calculated by the following equation:
A resistance ratio of 1 indicates that there is no resistance attributable to P-gp expression, while a resistance ratio > 1 indicates resistance that is reversible upon P-gp inhibition.41
HeLa cells transduced with chimeric, codon-optimized, and wild-type P-gp displayed a paclitaxel-resistant phenotype (Table 1). Co-incubation of transduced HeLa cells with paclitaxel and the P-gp inhibitor tariquidar abrogated resistance. HeLa cells transduced with wild-type and chimeric P-gp showed modest levels of paclitaxel resistance (resistance ratios of 14-52) compared to the drug-selected P-gp-expressing KB-8-5-11 cell line (resistance ratio of 929). This is most likely due to lower expression level achieved due to the absence of NaBT during viral transduction.
Table 1. Cytotoxicity assay of chimeric P-gp with the chemotherapeutic drug paclitaxela.
| Cell Line | IC50 Paclitaxel (nM) | IC50 Paclitaxel +TQR (nM) | Resistance Ratio |
|---|---|---|---|
| KB-8-5-11 | 3718 ± 193 | 4.0 ± 0.1 | 929 |
| HeLa | 9.2 ± 1.4 | 5.5 ± 1.7 | 1.67 |
| -HHHH_WT | 936 ± 81 | 19.4 ± 2.8 | 48.2 |
| -HHHH_Opti | 284 ± 19 | 11.9 ± 0.7 | 23.8 |
| -HHHM | 773 ± 22 | 12.0 ± 1.0 | 64.4 |
| -HMHH | 421 ± 126 | 14.0 ± 1.9 | 30.1 |
| -HMHM | 221 ± 7 | 15.3 ± 1.2 | 14.4 |
| -HHMM | 1049 ± 68 | 20.0 ± 0.6 | 52.5 |
Cytotoxicity assays of virus-transduced HeLa cells were carried out in the absence of 10 mM NaBT as described in Experimental Procedures. The ability of chimeras to confer a paclitaxel-resistant phenotype can be assessed by the increased IC50 values of HeLa cells expressing chimeras as compared to parental HeLa cells. Resistance to paclitaxel can be attributed to expression of chimeric P-gp, as resistance is reversed when cells are co-incubated with the P-gp inhibitor TQR (100 nM) during the course of the assay. Resistance ratios are shown to indicate the degree of P-gp-mediated resistance.
Substrates and Modulators Modulate the ATPase Activity of P-gp Chimeras
ATPase assays were carried out to determine differences in ATP hydrolysis between chimeric, codon-optimized and wild-type human P-gp. Crude membranes were isolated from insect cells infected with baculovirus genetically modified to express P-gp chimeras. All infections yielded equivalent expression of P-gp (Figure 6A). ATPase assays were completed with 1, 5, 10, 25, 50, and 100 μM verapamil (Figure 6B-F). Higher concentrations of verapamil inhibited P-gp ATPase activity and were therefore not tested. Dose-response curves were used to determine maximal fold-stimulation and concentration required for 50% stimulation. A side-by-side comparison of mouse and human P-gp verapamil-stimulated ATPase activity has been previously reported by this laboratory, and thus was not carried out here. 23
Figure 6.
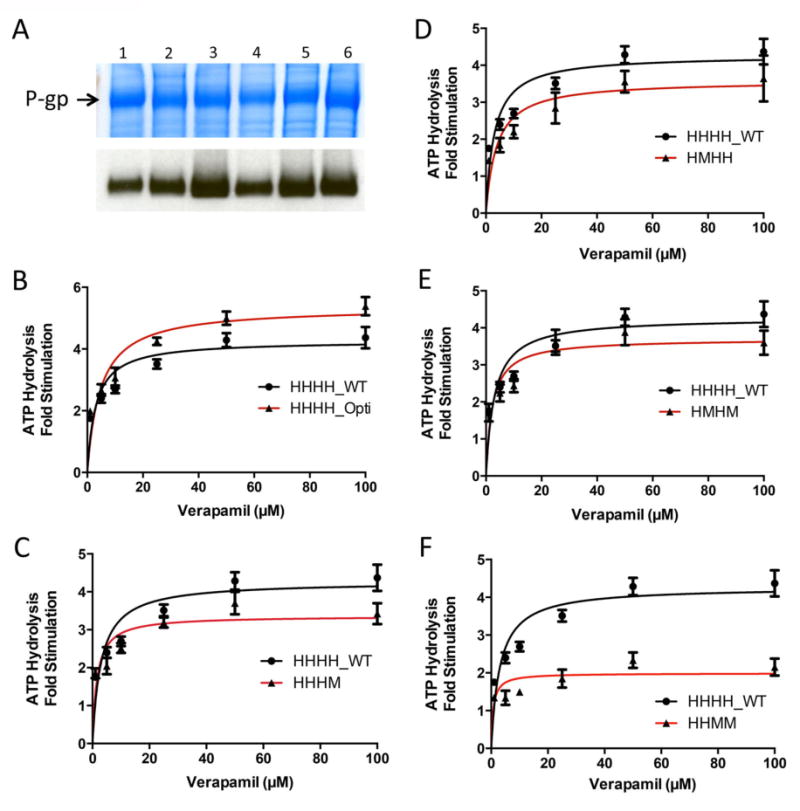
Stimulation of ATPase activity of P-gp chimeras by verapamil. (A) Colloidal blue stain of crude membranes isolated from infected insect cells (30 μg protein/lane). A band of over-expressed P-gp, indicated by the arrow, was seen in all crude membrane samples expressing WT or chimeric P-gp. Western blotting with the C219 antibody was completed to confirm that the overexpressed band seen in the colloidal blue stain was chimeric P-gp (1 μg protein/lane). Lanes for both the blue stain and Western blot are indicated: lane 1) HHHH_WT, 2) HHHH_Opti, 3) HHHM, 4) HMHH, 5) HMHM, and 6) HHMM. B-F, Dose response ATPase assays of (B) HHHH_Opti, (C) HHHM, (D) HMHH, (E) HMHM and (F) HHMM compared with wild-type human P-gp (HHHH_WT) in the presence of 0, 1, 5, 10, 25, 50 and 100 μM verapamil. ATPase activity is reported as the fold-change in the rate of ATP hydrolysis over basal levels. Data represent average and standard deviation of technical duplicates from three individual assays.
A range of maximal fold-stimulation values was observed, potentially suggesting altered function among chimeras. HHHH_Opti showed an approximately 30% increase in maximal fold stimulation as compared to wild-type P-gp. In contrast, the HHMM and HHHM chimeras displayed a 60% and 25% decrease in maximal fold stimulation, respectively, compared with wild-type P-gp. The HMHM and HMHH chimeras also showed smaller differences in maximal fold stimulation compared to wild-type P-gp, having a 16% and 18% decrease for HMHH and HMHM chimeras, respectively. While differences in maximal fold-stimulation were observed, values of 50% maximal fold stimulation were similar for all chimeras, ranging from 6.5 to 14 μM.
Several explanations may account for differences in maximal fold-stimulation observed. Firstly, chimeras may be differentially stimulated by verapamil due to changes in the drug-binding pocket. Changes in the pocket may arise directly, from chimerization of the TMDs as seen in the HHMM chimera, or indirectly, by subtle conformational changes that may have arisen from either the chimerization or codon optimization process even when both TMDs are comprised of human P-gp. Secondly, all chimeras contain a mixture between human and mouse TMDs and NBDs, a factor that may affect the way NBDs and TMDs communicate to hydrolyze ATP and transport substrates. Thirdly, chimerization of the NBDs may affect the rate of ATP hydrolysis, a characteristic of chimeras that could be detected in this assay system but not in whole-cell based flow cytometry assays. Lastly, this effect could be specific to the substrate verapamil.
To determine if differences in ATPase activity were specific to verapamil, a panel of eight known P-gp ATPase stimulators was tested with HHHH_WT and HHMM P-gp (Figure 7). The HHMM chimera was chosen for this analysis, as it displayed the greatest difference in maximal fold stimulation compared to wild-type human P-gp (Figure 6). The compounds cis-flupenthixol, CAM, nicardipine, paclitaxel, vincristine, Rh123, and loperamide were used to probe for differential ATPase activity between HHHH_WT and HHMM P-gp. No significant differences in ATPase stimulation between HHHH_WT and HHMM P-gp were seen with cis-flupenthixol or paclitaxel. However, CAM, nicardipine, vincristine, Rh123, and loperamide were all able to stimulate ATPase activity of HHHH_WT P-gp to a greater extent than the HHMM chimera. These data indicate that the differences seen previously were not specific to verapamil, but rather the ATPase activity of the HHMM chimera was less stimulated by various P-gp stimulators compared with wild-type human P-gp.
Figure 7.
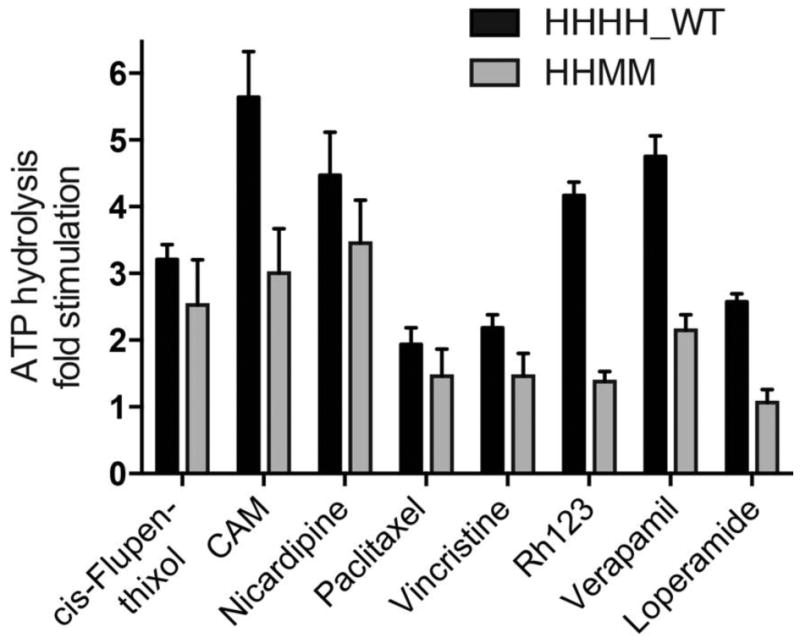
Effect of selected compounds on the ATPase activity of HHHH_WT versus HHMM P-gp. Eight compounds that are known substrates or modulators of P-gp were selected to determine if there were differences in ATPase activity of HHHH_WT and HHMM P-gp. The following concentrations were used for each assay: 20 μM cis-flupenthixol, 10 μM calcein-AM (CAM), 20 μM nicardipine, 20 μM paclitaxel, 20 μM vincristine, 500 μM Rh123, 25 μM verapamil, and 20 μM loperamide. To determine if differences in ATPase activity were significantly different between HHHH_WT and HHMM P-gp, multiple groups were analyzed using one-way ANOVA where significance was defined as p < 0.05 in GraphPad Prism.
[125I]-Iodoarylazidoprazosin (IAAP) Photo-Crosslinking
[125I]-IAAP photo-crosslinking was used to investigate the ability of chimeric and wild-type human and mouse P-gp to bind substrates at the primary drug-binding site. [125I]IAAP is a transport substrate of P-gp, binding to P-gp in the drug-binding pocket. [125I]IAAP can be covalently crosslinked to P-gp by exposure to UV light.42-44 Studies have shown that [125I]IAAP binding is displaced in human P-gp by various known P-gp substrates and modulators in a concentration-dependent manner.42, 43 We investigated the ability of the P-gp substrate CsA and inhibitors TQR, and zosuquidar (LY335979) to displace [125I]IAAP binding in human and mouse P-gp, and the HHHM, HMHH, HMHM, and HHMM chimeras (Figure 8).
Figure 8.
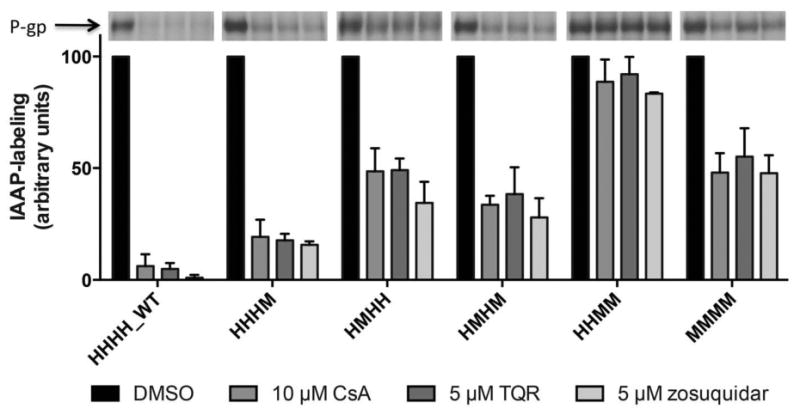
[125I]-IAAP photo-crosslinking of P-gp chimeras. Inhibition of [125I]IAAP-labeling of human (HHHH_WT), mouse (MMMM), and chimeric P-gps by CsA, TQR and zosuquidar is shown as a bar graph. The first bar (black) represents the control (100% binding) and contains 1% DMSO, which is the solvent used to prepare the drug solutions. The other bars represent the [125I]IAAP-labeling remaining upon inhibition by drugs. Representative autoradiograms are shown at the top of the figure. The experiments were carried out with crude membranes of High-Five insect cells expressing P-gp, as described in Experimental Procedures. Data represent mean values and standard deviation of at least three independent experiments, calculated in GraphPad Prism.
[125I]IAAP binding was fully or partially displaced with each modulator tested and with the HHHM, HMHH, and HMHM chimeras and human and mouse P-gp. In contrast, CsA, TQR, and zosuquidar were not able to displace [125I]IAAP binding in the HHMM chimera. While these data demonstrate that CsA, TQR, and zosuquidar do not interact with HHMM chimera at the primary drug-binding site, it is known from the flow cytometry efflux studies presented here that both CsA and TQR do, in fact, modulate the function of HHMM P-gp (Figure 4). Taken together, these data suggest that the conformation of the drug-binding pocket of the HHMM chimera is altered in a way that affects where specific P-gp modulators interact in the drug-binding pocket of P-gp. We recently observed that substrates and modulators bind at secondary or alternative sites when residues of the primary site are modified,45 and the data with the HHMM chimera are consistent with these studies.
To further understand these subtle changes in the drug-binding pocket of the HHMM chimera, molecular analysis of the central cavity of the HHMM chimera based on the recently published X-ray structure of mouse P-gp solved at 3.4 Å resolution (4Q9H.pdb) was carried out. This analysis revealed that at least five residues in TMD2 of mouse P-gp (S725, V726, V731, V732 and I977) are different from the human P-gp (A729, I730, I735, I736 and V981, respectively; human sequence numbering is +4 shifted with respect to mouse numbering) (Figure 9). These residues may play a critical role in the distinctive biochemical behavior of the HHMM chimera versus human P-gp. The residues S725 and V726 (A729 and I730 in human) are located in TM7, next to F724 (F728 in human), which is a residue demonstrated by us45 and others46 as a critical residue for drug binding in human P-gp. Residues V731 and V732 (I735 and I736 in human P-gp) are located in the deeper site of the cavity (i.e., at the vertex of the cone defined in the X-ray structures of mouse P-gp); some computational studies have shown this site as the site where many drugs are expected to bind P-gp.47 These residues are clearly situated in the drug translocation pathway of P-gp. Residue I977 (V981 in human) is located in TM12, next to V982, which is also a residue shown to be part of the drug-binding site of P-gp.45, 48, 49
Figure 9.
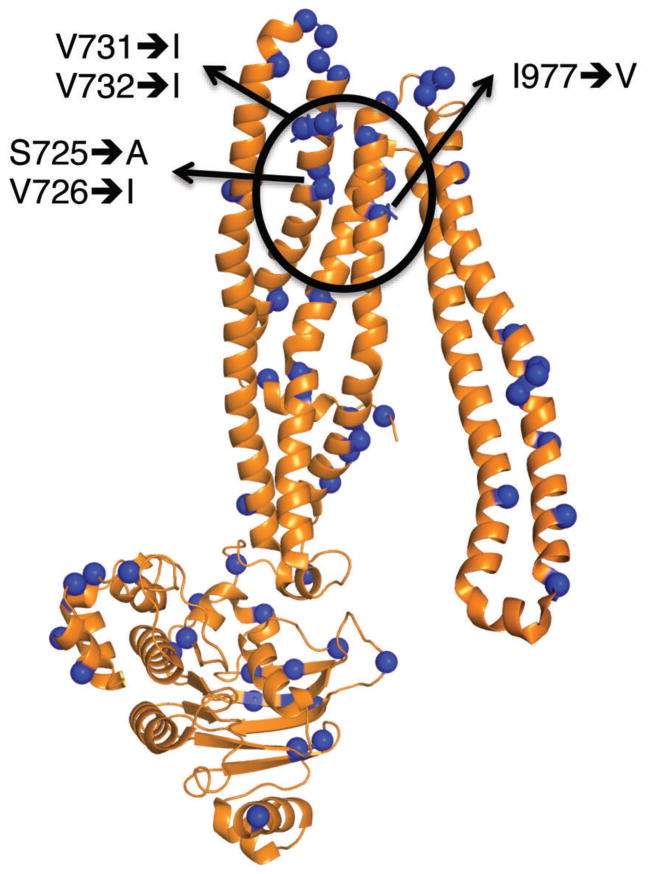
Molecular structure of the mouse TMD2-NBD2 domains of the HHMM P-gp chimera. The figure shows a cartoon model of the TMD2-NBD2 domains from the X-ray structure of mouse P-gp (4Q9H.pdb). The locations of the residues that are different from human P-gp are indicated as blue spheres. The drug-binding pocket is delineated with a black circle and the residues thought to be critical for the differences in drug binding between the HHMM chimera and human P-gp are indicated. Bolded arrows indicate the analogous residue present in human P-gp.
Interestingly, the substrates and modulators used in this study were unable to fully displace [125I]IAAP binding in mouse P-gp (the displacement was 40-50%). This finding most likely highlights interspecies differences in the conformation of the drug-binding pocket.
Discussion
The work presented here describes the generation and functional characterization of human-mouse P-gp chimeras. These studies were carried out to determine if human P-gp can accommodate significant structural changes and remain functional. Chimeras were generated by domain swapping between mouse and human P-gp (87% homologous), resulting in the generation of HHHM, HMHH, HMHM, and HHMM chimeras (Figure 1). Numerous assays were employed to obtain a thorough functional characterization of chimeras, including flow cytometry, cytotoxicity, Western blotting, ATPase, and [125I]IAAP photo-crosslinking assays. Chimeras were expressed in both mammalian and insect cells through BacMam- and baculovirus-mediated transient transduction, respectively; expression levels were similar in both experimental systems. No differences were detected in the ability of HeLa cells transduced with chimeras to efflux the fluorophores calcein-AM, rhodamine 123, and JC-1 as compared to wild-type P-gp. Additionally, the P-gp substrate cyclosporine A and inhibitors tariquidar and elacridar were able to reverse P-gp-mediated Rh123 efflux from cells equally in cells transduced with chimeric and wild-type P-gp. Lastly, chimeras were able to confer a paclitaxel-resistant phenotype to HeLa cells, which is characteristic of P-gp-mediated drug resistance.
Modest functional differences were observed with the HHMM chimera. HHMM P-gp showed decreased ATPase activity for six out of eight compounds tested and a 60% decrease in maximal fold-stimulation upon verapamil stimulation (Figures 6 and 7). Additionally, [125I]IAAP photo-crosslinking was unable to be displaced by the P-gp substrate CsA and modulators TQR, and zosuquidar (Figure 8). These findings indicate an altered drug-binding pocket in the HHMM chimera. While differences were observed in biochemical assays, whole cell-based assays such as flow cytometry and cytotoxicity assays did not indicate functional differences between HHMM and wild-type human P-gp. For example, while the HHMM chimera showed decreased P-gp ATPase activity to Rh123 and CAM, these substrates were also used as fluorophores in flow cytometry transport assays where Rh123 and CAM were not differentially effluxed from cells (Figure 3). These findings indicate that while differences in drug binding occur as a result of chimerization, these differences do not affect the ability of the HHMM chimera to efflux substrates or generate a MDR phenotype. These results are consistent with pharmacologic studies showing small differences in substrate specificities between human and mouse P-gp.10, 50 These findings also reinforce the earlier findings that substrates can be transported from secondary or alternative sites when the primary site is altered.45
Pharmacogenomic investigations into P-gp have characterized at least 50 SNPs naturally occurring in the human genome.51 While SNPs may contribute to subtly altered protein function, there are currently no known individuals harboring non-functional P-gp, indicating a variety of SNPs can be tolerated and result in functional P-gp. One interpretation is that non-functional P-gp is embryonically lethal. However, this is not supported by studies from mdr1a/mdr1b-/- knockout mice where P-gp is not necessary for murine survival.52 The studies presented here indicate that human P-gp can tolerate a variety of alterations and support the pharmacogenomic observation that, while P-gp is present with various SNPs and haplotypes within populations, these amino acid residue changes may alter substrate specificity to some extent, but seem to have little effect on the overall function of this transporter.
While human and mouse P-gps are considered highly homologous, numerous in vitro studies have shown interspecies differences between mouse and human P-gp homologs.10-13 Interestingly, this study also revealed differences in the ability of P-gp modulators to displace [125I]IAAP binding in wild-type human and mouse P-gp (Figure 8). Differences in modulation of [125I]IAAP photo-crosslinking between mouse and human P-gp support these previous findings, and indicate that a comprehensive functional characterization of mouse P-gp is necessary to fully understand how in vitro mouse models are translatable to the function of human P-gp. Additionally, these findings highlight the need for a high-resolution structure of human P-gp, as even closely related homologs differ in their substrate specificity and mechanisms of drug binding and recognition.
Despite attempts from various laboratories, a high-resolution crystal structure of human P-gp has remained elusive. However, several structures of mouse P-gp (87% homologous to human P-gp) have been recently reported.7-9, 30 It is currently not known why mouse P-gp can be crystallized but human P-gp cannot, despite their high sequence homology. Chimeragenesis can be used as a strategy to elucidate protein structure of membrane proteins recalcitrant to crystallization.53, 54 Future studies involving the chimeras generated here would include their heterologous protein expression, purification, and crystallization screening. By the introduction of mouse residues into human P-gp, this may result in P-gp chimeras containing domains of human P-gp that retain the favorable crystallographic properties of mouse P-gp. The studies presented here indicate that the exchange of homologous domains between mouse and human P-gp results in functional protein suitable for biochemical and structural investigations.
Supplementary Material
Acknowledgments
We would like to thank Lothar Esser for his assistance generating figures of the chimeras. We would also like to thank Dom Esposito and the Protein Expression Laboratory at the Frederick National Laboratory for Cancer Research for their assistance in BacMam- and baculovirus-mediated protein expression. A special thank you to George Leiman for editorial assistance.
Funding: This project was funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHN261200800001E, and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations
- P-gp
P-glycoprotein
- ABC
ATP-Binding Cassette
- MDR
multidrug resistance
- NBD
nucleotide-binding domain
- TMD
transmembrane domain
- Rh123
rhodamine 123
- CAM
calcein-AM
- 2-DG
2-deoxy-d-glucose
- NaBT
sodium butyrate
- IAAP
iodoarylazidoprazosin
- TQR
tariquidar
- CsA
cyclosporine A
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–7485. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 2.Kannan P, John C, Zoghbi SS, Halldin C, Gottesman MM, Innis RB, Hall MD. Imaging the function of P-glycoprotein with radiotracers: pharmacokinetics and in vivo applications. Clin Pharmacol Ther. 2009;86:368–377. doi: 10.1038/clpt.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin JH, Yamazaki M. Role of P-glycoprotein in pharmacokinetics: clinical implications. Clin Pharmacokinet. 2003;42:59–98. doi: 10.2165/00003088-200342010-00003. [DOI] [PubMed] [Google Scholar]
- 4.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 5.Sato T, Kodan A, Kimura Y, Ueda K, Nakatsu T, Kato H. Functional role of the linker region in purified human P-glycoprotein. FEBS J. 2009;276:3504–3516. doi: 10.1111/j.1742-4658.2009.07072.x. [DOI] [PubMed] [Google Scholar]
- 6.Chufan EE, Sim HM, Ambudkar SV. Molecular Basis of the Polyspecificity of P-Glycoprotein (ABCB1): Recent Biochemical and Structural Studies. Adv Cancer Res. 2015;125:71–96. doi: 10.1016/bs.acr.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li J, Jaimes KF, Aller SG. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szewczyk P, Tao H, McGrath AP, Villaluz M, Rees SD, Lee SC, Doshi R, Urbatsch IL, Zhang Q, Chang G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr D Biol Crystallogr. 2015;71:732–741. doi: 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ward AB, Szewczyk P, Grimard V, Lee CW, Martinez L, Doshi R, Caya A, Villaluz M, Pardon E, Cregger C, Swartz DJ, Falson PG, Urbatsch IL, Govaerts C, Steyaert J, Chang G. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci U S A. 2013;110:13386–13391. doi: 10.1073/pnas.1309275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim IW, Booth-Genthe C, Ambudkar SV. Relationship between drugs and functional activity of various mammalian P-glycoproteins (ABCB1) Mini Rev Med Chem. 2008;8:193–200. doi: 10.2174/138955708783744100. [DOI] [PubMed] [Google Scholar]
- 11.Takeuchi T, Yoshitomi S, Higuchi T, Ikemoto K, Niwa S, Ebihara T, Katoh M, Yokoi T, Asahi S. Establishment and characterization of the transformants stably-expressing MDR1 derived from various animal species in LLC-PK1. Pharm Res. 2006;23:1460–1472. doi: 10.1007/s11095-006-0285-7. [DOI] [PubMed] [Google Scholar]
- 12.Tang-Wai DF, Kajiji S, DiCapua F, de Graaf D, Roninson IB, Gros P. Human (MDR1) and mouse (mdr1, mdr3) P-glycoproteins can be distinguished by their respective drug resistance profiles and sensitivity to modulators. Biochemistry. 1995;34:32–39. doi: 10.1021/bi00001a005. [DOI] [PubMed] [Google Scholar]
- 13.Xia CQ, Xiao G, Liu N, Pimprale S, Fox L, Patten CJ, Crespi CL, Miwa G, Gan LS. Comparison of species differences of P-glycoproteins in beagle dog, rhesus monkey, and human using Atpase activity assays. Mol Pharm. 2006;3:78–86. doi: 10.1021/mp050034j. [DOI] [PubMed] [Google Scholar]
- 14.Dhir R, Gros P. Functional analysis of chimeric proteins constructed by exchanging homologous domains of two P-glycoproteins conferring distinct drug resistance profiles. Biochemistry. 1992;31:6103–6110. doi: 10.1021/bi00141a021. [DOI] [PubMed] [Google Scholar]
- 15.Bhatia A, Schafer HJ, Hrycyna CA. Oligomerization of the human ABC transporter ABCG2: evaluation of the native protein and chimeric dimers. Biochemistry. 2005;44:10893–10904. doi: 10.1021/bi0503807. [DOI] [PubMed] [Google Scholar]
- 16.Chen M, Abele R, Tampe R. Functional non-equivalence of ATP-binding cassette signature motifs in the transporter associated with antigen processing (TAP) J Biol Chem. 2004;279:46073–46081. doi: 10.1074/jbc.M404042200. [DOI] [PubMed] [Google Scholar]
- 17.Arora S, Lapinski PE, Raghavan M. Use of chimeric proteins to investigate the role of transporter associated with antigen processing (TAP) structural domains in peptide binding and translocation. Proc Natl Acad Sci U S A. 2001;98:7241–7246. doi: 10.1073/pnas.131132198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geillon F, Gondcaille C, Charbonnier S, Van Roermund CW, Lopez TE, Dias AM, Pais de Barros JP, Arnould C, Wanders RJ, Trompier D, Savary S. Structure-function analysis of peroxisomal ATP-binding cassette transporters using chimeric dimers. J Biol Chem. 2014;289:24511–24520. doi: 10.1074/jbc.M114.575506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saini P, Gaur NA, Prasad R. Chimeras of the ABC drug transporter Cdr1p reveal functional indispensability of transmembrane domains and nucleotide-binding domains, but transmembrane segment 12 is replaceable with the corresponding homologous region of the non-drug transporter Cdr3p. Microbiology. 2006;152:1559–1573. doi: 10.1099/mic.0.28471-0. [DOI] [PubMed] [Google Scholar]
- 20.Buschman E, Gros P. Functional analysis of chimeric genes obtained by exchanging homologous domains of the mouse mdr1 and mdr2 genes. Mol Cell Biol. 1991;11:595–603. doi: 10.1128/mcb.11.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou Y, Gottesman MM, Pastan I. Domain exchangeability between the multidrug transporter (MDR1) and phosphatidylcholine flippase (MDR2) Mol Pharmacol. 1999;56:997–1004. doi: 10.1124/mol.56.5.997. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Y, Gottesman MM, Pastan I. Studies of human MDR1-MDR2 chimeras demonstrate the functional exchangeability of a major transmembrane segment of the multidrug transporter and phosphatidylcholine flippase. Mol Cell Biol. 1999;19:1450–1459. doi: 10.1128/mcb.19.2.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pluchino KM, Esposito D, Moen JK, Hall MD, Madigan JP, Shukla S, Procter LV, Wall VE, Schneider TD, Pringle I, Ambudkar SV, Gill DR, Hyde SC, Gottesman MM. Identification of a Cryptic Bacterial Promoter in Mouse (mdr1a) P-Glycoprotein cDNA. PLoS One. 2015;10:e0136396. doi: 10.1371/journal.pone.0136396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akiyama S, Fojo A, Hanover JA, Pastan I, Gottesman MM. Isolation and genetic characterization of human KB cell lines resistant to multiple drugs. Somat Cell Mol Genet. 1985;11:117–126. doi: 10.1007/BF01534700. [DOI] [PubMed] [Google Scholar]
- 25.Currier SJ, Ueda K, Willingham MC, Pastan I, Gottesman MM. Deletion and insertion mutants of the multidrug transporter. J Biol Chem. 1989;264:14376–14381. [PubMed] [Google Scholar]
- 26.Shukla S, Schwartz C, Kapoor K, Kouanda A, Ambudkar SV. Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab Dispos. 2012;40:304–312. doi: 10.1124/dmd.111.042721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramachandra M, Ambudkar SV, Chen D, Hrycyna CA, Dey S, Gottesman MM, Pastan I. Human P-glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry. 1998;37:5010–5019. doi: 10.1021/bi973045u. [DOI] [PubMed] [Google Scholar]
- 28.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–514. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 29.Sauna ZE, Ambudkar SV. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci U S A. 2000;97:2515–2520. doi: 10.1073/pnas.97.6.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai J, Swartz DJ, Protasevich II, Brouillette CG, Harrell PM, Hildebrandt E, Gasser B, Mattanovich D, Ward A, Chang G, Urbatsch IL. A gene optimization strategy that enhances production of fully functional P-glycoprotein in Pichia pastoris. PLoS One. 2011;6:e22577. doi: 10.1371/journal.pone.0022577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ambudkar SV. Purification and reconstitution of functional human P-glycoprotein. J Bioenerg Biomembr. 1995;27:23–29. doi: 10.1007/BF02110327. [DOI] [PubMed] [Google Scholar]
- 33.van Den Elsen JM, Kuntz DA, Hoedemaeker FJ, Rose DR. Antibody C219 recognizes an alpha-helical epitope on P-glycoprotein. Proc Natl Acad Sci U S A. 1999;96:13679–13684. doi: 10.1073/pnas.96.24.13679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall MD, Brimacombe KR, Varonka MS, Pluchino KM, Monda JK, Li J, Walsh MJ, Boxer MB, Warren TH, Fales HM, Gottesman MM. Synthesis and structure-activity evaluation of isatin-beta-thiosemicarbazones with improved selective activity toward multidrug-resistant cells expressing P-glycoprotein. J Med Chem. 2011;54:5878–5889. doi: 10.1021/jm2006047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gribar JJ, Ramachandra M, Hrycyna CA, Dey S, Ambudkar SV. Functional characterization of glycosylation-deficient human P-glycoprotein using a vaccinia virus expression system. J Membr Biol. 2000;173:203–214. doi: 10.1007/s002320001020. [DOI] [PubMed] [Google Scholar]
- 36.Hrycyna CA, Ramachandra M, Ambudkar SV, Ko YH, Pedersen PL, Pastan I, Gottesman MM. Mechanism of action of human P-glycoprotein ATPase activity. Photochemical cleavage during a catalytic transition state using orthovanadate reveals cross-talk between the two ATP sites. J Biol Chem. 1998;273:16631–16634. doi: 10.1074/jbc.273.27.16631. [DOI] [PubMed] [Google Scholar]
- 37.Kapoor K, Bhatnagar J, Chufan EE, Ambudkar SV. Mutations in intracellular loops 1 and 3 lead to misfolding of human P-glycoprotein (ABCB1) that can be rescued by cyclosporine A, which reduces its association with chaperone Hsp70. J Biol Chem. 2013;288:32622–32636. doi: 10.1074/jbc.M113.498980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasudevan S, Tsuruo T, Rose DR. Mode of binding of anti-P-glycoprotein antibody MRK-16 to its antigen. A crystallographic and molecular modeling study. J Biol Chem. 1998;273:25413–25419. doi: 10.1074/jbc.273.39.25413. [DOI] [PubMed] [Google Scholar]
- 39.Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 40.Sauna ZE, Muller M, Peng XH, Ambudkar SV. Importance of the conserved Walker B glutamate residues, 556 and 1201, for the completion of the catalytic cycle of ATP hydrolysis by human P-glycoprotein (ABCB1) Biochemistry. 2002;41:13989–14000. doi: 10.1021/bi026626e. [DOI] [PubMed] [Google Scholar]
- 41.Pluchino KM, Hall MD, Goldsborough AS, Callaghan R, Gottesman MM. Collateral sensitivity as a strategy against cancer multidrug resistance. Drug Resist Updat. 2012;15:98–105. doi: 10.1016/j.drup.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV. Photoaffinity labeling of human P-glycoprotein: effect of modulator interaction and ATP hydrolysis on substrate binding. Methods Enzymol. 1998;292:318–328. doi: 10.1016/s0076-6879(98)92025-0. [DOI] [PubMed] [Google Scholar]
- 43.Greenberger LM. Identification of drug interaction sites in P-glycoprotein. Methods Enzymol. 1998;292:307–317. doi: 10.1016/s0076-6879(98)92024-9. [DOI] [PubMed] [Google Scholar]
- 44.Safa AR. Photoaffinity labels for characterizing drug interaction sites of P-glycoprotein. Methods Enzymol. 1998;292:289–307. doi: 10.1016/s0076-6879(98)92023-7. [DOI] [PubMed] [Google Scholar]
- 45.Chufan EE, Kapoor K, Sim HM, Singh S, Talele TT, Durell SR, Ambudkar SV. Multiple transport-active binding sites are available for a single substrate on human P-glycoprotein (ABCB1) PLoS One. 2013;8:e82463. doi: 10.1371/journal.pone.0082463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Loo TW, Bartlett MC, Clarke DM. Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochemical J. 2006;399:351–359. doi: 10.1042/BJ20060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dolghih E, Bryant C, Renslo AR, Jacobson MP. Predicting binding to p-glycoprotein by flexible receptor docking. PLoS Comp Biol. 2011;7:e1002083. doi: 10.1371/journal.pcbi.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loo TW, Clarke DM. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J Biol Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- 49.Loo TW, Clarke DM. Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J Biol Chem. 1997;272:31945–31948. doi: 10.1074/jbc.272.51.31945. [DOI] [PubMed] [Google Scholar]
- 50.Bakhsheshian J, Hall MD, Robey RW, Herrmann MA, Chen JQ, Bates SE, Gottesman MM. Overlapping substrate and inhibitor specificity of human and murine ABCG2. Drug Metab Dispos. 2013;41:1805–1812. doi: 10.1124/dmd.113.053140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999;39:361–398. doi: 10.1146/annurev.pharmtox.39.1.361. [DOI] [PubMed] [Google Scholar]
- 52.Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, Zijlmans JM, Fibbe WE, Borst P. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- 54.Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J. 2007;26:4005–4015. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.