

Abstract
DNA double-strand breaks (DSBs) are dangerous lesions that can lead to genomic instability and cell death. Eukaryotic cells repair DSBs either by nonhomologous end-joining (NHEJ) or by homologous recombination. We investigated the ability of yeast cells (Saccharomyces cerevisiae) to repair a single, chromosomal DSB by recombination at different stages of the cell cycle. We show that cells arrested at the G1 phase of the cell cycle restrict homologous recombination, but are able to repair the DSB by NHEJ. Furthermore, we demonstrate that recombination ability does not require duplicated chromatids or passage through S phase, and is controlled at the resection step by Clb–CDK activity.
Keywords: checkpoint, DNA repair, DNA replication, nonhomologous end-joining, yeast
Introduction
DNA can be damaged as a result of failures during replication or exposure to damage-inducing agents at any stage of the cell cycle. One of the most genotoxic lesions is a DNA double-strand break (DSB). Nonhomologous end-joining (NHEJ) and homologous recombination are the principal pathways responsible for repairing DSBs in eukaryotes. Defects in either repair pathway result in high frequencies of genomic instability (Ferguson et al, 2000; Kolodner et al, 2002), underscoring the importance of these processes as caretakers of the genome. NHEJ is a mechanism able to join DNA ends with no, or minimal, homology, whereas repair of DSBs by recombination requires a homologous sequence as a template. Choice between these genetically and enzymatically distinct repair pathways should be tightly regulated, but cellular variables determining pathway commitment have remained elusive. In addition, to ensure survival, DSB repair should be coordinated within the cell cycle. Progression within the eukaryotic cell cycle is primarily determined by the sequential action of cyclin-dependent kinases (CDKs). A single CDK, Cdc28, controls cell cycle in budding yeast. Association of Cdc28 with one of three G1 cyclins (CLNs) is necessary for entering the cell cycle (START) (Tyers et al, 1993). The Cln–CDK complex, in turn, promotes the transcription of the S and G2 cyclins (CLBs), responsible for the activation of a number of pathways, including DNA replication, bud emergence and spindle pole body duplication (Nasmyth, 1993). Premature entry into the S phase is precluded by high levels of Sic1, a stoichiometric inhibitor of Clb–CDK activity (Mendenhall, 1993). Upon destruction of Sic1 by the APC/cyclosome, Clb–CDK activity increases, shutting down Cln–CDK activity, thus triggering DNA replication (Lengronne and Schwob, 2002).
Here, we have investigated the ability of yeast cells to repair a single, chromosomal DSB at different stages of the cell cycle. We show that DSBs created at the G1 stage of the cell cycle are not repaired by homologous recombination. Instead, the broken chromosomes are repaired by NHEJ. Our analysis shows that homologous repair is not dependent on DNA replication, or the presence of duplicated chromatids. Rather, Clb–CDK activity is the essential feature required for recombinational repair of DSBs. Furthermore, we show that Clb–CDK activity is required to carry out resection, one of the earliest stages of the homologous recombination process.
Results and discussion
We have designed haploid strains of the yeast Saccharomyces cerevisiae that bear two copies of the URA3 gene; one of them, located on chromosome V, carries the recognition site for the yeast HO site-specific endonuclease (ura3-HOcs). The second copy, located ectopically on chromosome II, carries a single-base pair mutation that prevents HO recognition (ura3-HOcs-inc), flanked on either side by two restriction sites, BamHI and EcoRI. In these strains, the HO gene is under the transcriptional control of a galactose-inducible promoter. Upon transfer to galactose-containing medium, the enzyme creates a single DSB in each cell of the population. The broken chromosomes are then repaired by homologous recombination, during which the HOcs-inc and the flanking markers are copied, resulting in a gene conversion event (Figure 1A). During the repair, the donor chromosome remains unchanged and there is no loss of viability. There is no need for genetic selection of recombination products; instead, repair is monitored in the entire cell population (Aylon and Kupiec, 2003; Aylon et al, 2003).
Figure 1.
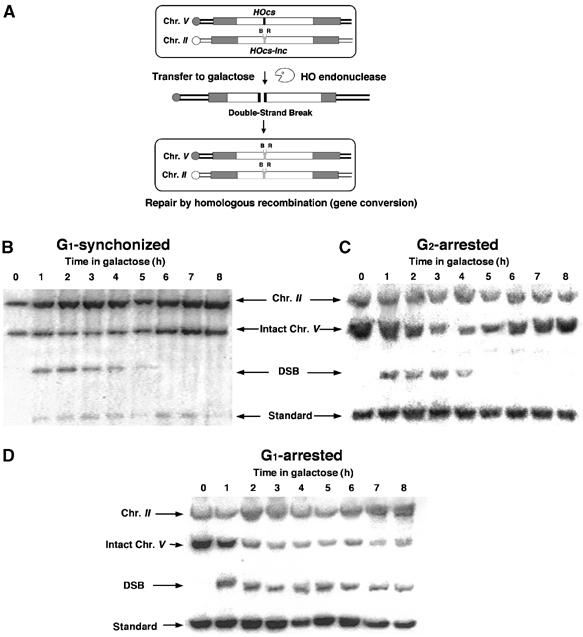
G1-arrested cells are unable to repair a DSB. (A) Schematic representation of our experimental system. Open rectangles represent the ura3 alleles on chromosomes II and V. A black box represents the HOcs; a grey box depicts the inactive HOcs-inc flanked by the BamHI (designated B) and EcoRI (designated R) restriction sites. Transfer of the cells to galactose-containing medium results in a DSB that is repaired by homologous recombination. (B) Southern blot analysis of cells synchronized in G1 with alpha-factor and released from the arrest at t=0 in medium containing galactose. The genomic DNA was digested with ClaI and probed with a 1.2 kb fragment containing the URA3 gene (open rectangles in (A)). The donor sequences on chromosome II, the intact chromosome V and one of the two bands created by the DSB are shown. A probe complementary to LEU2 sequences on chromosome III served as a loading standard. (C) Southern blot analysis of DNA from cells arrested at G2 with nocodazole and transferred to galactose-containing medium in the presence of nocodazole. (D) Southern blot analysis of DNA from cells arrested at G1 with alpha-factor and transferred to galactose-containing medium in the presence of alpha-factor.
In these strains, ectopic donor sequences are present throughout the cell cycle, thus enabling us to investigate homologous repair capability at different cell cycle stages. We arrested cells at G1 (with alpha-factor) or G2 (with nocodazole) and then transferred them to galactose to induce a DSB. In galactose, cells were either retained in the cell cycle arrest (blocked cells) or released to freely cycle (synchronized cells). The repair kinetics were monitored by Southern blot analysis (Figure 1B–D). Although chromosome V was broken with similar timing in all conditions, the ability of cells to undergo gene conversion was strikingly different at different stages of the cell cycle. Cycling cells synchronized at G1 or cells blocked at G2 repaired and re-accumulated intact chromosome V. Cells blocked at G1 did not repair the DSB. When arrested at this stage, the broken chromosome persisted throughout the experiment (8 h); nevertheless, approximately 40% of chromosome V was visualized as intact (Figure 1D). This is in marked contrast to what is observed in strains lacking components of the recombination machinery, in which the chromosome is broken and undergoes degradation, leaving no intact chromosome V (Aylon et al, 2003). This suggests that, in cells blocked at G1, there may be an alternate repair mechanism that can reinstate the HOcs.
The relative amount of intact chromosome V-carrying polymorphic restriction sites represents the portion of the population that has undergone homologous repair (Aylon and Kupiec, 2003; Aylon et al, 2003). Cells blocked and retained at the G1 phase of the cell cycle did not undergo gene conversion (only after prolonged incubation with alpha-factor a minimal amount of homologous repair is detected, probably reflecting a small fraction of cells that adapted to alpha-factor and escaped cell cycle blockage). In contrast, G2-arrested cells transferred the BamHI site and repaired the DSB in a highly efficient manner (Figure 2A and B). These results indicate that inability to undergo homologous recombination is not a function of imposed cell cycle arrest and that cells need not traverse the S phase with DNA damage in order to activate repair mechanisms. Cells avoid homologous recombination particularly in the G1 stage of the cell cycle. Consistently, cycling cells induced with a DSB at G2 completed recombination more rapidly than cells with a DSB at G1 (Figure 2B).
Figure 2.
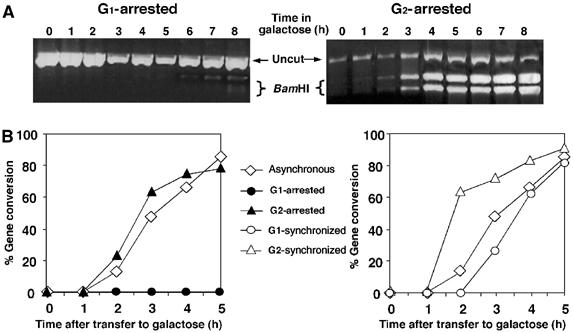
G1-arrested cells do not carry out gene conversion and cells synchronized at G1 delay gene conversion. (A) PCR-based assay for the detection of gene conversion. The region flanking the site of the DSB was amplified by PCR. The products were digested with BamHI and subjected to electrophoresis. The presence of a BamHI site demonstrates repair by gene conversion. (B) Kinetics of gene conversion of MK203 cells at different stages of the cell cycle. The ratio between BamHI-cut and total PCR products was plotted as a function of time.
When plated on medium containing glucose, the HO endonuclease is repressed, and cells can in principle repair the break either by NHEJ or by homologous recombination. Previously, we have defined commitment to gene conversion (CGC) as the stage at which the cells will complete repair by homologous recombination when plated on glucose-containing media (Aylon and Kupiec, 2003). In cycling cells, CGC occurs very rapidly, close to the time of DSB formation, and at least 90 min before actual conversion takes place. CGC probably represents a physical modification of the broken ends (e.g., resection) that precludes end-joining (Aylon and Kupiec, 2003; Aylon et al, 2003). Cycling cells committed to gene conversion very early and generated colonies carrying the polymorphic restriction sites (Figure 3A). In contrast, among cells arrested for 6 h at G1 and released from the arrest by plating on YEPD, as much as 30% of the cells had avoided commitment to gene conversion and retained intact HOcs (Figure 3A). Accordingly, 6 h after DSB induction, PCR products obtained from cells blocked at G1 could be digested neither by EcoRI nor BamHI, nor was inaccurate end-joining detected by DNA sequencing (data not shown). This indicates that in G1-blocked cells, the ends of the broken chromosomes are capable of simple re-ligation for prolonged periods of time after DSB induction, a phenomenon that has been observed previously (Frank-Vaillant and Marcand, 2002).
Figure 3.
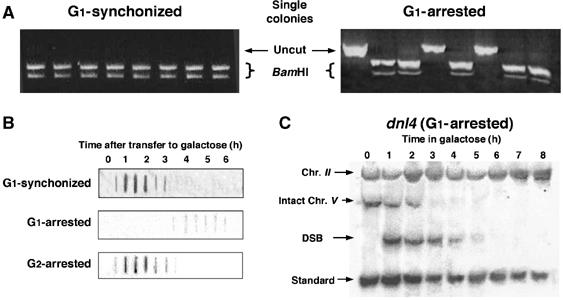
DSB ends remain unresected in G1-arrested cells and are repaired by NHEJ. (A) G1-synchronized and G1-arrested cells were incubated in galactose-containing medium for 6 h and plated on glucose-containing medium. The fate of the broken chromosomes in randomly picked individual colonies was analysed by PCR as described above. (B) Nondenaturing slot blot analysis was carried out using a 1.2 kb fragment of the URA3 gene (open rectangle in Figure 1A). Resected DNA hybridizes to the probe. (C) Southern blot analysis of genomic DNA from G1-arrested dnl4 cells in galactose.
Using nondenaturative dot-blot analysis and probes complementary to sequences flanking the DSB, the single-stranded DNA (ssDNA) repair intermediate can be measured (Aylon and Kupiec, 2003; Aylon et al, 2003). Both G1-synchronized and G2-arrested cells rapidly accumulated ssDNA (Figure 3B). In contrast, in G1-blocked cells, after prolonged incubation in galactose medium, ssDNA was detected only at extremely low levels. This indicates that DSB ends do not undergo processing at the G1 phase of the cell cycle.
Since unprocessed DSB ends are ideal substrates for end-joining, NHEJ is a plausible candidate pathway for DSB repair. The DNL4 gene is central to the NHEJ process. Unlike many other genes involved in end-joining, DNL4 has no additional phenotypes (telomeric or silencing defects) that might indirectly influence a cell's ability to repair a DSB (Teo and Jackson, 1997). We examined the repair kinetics of a G1-blocked or freely cycling dnl4 strain. Southern blot analysis of cycling cells showed that the efficiency of repair was indistinguishable between wild-type (wt) and dnl4 strains (data not shown). In contrast, in G1-arrested ligation-deficient cells, no intact chromosome V could be detected by 4 h after transfer to galactose (compare Figures 3C and 1D). This implies that the intact chromosome V that persists in G1-blocked wt cells is reconstituted through a mechanism that relies on Dnl4. Moreover, in the absence of both homologous recombination and ligation by the NHEJ machinery, the DSB ends were no longer stable and were degraded. When homologous recombination is impeded, the NHEJ pathway may have a competitive advantage and predominate.
Haploid yeast cells occur in two mating types, ‘a' and ‘alpha'. Through a highly controlled process, yeast cells at G1 undergo mating type switch (Haber, 1998). This process is induced by a single DSB created by the HO enzyme at the MAT locus, and is accomplished by directional gene conversion. Despite the similarities, genomic DSBs activate an emergency repair and checkpoint response that is not normally elicited in the developmentally controlled MAT switching process (Pellicioli et al, 2001). Our MK203-derived strains are unable to undergo switching because the endogenous HOcs has been mutated (MATa-inc). We reinstated the intact MATa allele and followed mating type switching with PCR primers specific for MATalpha. Despite the G1 block, cells were able to undergo MAT switching (Figure 4A), albeit with slightly delayed kinetics compared to asynchronous cultures (data not shown). Evidently, cells are enzymatically capable of performing gene conversion at G1, but genomic recombinational repair is restricted.
Figure 4.
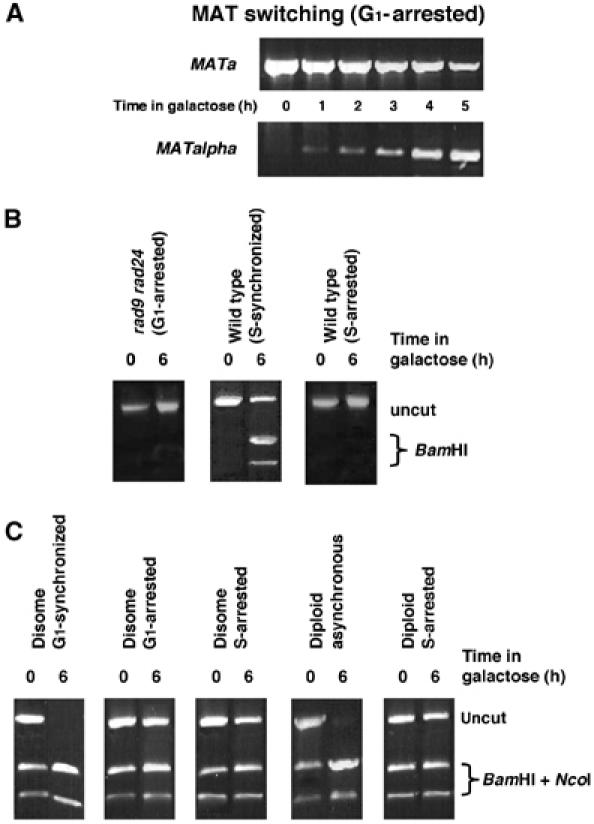
G1-arrested cells are able to carry out MAT switching; the inability to repair DSBs by homologous recombination in G1 is independent of checkpoint response and ploidy. (A) A MATa derivative of MK203 was retained at G1 (alpha-factor) and monitored for the ability to switch mating type with MATalpha-specific PCR primers. (B) PCR assays for gene conversion were carried out for rad9 rad24 synchronized or arrested in G1, and wt cells synchronized or arrested in S-phase with HU. (C) Disomic and diploid strains are unable to carry out gene conversion when arrested at G1 (alpha-factor) or S (HU). PCR was carried out as in (B) and PCR products were digested with BamHI and NcoI. The presence of uncut product at t=6 h indicates that no gene conversion occurred.
Genomic DSBs trigger a cell cycle arrest in a checkpoint-dependent manner. The DNA damage checkpoint also affects repair efficiency and pathway choice (Aylon and Kupiec, 2003). Deletion of RAD9 and RAD24 severely impairs the DNA damage response (de la Torre-Ruiz et al, 1998; Aylon and Kupiec, 2003). However, checkpoint activity is not required to prevent homologous recombination in G1, since rad9 rad24 mutants blocked at G1 also did not undergo gene conversion (Figure 4B).
The inability to carry out homologous recombination in G1 might have been particular to ectopic gene conversion. In diploids, allelic recombination between homologous chromosomes can occur, thus providing the means for homologous recombination at all stages of the cell cycle. Allelic recombination is also possible in genetically manipulated haploid yeast cells supplemented with an additional homologous chromosome (disomal strains). In these strains, gene conversion results in transfer of either NcoI from the allelic chromosome V or BamHI from the ectopic sequence on chromosome II. Since the allelic donor sequences are identical except the HOcs polymorphism, at t=0 h, half of the products can be digested with NcoI. In disomal strains synchronized at G1 and released, all cells had undergone either allelic or ectopic gene conversion by 6 h after transfer to galactose. In contrast, disomal cells blocked at G1 with alpha-factor executed neither ectopic nor allelic recombination (Figure 4C). Diploid a/alpha yeast cells do not respond to alpha-factor. To investigate the ability of diploid cells to carry out homologous recombination while blocked in the cell cycle, we used hydroxyurea (HU), a potent inhibitor of the enzyme ribonucleotide reductase, which arrests cells at the beginning of S phase (Pellicioli et al, 1999). No homologous recombination could be observed in HU-treated haploid or diploid cells (Figure 4B and C).
Taken together, these results indicate that the inhibition of recombination in G1/early S phase occurs irrespective of recombination pathway or cell ploidy. Although it has been recently reported that HU-arrested cells retain as much as 80% of their G1 levels of dNTPs (Koc et al, 2004), even such a mild reduction in dNTP levels by HU might have had a direct effect on recombination. In order to avoid this possibility, we carried out additional experiments with cells arrested before DNA replication. Moreover, by exploiting the wide variety of yeast mutants that have been characterized previously, we were able to determine whether recombination is dependent on the presence of duplicated DNA molecules or on other events that take place during S phase.
At restrictive temperatures, cdc4-1 mutants halt both replication and cell cycle progression through CDK inactivation (Goh and Surana, 1999), whereas cdc7-4 mutants prevent the initiation of DNA replication but are unable to prevent later cell cycle processes such as G2 progression and mitosis (Tercero et al, 2003). Replication mutants cdc4-1 and cdc7-4 incubated at 37°C for 3 h arrested with a 1C DNA content (data not shown and Figure 5A). The cultures were then transferred to galactose at either the permissive (25°C) or restrictive temperature (37°C). At the permissive temperature, both strains repaired the broken chromosome with wt kinetics (Figure 5B and C). As could be predicted for a strain that arrests at the G1/S transition, cdc4-1 mutants at the restrictive temperature did not undergo gene conversion. In contrast, a large portion of the cdc7-4 mutant population (approximately 40%) completed gene conversion, even at the restrictive temperature (Figure 5B). The ability of cdc7-4 cells to accomplish homologous repair in the absence of DNA replication suggests that competency for homologous recombination is controlled by the sequential action of CDK–cyclin complexes and not by the presence of duplicated DNA.
Figure 5.
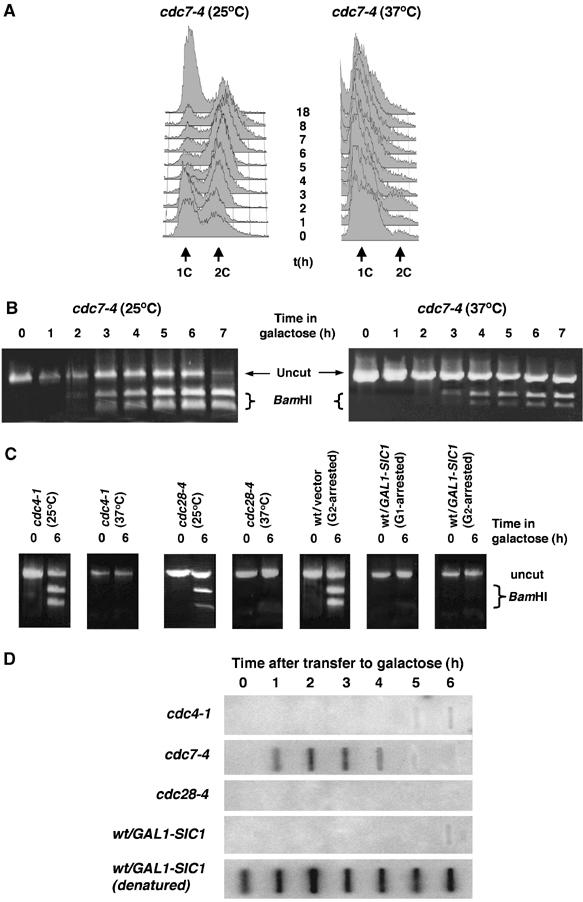
Homologous recombination is dependent on active Clb–Cdk and not on DNA replication. (A) cdc7-4 cells were arrested at the restrictive temperature and then incubated in galactose-containing medium at the permissive (25°C) or restrictive (37°C) temperatures. FACS analysis of the cdc7-4 cell culture confirmed that cells did not undergo DNA replication at the restrictive temperature. (B) PCR assays for gene conversion in cdc7-4 cells at the permissive and restrictive temperatures. (C) PCR assays for gene conversion were carried out for cdc4-1 and cdc28-4 cells at the permissive and restrictive temperatures, wt G2-arrested with vector only, or G1- or G2-arrested with GAL-SIC1 plasmid. (D) Nondenaturing slot blot analysis of cdc4-1, cdc7-4, cdc28-4 and wt cells overexpressing SIC1. As a positive control for hybridization, equivalent DNA samples were blotted after DNA denaturation (only wt/GAL-SIC is shown here).
Sic1 is a potent inhibitor of the Clb–CDK (Mendenhall, 1993; Peter and Herskowitz, 1994). We overexpressed SIC1 to inactivate the CDK (Peter and Herskowitz, 1994) in pre-replicative or post-replicative cells. Overexpression of SIC1 in both situations prevented repair by gene conversion (Figure 5C), reconfirming that Clb–CDK activity, and not the presence of duplicated DNA, is essential for homologous recombination. Consistently, inactivation of the CDK in cdc28-4 mutants at the restrictive temperature also prevented recombinational repair (Figure 5C).
In order to investigate the mechanism that controls homologous recombination during the cell cycle, we used nondenaturative dot-blot analysis to monitor ssDNA end resection in the different strains. Whereas cdc7-4 cells processed DSBs into ssDNA resection intermediates, all of the strains lacking Clb–CDK activity (cdc28-4, cdc4-1 and wt cells overexpressing SIC1) failed to undergo resection (Figure 5D). We thus conclude that Clb–CDK activity promotes homologous recombination by controlling the resection step of DSB repair.
Earlier studies in yeast have provided evidence that recombination proficiency is in fact independent of cell cycle stage (Fabre et al, 1984; Galli and Schiestl, 1998). However, most of these experiments were based on the selection of rare recombination products, which do not represent the majority of the cell population. Differential damage sensitivity of synchronized, cycling cells has been used to infer a preference for homologous recombination in G2 also in mammalian systems (Takata et al, 1998). In our experiments, a single, defined DSB was created uniformly in each cell and repair was monitored in the entire population. We were also able to retain the population at the specific cell cycle stages tested, and monitor the repair in real time while the cells remained arrested. Our experiments thus have uniquely defined the relationship between homologous repair, CDK activity and DNA replication in a eukaryotic system.
The ability of yeast cells to carry out the developmentally controlled gene conversion required for mating type switch at the G1/S stage demonstrates that the enzymatic machinery required for homologous recombination is present in the cells. Further work is required to find out the mechanism by which a DSB at MAT is recognized and processed by homologous recombination, whereas DSBs at other regions of the genome are not.
The lack of end processing has several important consequences. It preserves an appropriate substrate for NHEJ in G1 and defers initiation of recombination until G2, during which homologous sequences are present in the form of sister chromatids. In addition, since checkpoint activation is proportional to the extent of resected ssDNA (Melo et al, 2001; Zou and Elledge, 2003), the DNA damage checkpoint may not be triggered in G1-arrested cells (Pellicioli et al, 1999, 2001).
Our results clearly show that lack of Clb–CDK activity in G1 cells prevents homologous recombination by inhibiting ssDNA resection. This may be a direct effect (Clb–CDK directly phosphorylating a target protein involved in resection). An alternative model suggests that Clb–CDK may be needed to prevent end-joining, thus allowing the homologous recombination machinery to compete more efficiently for the broken ends. Future work will be aimed at identifying the CDK target(s) important for resection.
Restriction of homologous recombination from the G1/S phase of the cell cycle by lack of Clb–CDK activity has clear adaptive advantages for all eukaryotes. For organisms such as S. cerevisiae, which have a compact genome with a high gene density, it may be advantageous to rely upon homologous recombination, an error-free mechanism of repair. In contrast, in mammalian cells, inexactly ligated sequences are less likely to affect functional genes. Furthermore, the presence of numerous repeated sequences in the mammalian genome could make homologous recombination dangerous. Accordingly, DNA damage in the form of DSBs primarily elicit G1 checkpoints in mammalian cells and G2 checkpoints in yeast (Bartek et al, 1999). Our results provide a cellular mechanism for the control of eukaryotic DSB repair pathway choice.
Materials and methods
Yeast strains
All the yeast strains used in this study are isogenic derivatives of strain MK203 (MATa-inc ura3::HOcs lys2::ura3::HOcs-inc ade3::GALHO ade2-1 leu2-3,112 his3-11,15 trp1-1 can1-100) (Aylon et al, 2003), a derivative of W303. The ura3::HOcs allele on chromosome V was created by inserting a 39 bp oligonucleotide at the NcoI site of the URA3 gene. The ura3HOcs-inc allele on chromosome II was created by inserting a 1.2 kb ura3HOcs-inc HindIII fragment at a HpaI site within LYS2 sequences, as described (Inbar and Kupiec, 1999).
Deletion of genes was obtained by transformation of MK203 with a PCR product produced on the appropriate strain from the Saccharomyces Genome Deletion Project array.
W303 strains of the cdc4-1, cdc7-4 and cdc28-4 alleles were kindly provided by J Diffley and Y Kassir. MK203 derivatives carrying these alleles were generated by genetic crosses. The plasmid carrying GAL-SIC1 was generously provided by D Kornitzer and was introduced into MK203 by transformation. All chromosomal configurations were verified by Southern blot or PCR analysis after transformation.
Disomic strains were created by crossing cycloheximide-resistant papillae of MK203 bar1::LEU2 cells to mutants carrying a kar1-1 mutation (provided by Giora Simchen). Heterokaryons were plated on medium to select for the following phenotype: Cyhr Leu+ Ura+, and tested for additional markers (Lys− Trp− Ade− His− CANs), mating ability and high levels of papillation of 5-FOA and CAN medium, indicative of loss of heterozygozity of markers on chromosome V. In addition, heterozygosity for URA3 was verified using PCR, so that disomic strains carried one URA3 allele and one HOcs allele on each chromosome V (in addition to a ura3:: HOcs-inc on chromosome II).
Diploid MK235 strain was generated by crossing MK203 with MK228 (a LYS2 URA3 derivative of W303).
Media and growth conditions
S. cerevisiae strains were normally grown at 30°C. Temperature-sensitive strains were tested at 25°C (permissive) or 37°C (restrictive) temperatures. Standard YEP medium (1% yeast extract, 2% bacto peptone) supplemented with 3% glycerol (YEPGly), 2% galactose (YEPGal) or 2% dextrose (YEPD) was used for nonselective growth. 1.8% Bacto-Agar was added for solid media. For cell cycle arrest, 3.4 μM alpha-factor, 100 mM HU and 15 μg/ml nocodazole were used for G1, S and G2 block, respectively.
DSB induction experiments
Single colonies were resuspended in rich YEPGly medium, grown to logarithmic phase, centrifuged and resuspended in YEPGal. At timely intervals, samples were plated on YEPD plates to score viability and commitment to gene conversion, and DNA was extracted and subjected to the different assays. At least three independent experiments were carried out for each condition analyzed.
Fluorescent activated cell sorter (FACS) analysis
Samples of 107 cells were harvested and fixed in 1 ml 80% ethanol. Cells were washed twice with 1 ml 50 mM Tris–HCl (pH 8) and resuspended in the same buffer with 2 mg/ml RNAse A. Following overnight incubation at 37°C, the samples were washed twice with 1 ml 50 mM Tris–HCl (pH 8), resuspended in 50 μl 20 mg/ml Proteinase K and incubated for 1 h at 37°C. After another wash and brief sonication, 3 μl of 4 mg/ml propidium iodide was added and cells were appropriately diluted and analyzed.
Southern blot analysis
Southern blot analysis was carried out as described previously (Inbar and Kupiec, 1999).
PCR assays
Genomic DNA (5 ng) was amplified in each sample. Taq polymerase was used in standard reaction conditions. Primers OI9 and OI10 are complementary to sequences flanking the DSB on chromosome V. Primers MATa and MAT distal detect MATa configuration; MATalpha and MAT distal detect MATalpha configuration. Sequences are provided below:
- OI9:
5′ TTTTGCGAGGCATATTTATGGTGAAGG 3′
5′ TGTTACTTGGTTCTGGCGAGGTATTGG 3′
5′ AAATAAACGTATGAGATCTA 3′
5′ GCAGCACGGAATATGGGACT 3′
5′ ATGTGAACCGCATGGGCAGT 3′
- OI10:
5′ TGTTACTTGGTTCTGGCGAGGTATTGG 3′
5′ AAATAAACGTATGAGATCTA 3′
5′ GCAGCACGGAATATGGGACT 3′
5′ ATGTGAACCGCATGGGCAGT 3′
- MATa:
5′ AAATAAACGTATGAGATCTA 3′
5′ GCAGCACGGAATATGGGACT 3′
5′ ATGTGAACCGCATGGGCAGT 3′
- MATalpha:
5′ GCAGCACGGAATATGGGACT 3′
5′ ATGTGAACCGCATGGGCAGT 3′
- MAT distal:
5′ ATGTGAACCGCATGGGCAGT 3′
Nondenaturing slot blots
Equal amounts of nondenatured genomic DNA were directly spotted on nylon Hybond+ filters, or denatured by boiling and immediately spotted on filters. All denatured samples undergo hybridization, whereas hybridization of nondenatured DNA indicates that resection had occurred. A 1.2 kb HindIII URA3 fragment was used as a probe for ssDNA. Hybridization and exposure were carried out as in Southern blot analysis.
Survival
Survival was assayed by plating samples of cells grown on liquid YEPGal, onto YEPD plates at different times during a DSB induction experiment.
Commitment to gene conversion
At different times after transfer to galactose, cells were plated onto YPED and incubated 3 days at 30°C. In all, 30 individual colonies were subjected to either Southern blot analysis or PCR, to assay the transfer of information in individual colonies. The PCR reactions consisted of one cycle of 5 min at 96°C, followed by 30 cycles at standard conditions. The DNA thus obtained was then digested with BamHI or EcoRI and subjected to electrophoresis.
Quantitation of results
Southern blots and ethidium bromide-stained agarose gels were quantified using the TINA (v. 2.1) and NIHimage (v. 1.62) computer programs, respectively.
Acknowledgments
This work was supported by grants from the Israeli Science Fund, the Recanati Fund and the Ministry of Health to MK. We thank J Diffley, Y Kassir, G Simchen and D Kornitzer for generously providing strains and plasmids, and all members of the Kupiec lab for helpful discussions and support.
References
- Aylon Y, Kupiec M (2003) The checkpoint protein Rad24 of Saccharomyces cerevisiae is involved in processing double-strand break ends and in recombination partner choice. Mol Cell Biol 23: 6585–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Bitan-Banin G, Kupiec M (2003) Molecular dissection of mitotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol 23: 1403–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek J, Lukas J, Bartkova J (1999) Perspective: defects in cell cycle control and cancer. J Pathol 187: 95–99 [DOI] [PubMed] [Google Scholar]
- de la Torre-Ruiz MA, Green CM, Lowndes NF (1998) RAD9 and RAD24 define two additive, interacting branches of the DNA damage checkpoint pathway in budding yeast normally required for Rad53 modification and activation. EMBO J 17: 2687–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre F, Boulet A, Roman H (1984) Gene conversion at different points in the mitotic cycle of Saccharomyces cerevisiae. Mol Gen Genet 195: 139–143 [DOI] [PubMed] [Google Scholar]
- Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, DePinho RA, Alt FW (2000) The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc Natl Acad Sci USA 97: 6630–6633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Vaillant M, Marcand S (2002) Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell 10: 1189–1199 [DOI] [PubMed] [Google Scholar]
- Galli A, Schiestl RH (1998) Effects of DNA double-strand and single-strand breaks on intrachromosomal recombination events in cell-cycle-arrested yeast cells. Genetics 149: 1235–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh PY, Surana U (1999) Cdc4, a protein required for the onset of S phase, serves an essential function during G(2)/M transition in Saccharomyces cerevisiae. Mol Cell Biol 19: 5512–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber JE (1998) Mating-type gene switching in Saccharomyces cerevisiae. Annu Rev Genet 32: 561–599 [DOI] [PubMed] [Google Scholar]
- Inbar O, Kupiec M (1999) Homology search and choice of homologous partner during mitotic recombination. Mol Cell Biol 19: 4134–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koc A, Wheeler LJ, Mathews CK, Merrill GF (2004) Hydroxyurea arrests DNA replication by a mechanism that preserves basal dNTP pools. J Biol Chem 279: 223–230 [DOI] [PubMed] [Google Scholar]
- Kolodner RD, Putnam CD, Myung K (2002) Maintenance of genome stability in Saccharomyces cerevisiae. Science 297: 552–557 [DOI] [PubMed] [Google Scholar]
- Lengronne A, Schwob E (2002) The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1). Mol Cell 9: 1067–1078 [DOI] [PubMed] [Google Scholar]
- Melo JA, Cohen J, Toczyski DP (2001) Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev 15: 2809–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall MD (1993) An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science 259: 216–219 [DOI] [PubMed] [Google Scholar]
- Nasmyth K (1993) Control of the yeast cell cycle by the Cdc28 protein kinase. Curr Opin Cell Biol 5: 166–179 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lee SE, Lucca C, Foiani M, Haber JE (2001) Regulation of Saccharomyces Rad53 checkpoint kinase during adaptation from DNA damage-induced G2/M arrest. Mol Cell 7: 293–300 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter M, Herskowitz I (1994) Joining the complex: cyclin-dependent kinase inhibitory proteins and the cell cycle. Cell 79: 181–184 [DOI] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17: 5497–5508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo SH, Jackson SP (1997) Identification of Saccharomyces cerevisiae DNA ligase IV: involvement in DNA double-strand break repair. EMBO J 16: 4788–4795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- Tyers M, Tokiwa G, Futcher B (1993) Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2 and other cyclins. EMBO J 12: 1955–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA–ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]