

Abstract
Although gene-to-gene analyses identified genetic alterations such as APC, KRAS and TP53 mutations in colon adenomas, it is largely unknown whether there are any others in them. Mutational profiling of high-grade colon adenoma (HGCA) that just precedes colon carcinoma might identify not only novel adenoma-specific genes but also critical genes for its progression to carcinoma. For this, we performed whole-exome sequencing (WES) of 12 HGCAs and identified 11 non-hypermutated and one hypermutated (POLE-mutated) cases. We identified 22 genes including APC, KRAS, TP53, GNAS, NRAS, SMAD4, ARID2, and PIK3CA with non-silent mutations in the cancer Census Genes. Bi-allelic and mono-allelic APC alterations were found in nine and one HGCAs, respectively, while the other two harbored wild-type APC. Five HGCAs harbored either mono-allelic (four HGCAs) or bi-allelic (one HGCA) SMAD4 mutation or 18q loss that had been known as early carcinoma-specific changes. We identified MTOR, ACVR1B, GNAQ, ATM, CNOT1, EP300, ARID2, RET and MAP2K4 mutations for the first time in colon adenomas. Our WES data is largely matched with the earlier ‘adenoma-carcinoma model’ (APC, KRAS, NRAS and GNAS mutations), but there are newly identified SMAD4, MTOR, ACVR1B, GNAQ, ATM, CNOT1, EP300, ARID2, RET and MAP2K4 mutations in this study. Our findings provide resource for understanding colon premalignant lesions and for identifying genomic clues for differential diagnosis and therapy options for colon adenomas and carcinomas.
Keywords: colon, adenoma, high grade adenoma, mutation, whole-exome sequencing
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer in males, and the second most common in females worldwide [1]. Although there have been advances in the treatment of CRC and the mortality rate has declined in the past few decades, it remains the fourth most common cause of cancer-related deaths worldwide [2]. The multistep model in the adenoma-to-carcinoma sequence is well recognized in CRC development [3]. According to this model, most CRCs are considered to arise from pre-existing adenomas. Microscopically, conventional colorectal adenomas are classified on the basis of their architectural phenotype as either tubular or villous or tubulovillous type. According to the cellular atypia, they are classified as either low- or high-grade colon adenoma (HGCA) [4]. APC, KRAS and β-catenin mutations are considered key events in adenoma development while PIK3CA and TP53 mutations are considered key events in the progression to invasive CRC [5–7]. SMAD4 mutation is rarely detected in colon adenomas but occurs in intramucosal carcinoma and more commonly in invasive carcinomas with metastases [8, 9].
All of these mutations were first identified by conventional gene-to-gene analyses [10, 11]. With advances in sequencing technology (next-generation sequencing, NGS), a comprehensive molecular characterization of CRC has been studied using whole-exome (WES) and/or whole-genome sequencing (WGS) that allows for the interrogation of thousands of variants from multiple genes within a given tumor sample at the same time [12]. Although mutational landscapes of CRC have been reported several times, the genomic data of colon adenomas, especially those generated by high-throughput genome profiling technologies such as WES, is scarce. An earlier study analyzed a case of synchronous colon adenoma and CRC by WES [13]. Another study analyzed 82 nucleotide positions of 14 genes in colon adenomas with various types and grades by targeted sequencing [9]. Due to the small case number or narrow extent of genes analyzed in these reports, they did not reveal a general mutational landscape of colon adenomas. In this study, by using NGS-based WES for 12 HGCAs, we attempted to identify the mutational profiles of HGCA and found several mutations that had not been identified in colon adenomas in previous studies.
RESULTS
Whole-exome sequencing and somatic variants
To obtain the mutational profile of colorectal adenomas, we performed WES for the genome pairs of 12 HGCAs and their matched normals. Mean coverage of the sequencing depth was 98.0X (range: 30.1-149.7X) for normal samples and 115.9X (range: 90.8-150.6X) for HGCA samples (Supplementary Table 1). One HGCA (HGCA12) was found to be a hypermutated adenoma that harbored an exceptionally high incidence of somatic mutations (883 non-synonymous single nucleotide variants (SNVs) and 12 indels). It had a somatic mutation (p.E491K) in polymerase ε (POLE) gene that encodes the catalytic and proofreading subunits of the DNA polymerase ε [12]. Excluding this hypermutated HGCA, a total of 772 somatic non-silent mutations (752 non-synonymous SNVs and 20 indels) (average: 70.2, range: 36-102) were identified from the other 11 HGCAs (Figure 1A, Supplementary Table 2 and Supplementary Table 3). The C: G->T: A transitions were the most significant changes in the HGCA samples (Figure 1B and Supplementary Table 2), which was consistent with a previous report that analyzed a colon adenoma [13]. Based on the notion that DNA methylation frequently occurs within the context of CpG sites, the enrichment of C: G->T: A transitions in 5′-CpG-3′ dinucleotides might be related to extensive DNA methylation at 5′-CpG-3′ sites in CRC tumorigenesis [14, 15].
Figure 1. The mutational features of 11 non-hypermutated high-grade colon adenomas.
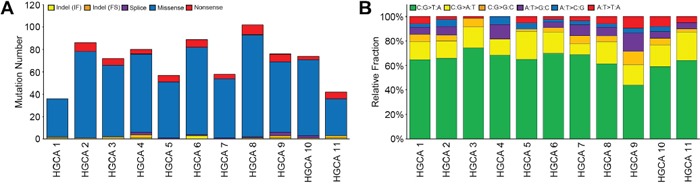
A. The numbers of non-silent somatic mutations are shown with the five functional categories indicated in the insets. B. Non-silent somatic mutations are classified according to both base context and sequence changes. Relative fraction of sequence-based mutation categories (y-axis) for each case is shown. A hypermutated HGCA (HGCA 12) was not shown in this figure.
To address whether the mutations found in our study were causally implicated in tumor development, we looked up the cancer Gene Census [16] and the Cancer Driver Database [17] with the CHASM [18] analysis. In this study, mutations that were significantly predicted as drivers (FDR < 0.2) in the CHASM analysis and overlapped the driver databases (the cancer Gene Census or the Cancer Driver Database) were considered potential driver mutations. Under this criterion, 17 genes were considered the candidate driver genes (APC, KRAS, SMAD4, GNAS, RET, NRAS, MTOR, ACVR1B, GNAQ, ATM, TP53, PIK3CA, ERBB2, TRRAP, MAP2K4, MAP3K4 and CNOT1). In addition, six genes with truncating mutations overlapped the driver databases were also included (APC, ARID2, ERBB4, TCF7L2, AMER1 and EP300). Overall, we defined 22 genes as candidate drivers (Table 1 and Figure 2). Mutations identified with WES were validated by either digital-polymerase chain reaction (PCR) or Sanger sequencing (Supplementary Figure 1). Nine genes with mutations identified in this study overlapped the CRC top 20 genes in the COSMIC database ( http://cancer.sanger.ac.uk/cosmic) (TP53, TCF7L2, PIK3CA, AMER1, APC, KRAS, SMAD4, GNAS and NRAS). Of note, most of the driver genes (16/22 genes; APC, KRAS, SMAD4, MTOR, TCF7L2, AMER1, TP53, GNAS, NRAS, ARID2, GNAQ, ATM, PIK3CA, ERBB2, EP300 and RET) were identified to harbor the same mutations at variant levels in the COSMIC mutation database as well. Among all of the mutated genes, 13 genes were recurrently mutated in more than three HGCA cases: APC (n=10), KRAS (n=7), TTN (n=7), OBSCN (n=5), CSMD1 (n=4), MXRA5 (n=4), SMAD4 (n=3), FAT2 (n=3), CELSR2 (n=3), CSMD2 (n=3), MAP2 (n=3), ANK2 (n=3) and HMCN1 (n=3) (Supplementary Table 3). As these genes besides APC, KRAS and SMAD4 are listed in neither of the cancer Census Genes nor the COSMIC CRC genes, they might be passenger mutations or HGCA-specific mutations that might not play an important role in HGCA progression to CRC.
Table 1. Non-silent somatic mutations identified in the cancer Census Genes.
| Gene | Case | Exonic function | cDNA Change | Amino acid |
|---|---|---|---|---|
| APC | HGCA 1 | frameshift | c.4385_4386delAG | p.K1462fs |
| HGCA 1 | missense | c.1958G>A | p.R653K | |
| HGCA 2 | nonsense | c.1660C>T | p.R554X | |
| HGCA 2 | nonsense | c.4189G>T | p.E1397X | |
| HGCA 3 | frameshift | c.4385_4386delAG | p.K1462fs | |
| HGCA 5 | nonsense | c.847C>T | p.R283X | |
| HGCA 5 | nonsense | c.4067C>A | p.S1356X | |
| HGCA 6 | nonsense | c.4348C>T | p.R1450X | |
| HGCA 6 | nonsense | c.3340C>T | p.R1114X | |
| HGCA 7 | nonsense | c.4222G>T | p.E1408X | |
| HGCA 7 | nonsense | c.3340C>T | p.R1114X | |
| HGCA 8 | nonsense | c.1690C>T | p.R564X | |
| HGCA 9 | nonsense | c.3093T>A | p.Y1031X | |
| HGCA 9 | nonsense | c.4348C>T | p.R1450X | |
| HGCA 11 | nonsense | c.3871C>T | p.Q1291X | |
| HGCA 11 | nonsense | c.2626C>T | p.R876X | |
| HGCA 12 | nonsense | c.694C>T | p.R232X | |
| HGCA12 | nonsense | c.2413C>T | p.R805X | |
| KRAS | HGCA 2 | missense | c.35G>T | p.G12V |
| HGCA 3 | missense | c.35G>A | p.G12D | |
| HGCA 5 | missense | c.35G>T | p.G12V | |
| HGCA 7 | missense | c.35G>A | p.G12D | |
| HGCA 8 | missense | c.34G>T | p.G12C | |
| HGCA 9 | missense | c.34G>A | p.G12S | |
| HGCA 10 | missense | c.38G>A | p.G13D | |
| SMAD4 | HGCA 1 | missense | c.1082G>A | p.R361H |
| HGCA 6 | missense | c.1212C>A | p.D404E | |
| HGCA 10 | missense | c.1156G>A | p.G386S | |
| ERBB4 | HGCA 4 | missense | c.1753G>T | p.D585Y |
| HGCA 9 | nonsense | c.2224G>T | p.E742X | |
| TCF7L2 | HGCA 5 | nonsense | c.70G>T | p.E24X |
| HGCA 11 | frameshift | c.463delA | p.K155fs | |
| AMER1 | HGCA 6 | nonsense | c.1072C>T | p.R358X |
| HGCA 9 | nonsense | c.1240C>T | p.Q414X | |
| TP53 | HGCA 9 | missense | c.524G>A | p.R175H |
| HGCA 11 | missense | c.747G>C | p.R249S | |
| GNAS | HGCA 2 | missense | c.601C>T | p.R201C |
| ARID2 | HGCA 2 | nonsense | c.3817C>T | p.R1273X |
| RET | HGCA 3 | missense | c.1267G>A | p.G423R |
| MTOR | HGCA 4 | missense | c.6644C>T | p.S2215F |
| NRAS | HGCA 4 | missense | c.183A>C | p.Q61H |
| ACVR1B | HGCA 4 | missense | c.101G>A | p.C34Y |
| GNAQ | HGCA 7 | missense | c.286A>T | p.T96S |
| ATM | HGCA 8 | missense | c.1010G>A | p.R337H |
| PIK3CA | HGCA 10 | missense | c.3140A>G | p.H1047R |
| ERBB2 | HGCA 12 | missense | c.2524G>A | p.V842I |
| TRRAP | HGCA 12 | missense | c.10751T>A | p.V3584E |
| MAP2K4 | HGCA 12 | missense | c.881G>A | p.G294E |
| MAP3K4 | HGCA 12 | missense | c.4577C>T | p.A1526V |
| CNOT1 | HGCA 12 | missense | c.7048G>A | p.E2350K |
| EP300 | HGCA12 | nonsense | c.3934C>T | p.R1312X |
HGCA: high-grade colon adenoma
Figure 2. Somatic mutations and copy number alterations of tumor-related genes in high-grade colon adenomas.
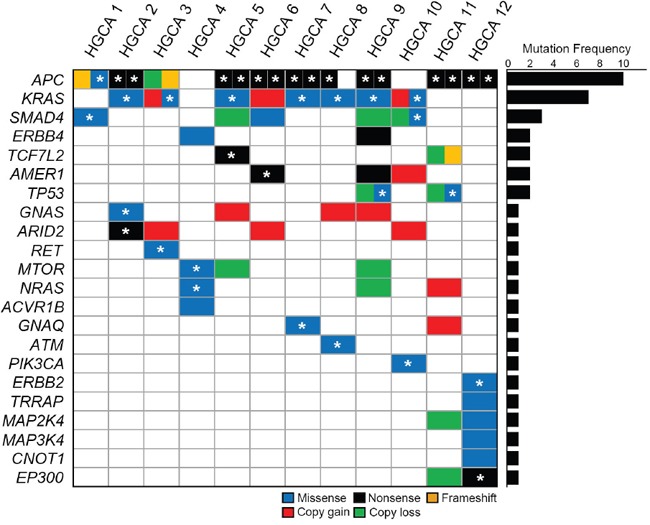
Twenty-two candidate driver genes are mutated in the adenomas. Block colors represent the functional categories of mutation and copy number alteration. Asterisks represent the somatic mutations that overlap the COSMIC database at variant level.
We compared our mutation data with those of two previous studies that had analyzed colon adenomas by either WES or targeted NGS [9, 13]. Of the mutated genes in our study, APC, KRAS and NRAS mutations were identified in the previous studies, but SMAD4 mutations were not. Not only the APC and KRAS mutations, but also TP53 (12.8% prevalence), NRAS (1.9%), AMER1 (6.9%), TCF7L2 (6.9%), MAP3K4 (3.4%), TRRAP (3.4%), ERBB2 (3.4%), ERBB4 (3.4%), TRRAP (3.4%), PIK3CA (1.3%) and GNAS (12.1%) mutations were catalogued in the COSMIC database of colon adenoma. In the COSMIC database, 0.8% (2/246) of SMAD4 mutations have been reported in colon adenomas. The COSMIC database, however, reported no ATM, EP300, MTOR, CNOT1, ARID2, RET, MAP2K4, ACVR1B and GNAQ mutations in colon adenomas. Their mutations were reported 8.9%, 6.0%, 5.4%, 5.1%, 4.9%, 3.4%, 2.4%, 1.7% and 0.9% in CRCs, respectively.
Next, we analyzed distribution of the tumor-related gene mutations in the 12 HGCAs. Non-hypermutated and hypermutated HGCA per case harbored 2-5 mutations and 7 mutations, respectively (Figure 2). APC gene was somatically mutated in all HGCAs except two cases, which harbored NRAS and MTOR mutations (HGCA4) and KRAS, SMAD4 and PIK3CA mutations (HGCA10).
Of the ten HGCA cases with APC mutations, eight harbored bi-allelic APC mutations, while two cases (HGCA3 and HGCA8) harbored mono-allelic APC mutations (Figure 2). All of the KRAS mutations were detected either in the amino acid residue p.G12 or p.G13 that is a mutational hotspot [19, 20]. Of the SMAD4 mutations (p.R361H, p.G386S and p.D404E), p.R361H and p.G386S have been found in many cancers, including colon, stomach and esophageal cancers [21], while p.D404E is a novel mutation that has not been reported. Missense mutations of TP53 (p.R175H and p.R249S) in this study have been reported in many cancers including colon adenoma [22] and p.R175H is one of the mutational hotspots in TP53 (the COSMIC database). Two HGCAs harbored nonsense mutations (p.R358X and p.Q414X) of AMER1, which encodes an X-linked negative regulator of WNT signaling [23]. Mutations of AMER1, TCF7L and MTOR have been reported in CRC [12, 24]. One case (HGCA4) harbored an NRAS hotspot mutation p.Q61H, which has been reported in many cancers, including thyroid, lung, skin and CRC [25]. PIK3CA mutation identified in APC wild-type case (HGCA10) was the hotspot mutation p.H1047R that is common in CRC as well [12]. ATM p.R337H mutation was the hotspot site and common in colon and liver cancers [26]. GNAQ p.T96S mutation has been reported in skin and prostate cancers [27, 28]. GNAS mutation identified in this study was the hotspot mutation p.R201C that is common in low-grade appendiceal mucinous neoplasm and pancreatic intraductal papillary mucinous neoplasm [29, 30]. Nonsense mutation of ARID2 p.R1273X in our study has been reported in esophageal and head/neck cancers [31, 32]. RET p.G423R mutation has been reported in hairy cell leukemias [33]. ERBB2 p.V842I and EP300 p.R1312X mutations have been reported in colon and ovary cancers, respectively [12, 34].
Copy number alterations
We analyzed somatic copy number alterations based on the read depth difference in the exome sequencing data between HGCA and matched normal tissues. Of the tumor-related gene mutations detected, 13 genes were accompanied by copy number alterations as well (Figure 2 and Supplementary Table 4). Copy number gains were found in six genes (KRAS, AMER1, GNAS, ARID2, NRAS and GNAQ) and copy number losses in eight genes (APC, SMAD4, TCF7L2, TP53, MTOR, NRAS, MAP2K4 and EP300). Of note, the case with mono-allelic APC mutation (HGCA3) exhibited copy loss as well, suggesting a bi-allelic alteration (mutation + copy loss). Likewise, two cases with mono-allelic TP53 mutation (HGCA9 and HGCA11) also exhibited copy loss in the locus. As for KRAS, in total, eight of twelve HGCAs (67%) exhibited mutation and/or copy gain. Similarly, SMAD4 gene showed mutation or copy loss in five of twelve HGCAs (42%).
Pathway analysis of mutated genes in the HGCA
The mutated genes identified in HGCAs were annotated through the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis using DAVID tools [35] and found that mutated genes in the HGCA were significantly associated with tumorigenesis-related gene functions (Supplementary Table 5).
DISCUSSION
Comprehensive analysis of genomic profiles is crucial for understanding tumorigenic mechanism in cancer development. Compared to CRC, mutational profiles of HGCA, a premalignant lesion that precedes invasive CRC, are not well studied. The aim of this study was twofold. First, we attempted to disclose the genomic profiles of HGCAs to find the mutational abundance compared to that of CRC especially with respect to the driver mutations. The next aim was to find cancer-related gene mutations that emerge from the HGCA stage rather than invasive CRC stage. Consistent with the previous report, total numbers and spectrum of somatic mutations identified in the HGCAs were not significantly different from those in CRCs [12]. We found 22 tumor-related gene mutations (APC, KRAS, SMAD4, ERBB4, TCF7L2, AMER1, TP53, GNAS, ARID2, RET, MTOR, NRAS, ACVR1B, GNAQ, ATM, PIK3CA, ERBB2, TRRAP, MAP2K4, MAP3K4, CNOT1 and EP300) from 12 HGCAs that harbored 2-7 of these mutations per case. Presence of APC, KRAS, NRAS, and GNAS mutations in HGCA well matched ‘classical adenoma-carcinoma model’ [20, 36]. As for TP53 and PIK3CA, their mutations are known to occur mainly in the late stage of the CRC model (invasive carcinoma) [20]. However, an earlier study [36] reported that TP53 mutations were fairly common in colon adenomas albeit less than CRC. PIK3CA mutations are rare in in colon adenoma, but often occur in advanced adenomas greater than 5 cm in diameter [37]. In our study, the HGCA10 with PIK3CA mutation was found in a large adenoma (3.7 cm in diameter) with a synchronous carcinoma component, suggesting that the HGCA case may be a full-grown late-stage one. SMAD4 mutation that had seldom been reported in HGCAs was identified in our study. Also, previously unreported mutations in colon adenomas (MTOR, ACVR1B, GNAQ, ATM, CNOT1, EP300, ARID2, RET and MAP2K4) were newly discovered in this study.
Earlier studies showed that SMAD4 mutation followed APC mutation and precedes TP53 mutation in CRC development [20, 38]. Our data on SMAD4 mutation in HGCAs is in agreement with the gene sequence (APC-SMAD4-TP53 mutations). However, it is not in agreement with the previous notion that SMAD4 mutations rarely occurred either in adenoma or intramucosal carcinoma, but was common in advanced CRCs [8, 38]. SMAD4 somatic mutations have been reported in 0.8% of colon adenomas (the COSMIC database), in 0% of low- and high-grade adenomas and 0% of adenomas and intramucosal carcinomas [8, 9], indicating SMAD4 mutation is a rare event in early CRC tumorigenesis. The patterns of SMAD4 alteration in our HGCAs are somewhat different from those described in advanced CRCs where SMAD4 mutations were usually accompanied by allelic loss of 18q where SMAD gene resides [8]. Of the five SMAD4 alterations four (HGCA1, HGCA5, HGCA6 and HGCA9) were either somatic mutation or 18q loss, indicating they were mono-allelic, while one (HGCA10) harbored both 18q loss and somatic mutation. Size of HGAC10 (3 cm in diameter) and with a synchronous CRC, suggesting that it may be a late-stage adenoma. Together, our data suggest that mono-allelic inactivation of SMAD4 either by mutation or copy loss may occur in HGCAs and that it may require additional hit during or after the progression to invasive or metastatic CRC.
The mutations (MTOR, ACVR1B, GNAQ, ATM, CNOT1, EP300, ARID2, RET and MAP2K4) newly detected in HGCAs in the present study have been reported in CRCs at low incidences (0.9%-8.9%) as well as other cancers [31–33]. The COSMIC database, however, shows that there is no mutation for these genes in 29 colon adenomas. Together with this, our data indicate that these gene mutations albeit low incidences may occur in adenomas, at least in HGCAs, and might possibly contribute to early CRC development.
Somatic mutations of POLE in the exonuclease domain are found in CRC and endometrial cancers that show association with hypermutability [39]. POLE somatic mutations in these cancers were recurrent in seven amino acids (p.P286R/H/L, p.S297F/Y, p.V411L, p.P436R, p.M444K, p.A456P and p.S459F), but the POLE p.E491K mutation in our result has not been reported. Association of germline POLE mutation with familial adenoma development suggests that POLE mutation might occur as an early event in sporadic tumors [40], but there has been no such data that support the hypothesis. In this study, we show data supporting that loss of proofreading activity by POLE mutation could play a role in early CRC development.
In summary, our study for the first time attempted to disclose mutational profiles of HGCAs by WES that had not been analyzed before except a study for one case. Our data is largely in agreement with the earlier ‘colorectal adenoma-carcinoma model’ that was made by gene-to-gene approaches [10, 11]. Our HGCA mutation list includes not only those in this classical model (APC and KRAS) but also those already known as adenoma genes (NRAS and GNAS). In addition, we newly identified SMAD4, MTOR, ACVR1B, GNAQ, ATM, CNOT1, EP300, ARID2, RET and MAP2K4 in HGCAs. Our data are based on the analysis of twelve HGCA genomes. The small size of the study set is due to the difficulties in obtaining histologically defined fresh HGCA tissues in a single institution since in most of the cases they are used up for diagnostic purpose. Further investigation in a larger cohort of HGCA may reveal valuable information to confirm our data, e.g., SMAD4 mutation, 18q loss and other newly discovered mutations. Also, such an approach would reveal additional information, e.g., discovery of potential additional driver mutations in HGCAs. In addition, meta-analysis of multiple cases with diverse ethnic backgrounds may be required to investigate whether such mutational contexts are population-specific. Our findings may provide a useful resource for understanding this premalignant disease and identifying genomic clues for differential diagnosis and therapy options for colon adenoma and carcinoma.
MATERIALS AND METHODS
Tumor specimen and DNA extraction
Fresh frozen normal and HGCA tissues from 12 Korean patients were obtained from the tissue banks of Seoul Saint Mary Hospital (Seoul, Korea), Guro Hospital of Korea University (Seoul, Korea) and Busan National University Hospital (Busan, Korea). All of the cases were sporadic, without any family history of CRC. Clinicopathologic features of the 12 HGCA patients are summarized in Table 2. We defined HGCA according to the World Health Organization [41] where is defined by architectural complexity and cytologic features including extent of nuclear stratification and severity of abnormal nuclear morphology. All of the samples from tumor and normal areas were frozen, cut, and stained with hematoxylin and eosin. A pathologist identified that HGCA lesions were in the samples (Supplementary Figure 2). All cases were tubulovillous adenomas (Table 2). We observed high grade dysplasia in both tubular and villous components in all cases. HGCA areas with rich tumor cell population (at least 70%) were selected and sliced from the frozen tissues with clean blades, and were subsequently used in the study. We also examined the histology by FFPE that showed definite HGCA. After the slicing, we examined again the histology of remnant frozen tissues under microscope and found no changes in the histologic diagnoses of HGCA. We used normal tissues as controls belonged to the same patients. For genomic DNA extraction, we used the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. The study protocol was reviewed and approved by the Institutional Review Board of the Catholic University of Korea, College of Medicine (MC14SISI0033).
Table 2. Clinicopathologic parameters of 12 high-grade colon adenoma patients.
| Case | Age/Sex | Size in diameter (cm) | Location in colon | Diagnosis | Accompanied malignant lesion | Tumor cell content (%) |
|---|---|---|---|---|---|---|
| HGCA 1 | 51/F | 3.8 | descending | Tubulovillous adenoma with high grade dysplasia | No | >70% |
| HGCA 2 | 71/M | 8.0 | descending | Tubulovillous adenoma with high grade dysplasia | No | >70% |
| HGCA 3 | 71/F | 3.2 | cecum | Tubulovillous adenoma with high grade dysplasia | No | >70% |
| HGCA 4 | 67/M | 4.0 | sigmoid | Tubulovillous adenoma with high grade dysplasia | No | >70% |
| HGCA 5 | 62/M | 4.0 | rectum | Tubulovillous adenoma with high grade dysplasia | Intraepithelial adenocarcinoma | >70% |
| HGCA 6 | 58/M | 2.5 | sigmoid | Tubulovillous adenoma with high grade dysplasia | Intraepithelial adenocarcinoma | >70% |
| HGCA 7 | 54/F | 3.0 | ascending | Tubulovillous adenoma with high grade dysplasia | No | >70% |
| HGCA 8 | 77/M | 1.0 | Descending | Tubulovillous adenoma with high grade dysplasia | No | >70% |
| HGCA 9 | 60/M | 1.5 | Sigmoid | Tubulovillous adenoma with high grade dysplasia | Invasive adenocarcinoma | >70% |
| HGCA 10 | 65/F | 3.7 | Hepatic flexure | Tubulovillous adenoma with high grade dysplasia | Invasive adenocarcinoma | >70% |
| HGCA 11 | 65/M | 3.0 | Sigmoid | Tubulovillous adenoma with high grade dysplasia | Invasive adenocarcinoma | >70% |
| HGCA 12 | 70/M | 1.2 | rectum | Tubulovillous adenoma with high grade dysplasia | No | >70% |
Whole-exome sequencing and copy number inference
WES was performed for genomic DNA from the HGCA and matched normal samples using Agilent SureSelect Human All Exome 50Mb kit (Agilent Technologies) and Illumina HiSeq2000 platform. The collection and processing of the sequencing data was performed as previously described [42]. The preparation of genomic DNA libraries and 101 bp paired-end sequencing reads was performed according to the manufacturer's instructions. Firstly, we used Burrows-Wheeler Alignment tool (BWA) to align the paired-end sequences onto the UCSC hg19 human reference genomes [43]. Local alignment and score recalibration of the sequencing data were performed, using the Genome Analysis ToolKit [44]. Additional downstream analyses, following alignment, were performed using Picard ( http://picard.sourceforge.net) and Samtools [45]. We used Mutect and SomaticIndelDetector to call somatic single nucleotide variants (SNVs) and small insertions/deletions (indels) by comparing the sequencing data from the adenoma samples with those from the matched normal tissue samples [44, 46]. ANNOVAR package was used for the analysis of mutations on coding sequences, and for the functional annotation of somatic variants [47]. For the identification of copy number alteration, we used VarScan 2 to obtain the read depth differences between the tumors and matched normal exome sequencing data [48]. The GC-corrected read depth was log2-transformed and segmented using circular binary segmentation algorithm [49].
Validation of the whole-exome sequencing
We validated mutations of 12 genes either by digital-PCR or Sanger sequencing as described previously [50]. Digital-PCR was performed using the TaqMan Genotyping assay and the QuantStudio 3D digital PCR system (Life Technologies) according to the manufacturer's protocol.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Acknowledgments
This study was supported by grants from National Research Foundation of Korea (2012R1A5A2047939, 2015R1D1A1A01057355, 2015057355 and 2013R1A1A2008110).
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Jung KW, Park S, Won YJ, Kong HJ, Lee JY, Seo HG, Lee JS. Prediction of cancer incidence and mortality in Korea, 2012. Cancer Res Treat. 2012;44:25–31. doi: 10.4143/crt.2012.44.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.Rubio CA, Nesi G, Messerini L, Zampi GC, Mandai K, Itabashi M, Takubo K. The Vienna classification applied to colorectal adenomas. J Gastroenterol Hepatol. 2006;21:1697–1703. doi: 10.1111/j.1440-1746.2006.04258.x. [DOI] [PubMed] [Google Scholar]
- 5.Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, White R, Vogelstein B. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 6.Thiagalingam S, Lengauer C, Leach FS, Schutte M, Hahn SA, Overhauser J, Willson JK, Markowitz S, Hamilton SR, Kern SE, Kinzler KW, Vogelstein B. Evaluation of candidate tumour suppressor genes on chromosome 18 in colorectal cancers. Nat Genet. 1996;13:343–346. doi: 10.1038/ng0796-343. [DOI] [PubMed] [Google Scholar]
- 7.Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, et al. Colorectal cancer: mutations in a signalling pathway. Nature. 2005;436:792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 8.Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M, Hishima T, Koike M, Shitara N, Iwama T, Utsunomiya J, Kuroki T, Mori T. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18:3098–3103. doi: 10.1038/sj.onc.1202642. [DOI] [PubMed] [Google Scholar]
- 9.Voorham QJ, Carvalho B, Spiertz AJ, Claes B, Mongera S, van Grieken NC, Grabsch H, Kliment M, Rembacken B, van de Wiel MA, Quirke P, Mulder CJ, Lambrechts D, et al. Comprehensive mutation analysis in colorectal flat adenomas. PLoS One. 2012;7:e41963. doi: 10.1371/journal.pone.0041963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leslie A, Stewart A, Baty DU, Mechan D, McGreavey L, Smith G, Wolf CR, Sales M, Pratt NR, Steele RJ, Carey FA. Chromosomal changes in colorectal adenomas: relationship to gene mutations and potential for clinical utility. Genes Chromosomes Cancer. 2006;45:126–135. doi: 10.1002/gcc.20271. [DOI] [PubMed] [Google Scholar]
- 11.Konda K, Konishi K, Yamochi T, Ito YM, Nozawa H, Tojo M, Shinmura K, Kogo M, Katagiri A, Kubota Y, Muramoto T, Yano Y, Kobayashi Y, et al. Distinct molecular features of different macroscopic subtypes of colorectal neoplasms. PLoS One. 2014;9:e103822. doi: 10.1371/journal.pone.0103822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou D, Yang L, Zheng L, Ge W, Li D, Zhang Y, Hu X, Gao Z, Xu J, Huang Y, Hu H, Zhang H, Zhang H, et al. Exome capture sequencing of adenoma reveals genetic alterations in multiple cellular pathways at the early stage of colorectal tumorigenesis. PLoS One. 2013;8:e53310. doi: 10.1371/journal.pone.0053310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, Wright FA, Feramisco JD, Peltomaki P, Lang JC, Schuller DE, Yu L, Bloomfield CD, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 15.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, In Lopez-Bigas N. silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015;27:382–396. doi: 10.1016/j.ccell.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Carter H, Samayoa J, Hruban RH, Karchin R. Prioritization of driver mutations in pancreatic cancer using cancer-specific high-throughput annotation of somatic mutations (CHASM) Cancer Biol Ther. 2010;10:582–587. doi: 10.4161/cbt.10.6.12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 20.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 21.Miyaki M, Kuroki T. Role of Smad4 (DPC4) inactivation in human cancer. Biochem Biophys Res Commun. 2003;306:799–804. doi: 10.1016/s0006-291x(03)01066-0. [DOI] [PubMed] [Google Scholar]
- 22.Sawyer EJ, Cerar A, Hanby AM, Gorman P, Arends M, Talbot IC, Tomlinson IP. Molecular characteristics of serrated adenomas of the colorectum. Gut. 2002;51:200–206. doi: 10.1136/gut.51.2.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Major MB, Camp ND, Berndt JD, Yi X, Goldenberg SJ, Hubbert C, Biechele TL, Gingras AC, Zheng N, Maccoss MJ, Angers S, Moon RT. Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science. 2007;316:1043–1046. doi: 10.1126/science/1141515. [DOI] [PubMed] [Google Scholar]
- 24.Li YY, Hanna GJ, Laga AC, Haddad RI, Lorch JH, Hammerman PS. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin Cancer Res. 2015;21:1447–1456. doi: 10.1158/1078-0432.CCR-14-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, Hauptschein R, Rejto PA, Fernandez J, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shain AH, Garrido M, Botton T, Talevich E, Yeh I, Sanborn JZ, Chung J, Wang NJ, Kakavand H, Mann GJ, Thompson JF, Wiesner T, Roy R, et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat Genet. 2015;47:1194–1199. doi: 10.1038/ng.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim TM, Jung SH, Baek IP, Lee SH, Choi YJ, Lee JY, Chung YJ, Lee SH. Regional biases in mutation screening due to intratumoural heterogeneity of prostate cancer. J Pathol. 2014;233:425–435. doi: 10.1002/path.4380. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Mody K, de Abreu FB, Pipas JM, Peterson JD, Gallagher TL, Suriawinata AA, Ripple GH, Hourdequin KC, Smith KD, Barth RJ, Colacchio TA, Jr, Tsapakos MJ, et al. Molecular profiling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin Chem. 2014;60:1004–1011. doi: 10.1373/clinchem.2014.225565. [DOI] [PubMed] [Google Scholar]
- 30.Reid MD, Saka B, Balci S, Goldblum AS, Adsay NV. Molecular genetics of pancreatic neoplasms and their morphologic correlates: an update on recent advances and potential diagnostic applications. Am J Clin Pathol. 2014;141:168–180. doi: 10.1309/AJCP0FKDP7ENVKEV. [DOI] [PubMed] [Google Scholar]
- 31.Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, Zhang F, Zhao ZR, Li ZT, Liu ZY, Zhao YD, Sun J, Zhou CC, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–1102. doi: 10.1038/ng.3076. [DOI] [PubMed] [Google Scholar]
- 32.India Project Team of the International Cancer Genome C. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. 2013;4:2873. doi: 10.1038/ncomms3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, Abaan OD, Davis SR, Kreitman RJ, Meltzer PS. High prevalence of MAP2K1 mutations in variant and IGHV4-34-expressing hairy-cell leukemias. Nat Genet. 2014;46:8–10. doi: 10.1038/ng.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 36.Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S, Vogelstein B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990;50:7717–7722. [PubMed] [Google Scholar]
- 37.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 38.Woodford-Richens KL, Rowan AJ, Gorman P, Halford S, Bicknell DC, Wasan HS, Roylance RR, Bodmer WF, Tomlinson IP. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc Natl Acad Sci U S A. 2001;98:9719–9723. doi: 10.1073/pnas.171321498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Briggs S, Tomlinson I. Germline and somatic polymerase epsilon and delta mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol. 2013;230:148–153. doi: 10.1002/path.4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, Church DN. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer. 2016;16:71–81. doi: 10.1038/nrc.2015.12. [DOI] [PubMed] [Google Scholar]
- 41.Hamilton SR BF, Boffetta P, Ilyas M, Morreau H, Nakamura SI, Quirke P, Riboli E, Sobin LH. Carcinoma of the colon and rectum. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, editors. WHO Clasification of Tumors of the Digestive System. 4th ed. Lyon, France: IARC Press; 2010. pp. 134–146. [Google Scholar]
- 42.Kim TM, Jung SH, An CH, Lee SH, Baek IP, Kim MS, Park SW, Rhee JK, Lee SH, Chung YJ. Subclonal Genomic Architectures of Primary and Metastatic Colorectal Cancer Based on Intratumoral Genetic Heterogeneity. Clin Cancer Res. 2015;21:4461–4472. doi: 10.1158/1078-0432.CCR-14-2413. [DOI] [PubMed] [Google Scholar]
- 43.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Genome Project Data Processing S. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 50.Jung SH, Shin S, Kim MS, Baek IP, Lee JY, Lee SH, Kim TM, Lee SH, Chung YJ. Genetic Progression of High Grade Prostatic Intraepithelial Neoplasia to Prostate Cancer. Eur Urol. 2016;69:823–830. doi: 10.1016/j.eururo.2015.10.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.