

INTRODUCTION
Increasing costs of drug development and ethical concerns about the risks of exposing humans and animals to novel chemical entities favor limited exposure clinical trials such as microdosing and other phase 0 trials. An increasing body of research supports the validity of extrapolation from the limited drug exposure of phase 0 approaches to the full, therapeutic exposure. An increasing number of applications and design options demonstrate the versatility and flexibility these approaches offer to drug developers.
BACKGROUND AND DEFINITIONS
The pursuit of alternative approaches for first‐in‐human (FIH) trials arose in response to a decline in drug development productivity and as an attempt to reduce the costs and time spent identifying failed drugs.1 Such alternative approaches, variously named Exploratory Investigational New Drug applications (eIND), phase 0, and, under the current guidelines, Exploratory Clinical Trials, were established by regulatory authorities to reduce the risks to humans by limiting drug exposure during FIH trials.2, 3 Reduction in exposure and expected risks led to a reduction in preclinical testing, associated time and costs, drug manufacturing requirements, and promises to provide valuable human‐based data prior to initiation of full‐fledged INDs.4, 5, 6 Just how much better informed and more efficient these approaches are than traditional ones depends on the type of information provided, associated time and costs, available alternatives, and the validity of extrapolating modeling from the limited exposure to full‐therapeutic dose exposure.
The current, internationally harmonized, regulatory framework defining and governing microdosing and other phase 0 clinical trials is the International Conference on Harmonization (ICH) M3 Guidelines3. Under this framework phase 0 trials are FIH trials where the exposure to the drug is less than in phase I studies (i.e., less than maximal tolerated dose [MTD]), have no therapeutic purpose, and are not intended to assess tolerability. The five phase 0 approaches described in the guidelines form a spectrum of exposure from single, minimal (microdose) exposure to multiple doses into the anticipated therapeutic range (Table 1). Other approaches that meet the spirit of the guidelines are possible and early consultation with local regulators is recommended to help identify the optimal approach.2 The 2006 the US Food and Drug Administration (FDA) eIND guidance emphasizes the inherent flexibility in the regulations: “Existing regulations allow a great deal of flexibility in the amount of data that needs to be submitted with an IND application, depending on the goals of the proposed investigation, the specific human testing proposed, and the expected risks. The Agency believes that sponsors have not taken full advantage of that flexibility and often provide more supporting information in INDs than is required by regulations”.2
Table 1.
Abbreviated definitions of phase 0 approaches (ICH M3)3
| Number/Duration of Doses | Maximum Dose | Preclinical Requirements | Genotoxicity/Dosimetry | |
|---|---|---|---|---|
| Approach 1 (microdosing) | 1 | 100 μg AND 1/100 of NOAEL | Extended single dose toxicity In rodent; GLP | No genotoxicity; PET dosimetry |
| Approach 2 (microdosing) | 5 (6 half‐lives between doses) | Each dose:100 μg AND 1/100 of NOAEL | 7‐day repeated‐dose toxicity In rodent; GLP | No genotoxicity; PET dosimetry |
| Approach 3 | 1 | Starting at subtherapeutic dose and moving into the anticipated therapeutic range but < ½ NOAEL | Extended single‐dose toxicity In rodent and non‐rodent; GLP | Ames assay; PET dosimetry |
| Approach 4 | Multiple<14 days | Starting dose:<1/50 of NOAEL AUC; Into the anticipated therapeutic range but< 10th preclinical AUC if no toxicity, or < NOAEL | 14‐day repeated‐dose toxicity in rodent and non‐rodent; GLP | Ames assay + chromosomal damage test; PET dosimetry |
| Approach 5 | Multiple<14 days | Starting dose: <1/50 NOAEL; Into the anticipated therapeutic range but< non‐rodent NOAEL AUC, or <½ rodent NOAEL AUC | 14‐day repeated‐dose toxicity in rodent and non‐rodent; GLP | Ames assay + chromosomal damage test; PET dosimetry |
NOAEL, No Observed Adverse Effect Level; AUC, area under the curve; GLP, good laboratory practice.
As a contribution to the body of definitions we propose the term “in‐humano” to describe the type of limited testing in “exploratory clinical trials.” The term was coined during discussions on the occasion of microdosing symposium at the American College of Clinical Pharmacology (ACCP) annual meeting in 2013. Although limited, such brief and/or local drug interactions within humans may generate data of mechanistic and conceptual value not otherwise available prior to phase I studies. The term “in humano” testing uses the Latin terminology similar to “in silico,” “in vitro,” and “in vivo” testing, to indicate preclinical testing in humans, meaning that no clinical, (i.e., therapeutic or toxic) effects are expected. It is a step on the spectrum from human in vitro tissues, to studies in intact preclinical species, to studies in isolated intact human organs or tissues, to systemic subtherapeutic exposure in humans and finally, systemic therapeutic exposure in humans.
In the case of microdosing (Approaches 1 and 2, Table 1) the dose is defined as no greater than 100 μg (for small molecules) or 1/100th of the No Observed Adverse Effect Level (NOAEL), whichever is the lower. With such low exposures no gross effects, therapeutic, toxic or radiotoxic when labeled with radioisotopes, are expected. As will be discussed later, however, pharmacological effects, both pharmacokinetic (PK) (e.g., absorption, distribution, metabolism, excretion [ADME]) and pharmacodynamic (PD) (e.g., receptor binding and displacement, production of intermediate metabolites, and modification of targets; Figure 1) may take place, and be detected with targeted approaches and sensitive analytical tools even if no gross effects are elicited in the organism as a whole. For microdosing approaches used to test proteins, a molar limit (30 nmol) is applied due to the large size of the molecules.2 The molar and mass definitions converge if the size of the test molecule is 3.3 kDa, when it is both 100 μg and 30 nmol. For any larger molecules the 100 μg definition of microdosing will be the more conservative one. As an illustration ‐ the TGN1412 monoclonal antibody, at 150 kDa, was given at 0.1 mg/kg to six healthy volunteers and caused a cytokine storm and near fatal multiple organ failure within 24 h7. For a 45 kg individual the resulting 4.5 mg meets the molar definition of a microdose study (30 nmol x 150 kDa = 4.5 mg). However, were the 100 μg definition used the resulting dose would have been 45‐fold lower.
Figure 1.
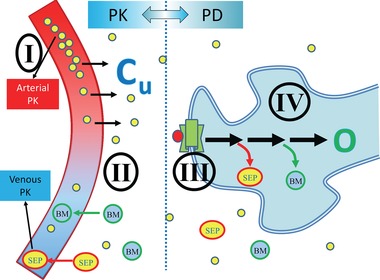
PKPD Continuum. phase 0/microdosing allows study of drug effects in the following domains: (I) – plasma PK; (II) – target PK; (III) – receptor binding and displacement; (IV) – pharmacological effects; biomarkers and/or clinical outcomes. PD, pharmacodynamics; PK, pharmacokinetics. Cu, concentration unbound in tissue; O, outcome; BM, biomarkers/metabolites; SEP, surrogate end points.
ANALYTICAL TOOLS
The limited systemic exposure of phase 0 studies may require more sensitive assays than conventional analytical tools. The three most commonly used techniques are liquid chromatography‐tandem mass spectrometry (LC‐MS/MS), positron emission tomography (PET), and accelerator mass spectrometry (AMS) (Table 2).4, 5, 8 Of these, AMS is by far the most sensitive, followed by PET and LC‐MS/MS. AMS and PET require radiolabelling, however unlike PET, AMS radiation exposure is minimal and typically does not require dosimetry studies.3, 9 The advantage of PET is the ability to provide continuous, dynamic information about drug effects in real time in the living human, in tissues of interest, including target receptor binding and occupancy; however, unlike AMS and LC‐MS/MS, PET imaging cannot distinguish metabolites from parent compound.10 LC‐MS/MS is the cheapest, most accessible, and associated with the least amount of processing while AMS and PET require the presence of specialized units for production of the radioisotopes, radiochemistry, processing, and analysis. While PET requires the presence of such facilities in close proximity to the clinical research site (with the exception of 18F and 124I that can be shipped due to their 100 min and 4‐day half‐life, respectively) AMS is usually processed in remote facilities with the long half‐life of 14C (∼5700 years) presenting no limitations on shipment schedules. The limited availability of specialized AMS and PET facilities is of little relevance since typically only one phase 0 study with six to 10 participants is required per drug development program.
Table 2.
Properties of analytical tools used in phase 0 studies. Adapted from Bauer et al.9
| AMS | PET | LC‐MS/MS | |
|---|---|---|---|
| Sensitivity | 10−16 to 10−18 g | 10−12 to 10−14 g | 10−12 g |
| Sample types | Mostly plasma but any samples may be used (e.g., biopsies, bronchial lavage, CSF, urine, feces, blister samples) | Real‐time imaging; dynamic, contemporaneous information from multiple tissues/targets | Mostly plasma but any samples may be used (e.g., biopsies, bronchial lavage, CSF, urine, feces, blister samples) |
| Sample frequency / duration | 6‐10 / h duration unlimited | Continuous / dynamic; duration limited by radioisotope half‐life | 6‐10 / h duration unlimited |
| Plasma sample volume | Typically 50 μL, but as little as 2 μL | N/A; continuous / dynamic “counting” of drug molecules per unit space | Typically 100 μL‐2 mL, but as little as 25 μL8, 11, 12 |
| Radiolabelling | 14C | 11C, 13N, 15O, 18F, and 124I | None |
| Radiation exposure | Very low | Low | None |
| Parent compound and metabolites | Discriminating parent compound from metabolites possible | No discrimination | Discriminating parent compound from metabolites possible |
| Administration | PO and IV | IV | PO and IV |
| Site of analysis | Can be outsourced | On‐site only | Can be outsourced |
| Costs per study13 | ∼ $ 400–600 k | ∼ $ 500–700 k | ∼ $ 80–140 k |
| Availability | Limited availability; ∼ six facilities dedicated to biomedical research worldwide | Available in specialized centers (e.g., tertiary‐care facilities) | Commonly available |
AMS, accelerator mass spectrometry; PET, positron emission tomography; LC‐MS/MS, liquid chromatography‐tandem mass spectrometry; CSF, cerebrospinal fluid; N/A, not applicable.
PHASE 0 VS. OTHER FIRST‐IN‐HUMAN (FIH) CLINICAL TRIALS (TABLE 3)
Table 3.
Comparison of phase 0/microdosing with traditional phase I approaches
| Phase 0/Microdosing (eIND) | Traditional Phase I (IND) | |
|---|---|---|
| Therapeutic intent | None | Possible |
| Study of systemic tolerability | None | Yes |
| Proof of Mechanism | Possible (e.g., PET receptor binding and displacement) | Possible |
| Preclinical Package | Limited, variable; depends on extent of exposure to the test article and experimental goals | Full requirements |
| in vitro models | Full requirement | Full requirements |
| toxicology | Limited, variable | Full requirements |
| genotoxicology | None or limited | Full requirements |
| GMP | Flexible, depending on available preclinical information and route of administration (e.g., sterility ensured for IV route) | Full requirements |
| Regulatory Review | 30‐day | 30‐day |
| Usual Duration of Program | 4‐12 months | 12‐24 months |
| Cost of Program | $ 0.5‐0.75 M | $ 1.5‐2.5 M |
| Studies | ||
| size (typical) | 4‐10 participants | 6‐30 participants |
| duration (per participant) | 1‐14 days* | 6‐60 days* |
| number of study sites | Single | Single/Multiple |
| maximal dose | <MTD | MTD |
| exposure | Limited (see Table 1) | Multiple doses allowed |
| population | Healthy volunteers or patientsVulnerable populations | Usually health volunteers (unless toxicity risk is high, e.g., in oncology trials) |
*on average, could be longer with longer half‐life drugs; MTD, maximum tolerated dose.
The conduct of a phase 0 study differs from other developmental clinical trials in several respects, most importantly the use of low doses, low plasma concentrations, and the highly sensitive analytic tools needed to analyze drug effects. The regulatory framework allows a quicker initiation of phase 0 studies due to reduced preclinical package requirements and simpler manufacturing process. In many other aspects, including the ethics, recruitment, volunteer management, phase 0 trials are similar to other FIH studies.
HUMAN‐BASED CONTRIBUTIONS TO DRUG DEVELOPMENT DECISION PROCESS (FIGURE 1)
Even though there is no therapeutic intent, phase 0 trials allow the study of key properties that are relevant to drug development “go‐no‐go” decision making (Figure 1). These properties, sometimes called “pillars of pharmacology,” include the following domains: (I) plasma PK, (II) target PK (for both efficacy and toxicity targets), (III) receptor binding and displacement, and (IV) PD (biomarkers and clinical outcomes).14 Of these, only clinical outcomes that depend on long‐term exposure cannot be studied with any of the phase 0 options. However, even with the most limited exposure of a single‐dose microdose (Approach 1, Table 1) the first three domains can be studied, and, as will be demonstrated in the following sections depending on the exposure, sampling (e.g., biopsies), analytical tools (e.g., PET), and approach (e.g., Intra‐Target Microdosing [ITM]), some PD information may also be obtained.15, 16, 17 All phase 0 approaches depend on the validity of extrapolation from the limited‐exposure scenario to the full exposure of therapeutic‐intent administration, to be discussed in detail below. However, in the ultimate analysis, phase 0 always provide previously unavailable information about test article effects in humans. This information reduces the uncertainty about the test article. Depending on the individual development scenario, such information may reach a meaningful threshold for a “go‐no‐go” decision or provide valuable guidance for the phase I and phase II programs that follow. Any information obtained, however, should justify the investment and time spent obtaining it. This was found to be the case in economic modeling and simulations of microdosing studies.13
PAST AND CURRENT VALIDATION EFFORTS AND APPLICATIONS
Since the introduction of the exploratory clinical trial framework more than a decade ago the main foci in the field have been testing of the validity of the approach and exploring potential applications.18 Validation efforts focused mainly on demonstration of linearity across the microdose to therapeutic dose spectrum and development of modeling and simulation tools to address circumstances when non‐linearity may be an issue. Application efforts focused on drug development scenarios where the requirement or convenience of safe testing and early arrival at informed “go‐no‐go” decision making has been particularly pronounced (Table 4). The validation efforts and research on the various and expanding number of applications is described in the following section and is an ongoing process.
Table 4.
Current and emerging applications of phase 0 / microdosing approaches. “Current applications” refers to phase 0 clinical studies that were part of new drug development, or those used in multiple research settings to obtain information on existing drugs in new populations or circumstances. “Emerging applications” denotes new and theoretical applications that are in the early stages of development and/or validation
| Examples and references | |
|---|---|
| Current applications | |
| PK and BA | Study of drug disposition (e.g., absorption, distribution, metabolism, excretion, bioavailability [ADME], TMDD);19 effectiveness‐related PK (i.e., PK parameters that directly impact on safety and efficacy).20 |
| DDI | Cocktail and cassette DDI studies.21, 22 |
| PD/Localization/Proof of mechanism | Phosphorylation in PBMCs.16 DNA adducts in PBMCs.17 Target tissue localization.23, 24, 25 |
| Vulnerable populations | Pediatric studies of drug disposition.26, 27, 28, 29 Future applications may apply also to women (including pregnant women), patients with hepatic impairment, renal impairment, poly‐pharmacy, comorbidity, and frail elderly patients.30 |
| Diagnostic radiopharmaceuticals | Due to lack of appropriate animal models, phase 0 used for selection amongst four 18F‐labelled PET amyloid imaging agents for assessment of β‐amyloid plaques in brains of patients with Alzheimer's disease.31 |
| Emerging applications | |
| PBPK, M&S | Modeling and simulations incorporating in‐silico, in vitro, PBPK/PD32 and economic parameters13 can all enhance the predictive value and feasibility of phase 0 studies.22 |
| Biologics | Small antibody (PET)33. Large protein (AMS).34 |
| Adaptive design phase 0/phase I | Microdosing/phase I adaptive design.34 |
| Intra‐Target Microdosing (ITM) – drug development in target. | Intra‐Arterial Microdosing (IAM) proof‐of‐concept in rats.15 Insulin injected intra‐arterially caused local effect (18F‐FDG uptake into muscle) while systemic exposure was at microdose levels with no effects. |
| Extreme environments | Space (micro‐gravity, radiation, altered chronobiology), north/south poles (cryo‐environments, altered chronobiology), hyper/hypobaric environments (e.g., high altitudes). Altered physiology and pharmacology may have drug efficacy and toxicity implications and requires testing of pharmaceuticals in the extreme environment setting. Lack of emergency facilities favors phase 0 approaches. |
| Individualized therapy phenotyping | Prediction of DDI in healthcare settings by using microdose probes prior to initiation of therapy.22 , 35, 36, 37, 38 |
| Environmental toxins | Describing the disposition of potential carcinogens (e.g., PAH) using nontoxic microdoses in humans.39, 40 |
PK, pharmacokinetics; BA, bioavailability; PD, pharmacodynamics; TMDD, target‐mediated drug disposition; DDI, drug‐drug interactions; PBPK, physiologically‐based pharmacokinetics; M&S, modeling and simulation; PBMCs, peripheral blood mononuclear cells; FDG, fluorodeoxyglucose; PAH, polycyclic aromatic hydrocarbon.
PK and bioavailability
The main application of phase 0/microdosing studies has been in the evaluation of PK and bioavailability of new drugs. There have been extensive efforts to predict PK of drugs in humans using preclinical methods, such as allometric scaling from experimental animals, physiologically based pharmacokinetics (PBPK) or extrapolation of study results obtained with in vitro systems (in vitro‐in vivo extrapolation [IVIVE]), using human liver microsomes.41 However, quantitative prediction of human PK can still be challenging, and phase 0/microdose studies can serve as a powerful complementary tool to existing approaches. Safely obtaining PK data is particularly valuable where a drug has a narrow therapeutic index (NTI), as is often the case with anticancer agents.37, 42, 43 The pivotal microdosing validation studies in the PK/bioavailability domain have been the CREAM (Consortium for Resourcing and Evaluating AMS Microdosing) and EUMAPP (European Microdosing accelerator mass spectrometry [AMS] Partnership Programme) trials in Europe, and the NEDO (New Energy and Industrial Technology Development Organization) trials in Japan.19, 44, 45 By now the results of more than 35 studies in both animals and humans have been reported comparing microdose vs. full dose. Of these, a microdose successfully predicted therapeutic dose PK in 79% of drugs administered orally (n = 27) and 100% of those administered IV (n = 12).30
Microdosing studies have been used in various drug development programs. One compared three analogues of diphenhydramine (DPH) with the aim of improving upon DPH's poor properties as a hypnotic, specifically, the delayed onset of action and residual wakeful hypnotic effects.20 DPH and an analogue that previously had failed in development both showed PK linearity between a microdose and full dose. With predictability demonstrated and assumed for the group of analogues, the candidate with the highest bioavailability, shortest t1/2, and least intersubject variability, deemed desired properties, was chosen for further development. This example is notable for the ability of the microdosing approach to detect specific and narrow PK parameters of direct relevance to drug clinical utility, both therapeutic and side effects, which could not otherwise be managed by simply modifying the dose or frequency of administration of DPH itself. Identifying such properties (or their absence) could reward the investment in a phase 0 study by leading to early termination of those analogues that did not demonstrate the desired PK properties. In this study each of the analogues was evaluated separately, using AMS, while in another study, using LC‐MS/MS, the oral bioavailability of three Ca‐channel blockers was assessed simultaneously by administering them as microdose cassette.46
Microdose absolute bioavailability studies
Absolute bioavailability (F), is a measure of the fraction of an extravascularly administered dose of drug reaching the systemic circulation intact. For small molecules the most common extravascular route is oral, for biologics it is subcutaneous. Phase 0 microdose absolute bioavailability studies consist typically of an oral, intravenous crossover design.47 In addition to F, this design provides a multitude of other preliminary pharmacokinetic data, including Cmax, tmax, t½, CL (clearance), and V (volume of distribution). The estimate of CL allows prediction as to whether it will be affected by such factors as blood flow and plasma protein binding, the anticipated loss of oral bioavailability on first passage through the liver during the absorption process, as well as facilitating PBPK model development (see below) and improving in vitro‐in vivo extrapolation of this parameter. V together with F guide the oral dose needed to ensure exposures within the anticipated therapeutic range.
Of the eight drugs reported where F was compared between a microdose and a therapeutic dose only two drugs, propafenone and sumatriptan, gave an anomalous result (propafenone: 5.8% and 13% sumatriptan: 7.6% and 20% for the microdose and therapeutic dose, respectively) while the other were within a twofold difference.30 Still, microdosing currently compares more favorably than allometry and PBPK, when both absolute bioavailability and shape of the concentration curve are considered.41
Evaluation of nonlinearity of drug disposition
If drug disposition pathways are linear, successful direct extrapolation of exposure from microdose to therapeutic dose is expected.18, 30, 41, 48 Common causes of nonlinear PK include intestinal solubility, metabolism in intestine or liver, membrane transport in intestine, liver, or kidney, and plasma protein binding.19 On rare occasions, with small molecular drugs, there is also a possibility of saturation of binding to drug target at the therapeutic dose, as observed with warfarin.19 These factors should be considered when designing phase 0 studies for the purpose of projecting human PK at therapeutic dose.
Even when nonlinear PK between microdose and therapeutic dose exist, model‐based simulations incorporating nonlinear processes may allow quantitative extrapolation of human exposure.22, 48 This may be possible if the non‐linear processes are identified in advance in preclinical IVIVE studies, suspected from similarly structured drugs, or suggested from the eIND study itself (see Intra‐Target Microdosing [ITM] below).15, 41, 48 Although not exclusive, the basic concept in analyzing saturation of metabolic enzymes or transporters can be expressed in Michaelis‐Menten equation:
where CL, Km, [S], v, and Vmax represent clearance, Michaelis‐Menten constant, concentration of a substrate S, velocity of reaction, and maximum reaction velocity, respectively. As demonstrated in Figure 2, if the level of exposure after therapeutic dose is below Km, the same clearance between microdose and therapeutic dose (i.e., direct extrapolation from microdose) is expected. If, on the other hand, the level of exposure after therapeutic dose is higher than Km, the clearance after microdose coupled with kinetic in vitro saturation information, especially when integrated within a PBPK model (see below), can still be used to predict the exposure level expected after therapeutic doses. Such extrapolation may need to account for auto‐induction or saturation of multiple pathways, as in the case following oral administration, when the drug concentrations in the intestine may saturate multiple enzymes and/or transporters.49
Figure 2.
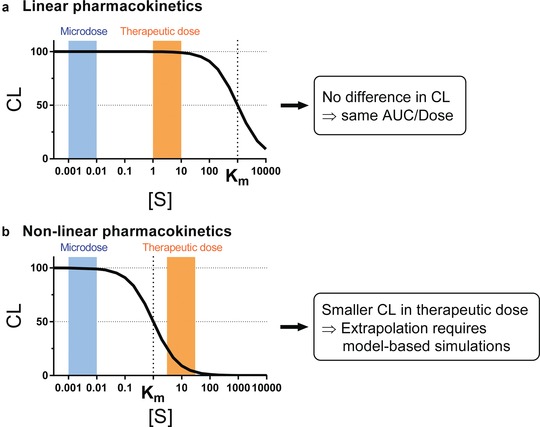
Conceptual representation of nonlinear drug disposition and extrapolation from microdose to therapeutic dose for (a) drugs showing linear PK and (b) drugs showing nonlinear (saturable) PK at therapeutic dose. Width of the blue and orange bars schematically represents ranges of substrate drug concentration after microdose and therapeutic dose, respectively. AUC, area under the concentration‐time curve; CL, clearance; Km, Michaelis‐Menten constant; [S], concentration of a substrate drug S.
Drug‐drug interactions (DDI)
The primary objective of DDI studies is to quantitatively characterize alterations in PK profiles of the substrate drug as a function of interaction with other xenobiotics. The DDI may result in abnormal increases or decreases of drug levels, potentially leading to safety or efficacy concerns, respectively. The most important DDI mechanisms are inhibition or induction of drug metabolic enzymes or transporters. Accurate evaluation of DDI effects is essential for appropriate use of new or existing medications.50
DDIs of PK origin are increasingly being predicted from in vitro experiments using PBPK. However, situations do arise, especially during candidate selection, where information on the compound is insufficient to allow confident DDI predictions to be made.51 Microdosing offers a powerful complementary tool to evaluate DDI PK susceptibility of new or existing drugs as a substrate, “victim” drug, with three key advantages. Firstly, at such low doses there are fewer safety concerns caused by the potentially higher exposure of victim drugs when coadministered with inhibitors of elimination pathways. This is particularly important when examining DDI effects in drugs with narrow therapeutic index. Secondly, when using “cocktail” strategies to evaluate drug effects on multiple elimination pathways, there is little risk of “cross‐reaction” when substrate drugs are administered simultaneously in a microdose.22 Thirdly, simultaneous, “cassette,” administration substantially reduces the variability in the results, hence increases the power of the studies and reduces the required sample size.
To demonstrate the validity of a DDI cocktail approach, which could be used to evaluate a potential perpetrator during drug development, a cocktail of four probe substrate drugs was administered as a microdose to evaluate DDI effects on different elimination pathways (midazolam for CYP3A, tolbutamide for CYP2C9, caffeine for CYP1A2, and fexofenadine for P‐glycoprotein [P‐gp]).52 As a result of co‐administration of ketoconazole and fluvoxamine, increases in the exposure of all four substrate drugs were observed. The magnitude of inhibition by these two perpetrators was consistent with previous observation after therapeutic doses of these four probe substrate drugs, suggesting that microdose studies can be directly and safely used to estimate the degree of DDIs in clinical settings. Similar DDI studies have been conducted with drugs subject to membrane transporters, such as metformin (substrate of organic cation transporter 2 [OCT2] and multidrug and toxin extrusion protein [MATEs] in the kidney)53 and ceriprolol (substrate of OATP2B1 and P‐gp in the intestine).38 These studies also demonstrated similar magnitude of DDIs between microdose and therapeutic dose.
To further extend the application of cocktail microdose studies, Sugiyama and colleagues conducted a cocktail‐dose clinical microdose DDI study to identify the rate‐determining process for the hepatic elimination of atorvastatin, a well‐established substrate of CYP3A and organic anion transporting polypeptide 1B (OATP1B) transporter.54 They compared the pharmacokinetic alteration of atorvastatin, pravastatin, and midazolam in the presence and absence of probe inhibitors, itraconazole for CYP3A and single dose rifampin for OATP1B. The pharmacokinetics of this cocktail was observed at baseline, after an oral dose of 600 mg rifampicin (OATP1B inhibitor), and after an intravenous dose of 200 mg itraconazole (CYP3A4 inhibitor). As a result, exposure of atorvastatin was increased dramatically with single dose rifampin coadministration, but not with itraconazole coadministration. Since the observed pharmacokinetic change was consistent with pravastatin, they concluded that OATP1B is the key determinant of atorvastatin systemic clearance.
PD/Proof of mechanism
The limited systemic exposure of research participants to administered compounds during phase 0 studies, even the so‐called “subpharmacological” levels of microdose studies, does not mean that no pharmacological effects have occurred, only that these effects are so limited that no therapeutic or toxic effects can be expected in the organism as a whole. With sensitive analytical tools and appropriate approaches certain PD effects of the test article may be observed. For example, PET imaging of labelled drug or labelled biomarker may identify receptor binding or biomarker accumulation, respectively, thus providing proof of mechanism of drug actions. AMS detection of 14C‐labelled DNA adducts or phosphorylated drug in PBMCs may do likewise. Intra‐Target Microdosing (ITM; see below) generates full exposure in a local target area by administering a microdose calculated on a full‐body basis directly into the target, thus enabling detection of therapeutic‐level exposure PD parameters in tissues of interest while keeping systemic exposure at subtherapeutic levels.15 These scenarios are described in detail in the following sections.
Drug‐DNA adducts, mitochondrial toxicity, and adverse drug reaction (ADRs) in anticancer and antiviral therapy
Formation of DNA adducts, the covalent binding of xenobiotics to DNA bases, is a crucial step in both carcinogenesis and cancer treatment.55 These formations can be detected with AMS if the drug or suspected carcinogen is labeled with 14C and administered in a microdose thus avoiding carcinogenic or cytotoxic risk.17, 56, 57
Lightfoot et al. conducted a study to examine the uptake of carcinogens into human breast tissue and their ability to bind to DNA to form DNA adducts.57 In the first such report, patients undergoing breast surgery were orally administered prior to surgery one of two known carcinogens (2‐Amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine [PhIP] and benzo[a]pyrene [B[a]P]) as microdoses, 20 μg of PhIP or 5 μg of B[a]P. At surgery, normal and tumor breast tissue were resected and tissue concentrations of carcinogen measured by liquid scintillation counting and DNA adduct levels by AMS. In both cases the carcinogen was shown to reach the target organ and form DNA adducts there (see also below under “Environmental toxins”).
Similarly, as mechanistic proof of anticancer cytotoxic effects, microdosing has been used to identify DNA mono‐adduct formation and repair after anticancer drug administration. Henderson et al. administered microdose 14C‐carboplatin to cancer patients followed by AMS assessment of carboplatin‐DNA mono‐adduct formation in peripheral blood mononuclear cells (PBMCs), an essential step in the therapeutic action of this drug. The study showed that 14C‐carboplatin microdoses had the same adduct formation and repair kinetics as therapeutic doses.17 It is also feasible to use such phase 0 studies to study drug‐resistance mediated by potential polymorphism in DNA‐repair, thus identifying patients best and least suited for carboplatin treatment.58
Vuong et al. applied microdosing to study zidovudine (ZDV) PK and uptake of active drug metabolites at the site of action and of DNA‐incorporation.16 In the study, AMS was used to measure 14C‐ZDV concentrations in PBMCs, the drug's site of action, following a 520 ng dose (less than one‐millionth of the standard daily dose). The study showed that ZDV is rapidly absorbed and eliminated, has one major metabolite, and is sequestered in PBMCs. Mass balance analysis of the 14C‐labeled dose indicated that a significant portion of ZDV remained after 96 h with a much prolonged elimination half‐life. The study also showed equivalent PK parameters for the microdose and therapeutic dose, supporting the usefulness of microdosing and AMS as a tool for studying PK and PD of drug substances.16
Phase 0 approaches could also be used to study adverse drug reactions (ADRs) such as cardiac mitochondrial dysfunction associated with doxorubicin mitochondrial DNA adduct formation, or potential for ZDV toxicity in neonates of mothers receiving antiviral therapy.16, 59 The value of such limited‐exposure phase 0 human studies is further highlighted by the poor prediction of cardiotoxicity and other adverse effects of drugs from preclinical in vitro and animal models.60, 61
Drug and biomarker localization
Determining appropriate target exposure is one of the key objectives of drug development (Figure 1).14 Due to methodological and technological limitations, however, drug PK has traditionally been investigated using plasma exposures. Recent application of imaging technologies in PK studies has enabled the quantitation of drug concentration in target organs.25, 62, 63 Among them, PET has been most frequently used to directly evaluate drug disposition at target organs and in the case of cancer therapeutics also allows determining cross‐reactivity with normal tissues.5, 23, 24, 33, 62, 64 Biodistribution, passage of membranes (e.g., BBB), tissue kinetics, receptor binding and occupancy, “surrogate end points” and inter‐subject variability, are all examples of data that can be obtained with PET and due to its high sensitivity (10−12 to 10−14 g; Table 2) can be obtained at microdose exposures.5, 9, 65
Examination of receptor binding occupancy was used to obtain mechanistic understanding of the interaction of a drug and its target, as in the development of ziprasidone as a central dopamine D2 receptor antagonist.64 In such a study, radiolabeled tracer specific to the receptor of interest is administered in the presence and absence of a test drug, and displacement of the radiotracer is quantified to evaluate the binding affinity of the test drug. Even though this study used therapeutic doses of ziprasidone, displacement methodology can be applied with subtherapeutic exposure when the displacing agent is a compound other than the test drug (e.g., known agonist or competitive antagonist for the receptor) and can be given in pharmacologically active doses. Such studies can provide not only target affinity but also dynamic information about local exposure.
PK at the site of action can be useful in evaluating a drug's potential to interact with PD targets. One representative example is in the development of a potential antiamyloid drug for treatment of Alzheimer's disease.23 The authors investigated the brain distribution of 11C‐labelled ST1859 with PET imaging to compare regions rich in amyloid‐beta and other regions. Although they observed rapid and high accumulation of this drug in brain, confirming passage across the blood‐brain‐barrier (BBB), they did not see regional distribution consistent with amyloid‐beta brain distribution, presumably due to a combination of non‐specific binding and relatively low binding affinity to amyloid‐beta.
PET imaging can also be used to directly observe PD effects by labelling the biomarker that is produced, modified, or mobilized by the test article. A proof‐of‐concept study with 18F‐labelled fluorodeoxyglucose (FDG) as a biomarker of insulin action is described in the “Intra‐Target Microdosing (ITM)” section.15 Another important application of PET imaging is the evaluation of drug interaction potential caused by the alteration in membrane transport functions. Major focus has been placed on the DDI effect on distribution into brain, liver, and kidney. For example, coadministration of P‐gp inhibitors (tariquidar and cyclosporine A) increased brain exposure of 11C‐labeled verapamil, suggesting the in vivo importance of P‐gp in limiting verapamil's brain distribution.66, 67, 68 For liver and kidney, because inhibition of basolateral uptake or apical efflux have different outcomes in terms of systemic and peripheral exposures, it is important to measure both of them when evaluating DDI effect.69 All of these in vivo peripheral exposure measurements are essential in building reliable PBPK models (see PBPK section) and IVIVE methodologies of drug disposition pathways, ultimately leading to quantitative prediction of not only systemic but also target organ exposures.
Vulnerable populations
Vulnerable populations include children, pregnant and lactating women, hepatically/renally impaired, frail elderly, comorbidity, poly‐pharmacy with potential for DDIs, and those with genotypes predisposing them to abnormal drug responses (e.g., through CYP mutations that increase/decrease drug metabolism, or genotypes predisposing to hypersensitivity reactions).70 Common to these populations is their exclusion from primary drug development programs due to safety concerns. Testing drugs in such populations is often restricted due to the risks associated with drug exposure.70, 71 However, prediction of drug response from adult pharmacological data in these populations is often impaired due to their unique pathophysiology, and in pediatric populations has been estimated to be reliable in less than 20% of cases.72 Even though the recent increase in use of PBPK modeling may increase the validity of extrapolation from adult and animal data, alternative drug disposition pathways in vulnerable populations may result in unexpected variability.72 An example in pediatrics is the switch from sulfation to glucuronidation as the primary metabolic pathway of acetaminophen.26 Lack of evidence‐based guidance to clinical management leads to extensive off‐label use and is associated with increased incidence of adverse drug reactions (ADRs).70 , 72, 73, 74 The safety inherent in phase 0 approaches is an attractive complement to extrapolation from adult data in these populations and use of PBPK simulations. Another advantage is that by the time the vulnerable populations are studied, data on linearity, or type of non‐linearity, are usually available from adult studies and can improve extrapolation. However, to date, phase 0 approaches have only been applied to the pediatric population.30
The main potential barriers to the application of phase 0 approaches in vulnerable populations, and in particular in children and pregnant women, are ethical in nature and are related to safety. The three principal safety concerns are exposure to the drug, burden of procedures, and exposure to radiation. Exposure to the drug in microdosing studies is sub‐therapeutic and hence identified as no more than minimal risk, constituting a major advantage over therapeutic‐dose studies in vulnerable populations.75 The burden of procedures in pediatric population is mainly related to the amount of blood sampling. European and World Health Organization (WHO) guidelines recommend restricting blood sampling volumes to no more than 3% of total blood volume (TBV) over 1 month, and 1% of TBV over a 24 h period. This equates to 1/100 of 240 ml = 2.4 ml in a 24 h period in a 3 kg neonate.76, 77 Such amount can easily satisfy the sampling requirements of LC‐MS/MS (∼ 100 μl per sample but as low as 25 μl).11 The much higher sensitivity of AMS further reduces sampling requirements (as little as 2 μl per sample, depending on drug concentration). Radiation exposure is low with PET and extremely low with AMS, consistent with normal background exposure.29 Finally, the ethics argument needs to be strengthened by scientific necessity and exposure to no more than minimal risk over that expected in usual daily activities. A recent FDA position statement supports the use of microdosing studies in patients but leaves the application in healthy children questionable.75
Several pediatric microdose studies have been conducted to date in the US and Europe.26, 27, 28, 29 Doses ranged from 3 to 30 ng/kg and sampling was 20 μL per sample where possible. Levels of radioactivity administered were kept extremely low (10‐100 Bq/kg or 0.3‐3 nCi/kg). These have been proof of concept studies with ursodiol,27 midazolam,29 and acetaminophen26, 28 demonstrating linearity across the microdose to therapeutic dose range thus paving the way to future applications with other drugs and other vulnerable populations. Of particular interest is elucidation of ontogeny of ADME pathways such as drug metabolism, transport, and renal function and the use of microdosing to phenotype such pathways.78
Development of diagnostic radiopharmaceuticals
In the past few years several pharmaceutical companies used eIND approaches for the development of diagnostic radiopharmaceuticals. An example is 18F‐AV‐45, a PET imaging agent to estimate β‐amyloid plaque density in patients assessed for Alzheimer's disease.31 The sponsor filed eINDs for four different candidates to be evaluated in healthy volunteers and in AD patients. The absence of appropriate animal models required a human study for selection among the four candidates thus favoring the safe and low‐cost phase 0 approach. The PET‐microdosing enabled demonstration of BBB passage and localization to regions of interest. The candidate with the appropriate PK, metabolism, target organ uptake, retention (in cortical brain regions known to be high in amyloid disposition), and clearance from non‐target brain tissue, was selected as the lead candidate for clinical development and eventually approved for marketing by the FDA.
EMERGING APPLICATIONS OF PHASE 0/MICRODOSING STUDIES
Modeling and simulations
Phase 0/microdosing data and PBPK models can complement and enhance each other. PBPK models comprise physiological and biochemical data, which are independent of the drug, and often called a systems property, onto which are overlaid drug specific data, often in the form of physicochemical and in vitro human data, to predict the likely PK in vivo under a variety of situations. PBPK models have now become an established part of both early and late stage drug development, and increasingly form part of the label information provided following approval of the drug by regulatory agencies.51, 79, 80 While the ability of PBPK to accurately predict human PK has improved over the years, uncertainty still exists when using in vitro data only, and often there is a need to update the model in the light of in vivo human data. Phase 0 data can enhance PBPK by providing valuable early human‐based data. Conversely, PBPK can enhance the extrapolation of phase 0 data to therapeutic dose levels. Coupling phase 0 data with PBPK is particularly helpful in situations where established methods such as allometry and in vitro‐in vivo extrapolation (IVIVE) are problematic, due to either large unexpected interspecies PK differences, or when IVIVE for one or more processes controlling the PK of the compound are not well established, respectively. Several approaches are being taken. One is to take advantage of the previously stated observation, that microdosing IV disposition PK of a drug is the same as that following therapeutic doses.19, 30, 44 Armed with the IV microdose data, the disposition kinetics of the PBPK model is defined, while it can also be used to inform and update the IVIVE scaling parameters controlling clearance and distribution. The expected oral input, and hence PK profile, is then simulated based on such in vitro data as solubility, particular size distribution, dissolution profile, intestinal permeability, and stability to intestinal and hepatic enzymes and transporters, taking into account nonlinearities if necessary. This helps not only to more accurately predict the likely oral dose needed to ensure adequate systemic exposure of drug, based on in vitro (or preclinical) pharmacodynamic information, but also the best sampling times, and likely inter‐subject variability. It also helps in the selection of the best candidate to meet therapeutic objectives (such as size of dose to ensure adequate exposure, dosing interval, and inter‐subject variability) when several are being considered prior to phase I testing.
PBPK models have been integrated with pharmacodynamic components to explain inter‐individual variability in response.32 For such approaches, it is important to support and validate the PBPK model with in vivo tissue concentration measurement whenever possible. As described earlier, PET‐based microdosing is a powerful tool to measure local exposure and transport of drugs in various tissues. These model‐based methodologies will allow better extrapolation of preclinical pharmacodynamic findings to the prediction of therapeutic outcomes.
For certain types of drugs, membrane transport can be the major determinant of tissue exposure. As a result, transporter‐mediated DDIs can cause altered exposure in the tissues leading to change in efficacy or safety profiles of drugs.69, 81 In many cases, however, changes in transport function are not reflected in systemic exposure, even for drug clearance organs such as liver or kidney, while they can affect tissue exposure of substrates.82 In order to predict complex outcomes of the change in membrane transporter functions, establishment of PBPK models with the support of in vivo tissue concentration measurement is critical.
Biologics
The specificity of modern biologic drugs for their targets favors them as candidates for phase 0 studies. The often high species specificity may render animal models poorly predictive of human PK and response. In addition, localization of biologics at their specific targets makes PET‐microdosing a useful tool in studying access and dynamic exposure at the target. With their PK and PD typically affected by target‐mediated drug disposition (TMDD), New Biological Entities (NBEs) lend themselves to the study of target dynamics, compartmental PK, and receptor binding and displacement using labeling with PET radioisotopes.83 Inter‐patient variability and cross‐reactivity between target (e.g., tumor) and non‐target (e.g., plasma, muscle) tissues can also be demonstrated.33 Finally, if the pharmacological target of the biologic is labeled (e.g., glucose [and 18F‐FDG] in the case of insulin) then the typically specific pharmacology of NBEs can be studied as well using PET‐microdosing.15 Many biologics are administered intravenously thereby bypassing potential non‐linear absorption, transport, and metabolism mechanisms that could render extrapolation from microdose to full‐dose less reliable.
In 2013, the first microdosing study of a biologic was reported, where PK, biodistribution, and tumor targeting of the mini antibody 124I‐F16SIP (80 kDa, 2 mg, 25 nmol), an antiangiogenesis drug for head and neck cancer, were studied using PET imaging.33 The study was able to demonstrate tumor targeting, near optimal bioavailability, and low interindividual variability in PK in the four patients studied, thereby supporting further development. In 2015, the first 14C‐labelled recombinant human protein (human recombinant placental alkaline phosphatase [hRESCAP], an endogenous anti‐inflammatory protein) was studied in a phase 0/phase I adaptive design study.34 Results demonstrated linear PK from microdose (53 μg) to therapeutic dose (5.3 mg). The study enabled the demonstration of a t1/2 greater than 2 days, a “go‐no‐go” developmental requirement for this compound, with the phase 0 component of the study prior to entry into the phase I component, thus allowing efficient allocation of resources. With this protein drug it appears that the elimination process is not saturable, and this may be the case for other classes of protein drugs, in contrast to monoclonal antibodies, which exhibit TMDD.84
A potential problem with biologics, especially those with potent binding to non‐target tissues, is that the drug administered in a microdose may “sink” into the non‐target tissue thus producing non‐linear observations in the target.85 This issue could be partly addressed by performing full‐body PET in an attempt to identify such “sink” tissues, or by using Intra‐Target Microdosing (ITM, see below), to temporarily expose the target to the entire dose prior to entry into the general circulation.15, 33 An issue here is the balance between the speed of distribution of the biologic from plasma to target, with subsequent binding, within the tissue and the transit time of plasma carrying the biologic away from the tissue. Clearly, the slower the former, the less the benefit of ITM.
Intra‐Target Microdosing (ITM) (Table 5)
Table 5.
Potential applications of Intra‐Target Microdosing (ITM)
| Drug | Organ / Tissue | Biomarker |
|---|---|---|
| Nitrates, inotropes, adrenergic, muscarinic, PDE5 inhibitors, neutral endopeptidase (NEP) inhibitors, natriuretic peptides | Peripheral vascular | Vasodilation, vasoconstriction, cGMP spillover measurement |
| Anesthetics, analgesics (e.g., Nav1.7 inhibitors) | Peripheral organ / tissue | Anesthesia, analgesia |
| Triptans | Blood vessels | Analgesia, substance P and CGRP levels |
| Neuromuscular blocking agents | Skeletal muscles | Muscle relaxation/paralysis |
| Chemotherapy | Liver, kidney, brain, breast | Receptor binding (with PET imaging of radiolabeled drug) |
| Anticoagulants, antiplatelet | Blood | Coagulation parameters, platelet aggregation |
| Immune modulators, antihistamines | Blood | Cytokines, allergic symptoms |
| Hypoglycemics, sodium glucose cotransporter‐2 (SGLT‐2) inhibitors, diuretics | Kidney | Glucose levels, reabsorption in proximal tubule (by 18F‐FDG) |
| Antiarrhythmics | Heart | ECG |
| CNS stimulants and depressants (e.g., hypnotics, sedatives, anxiolytics), NMDA antagonists | CNS | Neuronal activity (e.g., Wada Test) |
Due to the brief, local pharmacological‐level exposure ITM can be used to detect PD effects in drug classes that allow collection of biomarkers in the time frame of seconds to minutes. PDE5, phosphodiesterase type 5; cGMP, cyclic guanosine monophosphate; CGRP, Calcitonin Gene‐Related Peptide; SGLT, Sodium‐glucose transport; ECG, electrocardiogram; NMDA, N‐methyl‐D‐aspartate; CNS, Central Nervous System. Adapted from Burt et al.15
In this novel exploratory drug development approach the test article is administered directly into the target such that only a small fraction of tissue within the body is initially exposed to the test article and for a limited amount of time.15 The most readily available direct administration route is intra‐arterial but intra‐muscular, intra‐thecal, or even topical deliveries are possible. The test article then returns systemically through the venous system and is diluted by approximately the proportion of the target mass to the full‐body mass. If the target is about 1/100 of the body mass (e.g., the hand) and the test article is administered at the NOAEL then the rest of the body will receive microdose exposure. The advantage of the approach is that target exposure may be sufficient to detect human‐specific acute PD effects relevant to safety and efficacy while limiting systemic exposure to safe subpharmacological levels. Target exposure post‐ITM differs from systemic administration (e.g., PO or IV) in that the therapeutic‐level concentrations are maintained only for a brief time (seconds to minutes) after administration. Nevertheless, considerable variation is possible in the exposure profile depending on the rate and duration of administration for a given total dose, as well as the amount of fluid administered. Imaging of the labeled drug in the target, measurement of drug concentrations in the vein draining the target, and use of in vivo and PBPK models, could all inform the translation of the ITM information to the intended therapeutic route of administration. In effect, the approach combines two contemporaneous investigations: The local pharmacological exposure with a systemic microdose exposure, and allows comparing the two with minimal variability to examine the validity of extrapolation.
In a proof‐of‐concept rodent study Burt et al. hypothesized that after administering a microdose of insulin into the femoral artery effects on ipsilateral leg muscles would be similar to those seen after systemic administration of a therapeutic dose, while sparing the rest of the body any systemic effects.15 18F‐FDG uptake into tissues was chosen as the primary biomarker. Glucose and insulin levels were secondary outcomes. FDG which responds to insulin in the same manner as glucose, is taken up into tissues within minutes. After administering insulin into the femoral artery, 18F‐FDG uptake into ipsilateral leg muscles, as measured by the slope of the standard Uptake Value (SUV) plot, was similar (95% confidence interval [CI], 20.024 to 0.029, P = 0.7895) to that after systemic full‐dose administration. Likewise, glucose levels in the ipsilateral femoral vein were reduced. At the same time, contralateral and systemic effects remained at the microdose, subpharmacological, baseline level, statistically significantly less than the ipsilateral effects (CI, 20.045 to 20.007, P = 0.0147). These observations established the two proof‐of‐concept objectives, the first, to demonstrate that local exposure after intra‐arterial administration of subpharmacological doses (measured on a systemic basis) can produce pharmacological effects similar to those after systemic full‐dose administration and, second, that such local administration exposes the rest of the body to subpharmacological, microdose levels.
Extreme environments
This is an entirely new area of application that we anticipate will grow in demand. Humans are venturing beyond the narrow confines of the biosphere. Extreme environments include space (exposure to microgravity, radiation, altered chronobiology), poles (low temperatures, altered chronobiology), high altitudes, and underwater (hypo/hyperbaric). With the altered physical properties of the environment comes the potential for altered physiology (e.g., bone density, muscle strength, blood flow, and chronobiology), pathology, pharmacology, and even storage and handling of pharmaceuticals all potentially leading to altered response to pharmacotherapy. This calls for dedicated and controlled testing of xenobiotic substances at the extreme environment. For all practical purposes such studies would be akin to FIH studies in terms of the novelty of the expected response and implied risks. And with another feature of such extreme environment being the lack of readily accessible comprehensive emergency services, the inherent safety of phase 0 limited exposures is clearly an advantage.
Individualized therapy phenotyping
There is considerable inter‐individual variability in the mechanisms that affect drug disposition, and specifically, drug metabolism (e.g., by CYP450 enzymes), and transporter activity, that could lead to undesirable therapeutic consequences such as underdosing, overdosing, and drug‐drug interactions (DDI).35 The ability to predict such variability prior to initiation of pharmacotherapy, and to do so in a safe manner by using a microdose probe drug, is especially important for drugs that exhibit a narrow therapeutic index, where the population is known to exhibit variability in metabolic phenotypes, and in vulnerable populations.35 It is also important where concomitant polypharmacy may potentiate the probe drug as in the case of ketoconazole and fluvoxamine that can induce up to 12‐fold potentiation of midazolam exposure by inhibition of CYP3A4.52 Midazolam PK has been shown to be linear in the dose range of 100 ng to 3 mg, allowing the administration of a microdose midazolam probe to accurately predict full dose metabolism by CYP3A4 without the risk of adverse effects.36, 86 This was demonstrated following both PO and IV administrations allowing, through deconvolution, assessment of presystemic intestinal vs. hepatic CYP3A4 activity.
Environmental toxins
Phase 0 approaches offer the potential to study environmental and anthropogenic toxins in a controlled and safe manner.39, 40 In fact, exposure to most environmental toxins is normally in the microdose range, and this fact was used by the FDA as an argument against the practicality of performing routine genetic toxicology testing for microdosing studies.2 However, long term exposures to substances that do not display acute toxicities may lead to adverse health effects. This is most notable in the case of chemical carcinogens but may also apply to heavy metals and radioactive substances. While some of these substances have been studied in animals in high doses few controlled data are available in humans, where natural exposure is usually to small doses of complex mixtures.39 In the first such study, a single dose of 14C‐dibenzo[def,p]chrysene (DBC, 29 ng, 5 nCi), a polycyclic aromatic hydrocarbon (PAH), identified as a priority chemical of carcinogenic concern, was administered as a microdose to three female and six male healthy volunteers.40 The single‐dose exposure, equivalent to 28% of average daily dietary intake, was assessed to be “minimal risk” and approved by the local ethics committee. AMS was used to determine PK parameters in plasma samples. Such data can be used to determine the extent of metabolism, the type of metabolism used to activate the pro‐carcinogen, as well as the detoxifying type. And just as in the previously mentioned Henderson et al. study,17 formation of DNA mono‐adducts in PBMCs can be detected following these minute doses as potential “surrogate end points” of carcinogenicity.
RECOMMENDATIONS
Human is the best model of human
Animal models are notoriously poor predictors of human pathophysiology and treatment response.87, 88 This is especially true when interactions with multiple targets (e.g., transporters, enzymes, carrier proteins, target‐mediated drug disposition) take place, or when unique human targets or systems are engaged (e.g., neurodegenerative and psychiatric disorders). An example is the lack of appropriate animal models to study the role of amyloid plaques in pathogenesis of Alzheimer's disease and the aforementioned use of PET‐microdose study to study 18F‐labelled amyloid imaging agents and establish proof of concept in humans.31
Strategic timelines (Figure 3)
Figure 3.
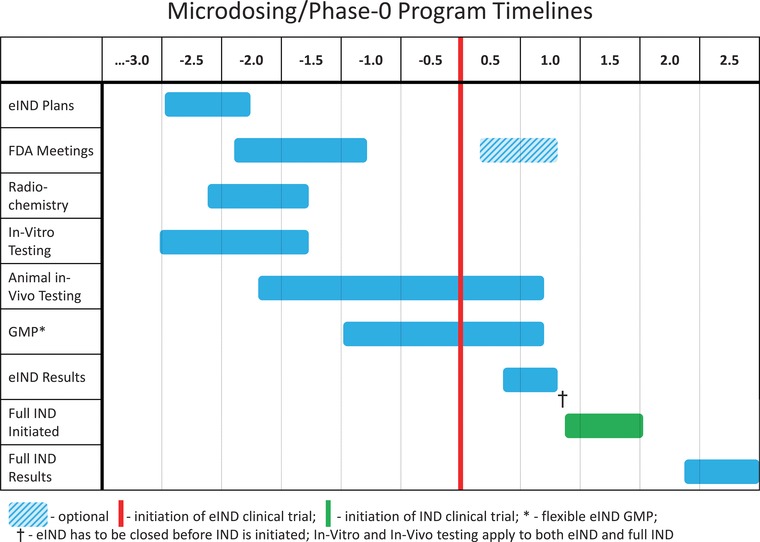
eIND Strategic Timelines. Timelines are indicated on a scale of years before and after initiation of eIND clinical trial (vertical red line). In vitro and in vivo testing in support of full IND and phase I testing may continue in parallel to the phase 0 program so that no delays are incurred should a “go” decision be taken.
Phase 0 approaches should be incorporated into the mainstream developmental process. It is important to start consideration of phase 0 approaches, especially if labelling is to be involved, as soon as possible, preferably 2‐2.5 years prior to anticipated entry into human testing. This period should include a pre‐IND type meeting with the regulatory authorities to discuss the goals of the proposed investigation, the specific human testing proposed, assessment of the expected risks, and determination of the amount and type of data that need to be submitted. This period should also anticipate the technical aspects of the eIND studies by initiating considerations of formulation and drug radiolabeling, and establishing the standards, capabilities, skills and expertise associated with the analytical methods.
Initially, DDI and vulnerable populations were not included as part of the framework of phase 0. However, finding information about DDI before phase I may play an important part in the decision to enter into full clinical development, especially if microdosing assessment of DDI is part of candidate selection among similarly performing preclinical candidates. In addition, there has been increasing pressure to obtain, as early as possible, evidence‐based data on pharmacotherapy for vulnerable populations, many of whom (e.g., frail elderly) are important consumers of healthcare.
Linearity and validity of extrapolation from limited exposure to full exposure
The potential for non‐linear mechanisms interfering with extrapolation of phase 0 results should be carefully examined prior to embarking on such approaches. Saturation of transporters, metabolism, protein binding, and receptor binding at anticipated pharmacologic doses should be studied using in vitro models and where possible with compounds from the same pharmacological class as reference compounds. It is important to note that non‐linearity by itself does not rule‐out limited‐exposure studies. As long as the nature of non‐linearity is correctly characterized it may allow accurate extrapolation of phase 0 study results to therapeutic dose levels.
Phase 0/phase I development program
In addition to the previously described phase 0/phase I adaptive study design34 there is the more general phase 0/phase I parallel program development. This means that the preclinical development, including animal testing, for the full IND submission for phase I testing continues in parallel to the phase 0 testing. The advantage of this approach is that the phase I development can be stopped at any point as soon as phase 0 testing indicates a “no‐go” decision while no time is lost in the phase I development if the results of the phase 0 testing indicate a “go” decision. In the latter case, the results of phase 0 (e.g., PK parameters, DDI, target exposure) can inform the design of the phase I program.
Government and organizational support
Regulators, professional organizations, industry, and academia should unite to provide formal guidance, incentives, and funding to promote research, education, and utilization of microdosing and other phase 0 approaches.
Collaboration
Drug developers should share not only data but also information on the decision‐making process, and the assumptions taken into consideration of drug development “go‐no‐go” decisions, while balancing collaboration benefits against confidentiality constraints. Specifically, the reasons for the under‐utilization of microdosing and other phase 0 approaches need to be understood and addressed. Maybe most important, all stakeholders should engage in continuous discussions on how to improve these methodologies and their applications. The multidisciplinary nature of this process should be embraced for its breakthrough collaborative potential, not available in each of the individual disciplines. Clinical pharmacologists, radiochemists, nuclear medicine and analytics experts, economists, clinical researchers, statisticians, and regulators, should not only gather together in task forces to tackle specific developmental challenges, but regularly work together to seek and identify new ones.
Culture
Drug development organizations should provide incentives to discover true failure early. Currently the bonus structure is sometimes based on advancing development forward. The implied aversion to failure may conflict with the main advantage of phase 0 approaches, i.e., the potential for early termination of development and reallocation of resources to the development of more promising candidates.89
Economics
An important element in the decision to utilize exploratory developmental approaches, the economic benefit, needs to be studied in more diverse developmental scenarios, applications, and details than previous reports.13, 90 To developers, the benefit will be the balance of the investment in resources vs. the savings in time and money when compared with full phase I programs. Of particular value to developers is the saving of time to meaningful developmental decisions as this could increase the patent‐life of back‐up compounds. To insurers and healthcare systems as a whole there may be an additional value in the earlier introduction of novel therapeutics that could reduce the burden of illness.
Ethics – risk/benefit assessment
The main ethical benefits of the exploratory approaches are in the reduction of risk in human testing, the early identification and rejection of ineffective or toxic compounds, thereby avoiding delays in development of successful back‐up compounds, and preventing unnecessary testing in humans and animals. The ethics of administering drugs to humans with no therapeutic intent was brought up in earlier reports as a challenge to phase 0 studies.91, 92, 93 However, such testing, no different from usual phase I testing in healthy volunteers where also there is no therapeutic intent, is justified by the scientific necessity and beneficence implied in a more efficient drug development process. Phase 0 approaches are also consistent with the 3Rs, reduction, refinement, and replacement, promoting ethical animal research.
Patient advocacy groups
These organizations are usually highly motivated to advance research, are especially concerned about safety of research participants, and are compelled by the potential for minimal exposure to experimental risk. They should be engaged early in the development process allowing time to become informed, involved in ethics discussion about their benefit/risk ratio, able to disseminate the information, and contribute to the recruitment process. They could also help advocating for such clinical research approaches with policy makers and potential funders.
An example of such productive collaboration is provided by the Dutch patient organization for congenital diseases (VSOP) that helped pave the way for the use of phase 0 trials in The Netherlands. This group was strongly aware of the need to conduct drug studies in children as well as the challenges, ethical, regulatory and practical, that are in the way of performing these studies. They understood and supported the concept of 14C‐labelled AMS microdosing. They supported the grant application with the Dutch Organization for Health Research and Innovation and provided a support letter, approved by the Ethics Board, to be given to parents at the time of the informed consent process.
SUMMARY
A number of drivers and developments suggest that microdosing and other phase 0 applications will experience increased utilization in the near‐to‐medium future. Increasing costs of drug development and ethical concerns about the risks of exposing humans and animals to novel chemical entities are important drivers in favor of these approaches, and can be expected only to increase in their relevance. An increasing body of research supports the validity of extrapolation from the limited drug exposure of phase 0 approaches to the full, therapeutic exposure, with modeling and simulations capable of extrapolating even non‐linear scenarios. An increasing number of applications and design options demonstrate the versatility and flexibility these approaches offer to drug developers including the study of PK, bioavailability, DDI, and mechanistic PD effects. PET microdosing allows study of target localization, PK and receptor binding and occupancy, while Intra‐Target Microdosing (ITM) allows study of local therapeutic‐level acute PD coupled with systemic microdose‐level exposure. Applications in vulnerable populations and extreme environments are attractive due to the unique risks of pharmacotherapy and increasing unmet healthcare needs. All phase 0 approaches depend on the validity of extrapolation from the limited‐exposure scenario to the full exposure of therapeutic intent, but in the final analysis the potential for controlled human data to reduce uncertainty about drug properties is bound to be a valuable addition to the drug development process.
Acknowledgments
K.Y. was supported in part by an appointment to the Research Participation Program at the Center for Drug Evaluation and Research, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the FDA. S.N.W. microdosing research is supported by a grant from the The Netherlands Organisation for Health Research and Development (project number 13202007).
Some of the content contained in this manuscript was presented at the symposium “Microdosing: Addressing the Challenges to Realize the Potential” during the 2013 American College of Clinical Pharmacology (ACCP) Annual Meeting, September 2013, Bethesda, MD, USA, and at the workshop “Microdosing in Drug Development: A Range of Clinical Trial Designs and Applications” during the 2015 ACCP Annual Meeting, September 2015, San Francisco, California, USA.
Conflict of Interest/Disclosure
T.B. holds patent for Intra‐Target Microdosing (ITM); K.Y., G.L., L.T.V., S.N.W., C.J., Y.S., and M.R. have declared no conflict of interest.
The contents of this manuscript reflect the views of the authors and should not be construed to represent the FDA's, or the other organizations’ views or policies. No official support or endorsement by the FDA or other organizations is intended or should be inferred. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the FDA or other organizations.
- 1. FDA . Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. http://wwwfdagov/oc/initiatives/criticalpath/whitepaperhtml, (2004).
- 2. FDA . Guidance for Industry, Investigators, and Reviewers Exploratory IND Studies. http://wwwfdagov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM078933pdf, (2006).
- 3. ICH . Guidance on Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals M3(R2). In: International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use 8–16 (ICH Secretariat, Geneve, Switzerland, 2009). [Google Scholar]
- 4. Lappin, G. & Garner, R.C. Big physics, small doses: the use of AMS and PET in human microdosing of development drugs. Nat. Rev. Drug Discov. 2, 233–240 (2003). [DOI] [PubMed] [Google Scholar]
- 5. Bergstrom, M. , Grahnen, A. & Langstrom, B. Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development. Eur. J. Clin. Pharmacol. 59, 357–366 (2003). [DOI] [PubMed] [Google Scholar]
- 6. Rowland, M. Microdosing and the 3Rs. In National Centre for the Replacement, Refinement & Reduction of animals in Research.
- 7. Suntharalingam, G. et al Cytokine storm in a phase I trial of the anti‐CD28 monoclonal antibody TGN1412. New Engl. J. Med. 355, 1018–1028 (2006). [DOI] [PubMed] [Google Scholar]
- 8. Yamane, N. , Tozuka, Z. , Kusama, M. , Maeda, K. , Ikeda, T. & Sugiyama, Y. Clinical relevance of liquid chromatography tandem mass spectrometry as an analytical method in microdose clinical studies. Pharm. Res. 28, 1963–1972 (2011). [DOI] [PubMed] [Google Scholar]
- 9. Bauer, M. , Wagner, C.C. & Langer, O. Microdosing studies in humans: the role of positron emission tomography. Drugs in R&D 9, 73–81 (2008). [DOI] [PubMed] [Google Scholar]
- 10. Tozuka, Z. et al Microdose study of 14C‐acetaminophen with accelerator mass spectrometry to examine pharmacokinetics of parent drug and metabolites in healthy subjects. Clin. Pharmacol. Ther. 88, 824–830 (2010). [DOI] [PubMed] [Google Scholar]
- 11. Bijleveld, Y. et al A simple quantitative method analysing amikacin, gentamicin, and vancomycin levels in human newborn plasma using ion‐pair liquid chromatography/tandem mass spectrometry and its applicability to a clinical study. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 951–952, 110–118 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Ieiri, I. et al Pharmacogenomic/pharmacokinetic assessment of a four‐probe cocktail for CYPs and OATPs following oral microdosing. Int. J. Clin. Pharmacol. Ther. 50, 689–700 (2012). [DOI] [PubMed] [Google Scholar]
- 13. Yamane, N. , Igarashi, A. , Kusama, M. , Maeda, K. , Ikeda, T. & Sugiyama, Y. Cost‐Effectiveness Analysis of Microdose Clinical Trials in Drug Development. Drug Metab. Pharmacokinet. (2012). [DOI] [PubMed] [Google Scholar]
- 14. Morgan, P. et al Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving phase II survival. Drug Discov. Today 17, 419–424 (2012). [DOI] [PubMed] [Google Scholar]
- 15. Burt, T. et al Intra‐Arterial Microdosing (IAM): A Novel Drug Development Approach, Proof‐of‐Concept PET Study in Rats. J. Nucl. Med. (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vuong, L.T. et al Use of accelerator mass spectrometry to measure the pharmacokinetics and peripheral blood mononuclear cell concentrations of zidovudine. J. Pharm. Sci. 97, 2833–2843 (2008). [DOI] [PubMed] [Google Scholar]
- 17. Henderson, P.T. et al A microdosing approach for characterizing formation and repair of carboplatin‐DNA monoadducts and chemoresistance. Int. J. Cancer 129, 1425–1434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bertino, J.S., Jr. , Greenberg, H.E. & Reed, M.D. American college of clinical pharmacology position statement on the use of microdosing in the drug development process. J. Clin. Pharmacol. 47, 418–422 (2007). [DOI] [PubMed] [Google Scholar]
- 19. Lappin, G. et al Use of microdosing to predict pharmacokinetics at the therapeutic dose: Experience with 5 drugs. Clin. Pharmacol. Ther. 80, 203–215 (2006). [DOI] [PubMed] [Google Scholar]
- 20. Madan, A. et al A Pharmacokinetic Evaluation of Five H1 Antagonists After an Oral and Intravenous Microdose to Human Subjects. Br. J. Clin. Pharmacol. 67, 288–298 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Minamide, Y. , Osawa, Y. , Nishida, H. , Igarashi, H. & Kudoh, S. A highly sensitive LC‐MS/MS method capable of simultaneously quantitating celiprolol and atenolol in human plasma for a cassette cold‐microdosing study. J. Sep. Sci. 34, 1590–1598 (2011). [DOI] [PubMed] [Google Scholar]
- 22. Maeda, K. & Sugiyama, Y. Novel stratergies for microdose studies using non‐radiolabeled compounds. Adv. Drug. Deliv. Rev. 63, 532–538 (2011). [DOI] [PubMed] [Google Scholar]
- 23. Bauer, M. et al A positron emission tomography microdosing study with a potential antiamyloid drug in healthy volunteers and patients with Alzheimer's disease. Clin. Pharmacol. Ther. 80, 216–227 (2006). [DOI] [PubMed] [Google Scholar]
- 24. Christian, B.T. et al Evaluation of cerebral pharmacokinetics of the novel antidepressant drug, BMS‐181101, by positron emission tomography. J. Pharmacol. Exp. Ther. 279, 325–331 (1996). [PubMed] [Google Scholar]
- 25. van der Veldt, A.A. et al Toward prediction of efficacy of chemotherapy: a proof of concept study in lung cancer patients using [(1)(1)C]docetaxel and positron emission tomography. Clin Cancer Res 19, 4163–4173 (2013). [DOI] [PubMed] [Google Scholar]
- 26. Mooij, M.G. et al Pediatric microdose study of [(14)C]paracetamol to study drug metabolism using accelerated mass spectrometry: proof of concept. Clin. Pharmacokinet. 53, 1045–1051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vuong, L.T. , Blood, A.B. , Vogel, J.S. , Anderson, M.E. & Goldstein, B. Applications of accelerator MS in pediatric drug evaluation. Bioanalysis 4, 1871–1882 (2012). [DOI] [PubMed] [Google Scholar]
- 28. Garner, C.R. et al Observational infant exploratory [(14)C]‐paracetamol pharmacokinetic microdose/therapeutic dose study with accelerator mass spectrometry bioanalysis. Br. J. Clin. Pharmacol. 80, 157–167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Turner, M.A. et al Pediatric microdose and microtracer studies using (14) C in Europe. Clin. Pharmacol. Ther. 98, 234–237 (2015). [DOI] [PubMed] [Google Scholar]
- 30. Lappin, G. , Noveck, R. & Burt, T. Microdosing and drug development: past, present and future. Expert Opin. Drug Metab. Toxicol. 9, 817–834 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Carpenter, A.P., Jr. , Pontecorvo, M.J. , Hefti, F.F. & Skovronsky, D.M. The use of the exploratory IND in the evaluation and development of 18F‐PET radiopharmaceuticals for amyloid imaging in the brain: a review of one company's experience. The quarterly journal of nuclear medicine and molecular imaging : official publication of the Italian Association of Nuclear Medicine (AIMN) [and] the International Association of Radiopharmacology (IAR), [and] Section of the So 53, 387–393 (2009). [PubMed] [Google Scholar]
- 32. Rose, R.H. , Neuhoff, S. , Abduljalil, K. , Chetty, M. , Rostami‐Hodjegan, A. & Jamei, M. Application of a Physiologically Based Pharmacokinetic Model to Predict OATP1B1‐Related Variability in Pharmacodynamics of Rosuvastatin. CPT Pharmacom. Syst. Pharmacol. 3, e124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heuveling, D.A. et al Phase 0 Microdosing PET Study Using the Human Mini Antibody F16SIP in Head and Neck Cancer Patients. J. Nucl. Med. (2013). [DOI] [PubMed] [Google Scholar]
- 34. Vlaming, M. et al Microdosing of a Carbon‐14 Labeled Protein in Healthy Volunteers Accurately Predicts Its Pharmacokinetics at Therapeutic Dosages. Clin. Pharmacol. Ther. 98, 196–204 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Hohmann, N. , Haefeli, W.E. & Mikus, G. Use of Microdose Phenotyping to Individualise Dosing of Patients. Clin. Pharmacokinet. 54, 893–900 (2015). [DOI] [PubMed] [Google Scholar]
- 36. Hohmann, N. , Kocheise, F. , Carls, A. , Burhenne, J. , Haefeli, W.E. & Mikus, G. Midazolam microdose to determine systemic and pre‐systemic metabolic CYP3A activity in humans. Br. J. Clin. Pharmacol. 79, 278–285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujita, K. et al A clinical pharmacokinetic microdosing study of docetaxel with Japanese patients with cancer. Cancer. Chemother. Pharmacol. 76, 793–801 (2015). [DOI] [PubMed] [Google Scholar]
- 38. Ieiri, I. et al Microdosing Clinical Study: Pharmacokinetic, Pharmacogenomic (SLCO2B1), and Interaction (Grapefruit Juice) Profiles of Celiprolol Following the Oral Microdose and Therapeutic Dose. J. Clin. Pharmacol. (2011). [DOI] [PubMed] [Google Scholar]
- 39. Kensler, T.W. & Groopman, J.D. Is it time to advance the chemoprevention of environmental carcinogenesis with microdosing trials? Cancer Prev Res (Phila Pa) 2, 1003–1007 (2009). [DOI] [PubMed] [Google Scholar]
- 40. Madeen, E. et al Human in vivo Pharmacokinetics of [C]Dibenzo[def,p]chrysene by Accelerator Mass Spectrometry Following Oral Microdosing. Chem. Res. Toxicol. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rowland, M. & Benet, L.Z. Lead PK commentary: Predicting human pharmacokinetics. J. Pharm. Sci. (2011). [DOI] [PubMed] [Google Scholar]
- 42. Paci, A. et al Review of therapeutic drug monitoring of anticancer drugs part 1–cytotoxics. Eur. J. Cancer. 50, 2010–2019 (2014). [DOI] [PubMed] [Google Scholar]
- 43. Lewis, D.F. Early human studies of investigational agents: dose or microdose. Br. J. Clin. Pharmacol. 67, 277–279 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lappin, G. et al Comparative pharmacokinetics between a microdose and therapeutic dose for clarithromycin, sumatriptan, propafenone, paracetamol (acetaminophen), and phenobarbital in human volunteers. Eur. J. Pharm. Sci. 43, 141–150 (2011). [DOI] [PubMed] [Google Scholar]
- 45. Sugiyama, Y. Effective use of microdosing and Positron Emission Tomography (PET) studies on new drug discovery and development. Drug Metab. Pharmacokinet. 24, 127–129 (2009). [DOI] [PubMed] [Google Scholar]
- 46. Yamashita, S. et al An Assessment of the Oral Bioavailability of Three Ca‐Channel Blockers Using a Cassette‐Microdose Study: A New Strategy for Streamlining Oral Drug Development. J. Pharm. Sci. 104, 3154–3161 (2015). [DOI] [PubMed] [Google Scholar]
- 47. Garner, R.C. & Lappin, G. The phase 0 microdosing concept. Br. J. Clin. Pharmacol. 61, 367–370 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bosgra, S. , Vlaming, M.L. & Vaes, W.H. To Apply Microdosing or Not? Recommendations to Single Out Compounds with Non‐Linear Pharmacokinetics. Clin. Pharmacokinet. (2015). [DOI] [PubMed] [Google Scholar]
- 49. Maeda, K. et al Nonlinear pharmacokinetics of oral quinidine and verapamil in healthy subjects: a clinical microdosing study. Clin. Pharmacol. Ther. 90, 263–270 (2011). [DOI] [PubMed] [Google Scholar]
- 50. Palleria, C. et al Pharmacokinetic drug‐drug interaction and their implication in clinical management. Journal of research in medical sciences : the official journal of Isfahan University of Medical Sciences 18, 601–610 (2013). [PMC free article] [PubMed] [Google Scholar]
- 51. Rowland, M. , Peck, C. & Tucker, G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu. Rev. Pharmacol. Toxicol. 51, 45–73 (2011). [DOI] [PubMed] [Google Scholar]
- 52. Croft, M. , Keely, B. , Morris, I. , Tann, L. & Lappin, G. Predicting Drug Candidate Victims of Drug‐Drug Interactions, using Microdosing. Clin. Pharmacokinet. 51, 237–246 (2012). [DOI] [PubMed] [Google Scholar]
- 53. Kusuhara, H. et al Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin. Pharmacol. Ther. 89, 837–844 (2011). [DOI] [PubMed] [Google Scholar]
- 54. Maeda, K. et al Identification of the rate‐determining process in the hepatic clearance of atorvastatin in a clinical cassette microdosing study. Clin. Pharmacol. Ther. 90, 575–581 (2011). [DOI] [PubMed] [Google Scholar]
- 55. Garner, R.C. The role of DNA adducts in chemical carcinogenesis. Mutat. Res. 402, 67–75 (1998). [DOI] [PubMed] [Google Scholar]
- 56. Turteltaub, K.W. et al Accelerator mass spectrometry in biomedical dosimetry: relationship between low‐level exposure and covalent binding of heterocyclic amine carcinogens to DNA. Proc. Natl. Acad. Sci. U. S. A. 87, 5288–5292 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lightfoot, T.J. , Coxhead, J.M. , Cupid, B.C. , Nicholson, S. & Garner, R.C. Analysis of DNA adducts by accelerator mass spectrometry in human breast tissue after administration of 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine and benzo[a]pyrene. Mutat. Res. 472, 119–127 (2000). [DOI] [PubMed] [Google Scholar]
- 58. Hah, S.S. , Sumbad, R.A. , de Vere White, R.W. , Turteltaub, K.W. & Henderson, P.T. Characterization of oxaliplatin‐DNA adduct formation in DNA and differentiation of cancer cell drug sensitivity at microdose concentrations. Chem. Res. Toxicol. 20, 1745–1751 (2007). [DOI] [PubMed] [Google Scholar]
- 59. Varga, Z.V. , Ferdinandy, P. , Liaudet, L. & Pacher, P. Drug‐induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart. Circ. Physiol. 309, H1453–H1467 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Madonna, R. et al Improving the preclinical models for the study of chemotherapy‐induced cardiotoxicity: a Position Paper of the Italian Working Group on Drug Cardiotoxicity and Cardioprotection. Heart Failure Rev. 20, 621–631 (2015). [DOI] [PubMed] [Google Scholar]
- 61. Santini‐Oliveira, M. & Grinsztejn, B. Adverse drug reactions associated with antiretroviral therapy during pregnancy. Expert Opinion Drug Saf. 13, 1623–1652 (2014). [DOI] [PubMed] [Google Scholar]
- 62. Willmann, J.K. , van Bruggen, N. , Dinkelborg, L.M. & Gambhir, S.S. Molecular imaging in drug development. Nat. Rev. Drug Discov. 7, 591–607 (2008). [DOI] [PubMed] [Google Scholar]
- 63. Cunha, L. , Szigeti, K. , Mathe, D. & Metello, L.F. The role of molecular imaging in modern drug development. Drug Discov. Today, (2014). [DOI] [PubMed] [Google Scholar]
- 64. Bench, C.J. et al The time course of binding to striatal dopamine D2 receptors by the neuroleptic ziprasidone (CP‐88,059‐01) determined by positron emission tomography. Psychopharmacology 124, 141–147 (1996). [DOI] [PubMed] [Google Scholar]
- 65. Wagner, C.C. & Langer, O. Approaches using molecular imaging technology – use of PET in clinical microdose studies. Adv. Drug. Deliv. Rev. 63, 539–546 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Muzi, M. et al Imaging of cyclosporine inhibition of P‐glycoprotein activity using 11C‐verapamil in the brain: studies of healthy humans. J. Nucl. Med. 50, 1267–1275 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kalvass, J.C. et al Why clinical modulation of efflux transport at the human blood‐brain barrier is unlikely: the ITC evidence‐based position. Clin. Pharmacol. Ther. 94, 80–94 (2013). [DOI] [PubMed] [Google Scholar]
- 68. Kusuhara, H. Imaging in the study of membrane transporters. Clin. Pharmacol. Ther. 94, 33–36 (2013). [DOI] [PubMed] [Google Scholar]
- 69. Shitara, Y. , Horie, T. & Sugiyama, Y. Transporters as a determinant of drug clearance and tissue distribution. Eur. J. Pharm. Sci. 27, 425–446 (2006). [DOI] [PubMed] [Google Scholar]
- 70. Roberts, D.J. & Hall, R.I. Drug absorption, distribution, metabolism and excretion considerations in critically ill adults. Expert Opin. Drug Metab. Toxicol. 9, 1067–1084 (2013). [DOI] [PubMed] [Google Scholar]
- 71. Roth‐Cline, M. & Nelson, R.M. Ethical Considerations in Conducting Pediatric and Neonatal Research in Clinical Pharmacology. Curr. Pharm. Des. 21, 5619–5635 (2015). [DOI] [PubMed] [Google Scholar]
- 72. Dunne, J. et al Extrapolation of adult data and other data in pediatric drug‐development programs. Pediatrics 128, e1242–e1249 (2011). [DOI] [PubMed] [Google Scholar]
- 73. Bellis, J.R. et al Adverse drug reactions and off‐label and unlicensed medicines in children: a nested case?control study of inpatients in a pediatric hospital. BMC Med. 11, 238 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kimland, E. & Odlind, V. Off‐label drug use in pediatric patients. Clin. Pharmacol. Ther. 91, 796–801 (2012). [DOI] [PubMed] [Google Scholar]
- 75. Roth‐Cline, M. & Nelson, R.M. Microdosing Studies in Children: A US Regulatory Perspective. Clin. Pharmacol. Ther. 98, 232–233 (2015). [DOI] [PubMed] [Google Scholar]
- 76. European Union . Ethical considerations for clinical trials on medicinal products conducted with the paediatric population. European journal of health law 15, 223–250 (2008). [DOI] [PubMed] [Google Scholar]
- 77. Howie, S.R. Blood sample volumes in child health research: review of safe limits. Bulletin of the World Health Organization 89, 46–53 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. de Wildt, S.N. , Ito, S. & Koren, G. Challenges for drug studies in children: CYP3A phenotyping as example. Drug Discov. Today 14, 6–15 (2009). [DOI] [PubMed] [Google Scholar]
- 79. Zhao, P. , Rowland, M. & Huang, S.M. Best practice in the use of physiologically based pharmacokinetic modeling and simulation to address clinical pharmacology regulatory questions. Clin. Pharmacol. Ther. 92, 17–20 (2012). [DOI] [PubMed] [Google Scholar]
- 80. Wagner, C. et al Predicting the effect of cytochrome P450 inhibitors on substrate drugs: analysis of physiologically based pharmacokinetic modeling submissions to the US Food and Drug Administration. Clin. Pharmacokinet. 54, 117–127 (2015). [DOI] [PubMed] [Google Scholar]
- 81. Yoshida, K. , Maeda, K. & Sugiyama, Y. Hepatic and intestinal drug transporters: prediction of pharmacokinetic effects caused by drug‐drug interactions and genetic polymorphisms. Annu. Rev. Pharmacol. Toxicol. 53, 581–612 (2013). [DOI] [PubMed] [Google Scholar]
- 82. Watanabe, T. , Kusuhara, H. & Sugiyama, Y. Application of physiologically based pharmacokinetic modeling and clearance concept to drugs showing transporter‐mediated distribution and clearance in humans. J. Pharmacokinet. Pharmacodyn. 37, 575–590 (2010). [DOI] [PubMed] [Google Scholar]
- 83. Zhao, L. , Shang, E.Y. & Sahajwalla, C.G. Application of pharmacokinetics‐pharmacodynamics/clinical response modeling and simulation for biologics drug development. J. Pharm. Sci. 101, 4367–4382 (2012). [DOI] [PubMed] [Google Scholar]
- 84. Rowland, M. Microdosing of protein drugs. Clin Pharmacol Ther 99, 150–152 (2016). [DOI] [PubMed] [Google Scholar]
- 85. Dijkers, E.C. et al Biodistribution of 89Zr‐trastuzumab and PET imaging of HER2‐positive lesions in patients with metastatic breast cancer. Clin. Pharmacol. Ther. 87, 586–592 (2010). [DOI] [PubMed] [Google Scholar]
- 86. Halama, B. , Hohmann, N. , Burhenne, J. , Weiss, J. , Mikus, G. & Haefeli, W.E. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin. Pharmacol. Ther. 93, 564–571 (2013). [DOI] [PubMed] [Google Scholar]
- 87. IOM . Improving the Utility and Translation of Animal Models for Nervous System Disorders: Workshop Summary (The National Academies Press, 2013). [PubMed] [Google Scholar]
- 88. van der Worp, H.B. et al Can animal models of disease reliably inform human studies? PLoS medicine 7, e1000245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cook, D. et al Lessons learned from the fate of AstraZeneca's drug pipeline: a five‐dimensional framework. Nat. Rev. Drug Discov. 13, 419–431 (2014). [DOI] [PubMed] [Google Scholar]
- 90. Wilding, I.R. & Bell, J.A. Improved early clinical development through human microdosing studies. Drug Discov. Today 10, 890–894 (2005). [DOI] [PubMed] [Google Scholar]
- 91. Kinders, R. et al Phase 0 clinical trials in cancer drug development: from FDA guidance to clinical practice. Mol. Interv. 7, 325–334 (2007). [DOI] [PubMed] [Google Scholar]
- 92. Kimmelman, J. Ethics at phase 0: clarifying the issues. J. Law. Med. Ethics. 35, 727–733, 514 (2007). [DOI] [PubMed] [Google Scholar]
- 93. Kurihara, C. Ethical, legal, and social implications (ELSI) of microdose clinical trials. Adv. Drug. Deliv. Rev. 63, 503–510 (2011). [DOI] [PubMed] [Google Scholar]