

ABSTRACT
FadR is a master regulator of fatty acid (FA) metabolism that coordinates the pathways of FA degradation and biosynthesis in enteric bacteria. We show here that a ΔfadR mutation in the El Tor biotype of Vibrio cholerae prevents the expression of the virulence cascade by influencing both the transcription and the posttranslational regulation of the master virulence regulator ToxT. FadR is a transcriptional regulator that represses the expression of genes involved in FA degradation, activates the expression of genes involved in unsaturated FA (UFA) biosynthesis, and also activates the expression of two operons involved in saturated FA (SFA) biosynthesis. Since FadR does not bind directly to the toxT promoter, we determined whether the regulation of any of its target genes indirectly influenced ToxT. This was accomplished by individually inserting a double point mutation into the FadR-binding site in the promoter of each target gene, thereby preventing their activation or repression. Although preventing FadR-mediated activation of fabA, which encodes the enzyme that carries out the first step in UFA biosynthesis, did not significantly influence either the transcription or the translation of ToxT, it reduced its levels and prevented virulence gene expression. In the mutant strain unable to carry out FadR-mediated activation of fabA, expressing fabA ectopically restored the levels of ToxT and virulence gene expression. Taken together, the results presented here indicate that V. cholerae FadR influences the virulence cascade in the El Tor biotype by modulating the levels of ToxT via two different mechanisms.
IMPORTANCE Fatty acids (FAs) play important roles in membrane lipid homeostasis and energy metabolism in all organisms. In Vibrio cholerae, the causative agent of the acute intestinal disease cholera, they also influence virulence by binding into an N-terminal pocket of the master virulence regulator, ToxT, and modulating its activity. FadR is a transcription factor that coordinately controls the pathways of FA degradation and biosynthesis in enteric bacteria. This study identifies a new link between FA metabolism and virulence in the El Tor biotype by showing that FadR influences both the transcription and posttranslational regulation of the master virulence regulator ToxT by two distinct mechanisms.
KEYWORDS: FadR, fatty acid, virulence, pathogenesis, ToxT
INTRODUCTION
Vibrio cholerae O1 is a natural inhabitant of aquatic environments and causes the diarrheal disease cholera. The expression of its two primary virulence factors, toxin-coregulated pilus (TCP) (1) and cholera toxin (CT), is activated by a transcriptional cascade involving a number of regulatory proteins (2). ToxT, an AraC family member, directly activates the expression of the tcp, ctx, and accessory colonization factor genes (3). The expression of ToxT, in turn, is activated by cooperation of two homologous transmembrane protein pairs, ToxRS and TcpPH (4–6). The induction of the virulence cascade by activation of the tcpPH promoter is dependent upon two additional regulators, AphA and AphB (7, 8). AphA is a winged helix transcription factor that facilitates the binding of the LysR-type regulator AphB to the promoter (9–11).
The expression of the V. cholerae virulence cascade is influenced by a wide variety of environmental stimuli such as pH, temperature, osmolarity, oxygen tension, bile, unsaturated fatty acids (UFAs), bicarbonate, c-di-GMP, and quorum sensing (2, 12–17). UFAs, which are a component of bile in the intestinal lumen, have been shown to inhibit virulence gene expression in V. cholerae (14). This occurs by direct binding of a UFA into a pocket in the N-terminal domain of ToxT, which prevents the protein from dimerizing and binding to DNA (18–20). The binding of UFAs to ToxT is thought to prevent the expression of virulence genes until the bacteria have penetrated the mucus of the intestine, where the concentrations of UFAs are presumably reduced (18). Thus, UFAs may serve as an in vivo signal to indicate that V. cholerae has progressed into the appropriate environment to initiate pathogenesis.
The pathways of fatty acid (FA) degradation and biosynthesis in enteric bacteria are coordinately controlled at the level of transcription by the master regulator FadR (21, 22). FadR from Escherichia coli is an extensively characterized member of the GntR family of transcriptional regulators with an N-terminal winged helix DNA binding domain and a C-terminal acyl coenzyme A (acyl-CoA) binding domain (23–25). In the absence of exogenous long-chain fatty acids (LCFAs), FadR represses transcription of the fad genes that encode proteins required for the transport, activation, and β-oxidation of LCFAs (Fig. 1A) (21). These genes include fadL, fadD, fadBA, fadE, and fadH (26). FadR also activates the expression of the fabA and fabB genes, which encode proteins required for the biosynthesis of UFAs (27, 28) as well as genes involved in saturated FA (SFA) biosynthesis (29, 30). When exogenous LCFAs are present (Fig. 1B), they diffuse across the outer membrane through FadL and are activated by the inner membrane-associated acyl-CoA ligase FadD (31) to produce long-chain fatty acyl-CoAs (LCFA-CoAs). These LCFA-CoAs bind directly to FadR and induce a conformational change that disrupts the FadR DNA complex (24, 32). This upregulates the expression of genes involved in FA degradation to utilize the LCFAs and downregulates the expression of genes involved in FA biosynthesis, which are no longer needed.
FIG 1.

Roles of FadR in V. cholerae. (A) In the absence of LCFAs, FadR represses the expression of genes involved in FA degradation and phospholipid biosynthesis by binding to sites in the promoters of the fadB, fadE, fadH, and plsB genes and activates the expression of genes involved in UFA and SFA biosynthesis by binding to sites in the promoters of the fabA, fabB, fabH, and fabF genes. (B) When exogenous LCFAs are present, they diffuse across the outer membrane through FadL and are activated to LCFA-CoAs by FadD. The resulting activated LCFA-CoAs bind to FadR, causing a conformational change that releases it from DNA and results in derepression and failure to activate its regulated promoters. The mechanism by which it controls virulence is the subject of this work.
In V. cholerae, FadR is also involved in regulating the pathways of FA degradation and UFA biosynthesis (33). However, unlike E. coli FadR, which has only a single binding site for acyl-CoA, FadR from V. cholerae has two distinct binding sites for acyl-CoA (33). One of these is structurally similar to the site in E. coli FadR, whereas the other is unique and comprised of residues from a 40-amino-acid insertion in the protein that is present only among Vibrionaceae (33, 34). Binding of ligand to both of these sites in V. cholerae FadR results in a more dramatic conformational change within the DNA binding domain than in E. coli FadR, which appears to more fully disrupt DNA binding (33). This is likely responsible for the enhanced expression of FA utilization genes in the presence of LCFAs in V. cholerae relative to other bacterial species (34). Since Vibrionaceae are natural inhabitants of aquatic environments, where they obtain FAs from the sediment, the acquisition of a second binding site in FadR may provide a more efficient mechanism for utilizing FAs in this environment.
In addition to its roles in FA degradation and UFA biosynthesis (33), FadR represses the expression of the plsB gene involved in phospholipid biosynthesis in V. cholerae (35) and, as shown here, activates the expression of two operons involved in SFA synthesis. Although prior studies in the classical biotype of V. cholerae did not reveal a role for FadR in the expression of the virulence cascade (36), the results here show that in the El Tor biotype of V. cholerae, FadR is also required for expression of TCP and CT. Examination of a V. cholerae strain C6706 ΔfadR mutant revealed a modest reduction in the transcription of ToxT. However, since FadR does not regulate expression from the toxT promoter directly, we assessed whether the ability of FadR to directly regulate the transcription of any of its known target genes indirectly influences ToxT. This was accomplished by individually preventing FadR-mediated regulation of each of its target genes by inserting a double point mutation into their FadR binding sites. Interestingly, disrupting FadR-mediated regulation of fabA, which encodes the enzyme that catalyzes the first step in UFA biosynthesis, decreased the levels of ToxT via a posttranslational mechanism. These results indicate that FadR influences the levels of ToxT in V. cholerae indirectly through two different mechanisms.
RESULTS
FadR influences the expression of the virulence cascade in the El Tor, but not in the classical, biotype of V. cholerae.
It has previously been shown that loss of FadR does not influence the expression of the virulence cascade in the classical biotype of V. cholerae (36). Since the conditions that induce the expression of the virulence cascade in the El Tor biotype (static growth in a peptone-based medium, AKI [37], at 37°C in the presence of bicarbonate) are different from those that induce its expression in the classical biotype, we assessed the loss of FadR in both biotypes on the expression of the virulence cascade under AKI conditions. As shown in Fig. 2A, in the classical biotype, both the wild-type and ΔfadR mutant strains showed high-level expression of a tcpA-lacZ fusion, indicating that FadR does not have an influence on the expression of the virulence cascade under this condition. Although the wild-type El Tor biotype fusion showed a 3-fold reduction in expression relative to the wild-type classical biotype fusion, consistent with the lowered expression of the virulence cascade in this biotype (38), the El Tor ΔfadR mutant showed an 8-fold reduction in expression relative to the wild type, indicating that FadR does have an influence on the expression of the virulence cascade in this biotype. Under AKI conditions, as well as when subjected to shaking, the growth rate of the El Tor biotype ΔfadR mutant (see Fig. S1 in the supplemental material) is only slightly reduced compared to that of the wild type. Introducing a plasmid expressing FadR into the El Tor ΔfadR mutant restored expression of tcpA-lacZ to wild-type levels (Fig. 2B). The expression of an El Tor biotype ctx-lacZ fusion was also reduced 8-fold by the ΔfadR mutation, and this effect was similarly complemented by the FadR expression plasmid (Fig. 2C). These findings indicate that FadR influences the expression of both tcpA and ctx in the El Tor biotype under AKI conditions. This effect is independent of quorum sensing, since a C6706 ΔfadR ΔhapR mutant still showed reduced expression of the virulence cascade (data not shown). Since the amino acid sequence of FadR is identical for classical and El Tor biotypes, the above-described findings indicate that the loss of FadR influences the expression of the virulence cascade differently in the two biotypes of V. cholerae.
FIG 2.

Influence of a ΔfadR mutation on the expression of tcpA and ctx promoter-lacZ fusions in V. cholerae. From left to right: MBN135, GK1536, KSK979, and GK1502 (*, P = 0.006) (A); KSK979, GK1502 (*, P = 0.003), GK1502/pKAS178 (*, P = 0.005), and GK1502/pWEL231 (B); KSK2325, GK1954 (*, P = 0.0006), GK1954/pKAS178 (*, P = 0.007), and GK1954/pWEL231 (C). Cultures were grown in AKI medium statically for 3.5 h.
Loss of FadR reduces the transcription of toxT but not that of tcpP or toxR.
Given that ToxT directly activates the expression of both tcpA and ctx in V. cholerae, the observation that the ΔfadR mutation reduced the expression of both of these genes suggested that FadR influences ToxT. Consistent with this finding, the El Tor biotype ΔfadR mutant strain does not produce detectable levels of ToxT by Western blotting (Fig. 3A). Since the expression of the virulence cascade is highly regulated at the level of transcription, we first examined the expression of a toxT-lacZ transcriptional fusion in wild-type and ΔfadR backgrounds. The ΔfadR mutation reduced the transcription of toxT approximately 3-fold under static incubation conditions (Fig. 3B). In contrast, the ΔfadR mutation did not significantly influence the transcription of either of its two activators, TcpP or ToxR, under these conditions (Fig. 3C and D). These results indicate that the expression of ToxT is the first point in the virulence cascade that is influenced by FadR. High-level CT production can be achieved in the El Tor biotype when static incubation is followed by an aerobic shaking phase (37). As shown in Fig. S2 in the supplemental material, both the wild-type and ΔfadR mutant strains showed a reduction in toxT expression after the aerobic phase of growth. However, the ΔfadR mutant still showed a 2-fold decrease in expression relative to the wild type that is statistically significant.
FIG 3.

A ΔfadR mutation reduces the transcription of toxT but not that of tcpP or toxR. (A) Western blot with strains C6706 str2, KSK1184, and GK1257. (B to D) Influence of ΔfadR on the transcription of toxT, tcpP, and toxR. From left to right: KSK1267, GK1499 (*, P < 0.0001), JAS273, and GK2136 (*, P < 0.0001) (B); KSK725 and WL982 (C); WL124 and GK1504 (D). Cultures were grown in AKI medium statically for 3.5 h.
It has recently been shown that the fatty acyl-CoA ligase FadD (Fig. 1) influences the virulence cascade in V. cholerae by promoting the localization of TcpP into the membrane (36). In the absence of FadD, induction of the σE-dependent extracytoplasmic stress response results in proteolysis of membrane-localized TcpP by the integral membrane protease RseP and reduces expression from the toxT promoter (39). To determine whether the effects of FadR on the virulence cascade shown here are independent of FadD and TcpP, we introduced a ΔrseP mutation into both the wild-type and ΔfadR toxT-lacZ fusions. As shown in Fig. 3B, the absence of RseP alone did not influence the expression of toxT and in the presence of the ΔfadR mutation did not restore the expression of toxT as it did in a ΔfadD mutant (39). These results suggest that FadR influences the virulence cascade in a manner independent of FadD and the σE-dependent extracytoplasmic stress response.
FadR directly regulates the expression of genes involved in UFA biosynthesis and FA degradation but not toxT.
We have previously shown that in V. cholerae, similar to the situation in E. coli, FadR represses the expression of the fadBA (VC2758-59), fadE (VC2231), and fadH (VC1993) genes, involved in FA degradation, and activates the expression of the fabA (VC1483) and fabB (VC2109) genes, involved in UFA biosynthesis (33). Each of these promoters contains a 17-bp palindromic motif (Fig. 4A) that matches the consensus sequence established for E. coli FadR binding (40–42). In contrast, a similar motif was not detected in the toxT promoter, and consistent with this, purified FadR was unable to bind to the toxT promoter (Fig. 4B), although it bound to the fadBA promoter (Fig. 4C). These findings suggest that the influence of FadR on toxT expression is indirect and may be due to its effects on genes that it directly regulates.
FIG 4.
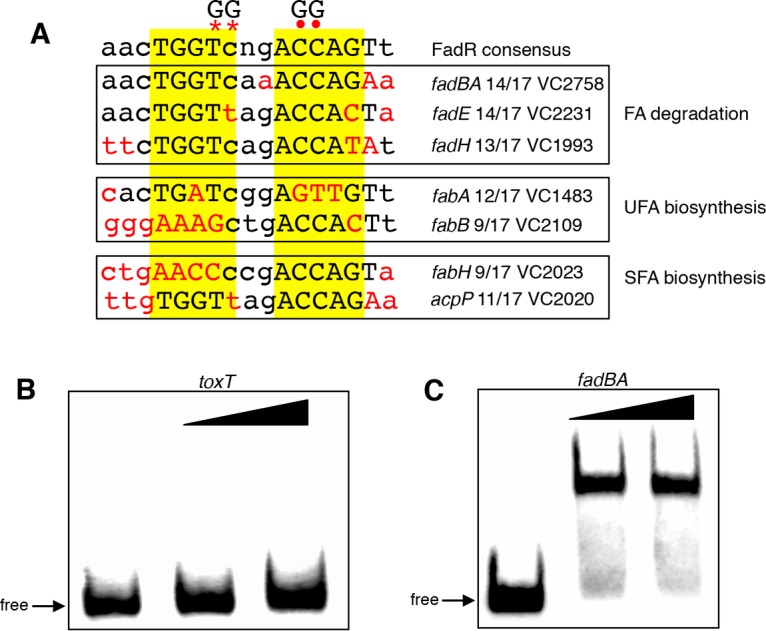
FadR binding sites in V. cholerae. (A) The FadR consensus determined from E. coli (40) is shown at the top. Within the consensus sequence, upper- and lowercase letters represent nucleotides found more and less frequently, respectively. Mismatches from the consensus are in red. Asterisks show the position of the GG mutations in the fadBA, fadE, fadH, fabA, fabH, and acpP promoters. Dots show the position of the GG mutation in the fabB promoter. (B, C) Binding of purified FadR to toxT and fadBA promoter fragments. The first lane in each set has no protein added, the second lane has 80 ng FadR, and the third lane has 160 ng FadR.
In an attempt to understand how FadR influences the expression of toxT, we wanted to determine whether the ability of FadR to directly regulate the transcription of any of its known target genes indirectly influences the toxT promoter. This was accomplished by individually preventing FadR-mediated regulation of each of its regulated promoters by inserting a double point mutation into their FadR binding sites (GG at positions 7 and 8 of the consensus, shown by asterisks in Fig. 4). This mutation has previously been shown in E. coli to eliminate FadR binding in vitro and prevent transcriptional regulation in vivo (40). Since the putative site at the fabB promoter, which shows a very poor match to the consensus (42), naturally contains a G at position 7 of the consensus, a different double point mutation (GG at positions 12 and 13 of the consensus, shown by dots in Fig. 4) was used to inactivate this binding site. As shown in Fig. 5, the ability of FadR to repress expression from the fadBA, fadE, and fadH promoters, as well as its ability to activate expression from the fabA and fabB promoters, was lost in the presence of the GG mutations. In addition, introduction of the ΔfadR mutation into each GG mutant strain did not further alter expression from the promoters, indicating that the FadR binding sites were no longer functional. These results indicate that FadR uses these binding sites to directly regulate the expression of these genes in V. cholerae.
FIG 5.

FadR regulates the expression of genes involved in FA degradation and UFA biosynthesis. The genetic organization of each of the genes is shown. The red boxes indicate the positions of the FadR binding sites (shown in Fig. 4), and the arrows show the putative transcriptional start sites. The positions of the GG mutations are shown by asterisks and dots. From left to right: WL1031, WL1035 (*, P = 0.003), WL1060 (*, P = 0.005), and WL1062 (*, P = 0.005) (A); WL1027, WL1029 (*, P = 0.002), WL1056 (*, P = 0.0001), and WL1058 (*, P = 0.002) (B); WL1040, WL1042 (*, P = 0.005), WL1064 (*, P = 0.01), and WL1066 (*, P = 0.0001) (C); WL1005, WL1007 (*, P = 0.0007), WL1020 (*, P = 0.002), and WL1022 (*, P = 0.002) (D); GK1609, GK1610 (*, P = 0.002), GK1669 (*, P = 0.02), and GK1672 (*, P = 0.02) (E). Cultures were grown in tryptone broth for 5 h with aeration.
FadR activates the expression of SFA biosynthesis genes in V. cholerae.
It has been shown that in addition to its roles in FA degradation and UFA biosynthesis, FadR activates the expression of genes involved in SFA biosynthesis in E. coli (29, 30). Unlike in mammals, where FAs are synthesized by a large multifunctional protein known as type I synthase (22, 43), bacteria produce a type II synthase composed of individual enzymes that catalyze discrete steps in the process, with each intermediate attached to the universal and highly conserved acyl carrier protein (ACP) (22, 43). Specifically, it has been shown that FadR activates the expression of the fabH promoter, which drives the expression of the fabHDG operon, encoding three enzymes involved in SFA biosynthesis, and shows a modest activation of the acpP promoter, which controls the expression of the gene encoding ACP, acpP, coexpressed with another SFA biosynthetic enzyme encoded by fabF (29). Since the arrangement of the SFA genes in V. cholerae is similar to that in E. coli (Fig. 6), we assessed whether these genes were also regulated by FadR. We identified a putative FadR binding site (Fig. 4) upstream of fabH (VC2023) within the V. cholerae plsX gene and, as shown in Fig. 6A, FadR activated a fabH-lacZ promoter fusion 3.6-fold. Inserting a GG mutation into the putative binding site reduced this activation to a level similar to that of the ΔfadR mutant. We also identified another putative FadR binding site (Fig. 4) upstream of the acpP and fabF (VC2020-VC2019) genes (Fig. 6B). FadR activated the expression of this promoter nearly 2-fold, and inserting a GG mutation into the putative binding site also reduced this activation to a level similar to that of the ΔfadR mutant. These results indicate that in addition to its roles in regulating the expression of genes involved in FA degradation and UFA biosynthesis, FadR activates the expression of two SFA biosynthesis operons in V. cholerae.
FIG 6.

FadR activates the expression of genes involved in SFA biosynthesis. Genetic organization of the fab-acpP locus. The red boxes indicate the positions of the FadR binding sites (shown in Fig. 4) and the arrows show the putative transcriptional start sites. The positions of the GG mutations are shown by asterisks. From left to right: GK2017, GK2020 (*, P = 0.01), GK2039 (*, P = 0.009), and GK2040 (*, P = 0.01) (A); GK1630, GK1632 (*, P = 0.02), GK2035 (*, P = 0.02), and GK2037 (*, P = 0.02) (B). Cultures were grown in tryptone broth for 5 h with aeration.
Altered FA content of FA biosynthesis FadR binding site mutants.
Only two enzymes, encoded by the fabA and fabB genes, are required for the biosynthesis of UFAs (44, 45). Since the expression of both of these genes is dependent on FadR, ΔfadR mutants in both E. coli and Vibrio vulnificus contain lower levels of UFAs (46, 47). To determine whether the levels of UFAs are reduced in the V. cholerae ΔfadR, fabAGG, and fabBGG mutants, these strains were analyzed for total FA content. As shown in Table 1, the levels of UFAs in wild-type C6706 were reduced from 59% to 43% by the ΔfadR mutation. In the fabAGG mutant, they were reduced to 39%, and in the fabBGG mutant, they were reduced to 23%. In addition, for each of these mutants, the levels of SFAs correspondingly increased (Table 1). These findings confirm that in V. cholerae, as in E. coli and V. vulnificus, the intracellular levels of UFAs are dependent upon FadR. In contrast, the intracellular levels of SFAs in V. cholerae do not appear to depend on FadR since the FA content of the fabHGG and acpPGG mutants was not appreciably different from that of the wild type (Table 1).
TABLE 1.
FA content of strains with GG mutations in FA biosynthesis promoters
| Strain | % unsaturated | % saturated | Ratio |
|---|---|---|---|
| C6706 str2 (wild type) | 59 ± 2 | 40 ± 2 | 1.4 |
| GK1257 (ΔfadR) | 43 ± 0.5 | 56 ± 0.8 | 0.8 |
| GK1689 (fabAGG) | 39 ± 0.2 | 60 ± 0.4 | 0.7 |
| GK1691 (fabBGG) | 23 ± 0.6 | 77 ± 0.6 | 0.3 |
| GK2043 (fabHGG) | 59 ± 0.2 | 40 ± 0.2 | 1.4 |
| GK2041 (acpPGG) | 60 ± 0 | 39 ± 0 | 1.5 |
Preventing FadR-mediated activation of UFA biosynthesis decreases the expression of the virulence cascade.
The above findings show that FadR directly regulates the expression of genes involved in FA degradation, UFA biosynthesis, and SFA biosynthesis in V. cholerae. To determine whether disruption of FadR-mediated regulation of any of these genes is responsible for the altered expression of the virulence cascade in the ΔfadR mutant, the GG-disrupted FadR binding sites in the promoters of these genes were individually introduced into a tcpA-lacZ fusion strain. As shown in Fig. 7A, the GG mutation in the fabA promoter strongly reduced the transcription of tcpA (approximately 8-fold) to a level similar to that of the ΔfadR mutant, whereas the GG mutations in the promoters of the genes involved in FA degradation or SFA biosynthesis did not. In addition, introduction of the fabAGG mutation into wild-type C6706 significantly reduced the levels of TcpA as observed by Western blotting (Fig. 7B). To confirm that a functional fabA gene is required for the expression of tcpA, a merodiploid fabAGG tcpA-lacZ mutant strain that contains a wild-type copy of the fabA gene and its promoter inserted at the lac locus was constructed. As shown in Fig. 7C, the wild-type copy of fabA in this strain fully restored the expression of tcpA. These results indicate that FadR-mediated activation of UFA biosynthesis influences the expression of the virulence cascade in V. cholerae.
FIG 7.

A fabAGG mutation reduces the expression of tcpA. (A) Influence of GG mutations on the expression of tcpA. From left to right: KSK979, GK1502 (*, P = 0.001), GK1675 (*, P = 0.0006), GK1657, GK1982 (*, P = 0.02), GK1687, GK1981 (*, P = 0.003), GK2022, and GK2016 (*, P = 0.03). (B) Western blot with strains C6706 str2, KSK1184, GK1257, and GK1689. (C) Ectopic expression of wild-type fabA restores the expression of tcpA to the fabAGG mutant. From left to right: KSK979, GK2207, GK1675 (*, P = 0.0008), and GK2209. Cultures were grown in AKI medium statically for 3.5 h.
We next determined whether the fabAGG mutation influences the expression of the virulence cascade in a manner similar to that of the ΔfadR mutant. Consistent with the reduced level of tcpA expression observed in the fabAGG mutant as described above, introduction of this mutation into wild-type C6706 prevented the production of ToxT (Fig. 8A), but unlike the ΔfadR mutation, it did not influence the transcription of toxT (Fig. 8B). This finding suggests that the fabAGG mutation influences ToxT posttranscriptionally, by reducing its translation and/or by decreasing its stability. To address the former possibility first, a translational fusion of ToxT to β-galactosidase (toxT‘-'lacZ) in the chromosome was made by fusing the first 44 amino acids of ToxT to amino acid 9 of β-galactosidase. Since this reporter is expressed under the control of the native toxT promoter, it measures transcription as well as translation. As shown in Fig. 8C, the ΔfadR mutation reduced the transcription of the toxT‘-'lacZ fusion similar to what is shown in Fig. 8B, whereas the fabAGG mutation did not influence either the transcription or the translation of ToxT. These findings suggest that preventing FadR from activating fabA expression reduces the levels of ToxT via a posttranslational mechanism. Taken together, the results shown here indicate that loss of FadR reduces the expression of the virulence cascade by two distinct mechanisms that influence ToxT. One is a reduction in the transcription of toxT, which occurs via an unknown process, and the second is a posttranslational reduction in the levels of ToxT due to an inability to activate fabA expression and UFA biosynthesis.
FIG 8.
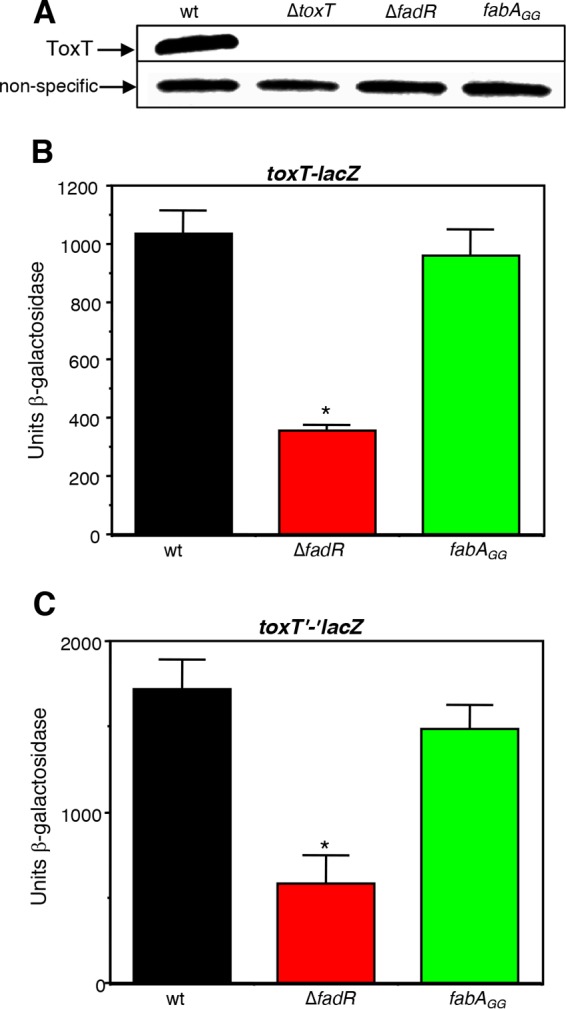
The fabAGG mutation reduces the levels of ToxT but does not influence either its transcription or its translation. (A) Western blot with strains C6706 str2, KSK1184, GK1257, and GK1689. (B and C) Left to right: transcriptional toxT-lacZ fusions in KSK1267, GK1499 (*, P = 0.0004), and WL1231 (B) and translational toxT‘-'lacZ fusions in WL1268, WL1271 (*, P < 0.0001), and GK1790 (C). Cultures were grown in AKI medium statically for 3.5 h.
DISCUSSION
FadR is present in a variety of bacterial species, where it plays a role in the transcriptional regulation of genes involved in FA metabolism (41, 42). Although no role for FadR in the regulation of virulence was observed in the classical biotype of V. cholerae (36), we show here that FadR is required for the expression of the cascade in the El Tor biotype. The influence of FadR on the expression of both the tcpA and ctx genes in this biotype suggested a possible effect on ToxT, the regulator that directly activates both of these genes. Since it has previously been shown that UFAs are capable of binding directly to ToxT and inhibiting its activity (18), it seemed possible that the altered regulation of UFAs in the ΔfadR mutant influences the activity of ToxT. However, as we show here, the ΔfadR mutation appears to influence ToxT at two different levels: (i) by reducing its transcription by an unknown mechanism and (ii) by reducing its levels posttranslationally due to an inability to activate the expression of fabA, encoding the enzyme that catalyzes the first step in the biosynthesis of UFAs.
FadR was originally identified in E. coli as a transcriptional repressor controlling the genes of FA metabolism (48) and later shown to also function as a transcriptional activator of genes involved in UFA biosynthesis (27, 28). We previously demonstrated that FadR also regulates both of these processes in V. cholerae (33) and have now extended these studies to show that sequences with similarity to the E. coli FadR binding site consensus are necessary for the FadR-dependent expression of these genes in V. cholerae. FadR has recently been shown to activate the expression of two operons involved in SFA biosynthesis in E. coli (29). We have similarly found that FadR also plays a role in activating the expression of these operons in V. cholerae. In E. coli, FabH is essential for the initiation of FA synthesis and is a regulated step thought to play a key role in determining the amount of FAs produced by the pathway (49). Despite the dependence of FadR on expression from the fabH and acpP promoters in V. cholerae, the GG mutations in these promoters did not strongly influence the intracellular levels of SFAs. One explanation to account for this is that the fabH operon (fabHDG) is transcribed from two different promoters; in addition to the FadR-dependent promoter within plsX, fabH is expressed from a FadR-independent promoter that is located further upstream (29, 50). A similar situation appears to occur with acpP. In addition to the FadR-dependent promoter immediately upstream of its gene, acpP has been shown to be coexpressed with fabG (51). Thus, other factors in addition to FA availability appear to be involved in regulating the levels of SFAs in bacteria (45).
To shed light on how FadR influences the virulence cascade in V. cholerae, the FadR binding site GG mutations that abolished regulation of its target genes were individually assessed in a tcpA-lacZ reporter strain. Only one of these, the fabAGG mutation, strongly reduced tcpA expression. The fabA gene encodes the enzyme β-hydroxydecanoyl-ACP dehydratase, which introduces a double bond at the C-10 level and is an essential step in the formation of UFAs (52, 53). The second step in the pathway, carried out by the fabB gene, encodes β-ketoacyl-ACP synthase I, which is capable of elongating the product synthesized by FabA (54). Null mutations in either one of these genes are lethal in E. coli, as they both encode enzymes that are essential for UFA biosynthesis (54). This appears to be the case in V. cholerae as well, since we were not able to isolate deletions of these genes. In view of the fact that both the V. cholerae fabAGG and fabBGG mutants have reduced levels of UFAs relative to the wild type, the finding that the former, but not the latter, exhibits reduced tcpA expression suggests that it is not the absolute level of UFAs in these mutants that influences the expression of the virulence cascade. Since the profiles of FA species produced in these two strains are not identical, it is possible that one or more of these differences are responsible for the effects on the virulence cascade. Experiments to assess this are currently in progress.
Since the fabAGG mutation reduced the expression of tcpA to a level similar to that of the ΔfadR mutation, it was surprising to find that, unlike the ΔfadR mutation, it did not influence the transcription of toxT. Since the fabAGG mutation also did not influence the translation of toxT, this suggests that the inability to activate fabA expression reduces the levels of certain UFAs in the cell that, in turn, reduce the levels of ToxT. ToxT has previously been shown to undergo proteolysis when V. cholerae is shifted to conditions that do not support virulence (i.e., from 30°C to 37°C, or when the pH increases from pH 6.5 to pH 8), but the specific proteases involved in this process are unknown (55). ToxT appears to be specifically cleaved in a recently structured region of the protein that lies in the N-terminal domain of the protein between amino acids 100 and 109 (56, 57). Moreover, it has also been shown that the addition of the UFA linoleic acid to ToxT prevents its proteolysis (56), possibly by promoting the “closed” conformation of ToxT that is unable to bind to DNA or to activate gene expression (18). These findings raise the possibility that in the absence of UFA biosynthesis in the ΔfadR and fabAGG mutants there is more ToxT present in the “open” conformation that lacks UFAs and this increases its susceptibility to proteolysis (see the model in Fig. 9). It is also possible that in the ΔfadR and fabAGG mutants there is an increase in the expression and/or activity of proteases that facilitates the degradation of ToxT. Further work is necessary in order to elucidate the specific mechanism involved in reducing the levels of ToxT in the fabAGG mutant.
FIG 9.

Model for the influence of FadR on the expression of the virulence cascade. (A) In wild-type V. cholerae, FadR activates the expression of fabA, encoding the enzyme that catalyzes the first step in UFA biosynthesis. In the presence of UFAs, ToxT forms a “closed complex” that is unable to activate gene expression or to be proteolyzed. (B) In a ΔfadR or fabAGG mutant, the levels of certain UFAs are reduced and ToxT is locked into the “open complex” that is capable of activating gene expression but is also able to be proteolyzed. In the ΔfadR and fabAGG mutants, there may also be an increase in the expression and/or activity of the proteases. Model based on that previously described (56). CTD, C-terminal domain; NTD, N-terminal domain.
It is still unclear how FadR influences the transcription of ToxT. The toxT promoter does not contain a FadR binding site that matches the consensus sequence observed in the promoter of its other regulated genes, and the purified protein does not appear to bind to it. The most likely explanation is that the influence of FadR on toxT transcription is indirect. However, none of the genes involved in FA degradation or UFA or SFA biosynthesis examined in this study strongly influenced the transcription of toxT when their expression was altered by FadR binding site mutations. Although we were unable to demonstrate repression of plsB by FadR in V. cholerae C6706, we ruled out a possible effect of this gene on virulence gene expression in this strain by inserting a GG mutation into the FadR binding site (35) and determined that it did not affect the expression of tcpA (data not shown). However, FadR clearly regulates the expression of additional genes in V. cholerae and one or more of these may influence the transcription of toxT. Since the toxT promoter is responsive to changes in central metabolism (58), it is also possible that global metabolic changes that occur in the ΔfadR mutant result in altered transcription of ToxT.
The strategy that we used here to try to elucidate why the ΔfadR mutant shows reduced expression from the toxT promoter in the El Tor biotype unexpectedly led to the discovery of a second route by which FadR impacts ToxT, via the activation of fabA expression. Since both ToxT and FadR are responsive to the presence of UFAs, it is possible that the intracellular levels of certain UFAs need to be optimal to promote the expression of the virulence cascade. High levels of exogenous UFAs bind directly to ToxT and inhibit its activity. In contrast, low levels of intracellular UFAs caused by the downregulation of fabA expression may promote the proteolysis of ToxT. In V. vulnificus, FadR was found to be essential for the organism to cause disease and the addition of the UFA oleate restored virulence to the ΔfadR mutant (47). However, the addition of oleate or other UFAs to either the V. cholerae ΔfadR or fabAGG mutants do not restore the levels of ToxT in these strains. We are currently in the process of trying to understand the reasons for this.
The work presented here identifies a new link between the expression of genes involved in UFA biosynthesis that are regulated by FadR and virulence in V. cholerae. Since the amino acid sequences of FadR from the classical and El Tor biotypes are identical, it is not yet clear why FadR influences the expression of the virulence cascade in the latter but not in the former. Previous transcriptome analyses have revealed a large number of differences in gene expression between the classical and El Tor biotypes (59). Thus, a novel way of regulating the expression of the virulence cascade in response to UFAs appears to have evolved in the El Tor biotype, and future work will be directed toward elucidating these mechanisms.
MATERIALS AND METHODS
Bacterial strains and media.
The bacterial strains used in this study are described in Table 2. Strains were maintained at −70°C in Luria-Bertani (LB) medium (60) containing 30% (vol/vol) glycerol. Cultures were grown in either AKI medium (37) or tryptone broth (60). Antibiotics were used at the following concentrations in LB medium: ampicillin, 100 μg/ml; kanamycin, 45 μg/ml; polymyxin B, 50 units/ml; streptomycin, 1 mg/ml. X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) was used in LB agar at 40 μg/ml.
TABLE 2.
Bacterial strains used in this study
| V. cholerae strain | Description/relevant genotype | Reference or source |
|---|---|---|
| C6706 str2 | El Tor Inaba; streptomycin resistant | Lab collection |
| GK1257 | C6706 ΔfadR | This work |
| GK1499 | KSK1267 ΔfadR | This work |
| GK1502 | KSK979 ΔfadR | This work |
| GK1504 | WL124 ΔfadR | This work |
| GK1536 | MBN135 ΔfadR | This work |
| GK1545 | KSK262 with lacZ from E. coli | 33 |
| GK1609 | GK1545 ΔlacZ::fabB-lacZ | 33 |
| GK1610 | GK1609 ΔfadR | 33 |
| GK1630 | GK1545 ΔlacZ::acpP-lacZ | This work |
| GK1632 | GK1630 ΔfadR | This work |
| GK1657 | KSK979 fabBGG | This work |
| GK1669 | GK1545ΔlacZ::fabBGG-lacZ | This work |
| GK1672 | GK1669 ΔfadR | This work |
| GK1675 | KSK979 fabAGG | This work |
| GK1687 | KSK979 fadEGG | This work |
| GK1689 | C6706 fabAGG | This work |
| GK1691 | C6706 fabBGG | This work |
| GK1790 | WL1268 fabAGG | This work |
| GK1954 | KSK2325 ΔfadR | This work |
| GK1981 | KSK979 fadHGG | This work |
| GK1982 | KSK979 fadBGG | This work |
| GK2016 | KSK979 acpPGG | This work |
| GK2017 | GK1545ΔlacZ::fabH-lacZ | This work |
| GK2020 | GK2017 ΔfadR | This work |
| GK2022 | KSK979 fabHGG | This work |
| GK2035 | GK1545 ΔlacZ::acpPGG-lacZ | This work |
| GK2037 | GK2035 ΔfadR | This work |
| GK2039 | GK1545ΔlacZ::fabHGG-lacZ | This work |
| GK2040 | GK2039 ΔfadR | This work |
| GK2041 | C6706 acpPGG | This work |
| GK2043 | C6706 fabHGG | This work |
| GK2136 | JAS273 ΔfadR | This work |
| GK2207 | KSK979 ΔlacZ::fabA | This work |
| GK2209 | GK1675 ΔlacZ::fabA | This work |
| JAS273 | KSK1267 ΔrseP | This work |
| KSK262 | C6706 str2 ΔlacZ3 | 8 |
| KSK725 | KSK262 tcpP-lacZ | 8 |
| KSK979 | KSK262 tcpA-lacZ | This work |
| KSK1184 | C6706 str2 ΔtoxT | This work |
| KSK1267 | KSK262 toxT-lacZ | This work |
| KSK2325 | KSK262 ctx-lacZ | This work |
| MBN135 | ΔtcpA-lacZ classical | 68 |
| WL124 | KSK262 toxR-lacZ | This work |
| WL982 | KSK725 ΔfadR | This work |
| WL1005 | KSK262 ΔlacZ::fabA-lacZ | 33 |
| WL1007 | WL1005 ΔfadR | 33 |
| WL1020 | KSK262 ΔlacZ::fabAGG-lacZ | This work |
| WL1022 | WL1020 ΔfadR | This work |
| WL1027 | GK1545 ΔlacZ::fadE-lacZ | 33 |
| WL1029 | WL1027 ΔfadR | 33 |
| WL1031 | GK1545 ΔlacZ::fadBA-lacZ | 33 |
| WL1035 | WL1031 ΔfadR | 33 |
| WL1040 | GK1545 ΔlacZ::fadH-lacZ | 33 |
| WL1042 | WL1040 ΔfadR | 33 |
| WL1056 | GK1545 ΔlacZ::fadEGG-lacZ | This work |
| WL1058 | WL1056 ΔfadR | This work |
| WL1060 | GK1545 ΔlacZ::fadBAGG-lacZ | This work |
| WL1062 | WL1060 ΔfadR | This work |
| WL1064 | GK1545 ΔlacZ::fadHGG-lacZ | This work |
| WL1066 | WL1064 ΔfadR | This work |
| WL1231 | KSK1267 fabAGG | This work |
| WL1268 | KSK1267 toxT’-‘lacZ | This work |
| WL1271 | WL1268 ΔfadR | This work |
Construction of lacZ fusion strains.
The various lacZ fusions were constructed by amplifying DNA fragments upstream and downstream of each gene from C6706 str2 chromosomal DNA using primers as follows (see Table S1 in the supplemental material): for ctx, CTX1a/CTX1b and CTX3/CTX4; for toxT, RT19/TX5 and TX6/TX7; and for toxR, MN27/TXR21 and MN30/TXR22. The fragments, together with a promoterless lacZ fragment from pVC200 (61), were inserted into either pKAS46 (62) or pKAS154 (63), and the resulting plasmids were used for allelic exchange (62) into V. cholerae strain KSK262 (8). The tcpA-lacZ and tcpP-lacZ fusions were previously described (8, 64). The toxT‘-'lacZ translational fusion was constructed by amplifying a DNA fragment from the toxT gene using RT12ET with TX14 and a DNA fragment from plasmid pVC200 (61) using T-LacZ10 with T-LacZ11. The fragments were ligated into pKAS154 (63), and the resulting plasmid was used for allelic exchange into V. cholerae strain KSK1267. The wild-type fabH-lacZ and acpP-lacZ fusions were constructed using primers VC2023FabH8/VC2023FabH2 and FabF1/FabF2, respectively. The resulting fragments were inserted into pWEL236 (33), and the fusions were introduced into the lacZ locus of KSK262 by allelic exchange. The construction of the wild-type fadBA, fadE, fadH, fabA, and fabB fusions were previously described (33).
Construction of FadR binding site mutations.
The various mutations in the FadR-regulated promoters were constructed by PCR amplifying two DNA fragments from C6706 str2 using primers (Table S1) as follows: fabA, FabA3/FabA6 and FabA4/FabA5; fabB, FabB15/FabB13 and FabB12/FabB14; fadBA, FadB4/FadB7 and FadB8/FadB9; fadE, FadE3/FadE4 and FadE5/FadE6; fadH, FadH6/FadH4 and FadH5/FadH7; fabH, FabH4/FabH5 and FabH6/FabH7; and acpP, FabF4/FabF5 and FabF6/FabF7. The fragments were inserted into pKAS154. The resulting GG mutant promoter plasmids were then used as a source of DNA for PCR amplification using the same primers that were initially used to construct the wild-type fusions (33): fabA, FabA1/FabA3; fabB, FabB3/FabB4; fadBA, FadB1/FadB4; fadE, FadE1/FadE3; fadH, FadH1/FadH3; fabH, VC2023FabH8/VC2023FabH2; and acpP, FabF1/FabF2. The fragments were inserted into either pKAS180 (65) or pWEL236 (33), and the resulting fusions were introduced into the lacZ locus of KSK262 by allelic exchange.
Construction of deletion mutations, expression plasmids, and merodiploid.
The ΔtoxT and ΔrseP mutations were constructed by amplifying DNA fragments upstream and downstream of the genes from C6706 str2 using RT19/TX5 and TX6/TX7 for the former and YaelA/YaelB and YaelC/YaelD for the latter (Table S1). The fragments were inserted into pKAS46 (62), and the resulting deletions were introduced into V. cholerae by allelic exchange. The ΔfadR mutation was constructed as previously described (33). The FadR overexpressing plasmid pWEL231 was constructed by amplifying fadR from C6706 str2 chromosomal DNA with primers FadR7 and FadR8 and ligating the resulting product into pKAS178 (66). The fabA merodiploid strain was constructed by PCR amplifying a DNA fragment containing the fabA gene along with upstream and downstream sequences using primers FabA9 and FabA10 and inserting this fragment into pGKK344, constructed similarly to pKAS180 (65) except that the fragments were ligated into pKAS154 (63). It was then introduced into V. cholerae by allelic exchange.
Fatty acid analysis.
Strains were grown in tryptone broth at 37°C with aeration for 5 h. Cells were pelleted by centrifugation, washed with fresh tryptone broth, and frozen on dry ice. Fatty acid content was determined by Microbial ID, Inc. (Newark, DE).
Immunoblot analysis.
Whole-cell extracts from the various cultures were prepared, and equivalent amounts of total protein as determined by the bicinchoninic acid (BCA) protein assay (Pierce) were analyzed on a 16% SDS-PAGE gel. Proteins were visualized by transferring to nitrocellulose and probing with either anti-ToxT antibody or anti-TcpA antibody using the ECL detection system (Amersham).
Gel mobility shift assays.
FadR was purified using the Impact-CN protein fusion and purification system (New England BioLabs) as previously described (33). The DNA fragments for the assays were amplified by PCR as follows: for fadBA, FadB1/FadB2; for toxT, TX11/TX12. The fragments were gel purified and end labeled with digoxigenin as described previously (67). Binding reactions for FadR were carried out with 20 mM Tris (pH 7.5), 10 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol (DTT), and 1 μg poly(dI-dC). The samples were applied to a 5% polyacrylamide gel and subjected to electrophoresis. The DNA was transferred to nylon membranes by electroblotting, probed with anti-digoxigenin-alkaline phosphatase antibody (Amersham Pharmacia), and visualized using chemiluminescence.
β-Galactosidase assays.
β-Galactosidase assays (60) were carried out by growing cultures either in AKI medium statically for 3.5 h at 37°C or in tryptone broth with aeration for 5 h at 37°C. Assays were done in duplicate for each culture, and the data are representative results from at least two separate experiments.
Supplementary Material
ACKNOWLEDGMENTS
We thank John Sutherland and Carolyn Lee for technical assistance, S. Almagro-Moreno and John Cronan for helpful discussions, and F. J. Kull for critical reading of the manuscript.
This work was supported by National Institutes of Health grants AI039654 (to R.K.T.; also supporting the efforts of G.K., W.L., and K.S.) and AI120068 (to K.S.; also supporting the efforts of G.K.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00762-16.
REFERENCES
- 1.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matson JS, Withey JH, DiRita VJ. 2007. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect Immun 75:5542–5549. doi: 10.1128/IAI.01094-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Champion GA, Neely MN, Brennan MA, DiRita VJ. 1997. A branch in the ToxR regulatory cascade of Vibrio cholerae revealed by characterization of toxT mutant strains. Mol Microbiol 23:323–331. doi: 10.1046/j.1365-2958.1997.2191585.x. [DOI] [PubMed] [Google Scholar]
- 4.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator ToxR is a transmembrane DNA binding protein. Cell 48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 5.Häse CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 95:730–734. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krukonis ES, Yu RR, DiRita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol 38:67–84. doi: 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 7.Skorupski K, Taylor RK. 1999. A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol Microbiol 31:763–771. doi: 10.1046/j.1365-2958.1999.01215.x. [DOI] [PubMed] [Google Scholar]
- 8.Kovacikova G, Skorupski K. 1999. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J Bacteriol 181:4250–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kovacikova G, Lin W, Skorupski K. 2004. Vibrio cholerae AphA uses a novel mechanism for virulence gene activation that involves interaction with the LysR-type regulator AphB at the tcpPH promoter. Mol Microbiol 53:129–142. doi: 10.1111/j.1365-2958.2004.04121.x. [DOI] [PubMed] [Google Scholar]
- 10.De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. 2005. Crystal structure of the virulence gene activator AphA from Vibrio cholerae reveals it is a novel member of the winged helix transcription factor superfamily. J Biol Chem 280:13779–13783. doi: 10.1074/jbc.M413781200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor JL, De Silva RS, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. 2012. The crystal structure of AphB, a virulence gene activator from Vibrio cholerae, reveals residues that influence its response to oxygen and pH. Mol Microbiol 83:457–470. doi: 10.1111/j.1365-2958.2011.07919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kovacikova G, Lin W, Skorupski K. 2010. The LysR-type virulence activator AphB regulates the expression of genes in Vibrio cholerae in response to low pH and anaerobiosis. J Bacteriol 192:4181–4191. doi: 10.1128/JB.00193-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta S, Chowdhury R. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun 65:1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatterjee A, Dutta PK, Chowdhury R. 2007. Effect of fatty acids and cholesterol present in bile on expression of virulence factors and motility of Vibrio cholerae. Infect Immun 75:1946–1953. doi: 10.1128/IAI.01435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abuaita BH, Withey JH. 2009. Bicarbonate induces Vibrio cholerae virulence gene expression by enhancing ToxT activity. Infect Immun 77:4111–4120. doi: 10.1128/IAI.00409-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tischler AD, Camilli A. 2005. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun 73:5873–5882. doi: 10.1128/IAI.73.9.5873-5882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller MB, Skorupski K, Lenz DH, Taylor RK, Bassler BL. 2002. Parallel quorum sensing systems converge to regulate virulence in Vibrio cholerae. Cell 110:303–314. doi: 10.1016/S0092-8674(02)00829-2. [DOI] [PubMed] [Google Scholar]
- 18.Lowden MJ, Skorupski K, Pellegrini M, Chiorazzo MG, Taylor RK, Kull FJ. 2010. Structure of Vibrio cholerae ToxT reveals a mechanism for fatty acid regulation of virulence genes. Proc Natl Acad Sci U S A 107:2860–2865. doi: 10.1073/pnas.0915021107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Childers BM, Cao X, Weber GG, Demeler B, Hart PJ, Klose KE. 2011. N-terminal residues of the Vibrio cholerae virulence regulatory protein ToxT involved in dimerization and modulation by fatty acids. J Biol Chem 286:28644–28655. doi: 10.1074/jbc.M111.258780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plecha SC, Withey JH. 2015. Mechanism for inhibition of Vibrio cholerae ToxT activity by the unsaturated fatty acid components of bile. J Bacteriol 197:1716–1725. doi: 10.1128/JB.02409-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cronan JE Jr, Subrahmanyam S. 1998. FadR, transcriptional co-ordination of metabolic expediency. Mol Microbiol 29:937–943. doi: 10.1046/j.1365-2958.1998.00917.x. [DOI] [PubMed] [Google Scholar]
- 22.Fujita Y, Matsuoka H, Hirooka K. 2007. Regulation of fatty acid metabolism in bacteria. Mol Microbiol 66:829–839. doi: 10.1111/j.1365-2958.2007.05947.x. [DOI] [PubMed] [Google Scholar]
- 23.van Aalten DMF, DiRusso CC, Knudsen J, Wierenga RK. 2000. Crystal structure of FadR, a fatty acid-responsive transcription factor with a novel acyl coenzyme A-binding fold. EMBO J 19:5167–5177. doi: 10.1093/emboj/19.19.5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Aalten DMF, DiRusso CC, Knudsen J. 2001. The structural basis of acyl coenzyme A-dependent regulation of the transcription factor FadR. EMBO J 20:2041–2050. doi: 10.1093/emboj/20.8.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Heath RJ, Li Z, Rock CO, White SW. 2001. The FadR-DNA complex. Transcriptional control of fatty acid metabolism in Escherichia coli. J Biol Chem 276:17373–17379. [DOI] [PubMed] [Google Scholar]
- 26.Clark DP, Cronan JE Jr. 1996. Two-carbon compounds and fatty acids as carbon sources, p 343–357. In Neidhardt FC. (ed), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC. [Google Scholar]
- 27.Henry MF, Cronan JE Jr. 1991. Escherichia coli transcription factor that both activates fatty acid synthesis and represses fatty acid degradation. J Mol Biol 222:843–849. doi: 10.1016/0022-2836(91)90574-P. [DOI] [PubMed] [Google Scholar]
- 28.Campbell JW, Cronan JE Jr. 2001. Escherichia coli FadR positively regulates transcription of the fabB fatty acid biosynthetic gene. J Bacteriol 183:5982–5990. doi: 10.1128/JB.183.20.5982-5990.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.My L, Rekoske B, Lemke JJ, Viala JP, Gourse RL, Bouveret E. 2013. Transcription of the Escherichia coli fatty acid synthesis operon fabHDG is directly activated by FadR and inhibited by ppGpp. J Bacteriol 195:3784–3795. doi: 10.1128/JB.00384-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.My L, Achkar NG, Viala JP, Bouveret E. 2015. Reassessment of the genetic regulation of fatty acid synthesis in Escherichia coli: global positive control by the functional dual regulator FadR. J Bacteriol 197:1862–1872. doi: 10.1128/JB.00064-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiRusso CC, Black PN. 2004. Bacterial long chain fatty acid transport: gateway to a fatty acid-responsive signaling system. J Biol Chem 279:49563–49566. doi: 10.1074/jbc.R400026200. [DOI] [PubMed] [Google Scholar]
- 32.DiRusso CC, Heimert TL, Metzger AK. 1992. Characterization of FadR, a global transcriptional regulator of fatty acid metabolism in Escherichia coli. Interaction with the fadB promoter is prevented by long chain fatty acyl coenzyme As. J Biol Chem 267:8685–8691. [PubMed] [Google Scholar]
- 33.Shi W, Kovacikova G, Lin W, Taylor RK, Skorupski K, Kull FJ. 2015. The 40-residue insertion in Vibrio cholerae FadR facilitates binding of an additional fatty acyl-CoA ligand. Nat Commun 6:6032. doi: 10.1038/ncomms7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iram SH, Cronan JE. 2005. Unexpected functional diversity among FadR fatty acid transcriptional regulatory proteins. J Biol Chem 280:32148–32156. doi: 10.1074/jbc.M504054200. [DOI] [PubMed] [Google Scholar]
- 35.Feng Y, Cronan JE. 2011. The Vibrio cholerae fatty acid regulatory protein, FadR, represses transcription of plsB, the gene encoding the first enzyme of membrane phospholipid biosynthesis. Mol Microbiol 81:1020–1033. doi: 10.1111/j.1365-2958.2011.07748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray S, Chatterjee E, Chatterjee A, Paul K, Chowdhury R. 2011. A fadD mutant of Vibrio cholerae is impaired in the production of virulence factors and membrane localization of the virulence regulatory protein TcpP. Infect Immun 79:258–266. doi: 10.1128/IAI.00663-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iwanaga M, Yamamoto K, Higa N, Ichinose Y, Nakasone N, Tanabe M. 1986. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol Immunol 30:1075–1083. doi: 10.1111/j.1348-0421.1986.tb03037.x. [DOI] [PubMed] [Google Scholar]
- 38.Kovacikova G, Skorupski K. 2000. Differential activation of the tcpPH promoter by AphB determines biotype specificity of virulence gene expression in Vibrio cholerae. J Bacteriol 182:3228–3238. doi: 10.1128/JB.182.11.3228-3238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chatterjee E, Chowdhury R. 2013. Reduced virulence of the Vibrio cholerae fadD mutant is due to induction of the extracytoplasmic stress response. Infect Immun 81:3935–3941. doi: 10.1128/IAI.00722-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gui L, Sunnarborg A, LaPorte DC. 1996. Regulated expression of a repressor protein: FadR activates iclR. J Bacteriol 178:4704–4709. doi: 10.1128/jb.178.15.4704-4709.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kazakov AE, Rodionov DA, Alm E, Arkin AP, Dubchak I, Gelfand MS. 2009. Comparative genomics of regulation of fatty acid and branched-chain amino acid utilization in proteobacteria. J Bacteriol 191:52–64. doi: 10.1128/JB.01175-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sadovskaya NS, Laikova ON, Mironov AA, Gelfand MS. 2001. Study of regulation of long-chain fatty acid metabolism using computer analysis of complete bacterial genomes. Mol Biol (Mosk) 35:1010–1014. (In Russian.) [PubMed] [Google Scholar]
- 43.Campbell JW, Cronan JE Jr. 2001. Bacterial fatty acid biosynthesis: targets for antibacterial drug discovery. Annu Rev Microbiol 55:305–332. [DOI] [PubMed] [Google Scholar]
- 44.Cronan JE Jr, Birge CH, Vagelos PR. 1969. Evidence for two genes specifically involved in unsaturated fatty acid biosynthesis in Escherichia coli. J Bacteriol 100:601–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magnuson K, Jackowski S, Rock CO, Cronan JE Jr. 1993. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol Rev 57:522–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nunn WD, Giffin K, Clark D, Cronan JE Jr. 1983. Role for fadR in unsaturated fatty acid biosynthesis in Escherichia coli. J Bacteriol 154:554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown RN, Gulig PA. 2008. Regulation of fatty acid metabolism by FadR is essential for Vibrio vulnificus to cause infection of mice. J Bacteriol 190:7633–7644. doi: 10.1128/JB.01016-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simons RW, Egan PA, Chute HT, Nunn WD. 1980. Regulation of fatty acid degradation in Escherichia coli: isolation and characterization of strains bearing insertion and temperature-sensitive mutations in gene fadR. J Bacteriol 142:621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai CY, Cronan JE. 2003. Beta-ketoacyl-acyl carrier protein synthase III (FabH) is essential for bacterial fatty acid synthesis. J Biol Chem 51:51494–51503. [DOI] [PubMed] [Google Scholar]
- 50.Podkovyrov SM, Larson TJ. 1995. Lipid biosynthetic genes and a ribosomal protein gene are cotranscribed. FEBS Lett 368:429–431. doi: 10.1016/0014-5793(95)00702-B. [DOI] [PubMed] [Google Scholar]
- 51.Rawlings M, Cronan JE Jr. 1992. The gene encoding Escherichia coli acyl carrier protein lies within a cluster of fatty acid biosynthetic genes. J Biol Chem 267:5751–5754. [PubMed] [Google Scholar]
- 52.Cronan JE Jr, Gelmann EP. 1973. An estimate of the minimum amount of unsaturated fatty acid required for growth of Escherichia coli. J Biol Chem 248:1188–1195. [PubMed] [Google Scholar]
- 53.Cronan JE Jr, Li WB, Coleman R, Narasimhan M, de Mendoza D, Schwab JM. 1988. Derived amino acid sequence and identification of active site residues of Escherichia coli β-hydroxydecanoyl thioester dehydrase. J Biol Chem 263:4641–4646. [PubMed] [Google Scholar]
- 54.Feng Y, Cronan JE. 2009. Escherichia coli unsaturated fatty acid synthesis. Complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J Biol Chem 284:29526–29535. doi: 10.1074/jbc.M109.023440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abuaita BH, Withey JH. 2011. Termination of Vibrio cholerae virulence gene expression is mediated by proteolysis of the major virulence activator, ToxT. Mol Microbiol 81:1640–1653. doi: 10.1111/j.1365-2958.2011.07798.x. [DOI] [PubMed] [Google Scholar]
- 56.Thomson JJ, Plecha SC, Withey JH. 2015. A small unstructured region in Vibrio cholerae ToxT mediates the response to positive and negative effectors and ToxT proteolysis. J Bacteriol 197:654–668. doi: 10.1128/JB.02068-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Wehmeyer G, Lovell S, Battaile KP, Egan SM. 2016. 1.65 Å resolution structure of the AraC-family transcriptional activator ToxT from Vibrio cholerae. Acta Crystallogr F Struct Biol Commun 72:726–731. doi: 10.1107/S2053230X1601298X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minato Y, Fassio SR, Wolfe AJ, Häse CC. 2013. Central metabolism controls transcription of a virulence gene regulator in Vibrio cholerae. Microbiology 159:792–802. doi: 10.1099/mic.0.064865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Beyhan S, Tischler AD, Camilli A, Yildiz FH. 2006. Differences in gene expression between the classical and El Tor biotypes of Vibrio cholerae O1. Infect Immun 74:3633–3642. doi: 10.1128/IAI.01750-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 61.Parsot C, Mekalanos JJ. 1990. Expression of ToxR, the transcriptional activator of the virulence factors in Vibrio cholerae, is modulated by the heat shock response. Proc Natl Acad Sci U S A 87:9898–9902. doi: 10.1073/pnas.87.24.9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skorupski K, Taylor RK. 1996. Positive selection vectors for allelic exchange. Gene 169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 63.Kovacikova G, Skorupski K. 2002. Regulation of virulence gene expression in Vibrio cholerae by quorum sensing: HapR functions at the aphA promoter. Mol Microbiol 46:1135–1147. doi: 10.1046/j.1365-2958.2002.03229.x. [DOI] [PubMed] [Google Scholar]
- 64.Lin W, Kovacikova G, Skorupski K. 2007. The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol Microbiol 64:953–967. doi: 10.1111/j.1365-2958.2007.05693.x. [DOI] [PubMed] [Google Scholar]
- 65.Kovacikova G, Skorupski K. 2002. The alternative sigma factor σE plays an important role in intestinal survival and virulence in Vibrio cholerae. Infect Immun 70:5355–5362. doi: 10.1128/IAI.70.10.5355-5362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kovacikova G, Lin W, Skorupski K. 2005. Dual regulation of genes involved in acetoin biosynthesis and motility/biofilm formation by the virulence activator AphA and the acetate-responsive LysR-type regulator AlsR in Vibrio cholerae. Mol Microbiol 57:420–433. doi: 10.1111/j.1365-2958.2005.04700.x. [DOI] [PubMed] [Google Scholar]
- 67.Kovacikova G, Skorupski K. 2001. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol Microbiol 41:393–407. doi: 10.1046/j.1365-2958.2001.02518.x. [DOI] [PubMed] [Google Scholar]
- 68.Nye MB, Pfau JD, Skorupski K, Taylor RK. 2000. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J Bacteriol 182:4295–4303. doi: 10.1128/JB.182.15.4295-4303.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.