

Abstract
The soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE) protein syntaxin‐1 adopts a closed conformation when bound to Munc18‐1, preventing binding to synaptobrevin‐2 and SNAP‐25 to form the ternary SNARE complex. Although it is known that the MUN domain of Munc13‐1 catalyzes the transition from the Munc18‐1/syntaxin‐1 complex to the SNARE complex, the molecular mechanism is unclear. Here, we identified two conserved residues (R151, I155) in the syntaxin‐1 linker region as key sites for the MUN domain interaction. This interaction is essential for SNARE complex formation in vitro and synaptic vesicle priming in neuronal cultures. Moreover, this interaction is important for a tripartite Munc18‐1/syntaxin‐1/MUN complex, in which syntaxin‐1 still adopts a closed conformation tightly bound to Munc18‐1, whereas the syntaxin‐1 linker region changes its conformation, similar to that of the LE mutant of syntaxin‐1 when bound to Munc18‐1. We suggest that the conformational change of the syntaxin‐1 linker region induced by Munc13‐1 initiates ternary SNARE complex formation in the neuronal system.
Keywords: Munc13, Munc18, neurotransmitter release, synaptic vesicle fusion, syntaxin
Subject Categories: Membrane & Intracellular Transport, Neuroscience
Introduction
In neurons, Ca2+‐triggered exocytosis, the fusion of synaptic vesicles with the plasma membrane, is mediated by the SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment receptor) proteins synaptobrevin‐2, syntaxin‐1 and SNAP‐25 (Südhof, 2013; Rothman, 2014; Rizo & Xu, 2015). Synaptobrevin‐2 is primarily localized to synaptic vesicles, while syntaxin‐1 and SNAP‐25 are localized to the plasma membrane. They form a trans ternary SNARE complex via their SNARE motifs, juxtaposing the two membranes and providing energy for membrane fusion in a zipper‐like fashion (Sutton et al, 1998; Weber et al, 1998; Pobbati et al, 2006). The neuronal SM (Sec1/Munc18‐like) protein Munc18‐1 is also essential for exocytosis, and it orchestrates SNARE complex formation through multiple interactions with the SNAREs (Verhage et al, 2000; Südhof & Rothman, 2009; Rizo & Südhof, 2012).
Among the neuronal SNAREs, syntaxin‐1 regulates SNARE activity at several distinct stages of exocytosis (Brunger, 2005; Südhof & Rothman, 2009). It consists of an N‐terminal sequence (the N‐peptide), a three‐helix bundle (the Habc domain), a linker region, a SNARE motif (the H3 domain), and a transmembrane region (Fig 1A, top panel) (Rizo & Xu, 2015). Prior to SNARE complex formation, Munc18‐1 locks syntaxin‐1 in a “closed” conformation that involves an interaction between the Habc domain and the H3 domain of syntaxin‐1, thus preventing SNARE‐mediated fusion by gating the entry of syntaxin‐1 into the ternary SNARE complex (Misura et al, 2000; Arunachalam et al, 2008; Südhof & Rothman, 2009). The crystal structure of the Munc18‐1/syntaxin‐1 heterodimeric complex revealed extensive interactions of syntaxin‐1 in the closed conformation with the concave surface formed by domains 1 and 3 of Munc18‐1 (Misura et al, 2000; see also Fig 1A), explaining the inability of the syntaxin‐1 H3 domain in this closed conformation to interact with SNAP‐25 and synaptobrevin‐2. In the late stage of exocytosis, Munc18‐1 also binds to the assembled ternary SNARE complex containing syntaxin‐1 in an “open” conformation (Shen et al, 2007; Deak et al, 2009; Xu et al, 2010; Hu et al, 2011), in a manner that requires an interaction of Munc18‐1 with the syntaxin‐1 N‐peptide. In this open conformation, the H3 domain of syntaxin‐1 interacts with its partner SNARE motifs to form a four‐helix bundle (Sutton et al, 1998), while the syntaxin‐1 Habc domain is apart from the H3 domain (Hanson et al, 1997; Nicholson et al, 1998). Therefore, ternary SNARE complex formation is accompanied by the transition of syntaxin‐1 from the closed to the open conformation.
Figure 1. Identification of the key residues in syntaxin‐1 that are essential for Munc13‐1 activity in ternary SNARE complex formation.
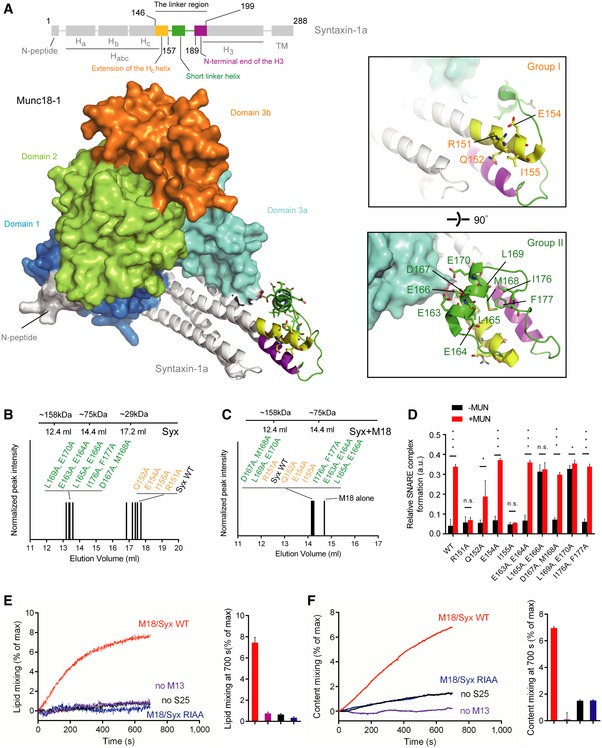
-
ADomain structure of syntaxin‐1 (top) and crystal structure of the closed Munc18‐1/syntaxin‐1 complex with annotations (bottom, PDB ID 3C98). The insets show two close‐up views of the syntaxin‐1 linker region. Labels correspond to residues that were mutated.
-
B, CSize‐exclusion chromatography peak elution volumes of mutants of the syntaxin‐1 linker region in the absence (B) and presence of Munc18‐1 (C) using a 24 ml Superdex 200 column.
-
DRelative efficiencies of ternary SNARE complex formation using syntaxin‐1 with selected mutations in the linker region. The reactions were performed in the presence of Munc18‐1 and/or the MUN domain and monitored by ensemble FRET efficiency between fluorescent dye‐labeled SNAP‐25 and synaptobrevin‐2. Shown are means ± SD; n.s., not significant (P > 0.05); *P < 0.05; ***P < 0.001, using two‐tailed Student's t‐test with n = 5 technical replicates.
-
E, FEnsemble lipid (E) and content mixing (F) between proteoliposomes with reconstituted Munc18‐1/syntaxin‐1 complex and proteoliposomes with reconstituted synaptobrevin‐2 and synaptotagmin‐1 in the presence of 1 mM Ca2+. Bar charts are means ± SD for n = 3 technical replicates. M18, Munc18‐1; M13, Munc13‐1 C1‐C2B‐MUN domain; S25, SNAP‐25; Syx, syntaxin‐1.
Munc13s belong to a large family of multidomain proteins that are abundant in the presynaptic terminal (Rizo & Xu, 2015) and they play a central function in synaptic vesicle priming (Richmond et al, 1999; Varoqueaux et al, 2002), a step prior to Ca2+‐triggered release. Moreover, in vitro studies have demonstrated that the Munc13‐1 MUN domain catalyzes the transit of syntaxin‐1 from the Munc18‐1/syntaxin‐1 heterodimeric complex to the ternary SNARE complex (Basu et al, 2005; Ma et al, 2011). This catalytic function of Munc13‐1 requires interactions between the MUN domain and the Munc18‐1/syntaxin‐1 complex, and it involves a highly conserved hydrophobic pocket (called the “NF” pocket) located at the midpoint of the MUN domain (Ma et al, 2011; Yang et al, 2015). However, this interaction is relatively weak in solution and it is unlikely to be able to dissociate syntaxin‐1 from the tight Munc18‐1/syntaxin‐1 complex (dissociation constant in the low nM range) (Misura et al, 2000; Burkhardt et al, 2008). Thus, it is unclear how the Munc13‐1 MUN domain achieves the transit of syntaxin‐1 from the Munc18‐1/syntaxin‐1 complex into the ternary SNARE complex.
The syntaxin‐1 linker region (residues 146–199) is a segment connecting the Habc domain and the H3 domain, and it is composed of a C‐terminal extension of the Hc helix (yellow), a short linker helix (green), and the N‐terminal end of the H3 domain (purple) (Fig 1A). The syntaxin‐1 linker region switches from a defined secondary structure conformation when syntaxin‐1 is in complex with Munc18‐1 to a random coil conformation when syntaxin‐1 is part of the ternary SNARE complex (Misura et al, 2000; Margittai et al, 2003). This conformational change of the linker region may represent an important step in the transition from the Munc18‐1/syntaxin‐1 complex to the ternary SNARE complex. In support of this notion, the L165A, E166A mutations in the linker region of syntaxin‐1 (also called the “LE mutant”) (DulubovaI et al, 1999) allow the LE mutant of syntaxin‐1 to transit from the closed Munc18‐1/syntaxin‐1 complex to the ternary SNARE complex in the absence of Munc13‐1 when synaptobrevin‐2 and SNAP‐25 are added in vitro (Ma et al, 2011; Yang et al, 2015). Moreover, the LE mutant of syntaxin‐1 partially rescues Munc13 knockout in C. elegans (Richmond et al, 2001), although Munc13‐deficient mice do not survive with knock‐in of the LE mutant of syntaxin‐1 (Gerber et al, 2008). These findings suggest that (i) the syntaxin‐1 linker region probably constitutes an accessible target for Munc13‐1 function, and (ii) Munc13‐1 may induce a conformational change in the linker region to initiate the transit of syntaxin‐1 from the Munc18‐1/syntaxin‐1 complex to the ternary SNARE complex.
To test this hypothesis and shed light on how syntaxin‐1 is activated for ternary SNARE complex formation in synaptic exocytosis, we have conducted in vitro reconstitution, in vivo electrophysiology, and single‐molecule fluorescence resonance energy transfer (smFRET) experiments. We identified two conserved residues R151 and I155 (referred to as RI) in the syntaxin‐1 linker region that are critical for the function of Munc13‐1, both in vitro and in vivo. Moreover, this function depends on an interaction between the NF pocket on the Munc13‐1 MUN domain and the RI residues of syntaxin‐1. In addition, smFRET experiments revealed that the MUN domain does not dissociate the closed Munc18‐1/syntaxin‐1 complex, but rather induces a conformational change in the syntaxin‐1 linker region via the NF/RI interaction, thus enabling syntaxin‐1 to transit into the ternary SNARE complex once synaptobrevin‐2 and SNAP‐25 are added.
Results
Mutational analysis of syntaxin‐1 linker region
As mentioned above, in the complex with Munc18‐1, the linker region between the Habc domain and the H3 domain of syntaxin‐1 forms a network of interactions via the extension of the Hc helix, the short linker helix, and the N‐terminal end of the H3 domain (Misura et al, 2000; see also Fig 1A). Since we hypothesized that there is a functional interplay between the MUN domain and the syntaxin‐1 linker region in catalyzing the transit of syntaxin‐1 into the ternary SNARE complex, we functionally tested a set of mutations (Fig 1A) in the linker region of syntaxin‐1.
We first characterized these mutations by size‐exclusion chromatography. The group‐I mutations (Fig 1A, upper inset) located in the extension of the Hc helix had similar elution volumes to wild‐type syntaxin‐1 (Syx WT) (Fig 1B and Table EV1), whereas the group‐II mutations (Fig 1A, lower inset) in the short linker helix and the flanking regions had substantially lower elution volumes than Syx WT (Fig 1B and Table EV1). This suggests that the group‐II mutations destabilize the linker region, and perhaps lead to H3 domain self‐association (Misura et al, 2001). However, all the mutations in both groups produced a heterodimeric complex with Munc18‐1 similar to Syx WT bound to Munc18‐1 as assessed by the elution volumes of the complexes (Fig 1C and Table EV2), suggesting that all of these mutations do not influence the closed conformation of syntaxin‐1 when bound to Munc18‐1.
We next tested for potential functional defects of the syntaxin‐1 mutations in the Munc13‐mediated catalysis of ternary SNARE complex formation beginning with the Munc18‐1/syntaxin‐1 complex using both native gel (Fig EV1) and ensemble FRET assays (Fig 1D) as described previously (Ma et al, 2011; Yang et al, 2015). Consistent with previous results (Ma et al, 2011), the LE (L165A, E166A) mutant of syntaxin‐1 allowed efficient formation of the ternary SNARE complex even in the absence of the MUN domain (Fig 1D, see also Fig EV1). We also found that mutation of an adjacent pair of residues (L169A, E170A) behaves similar to the LE (L165A, E166A) mutant of syntaxin‐1. In contrast, the R151A or I155A mutation of syntaxin‐1 abolished the catalytic activity of the MUN domain (Fig 1D, see also Fig EV1), implying an important role of the R151 and I155 residues (referred to as RI hereafter) of syntaxin‐1 in mediating the transit of syntaxin‐1 to the ternary SNARE complex. The adjacent Q152A mutation of syntaxin‐1 had a milder, but statistically significant, effect on reducing the function of the MUN domain (Fig 1D, see also Fig EV1). The defects in MUN function caused by the R151A, I155A (RIAA) and Q152A mutations are unlikely due to syntaxin‐1 misfolding since the mutant proteins exhibited similar elution volumes to Syx WT as determined by size‐exclusion chromatography (Fig 1B) and the mutations did not influence SNARE complex formation with syntaxin‐1, SNAP‐25, and synaptobrevin‐2 (i.e. in the absence of Munc18‐1 and the MUN domain) (Fig EV2). Moreover, the RI residues and intervening sequence are highly conserved among plasma membrane syntaxin isoforms in different species (Fig EV3).
Figure EV1. Relative ternary SNARE complex formation bearing the syntaxin‐1 linker region mutations in the presence of Munc18‐1 and/or the MUN domain as monitored by native gel electrophoresis.
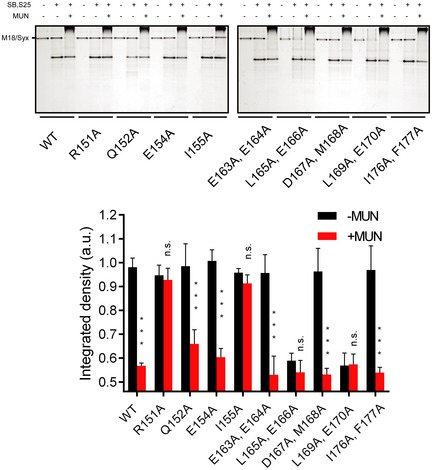
Integrated intensities of the bands of the Munc18‐1/syntaxin‐1 (M18/Syx) complex were used to determine relative ternary SNARE complex formation: The band of the M18/Syx complex gradually disappears with ternary SNARE complex formation in the presence of SNAP‐25, synaptobrevin‐2, and the MUN domain. The upper panel shows representative Coomassie brilliant blue‐stained native electrophoresis gels from one of three independent experiments. Data are processed by ImageJ (NIH) and shown as means ± SEM; n.s., not significant (P > 0.05); ***P < 0.001, using Student's t‐test with n = 3 technical replicates.
Figure EV2. The R151A, I155A mutations of syntaxin‐1 have no effect on ternary SNARE complex formation with syntaxin‐1, SNAP‐25, and synaptobrevin‐2 (i.e. in the absence of Munc18‐1 and the MUN domain).
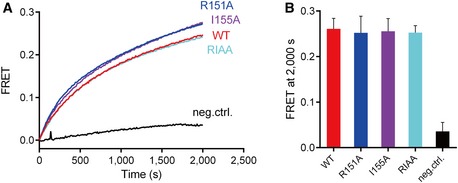
-
A, BTernary SNARE complex formation as monitored by ensemble FRET efficiency between fluorescent dye‐labeled SNAP‐25 and the cytoplasmic domain of synaptobrevin‐2 (A) and quantification of the results (B). Neg. ctrl. (negative control): The experiment was performed in the absence of syntaxin‐1. Shown are means ± SD for n = 3 technical replicates.
Figure EV3. Sequence alignment of the syntaxin‐1 linker region.

Sequence alignment was performed with Clustal Omega and interpreted with ESPript 3.0. Residues above 60% similarity are colored in red, and residues R151 and I155 are colored in cyan and indicated by asterisks on the top of the sequences. Secondary structure and relative solvent accessibility were calculated based on the crystal structure of the Munc18‐1/syntaxin‐1 complex (PDB entry 3C98): white, buried; blue, exposed; cyan, partially exposed. UniProt entries of each of the sequences are on the left. Rn, Rattus norvegicus; Mm, Mus musculus; Hs, Homo sapiens; Dr, Danio rerio; Dm, Drosophila melanogaster; Dp, Doryteuthis pealeii; Ce, Caenorhabditis elegans; Sc, Saccharomyces cerevisiae.
We further tested the importance of the RI residues with a proteoliposome fusion assay. Consistent with previous results (Ma et al, 2013; Yang et al, 2015), the C1‐C2B‐MUN fragment (which includes the C1 and C2B domains that bind to DAG and PIP2 containing membranes, respectively) promoted lipid mixing and content mixing between proteoliposomes reconstituted with the Munc18‐1/syntaxin‐1 complex and proteoliposomes reconstituted with synaptobrevin‐2 and synaptotagmin‐1 in the presence of SNAP‐25 and Ca2+ (Fig 1E and F). However, the catalytic activity of the C1‐C2B‐MUN fragment was much reduced when using Munc18‐1/syntaxin‐1 (RIAA) proteoliposomes (i.e. with syntaxin‐1 containing the R151A, I155A mutations) or, as negative control, in the absence of SNAP‐25 or the C1‐C2B‐MUN fragment, again suggesting a crucial function of the syntaxin‐1 RI residues for Munc13‐mediated ternary SNARE complex formation and subsequent membrane fusion beginning with the closed Munc18‐1/syntaxin‐1 complex.
The RI residues are crucial for synaptic vesicle priming
Considering the critical role of the syntaxin‐1 RI residues in vitro, we also tested the functional importance of these residues in neurotransmitter release in vivo by using a knockdown‐rescue approach (Zhou et al, 2013a) in cultured mouse hippocampal neurons. Endogenous syntaxin‐1 (syntaxin‐1a and syntaxin‐1b) expression was strongly suppressed by virally delivered shRNAs (see Materials and Methods), as previously reported (Zhou et al, 2013a). Expression of syntaxin‐1a rescued both the spontaneous mini inhibitory postsynaptic current (mIPSC) frequency and action potential‐evoked inhibitory postsynaptic current (evoked IPSC) amplitude and charge transfer (Fig 2A and B). In contrast, the syntaxin‐1a R151A or I155A mutation, individually or in combination, did not rescue these defects (Fig 2A and B). However, the mIPSC amplitude was unaltered in any condition (Fig 2A). Additional kinetic analyses for evoked IPSCs revealed no substantial difference in any condition that we tested, arguing against a major postsynaptic effect in these experiments (Appendix Fig S1A). However, the rise time of evoked IPSCs for syntaxin‐1a knockdown or for syntaxin‐1a R151A was more variable (Appendix Fig S1A–C), indicating a potential impairment of synchronous neurotransmitter release in these groups. Together, these data suggest that the syntaxin‐1 RI residues are essential for both spontaneous and Ca2+‐evoked release.
Figure 2. The syntaxin‐1 RI (R151, I155) residues are critical for synaptic vesicle priming and neurotransmitter release.
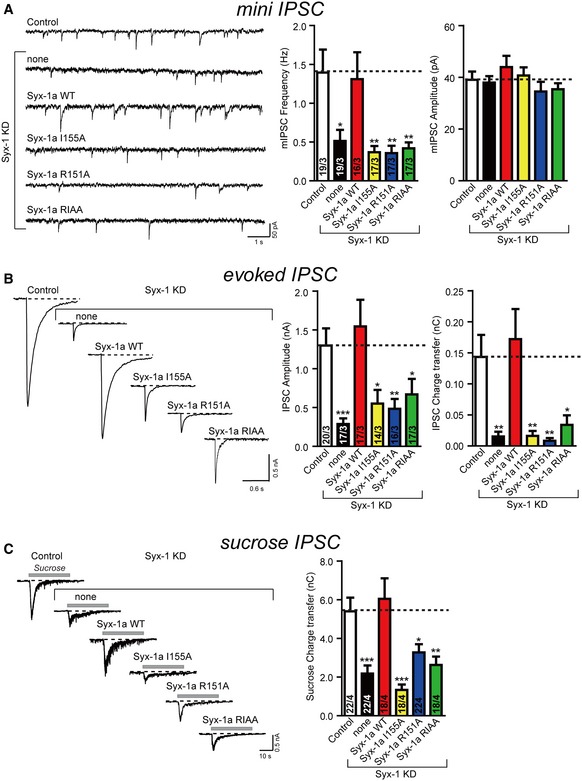
- Sample traces (left) and summary graphs (right) of mIPSCs recorded in WT hippocampal neurons that were infected with a control lentivirus (Control) or a lentivirus expressing only syntaxin‐1 shRNAs (none) or syntaxin‐1 shRNAs plus either full‐length syntaxin‐1a (Syx‐1a WT), or full‐length syntaxin‐1a with the I155 mutation (Syx‐1a I155A), full‐length syntaxin‐1a with the R151 mutation (Syx‐1a R151A), or full‐length syntaxin‐1a with the R151A, I155A mutations (Syx‐1a RIAA), respectively.
- Sample traces (left) and summary graphs (right) of action potential‐evoked IPSCs recorded in the infected neuronal cultures described in panel (A).
- Sample traces (left) and summary graphs (right) of IPSCs evoked by 0.5 M sucrose, recorded in the infected neuronal cultures described in panel (A).
We also characterized the size of the readily releasable pool (RRP) of vesicles from the synaptic responses induced by application of a hypertonic sucrose solution (Rosenmund & Stevens, 1996). The significant decrease in sucrose‐induced charge transfer in syntaxin‐1‐deficient neurons can be rescued by expressing syntaxin‐1a but not by the R151A or I155A mutant (Fig 2C). Thus, these results suggest that the syntaxin‐1 RI residues are also essential for priming of synaptic vesicles.
An essential pair of residues of the MUN domain has been previously identified (N1128, F1131, referred to as NF, located near the center of the MUN domain): mutation of the NF residues abolishes the catalytic activity of Munc13‐1 in vitro and the priming function of Munc13‐1 in neurotransmitter release in C. elegans (Yang et al, 2015). We also sought to validate the importance of the NF residues in mammals. Expression of either full‐length Munc13‐1 or the C1‐C2B‐MUN fragment in Munc13‐1‐deficient mouse hippocampal neurons restored mIPSCs and evoked IPSCs to levels comparable to those for WT neurons (Fig EV4A and B), suggesting that the C1‐C2B‐MUN fragment constitutes a basal requirement of Munc13‐1 for release in our experimental conditions. Note that in a more stringent condition (i.e. Munc13‐1/‐2 double knockout), the additional C2A and C2C domains flanking the C1‐C2B‐MUN fragment might also be indispensable for the full rescue of Munc13‐1 function (Zhou et al, 2013b; Liu et al, 2016). As expected, expression of the C1‐C2B‐MUN fragment containing the N1128A, F1131A (NFAA) mutations abolished both mIPSCs and evoked IPSCs (Fig EV4A and B), and led to a strongly reduced RRP size (Fig EV4C). Thus, consistent with the critical role in C. elegans (Yang et al, 2015), the Munc13‐1 NF residues are also essential for spontaneous and Ca2+‐evoked release and for synaptic vesicle priming in mammalian neurons.
Figure EV4. The NF residues in the MUN domain of Munc13‐1 are critical for synaptic vesicle priming and neurotransmitter release.
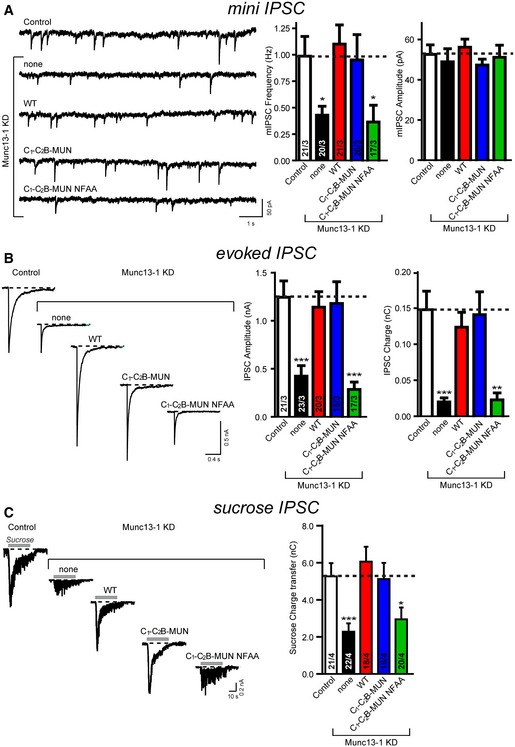
- Sample traces (left) and summary graphs (right) of mIPSCs recorded in WT hippocampal neurons that were infected with a control lentivirus (Control) or a lentivirus expressing only Munc13‐1 shRNAs (none) or Munc13‐1 shRNAs plus either full‐length Munc13‐1 (WT), or the C1‐C2B‐MUN fragment (C1‐C2B‐MUN) or the C1‐C2B‐MUN fragment containing the NFAA (N1128A, F1131A) mutations (C1‐C2B‐MUN NFAA), respectively.
- Sample traces (left) and summary graphs (right) of action potential‐evoked IPSCs recorded in the infected neuronal cultures described in panel (A).
- Sample traces (left) and summary graphs (right) of IPSCs evoked by 0.5 M sucrose, recorded in the infected neuronal cultures described in panel (A).
Interaction between the syntaxin‐1 RI and the MUN NF residues
We next performed liposome co‐flotation experiments to characterize a putative direct interaction between the syntaxin‐1 RI and Munc13‐1 NF residues. The MUN domain bound efficiently to liposomes containing reconstituted Munc18‐1/syntaxin‐1 complexes, but not to protein‐free liposomes (Fig 3A), consistent with previous observations (Yang et al, 2015). Intriguingly, either the MUN NFAA or the syntaxin‐1 RIAA mutations impaired such binding (Fig 3A), suggesting an interaction between regions involving the syntaxin‐1 RI and Munc13‐1 NF residues. To corroborate this notion, we synthesized a 15‐residue linker peptide (comprising residues 148–162 of syntaxin‐1, i.e. including the RI residues) and labeled it with Rhodamine B. We measured binding of the linker peptide to the MUN‐BC fragment (containing the B and C subdomains of the MUN domain, i.e. including the NF residues), as the MUN‐BC fragment retains the minimal catalytic activity of the MUN domain (Yang et al, 2015) and the MUN‐BC fragment can be concentrated to higher concentrations than the MUN domain in vitro. Using an ensemble fluorescence anisotropy assay, we found that the MUN‐BC fragment bound to the linker peptide in a dose‐dependent manner with a disassociation constant (K d) of 40.7 ± 14.4 μM (Fig 3B and Table EV3), suggesting a direct but weak interaction between the MUN domain and the syntaxin‐1 linker region. In addition, the cytoplasmic region of syntaxin‐1 (residues 2–253) competed with the interaction between the linker peptide and the MUN‐BC fragment (Fig 3B and Appendix Fig S2). As expected, the NFAA mutant of the MUN‐BC fragment and, as control, Munc18‐1 showed very little binding to the linker peptide (Fig 3B and C, and Table EV3).
Figure 3. Interaction between syntaxin‐1 and Munc13 is disrupted by the syntaxin‐1 RIAA (R151A, I155A) and Munc13‐1 NFAA (N1128A, F1131A) mutations.
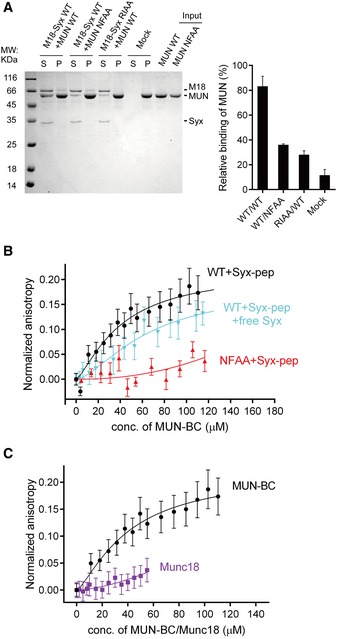
-
ACo‐flotation assay detecting the interaction between the MUN domain and proteoliposomes that reconstituted the Munc18‐1/syntaxin‐1 complex (left panel). Quantification of the results is shown in the right panel. “Mock” refers to the co‐flotation experiment of the MUN domain and plain (protein‐free) liposomes. S, supernatant; P, pellet. A representative Coomassie brilliant blue‐stained electrophoresis gel from one of three independent experiments is shown. Data were processed by ImageJ (NIH) and shown as mean ± SD for n = 3 technical replicates.
-
B, CInteraction of the fluorescent dye‐labeled syntaxin‐1 peptide (residues 148–162, referred to as Syx‐pep) with the MUN‐BC fragment (with or without the cytoplasmic region of syntaxin‐1, residues 2–253, referred to as free Syx), its NFAA mutant (B), and Munc18‐1 (C) as monitored by ensemble fluorescence anisotropy measurements. Nonlinear curve fits were performed by using the Hill equation without constraints. Shown are means ± SEM (n = 10).
Conformational changes in the syntaxin‐1 linker region
The above results suggest that the MUN domain catalyzes the transition from the closed Munc18‐1/syntaxin‐1 complex to the ternary SNARE complex through an interaction with the syntaxin‐1 linker region. We next investigated whether this interaction causes a conformational change in the syntaxin‐1 linker region by performing single‐molecule FRET (smFRET) experiments (Joo & Ha, 2012). We designed two FRET labeling pairs on syntaxin‐1: E35C and S249C (referred to as syntaxin‐1‐CC, Fig 4A) to monitor relative movements between the Habc domain and the H3 domain of syntaxin‐1; S95C and S171C (referred to as syntaxin‐1‐MN, Fig 4A) to monitor relative movements between the syntaxin‐1 linker region and the Habc domain. These pairs of mutations were introduced into a cysteine‐free cytoplasmic domain of syntaxin‐1 and labeled stochastically with Alexa 555 and Alexa 647 via maleimide covalent linkage. The dual‐labeled syntaxin‐1 molecules were tethered to a passivated surface consisting of BSA and phospholipids to mimic the environment of the plasma membrane (see Materials and Methods). To ensure that the observed changes in FRET efficiencies were due to genuine conformational changes and not due to photophysical effects or restriction of the rotational freedom of the attached fluorescent dye molecules upon binding of syntaxin‐1 with other proteins, we conducted ensemble fluorescence anisotropy and quantum yield measurements (Tables EV4 and EV5). Little or no change in fluorescence anisotropies and quantum yields was observed for both Alexa 555 and Alexa 647 labeled syntaxin‐1 regardless of the presence of other accessory proteins, suggesting that the observed FRET efficiency changes in the smFRET experiments reflect genuine conformational changes (Munro et al, 2014).
Figure 4. The MUN domain induces a conformational change in the syntaxin‐1 linker region bound to Munc18‐1 similar to that of the syntaxin‐1 LE mutant bound to Munc18‐1.
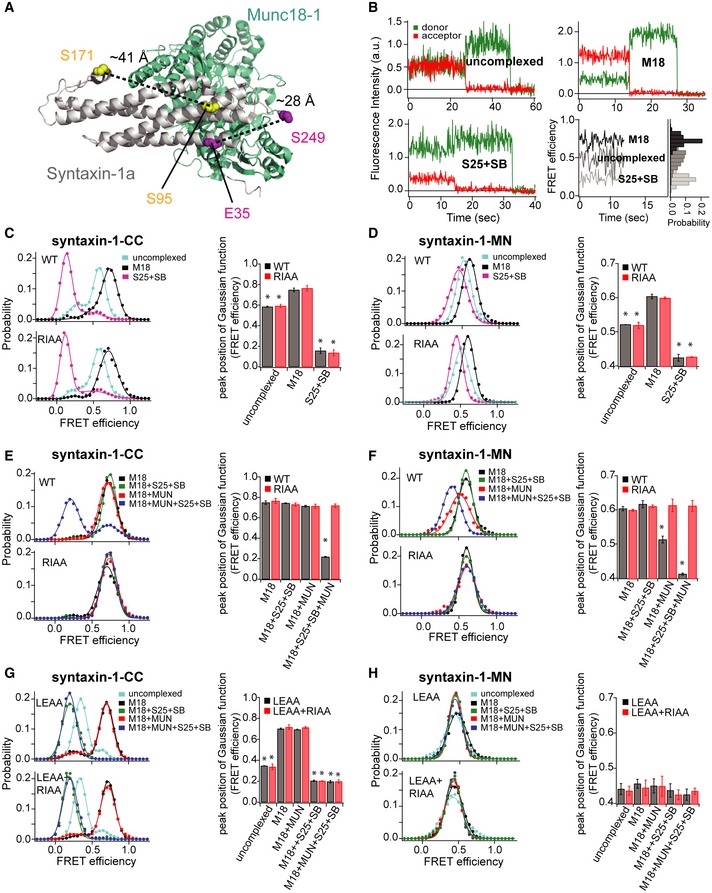
-
APositions of FRET label pairs in the crystal structure of the Munc18‐1/syntaxin‐1 complex (PDB ID 3C98): syntaxin‐1‐CC (E35/S249) and syntaxin‐1‐MN (S95/S171). Distances between the corresponding Cα positions are indicated as dashed lines.
-
BFluorescence intensity time traces of syntaxin‐1 alone with the syntaxin‐1‐CC label pair (upper left), in complex with Munc18‐1 (M18) (upper right), and within the ternary SNARE complex composed of the cytoplasmic domain of syntaxin‐1, SNAP‐25 (S25), and the cytoplasmic domain of synaptobrevin‐2 (SB) (lower left). Representative FRET efficiency time traces are shown in the lower right panel using the formula described in Materials and Methods.
-
C–HsmFRET efficiency histograms and FRET efficiency values (bar chart) corresponding to the peak positions of the Gaussian functions fit to the smFRET efficiency histograms for the syntaxin‐1‐CC and ‐MN label pairs and conditions. For the smFRET efficiency histograms of uncomplexed syntaxin‐1‐CC, two Gaussian functions were used to fit the histogram to distinguish the two observed FRET efficiency populations; otherwise, a single Gaussian function was used to extract the FRET efficiency value. The areas under the Gaussian functions were calculated, and the peak position of the Gaussian function with the higher area was used as the FRET efficiency value (see Appendix Table S1). The bar charts show mean values ± SD for the two subsets of an equal partition of the data that are comprised of the observed FRET efficiency values for all molecules for each different condition, *P < 0.05, using the two‐tailed Student's t‐test (see Appendix Tables S1–S3). Shown are smFRET efficiency histograms for uncomplexed syntaxin‐1 and its RIAA mutant, bound to Munc18‐1, and within the ternary SNARE complex (C and D); smFRET efficiency histograms for syntaxin‐1 and its RIAA mutant bound to Munc18‐1, upon the addition of SNAP‐25 and synaptobrevin‐2, upon the addition of the MUN domain alone, and upon the simultaneous addition of SNAP‐25, synaptobrevin‐2, and the MUN domain (E and F); smFRET efficiency histograms for the LE and the LE+RI mutants of uncomplexed syntaxin‐1, in complex with Munc18‐1, upon the addition of SNAP‐25 and synaptobrevin‐2, the MUN domain alone, and SNAP‐25, synaptobrevin‐2, and the MUN domain simultaneously (G and H).
Both the syntaxin‐1‐CC and ‐MN FRET label pairs exhibited high FRET efficiencies when bound to Munc18‐1 and low FRET efficiencies in the ternary SNARE complex (Fig 4B–D), consistent with the expected closed and open conformations of syntaxin‐1, respectively. The syntaxin‐1‐CC label pair produced larger changes in FRET efficiency compared to the syntaxin‐1‐MN label pair (Fig 4C and D), suggesting larger motions of the H3 domain relative to the Habc domain compared to the linker region relative to the Habc domain between the open and closed conformations of syntaxin‐1. Both uncomplexed syntaxin‐1‐CC and ‐MN exhibited intermediate FRET efficiency states (Fig 4C and D), along with occasional transitions between states, suggesting that uncomplexed, isolated syntaxin‐1 molecules sampled a range of conformations, including the open and closed conformations. As control, the syntaxin‐1 RIAA mutant did not influence the conformations of uncomplexed syntaxin‐1 (‐CC and ‐MN), when bound to Munc18‐1, or when it is part of the ternary SNARE complex (Fig 4C and D), consistent with observations shown in Figs 1 and EV2 that the RIAA mutant does not interfere with syntaxin‐1 folding, Munc18‐1 binding, and ternary SNARE complex formation.
Addition of the MUN domain to the Munc18‐1/syntaxin‐1 complex (‐CC and ‐MN), in the presence of SNAP‐25 and synaptobrevin‐2, shifted the main peak of the smFRET distribution from high to low FRET efficiency (Fig 4E and F), consistent with the transition of syntaxin‐1 from the closed to the open state that is required for ternary SNARE complex formation. Moreover, the RIAA mutant of syntaxin‐1 and the NFAA mutant of the MUN domain did not achieve the low FRET efficiency state of syntaxin‐1 (Fig 4E and F, see also Fig EV5), reinforcing that the RI residues of syntaxin‐1 and the NF residues of the MUN domain play key roles in catalyzing ternary SNARE complex formation beginning with the closed Munc18‐1/syntaxin‐1 complex.
Figure EV5. Effect of the MUN domain containing the NFAA (N1128A, F1131A) mutations on conformational changes of syntaxin‐1 bound to Munc18‐1.
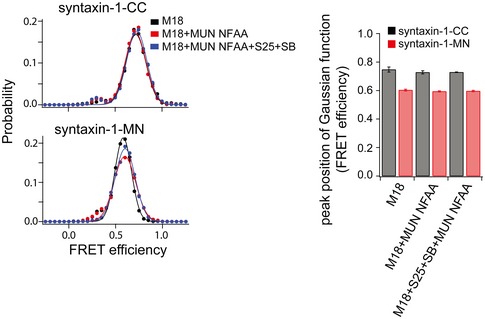
smFRET efficiency histograms and FRET efficiency values (bar chart) corresponding to the peak positions of the Gaussian functions fit to the smFRET efficiency histograms for the specified syntaxin‐1 label pairs and conditions, using the same procedure as in Fig 4. The bar charts show mean values ± SD for the two subsets of an equal partition of the data that are comprised of the observed FRET efficiency values for all molecules in each different condition (see Appendix Tables S2 and S3). Shown are smFRET efficiency histograms for the cytoplasmic domain of syntaxin‐1 bound to Munc18‐1, upon the addition of the NFAA mutant of the MUN domain, upon the simultaneous addition of SNAP‐25, the cytoplasmic domain of synaptobrevin‐2, and the NFAA mutant of the MUN domain. S25, SNAP‐25; SB, synaptobrevin‐2; M18, Munc18‐1.
We next measured the effect of the MUN domain on the closed Munc18‐1/syntaxin‐1 complex in the absence of SNAP‐25 and synaptobrevin‐2. The syntaxin‐1‐CC label pair showed identical FRET efficiency populations in the absence and presence of the MUN domain (Fig 4E), suggesting that the MUN domain is unable to dissociate the H3/Habc interaction within the closed Munc18‐1/syntaxin‐1 complex in the absence of SNAP‐25 and synaptobrevin‐2. However, when using the syntaxin‐1‐MN label pair, the addition of the MUN domain to the closed Munc18‐1/syntaxin‐1 complex generated an intermediate FRET efficiency population (red curve in Fig 4F), similar to that observed for uncomplexed syntaxin‐1‐MN (cyan curve in Fig 4D). Moreover, the emergence of this intermediate FRET efficiency population was abolished when using the syntaxin‐1 RIAA mutant or the MUN NFAA mutant (Figs 4F and EV5). These results suggest that the MUN domain stably interacts with the Munc18‐1/syntaxin‐1 complex and forms a tripartite Munc18‐1/syntaxin‐1/MUN assembly where the MUN domain specifically induces a conformational change in the syntaxin‐1 linker region, and that this tripartite complex is sensitive to the mutations of the syntaxin‐1 RI and MUN NF residues.
Since the MUN domain induces a conformational change in the syntaxin‐1 linker region in the presence of Munc18‐1, the linker region may control the catalysis of the transit of syntaxin‐1 into the ternary SNARE complex. To further corroborate this notion, we performed smFRET studies of the syntaxin‐1 LE mutant (containing the L165A, E166A mutations, referred to as syntaxin‐1LE), considering that this mutant can bypass the requirement of the MUN domain in ternary SNARE complex formation beginning with the closed Munc18‐1/syntaxin‐1 complex (Ma et al, 2011). The syntaxin‐1LE‐CC and ‐MN label pairs of the uncomplexed syntaxin‐1 LE mutant displayed much lower FRET efficiency populations than WT syntaxin‐1 (compare the cyan curves in Fig 4G and H with Fig 4C and D), but comparable to those observed for the syntaxin‐1‐CC and ‐MN label pairs within the ternary SNARE complex (the purple curves in Fig 4C and D). These results suggest that uncomplexed syntaxin‐1LE is mostly in an open conformation (Appendix Table S1), consistent with a previous observation that SNARE complex formation (i.e. in the absence of Munc18‐1 and the MUN domain) beginning with uncomplexed, isolated syntaxin‐1LE is faster than that observed with WT syntaxin‐1 (Ma et al, 2011).
Addition of Munc18‐1 to syntaxin‐1LE‐CC produced a high FRET efficiency population (Fig 4G), comparable to that observed for syntaxin‐1‐CC bound to Munc18‐1 (Fig 4C). Together with a previous SAXS study (Colbert et al, 2013) and our size‐exclusion chromatography results (Fig 1C), this suggests that syntaxin‐1LE adopts a closed conformation when bound to Munc18‐1 similar to WT syntaxin‐1 bound to Munc18‐1. As expected, further addition of SNAP‐25 and synaptobrevin‐2 with or without the MUN domain led to low FRET efficiencies for syntaxin‐1LE‐CC (Fig 4G), confirming that the MUN domain is not required for the transit of syntaxin‐1LE from Munc18‐1 binding to the ternary SNARE complex. In contrast, no difference in the smFRET efficiency histogram was observed for the syntaxin‐1LE‐MN label pair upon the addition of Munc18‐1, SNAP‐25, synaptobrevin‐2, and/or the MUN domain (Fig 4H), implying that the linker region (bearing the LE mutation) assumes a consistently open conformation. Notably, syntaxin‐1LE‐MN bound to Munc18‐1 exhibited a similar FRET efficiency as that induced by the MUN domain when binding to the WT Munc18‐1/syntaxin‐1 complex (compare Fig 4H with the red curve in the upper panel of Fig 4F). Thus, we predicted that disruption of the MUN/syntaxin‐1 interaction by the syntaxin‐1 RIAA mutations should not affect the conformations of syntaxin‐1LE in all complexes, which is indeed the case (compare the upper and lower panels in Fig 4G and H).
Together, these results suggest the existence of a tripartite Munc18‐1/syntaxin‐1/MUN complex that is essential for initiating ternary SNARE complex formation. In this complex, syntaxin‐1 adopts an intermediate, still closed (i.e. Habc still interacts with H3), but “activated” conformation. This conformation is reminiscent to the conformation of the syntaxin‐1 LE mutant bound to Munc18‐1 in the absence of the MUN domain, as suggested by our smFRET experiments.
Discussion
Syntaxin‐1 transitions between different conformations that are essential for its function in synaptic vesicle exocytosis. Generally, syntaxin‐1 transitions from a closed conformation that is tightly complexed with Munc18‐1 to an open conformation when it is part of the ternary SNARE complex. This transition is mediated by Munc13s for exquisite regulation of synaptic vesicle exocytosis, but the underlying molecular mechanism remains poorly understood. Here, we found that the MUN domain of Munc13‐1 binds to the Munc18‐1/syntaxin‐1 complex via a functionally important interaction with the conserved RI (R151 and I155) residues that are located in the linker region between the Habc domain and the H3 domain of syntaxin‐1. This interaction results in a conformational change of the linker region that enables syntaxin‐1 to transit into the ternary SNARE complex upon the addition of synaptobrevin‐2 and SNAP‐25.
The functional role of the syntaxin‐1 RI residues appears to be related to that of the NF residues in the Munc13‐1 MUN domain for ternary SNARE complex formation and synaptic vesicle priming (Yang et al, 2015). The extension of the Hc helix that contains the RI residues interacts with the short linker helix and the N‐terminal end of the H3 domain to form a structural unit that protrudes from the Munc18‐1/syntaxin‐1 complex (Fig 1A). We speculate that this protruding unit may provide a binding platform for the MUN NF pocket, a notion that is supported by impairment of binding between the MUN domain and the Munc18‐1/syntaxin‐1 complex on membranes when either the RIAA or NFAA mutations are introduced (Fig 3A). Additionally, other Munc18‐1/MUN interactions may help to position the MUN NF pocket close to the syntaxin‐1 linker region, thereby increasing the RI/NF binding affinity and enhancing the catalytic activity of the MUN domain.
Our smFRET experiments suggest the existence of a tripartite Munc18‐1/syntaxin‐1/MUN complex in the absence of SNAP‐25 and synaptobrevin‐2, in which syntaxin‐1 still adopts a closed conformation tightly bound to Munc18‐1, while the syntaxin‐1 linker region changes its conformation due to the RI/NF interaction (Fig 4E and F). This local conformational change in the Munc18‐1/syntaxin‐1/MUN complex (i.e. the conformational change in the syntaxin‐1 linker region) may expose the N‐terminal end of the H3 domain of syntaxin‐1, thereby providing a nucleation site for SNAP‐25 and synaptobrevin‐2 binding (Fig 5). In support of this model, the addition of SNAP‐25 and synaptobrevin‐2 to this tripartite complex resulted in a fully open conformation of syntaxin‐1 (Fig 4E and F) and ternary SNARE complex formation. Propagation of the four‐helical SNARE bundle toward the C‐terminal end would eventually dissociate the H3 domain from the closed Munc18‐1/syntaxin‐1 complex (Fig 5).
Figure 5. Working model of Munc13 in initiating ternary SNARE complex formation in synaptic exocytosis.
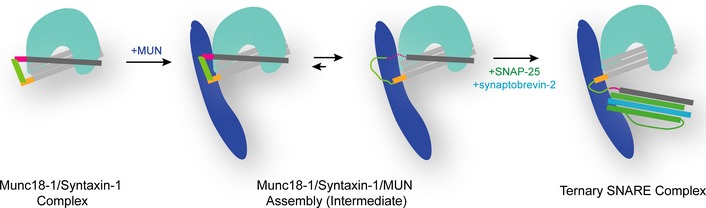
Colors are as follows: syntaxin‐1 Habc domain, light gray; the H3 domain, dark gray; the C‐terminal extension of the Hc helix, yellow; the short linker helix, light green; the N‐terminal end of the H3 domain, magenta; Munc18‐1, cyan; the Munc13‐1 MUN domain, dark blue; SNAP‐25, green; synaptobrevin‐2, blue. The unstructured regions of the SNARE proteins are depicted by lines, and the structured regions are displayed by bold lines. The N‐terminal end of the H3 domain in the “intermediate” state is shown as a dashed line, implying a nucleation site for ternary SNARE complex formation.
Our data rectify a previous hypothesis that the syntaxin‐1 H3 domain predissociates from Munc18‐1 and/or the Habc domain binding for initiating SNARE complex formation (Sassa et al, 1999; Ma et al, 2011; Christie et al, 2012). Rather, the linker region disturbance in the Munc18‐1/syntaxin‐1 complex is sufficient for syntaxin‐1 activation and initiation of subsequent ternary SNARE complex formation. Moreover, our data are in good agreement with SAXS data showing that the syntaxin‐1 LE mutant adopts a closed conformation bound to Munc18‐1 that is globally similar to that of the WT Munc18‐1/syntaxin‐1 complex (Colbert et al, 2013). Our smFRET data show that, in this closed conformation, the linker region of the syntaxin‐1 LE mutant assumes a conformation similar to that observed for the tripartite Munc18‐1/syntaxin‐1/MUN complex (Fig 4F and H), explaining why the LE mutant, albeit in tight association with Munc18‐1 (Burkhardt et al, 2008), efficiently transits into the ternary SNARE complex upon the addition of synaptobrevin‐2 and SNAP‐25 in vitro (Ma et al, 2011; Yang et al, 2015). In other previous work, it was proposed that the function of the MUN domain involves a transient “extraction” of the H3 domain from the Munc18‐1/syntaxin‐1 complex (via binding of the MUN domain to the syntaxin‐1 H3 R210 residue) (Ma et al, 2011). However, our new data obtained with both ensemble FRET and native gel assays now argue against this possibility because the R210 mutation does not affect the catalytic activity of the MUN domain (Appendix Fig S3).
It has been proposed that the domain 3a of Munc18‐1 undergoes a significant conformational change, switching from a “bent” structure observed in the closed Munc18‐1/syntaxin‐1 complex to an “extended” conformation that promotes SNARE complex formation (Hu et al, 2011). This notion is further supported by the crystal structure of the complex between Vps33 and Nyv1 (Munc18‐1 and synaptobrevin‐2 homologs in yeast, respectively) (Baker et al, 2015). Given that the linker region is structurally adjacent to the domain 3a in the Munc18‐1/syntaxin‐1 complex (see Fig 1A), the domain 3a of Munc18‐1 may adjust its conformation for accommodating an intermediate to promote ternary SNARE complex formation (Baker et al, 2013, 2015; Parisotto et al, 2014; Shen et al, 2015) in coordination with the Munc13‐mediated conformational changes of the syntaxin‐1 linker region.
Our results suggest that the activation of the syntaxin‐1 linker region by Munc13‐1 initiates ternary SNARE complex formation upon the addition of syntaxin‐1 and synaptobrevin‐2. The Munc18‐1/syntaxin‐1/Munc13‐1 network of interactions may allow exquisite regulation of synaptic exocytosis. Indeed, Munc13‐1 and Munc18‐1 cooperate to properly assemble ternary SNARE complexes by suppressing anti‐parallel and other nonproductive configurations (Y. Lai, U.B. Choi, H.J. Rhee, J. Leitz, M. Zhao, Y. Zhang, R. Pfuetzner, A. Wang, J. Rhee, A.T. Brunger, submitted), and the Munc18‐1/syntaxin‐1/Munc13‐1 interactions would prevent the formation of the syntaxin‐1/SNAP‐25 (2:1) “dead‐end” complex, promoting the formation of parallel ternary SNARE complex with proper 1:1:1 stoichiometry.
Materials and Methods
Expression constructs and protein purification
The rat Munc13‐1 MUN domain (residues 859–1,407–EF–1,453–1,531), rat SNAP‐25A (residues 1–206), and the cytoplasmic domain of rat synaptobrevin‐2 (residues 1–96) were cloned into the pTEV5 vector that includes an N‐terminal TEV cleavable hexa‐histidine tag. WT rat Munc18‐1 (residues 1–594) was cloned into the pPROExHTa vector that includes an N‐terminal TEV cleavable hexa‐histidine tag. Full‐length rat synaptotagmin‐1 (with its native cysteines mutated to alanines, except for residue 277) was cloned into the pET28a vector (Novagen) with a C‐terminal hexa‐histidine tag. Rat Munc18‐1 and full‐length syntaxin‐1A were cloned into the pETDuet‐1 vector (Novagen) with an N‐terminal hexa‐histidine tag on Munc18‐1. The cytoplasmic domain of syntaxin‐1A (residues 2–253), the Munc13‐1 MUN‐BC fragment (residues 1,011–1,407), the cytoplasmic domain of synaptobrevin‐2 (residues 29–93), and full‐length synaptobrevin‐2 were cloned into the pGEX‐KG vector. The Munc13‐1 C1‐C2B‐MUN fragment was cloned into the pFast‐Bac™HT B vector (Invitrogen).
For the smFRET experiments, the construct of the cytoplasmic domain of syntaxin‐1A (residues 1–265) was fused to a C‐terminal biotinylation sequence (GLNDIFEAQKIEWHE) and cloned into the pTEV5 vector that includes an N‐terminal TEV cleavable hexa‐histidine tag (Rocco et al, 2008). Starting from this construct, the mutants syntaxin‐1‐CC (E35C/S249C) and syntaxin‐1‐MN (S95C/S171C) were generated by site‐directed mutagenesis (QuickChange Kit, Agilent). Biotinylation was performed in vivo by co‐expression with a BirA gene cloned into the pACYC184 vector (Avidity, Aurora, CO) and induced with 0.5 mM IPTG at 25°C in the presence of 0.1 mM biotin for 16–20 h.
The constructs for the ensemble FRET assays (i.e. the cytoplasmic domain of synaptobrevin‐2 (residues 29–93, S61C) and SNAP‐25A (Q197C)) were generated by site‐directed mutagenesis (QuickChange Kit, Agilent).
The Munc13‐1 C1‐C2B‐MUN was expressed in Sf9 cells as previously described (Ma et al, 2013). All other proteins were expressed in E. coli BL21 (DE3) by growing the cells to an optimal density (OD600 of 0.6–0.8) at 37°C and then induced with 0.5 mM IPTG for 16–20 h at 25°C.
GST‐fused proteins and full‐length rat synaptotagmin‐1 were purified as previously described (Ma et al, 2011; Lai et al, 2014; Yang et al, 2015; Wang et al, 2016). For hexa‐histidine tagged proteins, cell pellets from 1 l culture were harvested and suspended in lysis buffer [20 mM Tris, 300 mM NaCl, 2 mM DTT, 0.5 mM PMSF, pH 8.0 supplemented with EDTA free Complete Protease Inhibitor Cocktail tablets (1 protease inhibitor tablet per 50 ml (Roche, Basel, Switzerland))]. Cells were lysed by sonication and centrifuged at 186,000 g using a Ti‐45 rotor (Beckman Coulter, Brea, CA) for 45 min in order to remove inclusion bodies. To separate the hexa‐histidine tagged proteins from impurities, the supernatant was bound to Nickel‐NTA agarose beads (Qiagen, Hilden, Germany) for 1 h on a rotating platform at 4°C, washed extensively with lysis buffer supplemented with 20 mM imidazole and then eluted with lysis buffer containing 400 mM imidazole. To cleave off the N‐terminal hexa‐histidine tags, the eluted proteins were dialyzed overnight in 20 mM Tris, 50 mM NaCl, 0.5 mM EDTA, 1 mM DTT, pH 8.0 in the presence of 100 μg of TEV protease. The protease and the cleaved proteins were separated by using a Mono Q (4.6/100) ion exchange column (GE Healthcare, Uppsala, Sweden) with a linear gradient of 50 mM to 600 mM NaCl in 20 mM Tris, 0.5 mM TCEP, pH 7.5, except for the cytoplasmic domain of synaptobrevin‐2 which was purified using a Superdex 75 size exclusion column (GE Healthcare Bio‐Sciences, Piscataway, NJ) in 20 mM Tris, 150 mM NaCl, 0.5 mM TCEP, pH 7.5. The protein purity was checked using SDS–PAGE electrophoresis gels (> 95%). The concentration of proteins was determined by UV absorbance at 280 nm, and aliquots were frozen in liquid nitrogen.
Ensemble FRET assays
General procedures were described previously (Yang et al, 2015). Briefly, the cytoplasmic domain of synaptobrevin‐2 (residues 29–93, with the designed mutation S61C) was labeled with the FRET‐donor dye BODIPY FL (Molecular Probes) and SNAP‐25 (with native cysteines mutated to serines, and with the designed mutation Q197C) was labeled with the FRET‐acceptor dye 5‐tetramethylrhodamine (Molecular Probes). The experiments were carried out on a PTI QM‐40 spectrophotometer with an excitation wavelength of 485 nm and an emission wavelength of 513 nm at 30°C.
Native gel assays
Native electrophoresis gels were performed as previous described in Yang et al (2015). Briefly, 2.0 μM of the complex of Munc18‐1 and the cytoplasmic domain of syntaxin‐1 (WT or its mutants) was mixed with 10 μM SNAP‐25, 10 μM cytoplasmic domain of synaptobrevin‐2 (residues 29–93), and 30 μM Munc13‐1 MUN domain. After incubation at 37°C for 3 h, samples were loaded into native gels consisting of 15% poly‐acrylamide in the separating gel (pH 8.4) and 5% in the stacking gel (pH 6.8) (all SDS‐free). Electrophoresis was done in a buffer containing 25 mM Tris base and 250 mM glycine (pH 8.3) overnight at 80 V, 4°C.
Ensemble liposome fusion assays
Proteoliposomes were prepared as previously described (Ma et al, 2013; Yang et al, 2015; Wang et al, 2016). Lipid powder (all from Avanti Polar Lipids) was dissolved in chloroform at a concentration of 10 mg/ml and stored at −20°C. Brain PI(4,5)P2 (PIP2) was dissolved in chloroform: methanol: water (20: 9: 1) at 1 mg/ml. Lipids were mixed at the desired ratio (see below) to a final concentration of 5 mM and dried under a nitrogen fast flow followed by vacuum for at least 3 h at room temperature. Lipid films were rehydrated by buffer H (25 mM HEPES pH 7.4, 150 mM KCl and 10% (v/v) glycerol) containing 0.2 mM Tris (2‐carboxyethyl) phosphine (TCEP, Sigma‐Aldrich) and 1% CHAPS (w/v, Amresco) and vortexed for 5 min at room temperature. Purified proteins that were dissolved in buffer H supplemented with 0.2 mM TCEP and 1% CHAPS (w/v) were added into the dissolved lipid mixtures to a final protein‐to‐lipid ratio of 1:800 (t‐liposome) and 1:500 (v‐liposome), respectively; T‐liposomes (36% POPC, 20% POPE, 20% DOPS, 2% DAG, 2% PI(4,5)P2, 20% cholesterol) with reconstituted full‐length Munc18‐1/syntaxin‐1 complex and v‐liposomes (45% POPC, 20% POPE, 15% DOPS, 20% cholesterol) with reconstituted full‐length synaptobrevin‐2 & synaptotagmin‐1 (at a ratio of 4:1) were all prepared by dialysis against buffer H containing 0.2 mM TCEP three times at 4°C supplemented with 0.5 g/l Bio‐beads SM2 (Bio‐Rad). For lipid mixing, 1.5% NBD‐POPE and 1.5% rhodamine‐POPE were incorporated into v‐liposomes, and donor (NBD) fluorescence was monitored on a PTI QM‐40 fluorescence spectrophotometer with an excitation wavelength of 460 nm and an emission wavelength of 538 nm. For content mixing, 40 mM sulforhodamine B (Sigma) was loaded into v‐liposomes during the first dialysis procedure, and fluorescence was monitored on a PTI QM‐40 fluorescence spectrophotometer with an excitation wavelength of 565 nm and an emission wavelength of 580 nm. Fluorescence intensities were normalized to the fluorescence intensity of the sample in the presence of 0.1% Triton X‐100.
Cell culture
HEK293T cells (CRL‐11268, ATCC) were grown at 37°C and 5% CO2 in a humidified atmosphere incubator (Thermo). The culture medium contained Dulbecco's modified Eagle's medium (Gibco), 10% fetal bovine serum, and penicillin–streptomycin (50 and 50 μg/ml).
The dissociated hippocampal neurons were prepared from randomly chosen P0 pups, as described previously (Yang et al, 2010). Briefly, mouse hippocampi were dissected from postnatal day 0 (P0) of WT mice, dissociated by 0.25% trypsin digestion for 12 min at 37°C, plated at a density of 80,000 cells per 8‐mm × 8‐mm cover slip coated with poly‐lysine (Sigma), and cultured in MEM (GIBCO) supplemented with 2 v/v% B27 (GIBCO), 0.5 w/v% glucose, 100 mg/l transferrin, 5 v/v% fetal bovine serum, and 2 mM Ara‐C (Sigma).
Lentiviruses preparation
HEK293 cells were co‐transfected with lentiviral expression vectors and three helper plasmids (pRSV‐REV, pMDLg/pRRE, and pVSVG) at the ratio pL309:pVSVg:RRE:REV = 3:2 by using polyethyleneimine (PEI) to produce lentiviruses. The virus‐containing medium was precleaned by centrifugation at 3,000 g and a 0.45 μm filtration (Millipore) after transfection. The virus was then concentrated in a sucrose‐containing buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 0.5 mM ethylene diaminetetraacetic acid [EDTA]) at a 4:1 v/v ratio and centrifuged at 4°C. The precipitate was carefully collected after centrifugation. For re‐suspension of the virus, phosphate‐buffered saline (PBS) was added to the tube at 4°C overnight. Level II biosafety conditions were maintained at all times. Cultured neurons were infected with lentiviruses at DIV5–6 and recorded at DIV13–14.
Electrophysiological recordings
Electrophysiological recordings were monitored as previously described (Zhou et al, 2013a). Whole‐cell voltage clamp mode was used for all electrophysiological recordings. A bipolar electrode was placed 100–150 mm from the soma of neurons for stimulating evoked synaptic responses. Borosilicate glass capillary tubes (World Precision Instruments, Inc.) were used for pulling patch pipettes by a P‐97 pipette puller. The resistance of pipettes varied between 3 and 5 MOhm after filling with intracellular solution, and the series resistance was adjusted to 8–10 MOhm after formation of the whole‐cell configuration and equilibration of the intracellular pipette solution. Synaptic currents were measured by an EPC10 amplifier (HEKA). Single extracellular stimulations (90 μA, 1 ms) were controlled with a Model 2100 Isolated Pulse Stimulator (A‐M Systems, Inc.). The whole‐cell pipette solution contained 120 mM CsCl, 10 mM HEPES, 10 mM EGTA, 0.3 mM Na‐GTP, 3 mM Mg‐ATP, and 5 mM QX‐314 (pH 7.2, adjusted with CsOH). The bath solution contained 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES‐NaOH, and 10 mM glucose (pH 7.4). CNQX (20 μM) and AP‐5 (50 μM) were added to the extracellular solution to pharmacologically isolate IPSCs. To block action potentials, miniature IPSCs were monitored in a bath solution supplemented with 1 μM tetrodotoxin (TTX). Miniature events were analyzed in Clampfit 10 (Molecular Devices) using a template matching search. Events smaller than 5 pA were rejected by an experimenter who had no knowledge of the recording condition. Sucrose‐evoked release was triggered by an application of bath solution that contained 0.5 M sucrose and AP‐5, CNQX, and TTX for 30 s.
Single‐molecule FRET experiments
Syntaxin‐1‐CC (E35C/S249C) and ‐MN (S95C/S171C) were labeled stochastically with Alexa 555 and 647 via maleimide linkage (Invitrogen, Carlsbad, CA) in TBS (20 mM Tris, 150 mM NaCl, pH 7.5 with 0.5 mM TECP) on a rotating platform overnight at 4°C. Free dyes were removed by a column packed with Sephadex G50 resin (GE Healthcare, Piscataway, NJ) in TBS.
The labeled syntaxin‐1 molecules were surface‐tethered on a biotinylated BSA surface through biotin–streptavidin linkage (Choi et al, 2012). The BSA surface was exposed to small (50 nm diameter) egg phosphatidylcholine liposomes which will fuse with exposed regions of the glass surface and thereby create an environment where BSA molecules are surrounded by phospholipids (Choi et al, 2011, 2012). This method prevents nonspecific binding to the surface, and it mimics an environment where the tethered syntaxin‐1 molecules are surrounded by lipids (Choi et al, 2011). To achieve single‐molecule conditions, labeled syntaxin‐1 molecules were diluted to about 50 pM for surface tethering, producing a density of about 200–300 molecules per 45 × 90 μm2 field of view.
After removal of free syntaxin‐1 molecules in solution by extensive washing, 1 μM of Munc18‐1 was incubated for 5 min to form the Munc18‐1/syntaxin‐1 complex, or 1 μM of SNAP‐25 and the cytoplasmic domain of synaptobrevin‐2 to form the ternary SNARE complex. Free proteins that did not form complexes were washed away before smFRET measurements are performed. To measure the effect of the catalytic activity of the MUN domain, 10 μM of the MUN domain in the presence and absence of 1 μM SNAP‐25 and the cytoplasmic domain of synaptobrevin‐2 were incubated for 5 min. For all smFRET experiments, protein‐free observation buffer (1% w/v glucose, 20 mM Tris, 150 mM NaCl, pH 7.5) contained oxygen scavenger (20 units/ml glucose oxidase, 1,000 units/ml catalase) and triplet‐state quencher (100 μM cyclooctatetraene) to prevent fast photo‐bleaching and blinking of the dye molecules (Choi et al, 2016).
Details of the single‐molecule fluorescence microscopy setup have been described previously (Choi et al, 2012, 2016). Briefly, single‐molecule fluorescence intensities were recorded with a custom built prism‐type total internal reflection fluorescence (TIRF) microscope using a 532 nm laser (CrystaLaser, Reno, NV) excitation and detected by an Andor iXon EMCCD camera (Andor Technology, South Windsor, CT) at a time resolution of 100 ms per frame (Choi et al, 2016). The donor and the acceptor fluorescence intensities were separated using a 640 nm single‐edge dichroic beam‐splitter (Semrock, Rochester, NY) and were recorded on the camera using the smCamera software developed from Taekjip Ha's group at University of Illinois and analyzed with scripts written for MATLAB (Mathworks). For the smFRET experiments shown in Figs 4 and EV5, the fluorescence intensity histograms were plotted by accumulating 50 frames from individual fluorescence intensity time traces (representative examples in Fig 4B) and converted to FRET efficiencies by
where αI A corrects for leakage of acceptor emission into donor channel and βI D corrects for leakage of donor emission into acceptor channel (McCann et al, 2010). α was measured to be 16.5% and β was measured to be 1.7%.
Ensemble fluorescence anisotropy measurements
Ensemble fluorescence anisotropy measurements (Table EV3) of the interaction between the MUN‐BC fragment and the syntaxin‐1 peptide (residues 148–162, with rhodamine B (Scilight Biotechnology LLC, Beijing, China) covalently linked to the N‐terminus) were performed on a PTI QM‐40 fluorescence spectrophotometer equipped with adjustable polarizing films with an excitation/emission wavelengths of 532/565 nm. Nonlinear curve fits were performed using the Hill equation (Table EV3).
For the isotropic fluorophore tumbling measurements (Table EV4), ensemble fluorescence anisotropies were measured using Alexa 555 or Alexa 647 double‐labeled syntaxin‐1 (CC and MN) in the presence of the specified protein. All measurements were performed on a PTI QM‐40 fluorescence spectrophotometer equipped with adjustable polarizing films with an excitation/emission wavelengths of 532/565 nm (for Alexa 555) and 633/670 nm (for Alexa 647) at 25°C, respectively.
Ensemble fluorescent quantum yield measurements
All fluorescent dye‐labeled samples were adjusted to 0.05 a.u. on a SHIMADZU ultraviolet–visible spectrometer (UV‐2450) at the desired wavelengths. Ensemble fluorescence quantum yields were measured on a PTI QM/TM system. For a fluorescent quantum yield standard (Karstens & Kobs, 1980), a time‐resolved fluorescent spectrum of Rhodamine 101 (Aladdin, Shanghai, China) was measured using an excitation wavelength of 481 nm and emission wavelengths of 585 nm in 100% ethanol. For Alexa 555 labeled samples, time‐resolved fluorescent spectra were measured using an excitation wavelength of 481 nm and emission wavelengths of 565 nm in buffer H. Raw data were analyzed using a home‐written software compiled with MATLAB (Mathworks). Quantum yields were calculated as:
where QY is the quantum yield of the fluorescent sample; Abs is the absorbance (a.u.) of the fluorescent sample at desired wavelength; I is the fluorescence intensity (a.u.); t is the time; Subscripts represent the determinant (d) and the standard sample (s, rhodamine 101), respectively.
Sequence alignment
Sequence alignment was performed with Clustal Omega and analyzed with ESPript 3.0.
Data analysis
Prism 6.01 (GraphPad) and ImageJ (NIH) were used for graphing and statistical tests, all of which are described in the figure legends.
Author contributions
SW, YL, and XiaoyY generated all mutants used in this study and performed the in vitro SNARE complex formation and liposome fusion experiments; JG performed the in vivo electrophysiology experiments; UBC and ALW performed single‐molecule FRET experiments; XiaofY, ATB, and CM conceived the project and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File
Acknowledgements
We thank Xin Wen, Hao Zhou, Ziqing Wei, Le Zhu, and Yaru Hu for protein expression and purification; Min Li and Yifan Xia for initial efforts with smFRET experiments; and Tao Xu, Josep Rizo, Jeremy Leitz, and William Weis for insightful discussions and comments on the manuscript. This work was supported by the National Science Foundation of China (31322034 and 31370819 to C.M., 31670850 and 31300892 to X.Y.), the National Key Basic Research Program of China (2015CB910800 and 2014CB910203 to C.M.), the National Institutes of Health (R37‐MH63105 to A.T.B.), and Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT: IRT13016).
The EMBO Journal (2017) 36: 816–829
Contributor Information
Xiaofei Yang, Email: sunlittlefly@hotmail.com.
Axel T Brunger, Email: brunger@stanford.edu.
Cong Ma, Email: cong.ma@hust.edu.cn.
References
- Arunachalam L, Han L, Tassew NG, He Y, Wang L, Xie L, Fujita Y, Kwan E, Davletov B, Monnier PP, Gaisano HY, Sugita S (2008) Munc18‐1 is critical for plasma membrane localization of syntaxin1 but not of SNAP‐25 in PC12 cells. Mol Biol Cell 19: 722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Jeffrey PD, Hughson FM (2013) Crystal structures of the Sec1/Munc18 (SM) protein Vps33, alone and bound to the homotypic fusion and vacuolar protein sorting (HOPS) subunit Vps16*. PLoS ONE 8: e67409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM (2015) A direct role for the Sec1/Munc18‐family protein Vps33 as a template for SNARE assembly. Science 349: 1111–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, Rizo J (2005) A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol 12: 1017–1018 [DOI] [PubMed] [Google Scholar]
- Brunger AT (2005) Structure and function of SNARE and SNARE‐interacting proteins. Q Rev Biophys 38: 1–47 [DOI] [PubMed] [Google Scholar]
- Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D (2008) Munc18a controls SNARE assembly through its interaction with the syntaxin N‐peptide. EMBO J 27: 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi UB, Xiao S, Wollmuth LP, Bowen ME (2011) Effect of Src kinase phosphorylation on disordered C‐terminal domain of N‐methyl‐D‐aspartic acid (NMDA) receptor subunit GluN2B protein. J Biol Chem 286: 29904–29912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi UB, Weninger KR, Bowen ME (2012) Immobilization of proteins for single‐molecule fluorescence resonance energy transfer measurements of conformation and dynamics. Methods Mol Biol 896: 3–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi UB, Zhao M, Zhang Y, Lai Y, Brunger AT (2016) Complexin induces a conformational change at the membrane‐proximal C‐terminal end of the SNARE complex. eLife 5: e16886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MP, Whitten AE, King GJ, Hu SH, Jarrott RJ, Chen KE, Duff AP, Callow P, Collins BM, James DE, Martin JL (2012) Low‐resolution solution structures of Munc18: Syntaxin protein complexes indicate an open binding mode driven by the Syntaxin N‐peptide. Proc Natl Acad Sci USA 109: 9816–9821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert KN, Hattendorf DA, Weiss TM, Burkhardt P, Fasshauer D, Weis WI (2013) Syntaxin1a variants lacking an N‐peptide or bearing the LE mutation bind to Munc18a in a closed conformation. Proc Natl Acad Sci USA 110: 12637–12642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak F, Xu Y, Chang WP, Dulubova I, Khvotchev M, Liu X, Südhof TC, Rizo J (2009) Munc18‐1 binding to the neuronal SNARE complex controls synaptic vesicle priming. J Cell Biol 184: 751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Südhof TC, Rizo J (1999) A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J 18: 4372–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber SH, Rah JC, Min SW, Liu X, de Wit H, Dulubova I, Meyer AC, Rizo J, Arancillo M, Hammer RE, Verhage M, Rosenmund C, Südhof TC (2008) Conformational switch of syntaxin‐1 controls synaptic vesicle fusion. Science 321: 1507–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE (1997) Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick‐freeze/deep‐etch electron microscopy. Cell 90: 523–535 [DOI] [PubMed] [Google Scholar]
- Hu SH, Christie MP, Saez NJ, Latham CF, Jarrott R, Lua LH, Collins BM, Martin JL (2011) Possible roles for Munc18‐1 domain 3a and Syntaxin1 N‐peptide and C‐terminal anchor in SNARE complex formation. Proc Natl Acad Sci USA 108: 1040–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo C, Ha T (2012) Single‐molecule FRET with total internal reflection microscopy. Cold Spring Harb Protoc 2012: 1104–1108 [DOI] [PubMed] [Google Scholar]
- Karstens T, Kobs K (1980) Rhodamine B and rhodamine 101 as reference substances for fluorescence quantum yield measurements. J Phys Chem 84: 1871–1872 [Google Scholar]
- Lai Y, Diao J, Cipriano DJ, Zhang Y, Pfuetzner RA, Padolina MS, Brunger AT (2014) Complexin inhibits spontaneous release and synchronizes Ca2+‐triggered synaptic vesicle fusion by distinct mechanisms. eLife 3: e03756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Seven AB, Camacho M, Esser V, Xu J, Trimbuch T, Quade B, Su L, Ma C, Rosenmund C, Rizo J (2016) Functional synergy between the Munc13 C‐terminal C1 and C2 domains. eLife 5: e13696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Li W, Xu Y, Rizo J (2011) Munc13 mediates the transition from the closed syntaxin‐Munc18 complex to the SNARE complex. Nat Struct Mol Biol 18: 542–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Su L, Seven AB, Xu Y, Rizo J (2013) Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 339: 421–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margittai M, Fasshauer D, Jahn R, Langen R (2003) The Habc domain and the SNARE core complex are connected by a highly flexible linker. Biochemistry 42: 4009–4014 [DOI] [PubMed] [Google Scholar]
- McCann JJ, Choi UB, Zheng L, Weninger K, Bowen ME (2010) Optimizing methods to recover absolute FRET efficiency from immobilized single molecules. Biophys J 99: 961–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI (2000) Three‐dimensional structure of the neuronal‐Sec1‐syntaxin 1a complex. Nature 404: 355–362 [DOI] [PubMed] [Google Scholar]
- Misura KM, Scheller RH, Weis WI (2001) Self‐association of the H3 region of syntaxin 1A. Implications for intermediates in SNARE complex assembly. J Biol Chem 276: 13273–13282 [DOI] [PubMed] [Google Scholar]
- Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB III, Kwong PD, Blanchard SC, Mothes W (2014) Conformational dynamics of single HIV‐1 envelope trimers on the surface of native virions. Science 346: 759–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson KL, Munson M, Miller RB, Filip TJ, Fairman R, Hughson FM (1998) Regulation of SNARE complex assembly by an N‐terminal domain of the t‐SNARE Sso1p. Nat Struct Biol 5: 793–802 [DOI] [PubMed] [Google Scholar]
- Parisotto D, Pfau M, Scheutzow A, Wild K, Mayer MP, Malsam J, Sinning I, Sollner TH (2014) An extended helical conformation in domain 3a of Munc18‐1 provides a template for SNARE (soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor) complex assembly. J Biol Chem 289: 9639–9650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobbati AV, Stein A, Fasshauer D (2006) N‐ to C‐terminal SNARE complex assembly promotes rapid membrane fusion. Science 313: 673–676 [DOI] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM (1999) UNC‐13 is required for synaptic vesicle fusion in C. elegans . Nat Neurosci 2: 959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Weimer RM, Jorgensen EM (2001) An open form of syntaxin bypasses the requirement for UNC‐13 in vesicle priming. Nature 412: 338–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Südhof TC (2012) The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices–guilty as charged? Annu Rev Cell Dev Biol 28: 279–308 [DOI] [PubMed] [Google Scholar]
- Rizo J, Xu J (2015) The synaptic vesicle release machinery. Annu Rev Biophys 44: 339–367 [DOI] [PubMed] [Google Scholar]
- Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante‐Semerena JC (2008) Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli . Plasmid 59: 231–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16: 1197–1207 [DOI] [PubMed] [Google Scholar]
- Rothman JE (2014) The principle of membrane fusion in the cell (Nobel lecture). Angew Chem Int Ed 53: 12676–12694 [DOI] [PubMed] [Google Scholar]
- Sassa T, Harada S, Ogawa H, Rand JB, Maruyama IN, Hosono R (1999) Regulation of the UNC‐18‐Caenorhabditis elegans syntaxin complex by UNC‐13. J Neurosci 19: 4772–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ (2007) Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell 128: 183–195 [DOI] [PubMed] [Google Scholar]
- Shen C, Rathore SS, Yu H, Gulbranson DR, Hua R, Zhang C, Schoppa NE, Shen J (2015) The trans‐SNARE‐regulating function of Munc18‐1 is essential to synaptic exocytosis. Nat Commun 6: 8852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323: 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC (2013) Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80: 675–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395: 347–353 [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Sigler A, Rhee JS, Brose N, Enk C, Reim K, Rosenmund C (2002) Total arrest of spontaneous and evoked synaptic transmission but normal synaptogenesis in the absence of Munc13‐mediated vesicle priming. Proc Natl Acad Sci USA 99: 9037–9042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287: 864–869 [DOI] [PubMed] [Google Scholar]
- Wang S, Li Y, Ma C (2016) Synaptotagmin‐1 C2B domain interacts simultaneously with SNAREs and membranes to promote membrane fusion. eLife 5: e14211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE (1998) SNAREpins: minimal machinery for membrane fusion. Cell 92: 759–772 [DOI] [PubMed] [Google Scholar]
- Xu Y, Su L, Rizo J (2010) Binding of Munc18‐1 to synaptobrevin and to the SNARE four‐helix bundle. Biochemistry 49: 1568–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kaeser‐Woo YJ, Pang ZP, Xu W, Südhof TC (2010) Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron 68: 907–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Wang S, Sheng Y, Zhang M, Zou W, Wu L, Kang L, Rizo J, Zhang R, Xu T, Ma C (2015) Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic‐vesicle priming. Nat Struct Mol Biol 22: 547–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P, Pang ZP, Yang X, Zhang Y, Rosenmund C, Bacaj T, Südhof TC (2013a) Syntaxin‐1N‐peptide and Habc‐domain perform distinct essential functions in synaptic vesicle fusion. EMBO J 32: 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Stawicki TM, Goncharov A, Jin Y (2013b) Position of UNC‐13 in the active zone regulates synaptic vesicle release probability and release kinetics. eLife 2: e01180 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File